


Abstract
Prion proteins (PrPs) cause prion diseases, such as bovine spongiform encephalopathy. The conversion of a normal cellular form (PrPC) of PrP into an abnormal form (PrPSc) is thought to be associated with the pathogenesis. An RNA aptamer that tightly binds to and stabilizes PrPC is expected to block this conversion and to thereby prevent prion diseases. Here, we show that an RNA aptamer comprising only 12 residues, r(GGAGGAGGAGGA) (R12), reduces the PrPSc level in mouse neuronal cells persistently infected with the transmissible spongiform encephalopathy agent. Nuclear magnetic resonance analysis revealed that R12, folded into a unique quadruplex structure, forms a dimer and that each monomer simultaneously binds to two portions of the N-terminal half of PrPC, resulting in tight binding. Electrostatic and stacking interactions contribute to the affinity of each portion. Our results demonstrate the therapeutic potential of an RNA aptamer as to prion diseases.
INTRODUCTION
Prion protein (PrP) is almost ubiquitously expressed and is highly conserved in mammals. PrP exhibits two alternative forms; a normal cellular form (PrPC), which is a soluble α-helix-rich isoform, and an abnormal form (PrPSc), which is an insoluble β-sheet-rich isoform and resistant to cleavage by proteinase K (1,2). The details of the structure of PrPSc and the mechanism underlying the conversion of PrPC to PrPSc remain unknown. Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are invariably fatal neurodegenerative disorders of mammals characterized by the accumulation of PrPSc in the central nervous system (1–7). These disorders include Creutzfeldt–Jakob disease in humans, bovine spongiform encephalopathy in cattle and scrapie in sheep.
Considerable efforts have been made to develop a compound that inhibits the accumulation of PrPSc in prion-infected cells (8–17). An RNA aptamer that tightly binds to PrPC is expected to stabilize PrPC and, thus, to block the conversion to PrPSc. Therefore, such an RNA aptamer may prevent prion diseases. However, attempts in this context have been limited, and a structural basis of the binding of an RNA aptamer to PrPC, which would facilitate such an application, has not been available. We identified RNA aptamers that tightly bind to bovine PrPC (bPrPC) (18,19). We revealed that the RNA aptamer specifically binds to PrPC among various kinds of proteins present in bovine brain homogenate (18). Surprisingly, among the RNA aptamers, a short RNA comprising only 12 residues, r(GGAGGAGGAGGA) (R12), binds to bPrP strongly, the dissociation constant being 8.5 × 10−9 M (18). We already reported the unique quadruplex structure of R12 (20). Here, we have revealed that R12 actually exhibits anti-prion activity on the basis of the assay with mouse neuronal cells. Nuclear magnetic resonance (NMR) analysis elucidates the structural basis of tight binding of R12 with PrPC that causes anti-prion activity.
MATERIALS AND METHODS
Sample preparation
R12 [r(GGAGGAGGAGGA)], D12 [d(GGAGGAGGAGGA)] and U12 [r(U)12], synthesized, purified by high-performance liquid chromatography and desalted, were purchased from Sigma-Aldrich and Nippon Seihun, respectively. P1 (residues 25–35 of bPrP: NH2-SKKRPKPGGGWN-COOH), P16 (residues 108–119: NH2-GQWNKPSKPKTN-COOH) and mutant P16 peptides (K5A, K8A, K10A and W3A, respectively), synthesized and purified by high-performance liquid chromatography, were purchased from Sigma-Aldrich or Hipep Laboratories. Wild-type bPrP-N (residues 25–131) and mutant bPrP-N (residues 25–131, 5WS) were expressed with an Escherichia coli system and purified using a reported protocol (21). Full-length bPrP (residues 25–241) was purchased from Alicon.
Evaluation of anti-prion activity
The anti-prion activity of either R12 or D12 was examined using mouse neuronal cells (GT1–7) persistently infected with the human TSE agent (Fukuoka-1 strain), designated as GT + FK, as described previously (14,16). The cells were grown and maintained at 37°C under 5% of CO2 in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% of fetal bovine serum (Equitech-bio), 50 U/ml of penicillin G sodium and 50 μg/ml of streptomycin sulphate (Invitrogen). Approximately 1.5 × 105 cells were plated in each well of a six-well plate, and treatment with either R12 or D12 was started 15 h later. R12 (0.25 mM) and D12 (0.35 mM) were dissolved in a buffer solution containing 10 mM of K-phosphate (pH 6.2) and 100 mM of KCl, respectively. Either 80 μl of the R12 solution or 57 μl of the D12 solution was added to 2 ml of the medium, with the final concentration of both R12 and D12 being 10 μM. As a control, either 80 or 57 μl of the buffer solution was added to the medium. For the conditions with an RNase inhibitor, 800 U of RNasin (Promega) was added to the medium 5 min before the addition of the R12 solution. As a reference nucleic acid, U12 was added to the medium to the final concentration of 10 μM after the addition of the RNase inhibitor. After 72 h of treatment, cells were lysed in 150 μl of 1× Triton X-100-deoxycholate lysis buffer [150 mM of NaCl, 0.5% of Triton X-100, 0.5% of sodium deoxycholate, 50 mM of Tris–HCl (pH 7.5)], and the supernatant was collected. Samples were digested with 20 μg/ml of proteinase K for 30 min at 37°C. Western blotting for PrPSc was performed as described previously (22). As the primary antibody, PrP M-20 antibody (Santa Cruz Biotechnology) was used to detect PrPSc. The signals were visualized with Super-Signal (Pierce Biotechnology) and scanned using a LAS-1000 UV mini analyser (Fuji Film). The density of PrPSc in each solution was measured and compared with that in the control treated with just the buffer solution. Each assay with either R12 or D12 was repeated eight times.
