

Abstract
The histone methyltransferase EZH2 regulates cell proliferation and differentiation by silencing Polycomb group target genes. NIPP1, a nuclear regulator of serine/threonine protein phosphatase 1 (PP1), has been implicated in the regulation of EZH2 occupancy at target loci, but the underlying mechanism is not understood. Here, we demonstrate that the phosphorylation of EZH2 by cyclin-dependent kinases at Thr416 creates a docking site for the ForkHead-associated domain of NIPP1. Recruited NIPP1 enables the net phosphorylation of EZH2 by inhibiting its dephosphorylation by PP1. Accordingly, a NIPP1-binding mutant of EZH2 is hypophosphorylated, and the knockdown of NIPP1 results in a reduced phosphorylation of endogenous EZH2. Conversely, the loss of PP1 is associated with a hyperphosphorylation of EZH2. A genome-wide promoter-binding profiling in HeLa cells revealed that the NIPP1-binding mutant shows a deficient association with about a third of the Polycomb target genes, and these are enriched for functions in proliferation. Our data identify PP1 as an EZH2 phosphatase and demonstrate that the phosphorylation-regulated association of EZH2 with proliferation-related targets depends on associated NIPP1.
INTRODUCTION
EZH2 is the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2), which contains EED, SUZ12 and RbAp48 as its core regulatory subunits (1). In addition, the PRC2 complex can make (transient) interactions with a host of other proteins or non-coding RNAs that modulate its activity or association with Polycomb target loci. The EZH2-catalyzed trimethylation of histone H3 at Lys 27 (H3K27) contributes to the silencing of Polycomb targets and thereby regulates cell proliferation and differentiation. A deficiency of EZH2 is associated with aberrant developmental patterning and a loss of stem-cell pluripotency, whereas an excess of EZH2 has been linked to cancer progression (2,3).
The chromatin targeting and activity of EZH2 are regulated by multiple protein kinases. Phosphorylation of EZH2 at Ser21 (mouse residue numbers used throughout this manuscript) by protein kinase B (PKB/Akt) results in the dissociation of EZH2 from chromatin, a decline in H3K27me3 levels and an activation of target genes (4). In contrast, the phosphorylation of Thr367 by the p38 MAP kinase creates a binding site for the transcription factor YY1, which recruits the PRC2 complex to repress the lineage marker Pax7 in differentiating muscle cells (5). EZH2 is also phosphorylated at Thr345 and Thr487 by a PRC2-associated pool of the cyclin-dependent kinases (CDK) 1 and 2 (6–9). Phosphorylation at Thr345 is required for the binding of EZH2 to chromatin (7) and non-coding RNAs (8). Wei et al. (9) reported that the phosphorylation of Thr487 prevents the binding of EZH2 to its co-activators EED and SUZ12, leading to reduced H3K27me3 levels, but these results were not confirmed in a subsequent study using a phosphomimetic mutant (8). Finally, Wu and Zhang (6) showed that the CDK-mediated phosphorylation at Thr345 and Thr487 leads to the ubiquitylation and degradation of EZH2. Although it is now firmly established that EZH2 is an in vivo substrate for phosphorylation by CDKs, the counteracting phosphatase and its regulation are not yet known.
We have previously demonstrated that the protein phosphatase 1 (PP1) interactor NIPP1 is associated with a subset of established Polycomb target genes (10,11). Also, NIPP1 functions as a PRC2-dependent transcriptional repressor in reporter assays and interacts directly and independently with the PRC2 core components EZH2 and EED (12,13). Consistent with a role for NIPP1 in PRC2 signaling, mouse NIPP1−/− blastocyst outgrowths show a deficient trimethylation of H3K27 (11,14). Moreover, the knockdown of NIPP1 in cancer cells results in the dissociation of EZH2 from a subset of target genes (11), whereas the overexpression of NIPP1 causes a redistribution of EZH2 between target loci (10).
Here, we identify Thr416 of EZH2 as a novel CDK phosphorylation site in intact cells and show that phosphorylated Thr416 functions as a docking site for the ForkHead-associated (FHA) domain of NIPP1. The recruitment of NIPP1 is essential to maintain the CDK-mediated phosphorylation of EZH2 at TP-dipeptide motifs by opposing their dephosphorylation by PP1. Finally, we show that this regulation by NIPP1 is required for the association of EZH2 with a large number of proliferation-related target loci.
MATERIALS AND METHODS
Antibodies
For immunoprecipitation of endogenous EZH2, an antibody was raised by immunizing rabbits with the non-phosphorylated TP6 dodecapeptide. EZH2 (3147, clone AC-22), and pan-phospho-Threonine-Proline (pTP) (9391) antibodies were purchased from Cell Signaling. EGFP (SC-8334), PP1α (SC-6104) and PP1γ (SC-6108) antibodies were obtained from Santa Cruz. SUZ12 (clone 3C1.2), RbAp48 (ab488) and αTubulin (clone B-5-1-2) antibodies were delivered by Millipore, Abcam and Sigma-Aldrich, respectively. A mouse monoclonal NIPP1 antibody (mAb 15B8C11) was raised as described by (15). Human recombinant polyhistidine-tagged EED was used to raise antibodies in rabbits, as previously described (13). A monoclonal non-isoform-specific PP1 antibody was a kind gift of Dr. J. Vandenheede (University of Leuven, Belgium). For visualization, HRP-conjugated swine anti-rabbit IgG (P0217) and rabbit anti-mouse IgG (P0260) were purchased from Dako. Antibodies specific for EZH2 phosphorylated at Thr416 were raised by immunizing rabbits with the phosphorylated TP5 peptide (residues 410-421) coupled via an additional N-terminal Cys residue to keyhole limpet hemocyanin. The polyclonal antibody was affinity purified by consecutive chromatography on TP5-coupled and pTP5-coupled bovine serum albumin (BSA), linked to cyanogen bromide (CNBr)-activated Sepharose-4B (GE Healthcare). Polyclonal rabbit anti-mouse IgG antibodies (Dako) were used as a negative control for immunoprecipitations. The antibodies for the EZH2 chromatin immunoprecipitation (ChIP) experiments (AC22) are from Millipore.