Microchip electrophoresis assay
A Hitachi SV1210 microchip CE system (Hitachi Electronics Engineering) was used for microchip electrophoresis (ME) analysis, as described previously (19,20). R12 was incubated in the binding buffer comprising 20 mM of Tris–HCl (pH 7.5) and 10 mM of KCl under the re-folding conditions: 90°C for 2 min, 72°C for 5 min, 55°C for 5 min and 37°C for 2 min and then kept at 25°C. After the addition of 10 nM of Cy5-2′-deoxyuridine 5′-triphosphate (dUTP) (Amersham Biosciences), the internal standard, R12 (100 nM), was incubated with a peptide (0 and 100 μM) in the binding buffer of 10 μl for 15 min and was then analysed by ME (20). The observed peak intensity of R12 was divided by the peak intensity of the internal standard, which gave the relative peak intensity. The relative peak intensity value of R12 was normalized by dividing by the relative peak intensity of R12 in the absence of peptide, which gave the normalized peak intensity. The relative binding of R12 was defined as 1 (the normalized peak intensity) and was calculated for the wild-type and mutant P16 peptide (Figure 3H).
Figure 3.
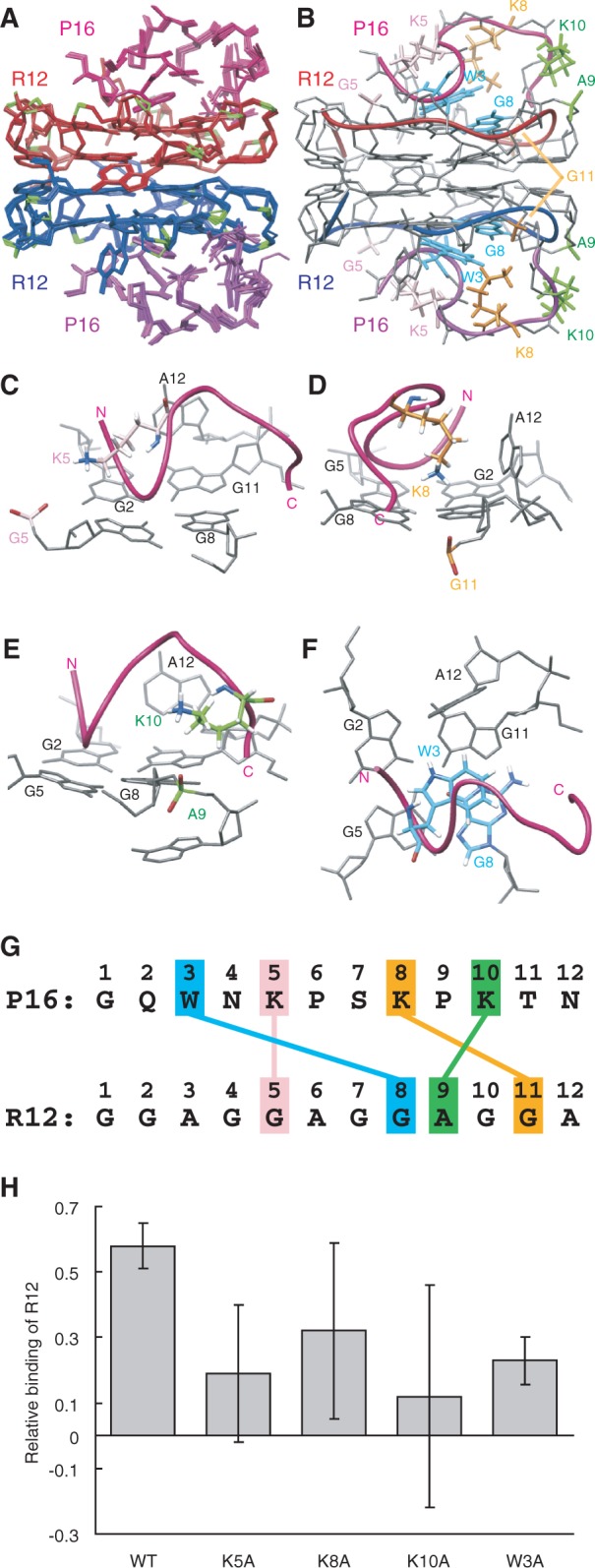
The structure of the R12:P16 complex. (A) NMR structure ensemble of the R12:P16 complex. The two R12 monomers are coloured red and blue, respectively, phosphate groups being coloured green. The two P16 monomers are coloured magenta and purple, respectively. (B) The representative structure with the lowest energy. The chains connecting the C3′ and C4′ atoms of each R12 monomer are indicated by red and blue tubes, respectively. The chains connecting the Cα atoms of each P16 monomer are indicated by magenta and purple tubes, respectively. The pairs K5 and G5, K8 and G11, K10 and A9 and W3 and G8 are coloured pink, orange, green and blue, respectively. (C–F) Close-up views of intermolecular interactions between K5 of P16 and G5 of R12 (C), K8 and G11 (D), K10 and A9 (E) and W3 and G8 (F), respectively. (G) Summary of interactions. The electrostatic interactions between K5 of P16 and G5 of R12, K8 and G11 and K10 and A9 and the stacking interaction between W3 and G8 are indicated. (H) The relative binding is shown for wild-type P16 and four mutant P16, in which either K5, K8, K10 or W3 was replaced by an alanine residue, respectively.
Filter binding assay
The 5′-end γ-32P-labelled R12 (<10 nM) was mixed with varying concentrations of either a peptide or protein to give a total volume of 25 μl in a solution comprising 20 mM of Tris–HCl (pH 7.5) and 10 mM of KCl. After 20 min incubation, each mixture was passed through a nitrocellulose filter and washed with 500 μl of the solution. The amount of bound RNA was measured with BAS 2500 (Fuji Film), and binding activities were calculated as the percentage of input RNA in the peptide- or protein-RNA complex retained on the filter. We determined the dissociation constant (Kd) using GraphPad PRISM and non-linear regression curve fitting, and a one site-binding hyperbola equation {RNA binding (%) = Bmax × (peptide or protein)/[Kd + (peptide or protein)], where Bmax is the maximum bound at the saturating peptide or protein concentration} (18).
Titration and structure determination
In titration experiments, either P1, P16 or bPrP-N(25–131, 5WS) was added step-by-step to R12 in a solution comprising 10 mM of K-phosphate (pH 6.2) and 10 mM of KCl at 30°C. Structural determination of the R12:P16 complex was carried out under the same conditions. NMR spectra, nuclear overhauser effect spectroscopy (NOESY), total correlation spectroscopy (TOCSY), double-quantum filtered correlation spectroscopy (DQF-COSY), 1H-13C heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond coherence with jump and return solvent suppression (JRHMBC) were recorded with Bruker DRX600 and DRX800 spectrometers equipped with a cryoprobe. Distance and dihedral constraints were obtained as described previously (20) (Supplementary Table S1). Structural calculations were carried out using these constraints and a simulated annealing protocol supplied with XPLOR-NIH v. 2.26 (23,24), as described previously (20,25). The R12:P16 complex comprises two R12 monomers and two P16 monomers, as explained in the main text. Ten final structures were selected from 100 calculations on the basis of the criterion of the smallest residual energy term. The statistics are shown in Supplementary Table S1. The final ensemble of ten structures of the R12:P16 complex has 13.1 and 86.9% in the most favoured and additional allowed regions, respectively, for the P16 peptide. Molecular images were generated with MOLMOL (http://www.mol.biol.ethz.ch/groups/wuthrich_group/software) and University of California, San Francisco (UCSF) Chimera (http://www.cgl.ucsf.edu/chimera/).
RESULTS
Anti-prion activity of R12
The anti-prion activity of R12 has been examined using mouse neuronal cells (GT1-7) persistently infected with the human TSE agent (Fukuoka-1 strain), designated as GT + FK. R12 was found to inhibit the PrPSc formation in GT + FK at 10 μM (second and third lanes in Figure 1). R12 reduced the PrPSc level to 65.0% of that in the control (Table 1, P < 0.001). When an RNase inhibitor, RNasin, was added to the medium, R12 reduced the PrPSc level even more, that is, to 49.4% (eighth and ninth lanes in Figure 1 and Table 1, P < 0.001). The observed difference between in absence and presence of an RNase inhibitor (Table 1, P < 0.05) is supposed to be because of the repressed degradation of R12 in the medium caused by the RNase inhibitor. No reduction of the PrPSc level was observed for U12 even in the presence of the RNase inhibitor (Table 1). Therefore, the reduction is specific to R12. The DNA version of R12, d(GGAGGAGGAGGA) (D12), also tightly binds to bPrPC, the dissociation constant being 6.4 × 10−8 M (18). D12 reduced the PrPSc level to 75.5% (fifth and sixth lanes in Figure 1 and Table 1, P < 0.01), although its inhibitory effect is less than that of R12. Thus, we have demonstrated that R12, and also D12 to a lesser extent, exhibits anti-prion activity towards cells persistently infected with the TSE agent.
Figure 1.
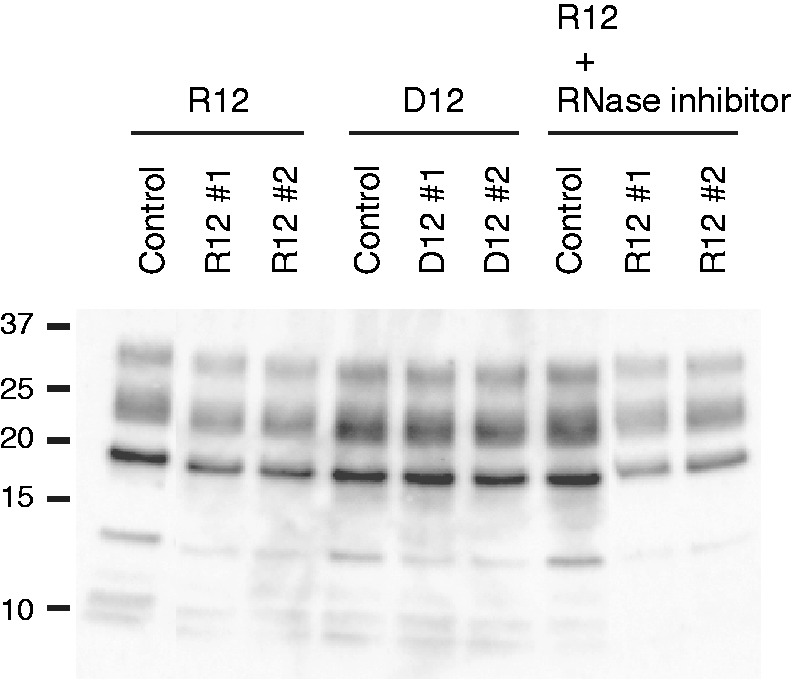
Anti-prion activity of r(GGAGGAGGAGGA) (R12) and d(GGAGGAGGAGGA) (D12). Western blotting of PrPSc in GT + FK cells after treatment with either 10 μM R12 or D12. Two independent experiments, #1 and #2, are shown. The control was treated with just the buffer solution. The treatment with R12 was also performed in the presence of an RNase inhibitor, RNasin. Molecular mass markers are shown at the left.
Table 1.
Anti-prion activities of R12 and D12*
 |
Structure of R12 in complex with the binding peptide of bPrP
Then, we tried to elucidate the structural basis of the tight binding of R12 to bPrPC. The structure of bPrPC is available (21); the C-terminal half of bPrPC is composed of three α-helices and two short β-strands, whereas the N-terminal half is basically flexible and disordered. Two portions of bPrP were identified as binding sites for R12 by epitope mapping using ME (20). They are residues 25–35 (SKKRPKPGGGWN, designated as P1) and 108–119 (GQWNKPSKPKTN, designated as P16) of bPrP. Both binding sites commonly include three Lys residues and one Trp residue. The dissociation constants for the R12:P1 and R12:P16 complexes are 1.7 × 10−5 M and 1.0 × 10−5 M, respectively, being similar to each other. Chemical shift perturbation of each imino proton of R12 was traced during the course of titration with P16 (Figure 2A). The perturbation reached a plateau when the P16/R12 molar ratio was 3.0. Thus, the perturbation at each molar ratio was divided by the perturbation at the molar ratio of 3.0 and defined as a normalized perturbation for each imino proton. Then, the normalized perturbation was plotted against the P16/R12 molar ratio (Figure 2B). The initial slope of the plot almost coincides with that expected for the formation of the R12:P16 = 1:1 complex. The deviation of the experimental points from the initial slope for larger P16/R12 molar ratios is rationally understood, when the dissociation constant of 1.0 × 10−5 M and the concentrations of P16 and R12 applied for each point are taken into account, which also supports the formation of the 1:1 complex. As described later in the text, R12 forms a dimer in the complex. Therefore, the complex should comprise two R12 monomers and two P16 monomers.
Figure 2.

NMR analysis of the R12:P16 complex. (A) The imino proton spectra of R12 in the course of the titration with P16, the assignments being indicated by residue numbers. The numbering of R12 is G1G2A3G4G5A6G7G8A9G10G11A12. The molar ratio is indicated at the left. (B) Normalized chemical shift perturbation for each imino proton of R12 in the course of the titration with P16. (C) Imino proton spectra of R12, the R12:P1 complex and the R12:P16 complex with the assignments. (D and E) The G:G:G:G tetrad (D) and G(:A):G:G(:A):G hexad (E) planes. The observed NOESY cross peaks and hydrogen bonds are indicated by solid and dotted lines, respectively. (F and G) Intermolecular NOESY cross peaks observed for the K5 (F) and W3 (G) residues of P16, respectively. The numbering of P16 is G1Q2W3N4K5P6S7K8P9K10T11N12.
The imino proton spectrum of the R12:P1 complex is similar to that of the R12:P16 complex (Figure 2C). In fact, the chemical shift perturbation of R12 on binding of P1 is similar to that on binding of P16, except for that for a G11 residue (Supplementary Figure S1). This suggests that the interaction of R12 with P1 is similar to that with P16, although some difference in interaction and/or structure may be present around G11. The similar dissociation constants obtained for the R12:P1 and R12:P16 complexes are consistent with this idea. Thus, to elucidate the interaction of R12 with bPrP, we have determined the structure of R12 in a complex with P16 as a representative.
The resonance assignments of R12 in the R12:P16 complex were made (Supplementary Figure S2A and B) in the same way as reported for free R12 (20). Then, the G:G:G:G tetrad and G(:A):G:G(:A):G hexad structures were identified on the basis of the observation of characteristic NOESY cross peaks (Figure 2D and E), as for free R12 (20). The distance between H2 and H1′ of an adenosine residue is larger than 4.5 Å for any conformation (26). Therefore, observation of strong NOESY cross peaks between H2 and H1′ for an A3 residue and also for an A9 residue (Supplementary Figure S2C) is an indication that R12 in the complex forms a dimer, as free R12 does (20). The dimer formation of R12 in the complex was supported by many other NOESY cross peaks as well. Resonance assignments of P16 in the R12:P16 complex were also made (Supplementary Figure S2D) using a standard protocol (26). Then, many intermolecular NOESY cross peaks were identified on the basis of the resonance assignments (Figure 2F and G).
The structure of the R12:P16 complex comprising two R12 monomers and two P16 monomers was determined on the basis of distance and dihedral angle constraints (Figure 3). The structure of the complex reveals the origin of the high affinity. Three Lys residues of P16 are involved in the electrostatic interactions with the phosphate groups of R12; K5 of P16 with G5 of R12, K8 with G11 and K10 with A9, respectively (Figure 3C–E and G). The average of the closest distance for 10 final structures between an Hζ atom of the Lys residue and an oxygen atom of a phosphate group is 3.71 Å, 3.67 Å and 4.65 Å, respectively. In addition, a stacking interaction was found between the rings of W3 of P16 and G8 of R12 (Figure 3F and G). These interactions were biochemically confirmed by ME using mutant P16 peptides. A decrease in affinity to R12 was observed for four mutant P16 peptides in which either K5, K8, K10 or W3 was replaced by an alanine residue, respectively (Figure 3H).
Overall architecture responsible for tight trapping of bPrP by R12
As described earlier in the text, R12 is suggested to interact with P1 in a similar way as it does with P16, the interaction involving three lysine residues and one tryptophan residue, which both P1 and P16 commonly possess. Therefore, it is likely that in the R12:bPrP complex, one monomer of the R12 dimer interacts with P1 and the other monomer with P16 (Figure 4A). To confirm this, a titration experiment on the N-terminal region of bPrP, bPrP-N(25–131), which contains the two binding sites, P1 (residues 25–35) and P16 (residues 108–119), was performed. bPrP-N(25–131) tightly binds to R12. The dissociation constant for the R12:bPrP-N(25-131) complex was 2.1 × 10−8 M, which is roughly comparable with that for the R12: full-length bPrP(25–241) complex, 8.5 × 10−9 M. Unfortunately, precipitation was observed in the course of the titration with bPrP-N(25–131), probably because of the low solubility of the complex, which hinders detailed analysis. To improve its solubility, five tryptophan residues of the octapeptide repeat region of bPrP-N(25–131), Trp 60, Trp 68, Trp76, Trp 84 and Trp 92 were replaced by serine residues, resulting in bPrP-N(25–131, 5WS). It should be noted that the tryptophan residues that are involved in P1 and P16 and interact with R12 are Trp 34 and Trp 110, respectively, and that they are not replaced. In fact, it was found that bPrP-N(25–131, 5WS) also tightly binds to R12, the dissociation constant being 5.5 × 10−8 M, which is almost comparable with that for wild-type bPrP-N(25–131). Precipitation was not observed during the titration with bPrP-N(25–131, 5WS). The chemical shift perturbation of each imino proton of R12 in the course of the titration with bPrP-N(25–131, 5WS) was traced. The perturbation reached a plateau when the bPrP-N/R12 molar ratio was 0.56. Thus, the perturbation at each molar ratio was divided by the perturbation at the molar ratio of 0.56 and defined as a normalized perturbation for each imino proton. The normalized perturbation was plotted against the molar ratio (Figure 4B). The initial slope of the plot almost coincides with that expected for the formation of the R12:bPrP-N(25–131, 5WS) = 1:0.5 complex. Because of the dimer formation of R12, this indicates that the complex comprises two R12 monomers and one bPrP-N(25–131, 5WS) molecule. This is consistent with the idea aforementioned that one monomer of the R12 dimer interacts with P1 of bPrP and that the other monomer with P16 of the same bPrP molecule (Figure 4A). The deduced simultaneous binding of the R12 dimer with two binding sites of a single bPrP molecule must also contribute to the high affinity because the double binding could ideally increase the affinity by as much as the square of the binding constant if each interaction is perfectly achieved, respectively (27).
Figure 4.
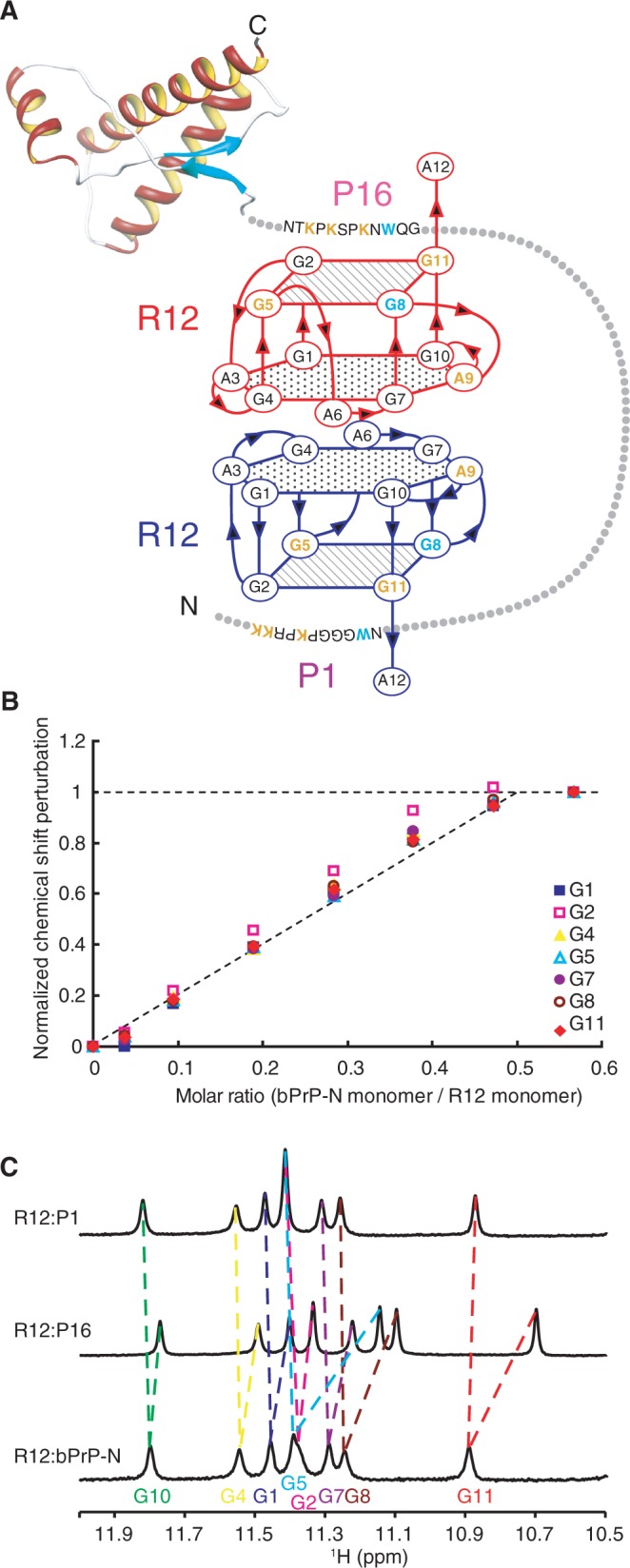
Overall architecture of tight trapping of bPrP by R12. (A) Overall architecture of the complex between bPrP and R12. The tetrad and hexad planes of R12 are indicated by a square and a hexagon, respectively. The lysine and guanosine residues involved in the electrostatic interaction are coloured orange, and the tryptophan and guanosine residues involved in the stacking interaction are coloured blue, respectively. The structure of the C-terminal region of bPrP is drawn on the basis of the coordinates under accession number 1DX0 in the Protein Data Bank (21). (B) Normalized chemical shift perturbation for each imino proton of R12 in the course of the titration with bPrP-N(25–131, 5WS). (C) Imino proton spectra of the R12:P1 complex, the R12:P16 complex and the R12:bPrP-N(25–131, 5WS) complex with the assignments.
Only one resonance is observed for each imino proton of R12 in complex with bPrP-N(25–131, 5WS) (Figure 4C), although two imino protons of each monomer of R12 could potentially be non-equivalent in the complex. This observation suggests that each R12 monomer switches binding site, P1 and P16 and vice versa, in fast exchange regime on an NMR time scale. It is consistent with this idea that each imino proton resonance of R12 in the complex with bPrP-N(25–131, 5WS) appears between the corresponding resonances of R12 in the complex with P1 and that with P16, except for a G11 residue (Figure 4C).
DISCUSSION
We have demonstrated that R12 reduces the PrPSc level of mouse neuronal cells persistently infected with the human TSE agent. It is supposed that R12, which tightly binds to and stabilizes PrPC, blocked the conversion into PrPSc. The amino acid identity and similarity between the bovine and mouse PrPs are high, 89 and 93%, respectively. Moreover, the two sites of bPrP that are responsible for binding of R12 are located in the N-terminal unfolded region, as described later in the text, and the amino acid sequences of these two sites are almost identical for bovine and mouse PrPs. Therefore, it is likely that R12 selected as to bPrP also functioned for mouse PrP. Moreover, the amino acid sequences of these two sites for human PrP are also almost identical to those for bovine and mouse PrPs. There is the possibility that R12 may function for human PrP.
Recently, it has been reported that the segment 100–104 of mouse PrPC plays a key role in the conversion after binding to mouse PrPSc (28). This segment corresponds to the 112–116 segment of bPrP, which is actually the K5 to P9 segment of a P16 peptide used in this work. Therefore, our observation that R12, which tightly binds to P16 (and P1) of bPrP, reduces the PrPSc level is consistent with this report.
It has been revealed this time that the unique R12 structure observed in a free form is preserved also in a complex form with bPrP. The structure determination of the complex with the binding peptide of bPrP has provided the detailed information on the interaction. It has been identified for the first time that the phosphate groups of G5, A9 and G11 of R12 are involved in the electrostatic interaction. Pairing for the interaction with one of three Lys residues of P16 has also been found. The structure has also revealed that the guanine base that makes stacking interaction with a Trp residue of P16 is that of G8 of R12. These kinds of information cannot be obtained without the structure determination of the complex. Then, the high affinity of R12 has been attributed to the simultaneous dual binding. For therapeutic application of R12 in future, chemical modification of either sugars or backbone may be required to increase the RNase resistance. In this case, however, the chemical modifications on R12 should not interfere with the R12–PrP interactions. The information on the interactions obtained from both the detailed complex structure and the overall architecture must be helpful to determine the position and the kind of modification.
We previously reported the structure of D12 (29). D12 forms a similar quadruplex structure to R12. In addition to the common G:G:G:G tetrad, the G(:A):G(:A):G(:A):G heptad is formed for the D12 quadruplex, instead of the G(:A):G:G(:A):G hexad for the R12 quadruplex. D12 also forms a similar dimer structure to R12, although the direction of the dyad axis differs between the D12 and R12 dimers. Therefore, it is likely that D12 interacts with bPrP in a similar way to R12. Some structural differences between the R12 and D12 quadruplexes may be responsible for the difference in affinity to bPrP. The lower anti-prion activity of D12 is supposed to be because of the lower affinity to and less stabilization of bPrPC. Nonetheless, the finding of anti-prion activity for D12 is still interesting.
Compounds that inhibit the accumulation of PrPSc in prion-infected cells have been developed. Among them, GN8 binds to the hot spots in the C-terminal region of PrP (14), and a lysosomotropic agent, quinacrine, to the third helix in the C-terminal region of PrP (13). Pentosan polysulphate is reported to bind to the octapeptide repeats in the N-terminal region of PrP (15). Recently, Congo red was reported to bind to the lysine residue in the groove formed in Heterokaryon incompatibility protein s (HET-s) amyloid (17). The site and mode of interaction of R12 with PrP are different from those of these compounds. Therefore, the modulation of PrP conformational conversion by R12, and probably by D12 also, would be distinct from that by pre-existing anti-prion agents.
Recently, PrPC has also been suggested to be a mediator of the amyloid-β-oligomer-induced synaptic dysfunction linked to Alzheimer’s disease (30). It was revealed that anti-PrP antibodies prevent amyloid-β-oligomer binding to PrPC, and that the antibodies rescue synaptic plasticity in hippocampal slices from oligomeric amyloid-β. An RNA aptamer also tightly binds to PrPC like antibodies; hence, the RNA aptamer may also rescue synaptic plasticity by preventing amyloid-β-oligomer binding to PrPC. Therefore, an RNA aptamer for PrPC may have therapeutic potential not only for prion diseases but also for Alzheimer’s disease.
In conclusion, we have demonstrated that a short RNA aptamer for PrPC that comprises only 12 residues, R12, exhibits anti-prion activity in mouse neuronal cells persistently infected with the TSE agent. There is the possibility that R12 is also effective for bovine spongiform encephalopathy. The structural basis of the tight binding of R12 to PrPC has been elucidated. These findings may be used to develop RNA aptamer-based drugs against the prion and Alzheimer’s diseases.
ACCESSION NUMBERS
Protein Data Bank: Coordinates and restraints for the final ensemble of 10 structures of the R12:P16 complex have been deposited under the accession code 2rsk.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1 and 2.
FUNDING
Ministry of Education, Culture, Sports, Science and Technology in Japan [23657072, 24121714 to M.K.; 23570146, 24113710 to T.N.]; JST [SENTAN and CREST]; Sumitomo-Denko Foundation; Iwatani Foundation. JSPS Research Fellow Program (to M.T.). Funding for open access charge: Ministry of Education, Culture, Sports, Science and Technology in Japan.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank M. Imamura and T. Yokoyama for giving them the plasmid containing the bPrP gene, and T. Saeki for the technical help in evaluation of the anti-prion activity.
REFERENCES
- 1.Prusiner SB. Prions. Proc. Natl Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl Acad. Sci. USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner SB. Molecular biology and transgenetics of prion diseases. Crit. Rev. Biochem. Mol. Biol. 1991;26:397–438. doi: 10.3109/10409239109086789. [DOI] [PubMed] [Google Scholar]
- 6.Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 7.Sailer A, Büeler H, Fischer M, Aguzzi A, Weissmann C. No propagation of prions in mice devoid of PrP. Cell. 1994;77:967–968. doi: 10.1016/0092-8674(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 8.Cashman NR, Caughey B. Prion diseases—close to effective therapy? Nat. Rev. Drug. Disc. 2004;3:874–884. doi: 10.1038/nrd1525. [DOI] [PubMed] [Google Scholar]
- 9.Mallucci G, Collinge J. Rational targeting for prion therapeutics. Nat. Rev. Neurosci. 2005;6:23–34. doi: 10.1038/nrn1584. [DOI] [PubMed] [Google Scholar]
- 10.Sim VL, Caughey B. Recent advances in prion chemotherapeutics. Infect. Disord. Drug Targets. 2009;9:81–91. doi: 10.2174/1871526510909010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proske D, Gilch S, Wopfner F, Schätzl HM, Winnacker EL, Famulok M. Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem. 2002;3:717–725. doi: 10.1002/1439-7633(20020802)3:8<717::AID-CBIC717>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Rhie A, Kirby L, Sayer N, Wellesley R, Disterer P, Sylvester I, Gill A, Hope J, James W, Tahiri-Alaoui A. Characterization of 2′-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J. Biol. Chem. 2003;278:39697–39705. doi: 10.1074/jbc.M305297200. [DOI] [PubMed] [Google Scholar]
- 13.Vogtherr M, Grimme S, Elshorst B, Jacobs DM, Fiebig K, Griesinger C, Zahn R. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J. Med. Chem. 2003;46:3563–3564. doi: 10.1021/jm034093h. [DOI] [PubMed] [Google Scholar]
- 14.Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, Nakamura HK, Kimura K, Kawasaki M, Takakura Y, et al. Hot spots in prion protein for pathogenic conversion. Proc. Natl Acad. Sci. USA. 2007;104:11921–11926. doi: 10.1073/pnas.0702671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taubner LM, Bienkiewicz EA, Copie V, Caughey B. Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J. Mol. Biol. 2010;395:475–490. doi: 10.1016/j.jmb.2009.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura T, Hosokawa-Muto J, Kamatari YO, Kuwata K. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg. Med. Chem. Lett. 2011;21:1502–1507. doi: 10.1016/j.bmcl.2010.12.132. [DOI] [PubMed] [Google Scholar]
- 17.Schütz AK, Soragni A, Hornemann S, Aguzzi A, Ernst M, Böckmann A, Meier BH. The amyloid-Congo red interface at atomic resolution. Angew. Chem. Int. Ed. Engl. 2011;50:5956–5960. doi: 10.1002/anie.201008276. [DOI] [PubMed] [Google Scholar]
- 18.Murakami K, Nishikawa F, Noda K, Yokoyama T, Nishikawa S. Anti-bovine prion protein RNA aptamer containing tandem GGA repeat interacts both with recombinant prion protein and its β isoform with high affinity. Prion. 2008;2:73–78. doi: 10.4161/pri.2.2.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishikawa F, Murakami K, Matsugami A, Katahira M, Nishikawa S. Structural studies of an RNA aptamer containing GGA repeats under ionic conditions using microchip electrophoresis, circular dichroism, and 1D-NMR. Oligonucleotides. 2009;19:179–190. doi: 10.1089/oli.2008.0167. [DOI] [PubMed] [Google Scholar]
- 20.Mashima T, Matsugami A, Nishikawa F, Nishikawa S, Katahira M. Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 2009;37:6249–6258. doi: 10.1093/nar/gkp647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García FL, Zahn R, Riek R, Wüthrich K. NMR structure of the bovine prion protein. Proc. Natl Acad. Sci. USA. 2000;97:8334–8339. doi: 10.1073/pnas.97.15.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J. Virol. 2000;74:320–325. doi: 10.1128/jvi.74.1.320-325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brünger AT. X-PLOR Version 3.1: A System for X-ray Crystallography and NMR. Yale University Press: New Haven; 1993. [Google Scholar]
- 24.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 25.Furukawa A, Nagata T, Matsugami A, Habu Y, Sugiyama R, Hayashi F, Kobayashi N, Yokoyama S, Takaku H, Katahira M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009;28:440–451. doi: 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wüthrich K. NMR of Protein and Nucleic Acids. New York, NY: John Wiley & Sons; 1986. [Google Scholar]
- 27.Campisi DM, Calabro V, Frankel AD. Structure-based design of a dimeric RNA-peptide complex. EMBO J. 2001;20:178–186. doi: 10.1093/emboj/20.1.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hara H, Okemoto-Nakamura Y, Shinkai-Ouchi F, Hanada K, Yamakawa Y, Hagiwara K. Mouse prion protein (PrP) segment 100 to 104 regulates conversion of PrPC to PrPSc in prion-infected neuroblastoma cells. J. Virol. 2012;86:5626–5636. doi: 10.1128/JVI.06606-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsugami A, Ouhashi K, Kanagawa M, Liu H, Kanagawa S, Uesugi S, Katahira M. An intramolecular quadruplex of (GGA)4 triplet repeat DNA with a G:G:G:G tetrad and a G(:A):G(:A):G(:A):G heptad, and its dimeric interaction. J. Mol. Biol. 2001;313:255–269. doi: 10.1006/jmbi.2001.5047. [DOI] [PubMed] [Google Scholar]
- 30.Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.