siRNAs
Duplexes of siRNA against human PP1α (5′-CCGCATCTATGGTTTCTAC-3′) and PP1γ (5′-GCATGATTTGGATCTTATA-3′) were obtained from Dharmacon. Duplexes of siRNA against human NIPP1 (5′-GGAACCTCACAAGCCTCAGCAAATT-3′) and a scrambled control (5′-AATTGTTCGTGGAGGTTCGAGTTCC-3′) siRNA were purchased from Invitrogen.
Peptides
Chemically synthesized protein dodecapeptides of mouse EZH2 were purchased from EZBiolab. Phosphorylated (p) and non-phosphorylated peptide sequences were EZH2339−351: TAERIK(p)TPPKRPG, EZH2410−421: ANSRCQ(p)TPIKMK and EZH2483−495: EDVD(p)TPPRKKKRK.
Cell culture conditions and transfection
HEK293T and HeLa cells were cultured as described previously (10). siRNA-mediated knockdowns were performed using RNAiMAX transfection reagent (Invitrogen). Transfection with plasmid DNA was carried out using Jetprime reagent (Polyplus). Mouse EZH2 coding sequences were cloned into the BsrGI/XhoI digested pCHMWS-EGFP-IRES-puro expression vector (16) using the In-Fusion HD Cloning Kit (Clontech), following the manufacturer’s instructions. Mutations were introduced using the Quickchange Mutagenesis Kit (Agilent Technologies). Plasmids encoding NIPP1-EGFP have been described previously (17). The pFastBac vector-containing mouse EZH2 for His-tagged protein expression and purification from Sf9 insect cells was a kind gift of Dr. C. Helin (BRIC, University of Copenhagen, Denmark).
Immunoprecipitation and immunoblotting
Cell lysis, isolation of the chromatin-enriched fraction, immunoprecipitations and EGFP traps were performed as described in (10,12). Lysis buffers were supplemented with 20 mM NaF, 5 µM leupeptin, 0.5 mM phenylmethanesulfonyl fluoride and 0.5 mM benzamidine, unless stated otherwise. For elution of EGFP-tagged proteins from the beads that were used for the EGFP traps, the beads were incubated for 90 s in 100 µl glycine (0.2 M at pH 2.5). The eluates were immediately neutralized by the addition of 10 µl Tris/HCl (1 M at pH 10.8). For in vitro dephosphorylation with lambda phosphatase, samples were incubated for 30 min at 30°C with 400 units of enzyme (Santa cruz) in a buffer containing 20 mM Tris/HCl at pH 7.5, 5 mM DTT and 2 mM MnCl2. For dephosphorylation with PP1, samples were incubated for 30 min at 30°C in a buffer containing 50 mM Tris/HCl at pH 7.5, 1 mM DTT and 0.1 mg/ml BSA. For in vitro phosphorylation, samples were incubated for 60 min at 30°C with recombinant cyclinA/CDK2 in a buffer containing 20 mM Tris/HCl at pH 7.5, 2 mM DTT, 0.1 mg/ml BSA, 2 mM MgAc, and 100 µM ATP with or without γ32P-ATP. Immunoblots were visualized using eCL reagent (Perkin Elmer) in a ImageQuant LAS4000 imaging system (GE Healthcare) and were quantified using ImageQuant TL software (GE Healthcare).
ChIP
ChIP assays were performed as described previously (10), with slight modifications. A double cross-linking strategy was used to stabilize protein–protein interactions (18). Cells were treated with 2 mM disuccinimidyl glutarate (DSG, Pierce) for 45 min before formaldehyde fixation.
Recombinant protein production
N-terminally glutathione-S-transferase (GST)-tagged proteins were produced by transforming Escherichia coli BL21 (DE3) pLysE cells with pGEX-2TK expression vectors containing NIPP11−142 wild-type (WT) (FHA), NIPP11−142 S68A/R69A/V70A/H71A (FHAm) or the empty vector as GST control. Protein production was induced for 4 h at 37°C with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). After centrifugation (10 min, 3200g), cells were lysed in phosphate buffered saline using sonication, and proteins were extracted with GST SpinTrap columns (GE Healthcare) following the manufacturer’s instructions. His-EZH2 and skeletal muscle PP1 were purified as described in (19) and (20), respectively. The pET16b-CyclinA2 and pGEX3C-CDK2 vectors for the expression of cyclinA and CDK2, respectively, were a kind gift of Dr. Tim Hunt (London research institute, Great Britain), and purification was performed as described in (21).
For purification of the NIPP1 FHA domain for Nuclear Magnetic Resonance (NMR) studies, the fragment was subcloned into pETM30-MBP. Freshly transformed E. coli BL21-Codon-Plus (DE3)-RIL (Agilent) cells were grown at 37°C to an OD600 of ∼0.6, and expression was induced with 1 mM IPTG. Proteins were expressed for 18 h at 18°C. Cultures were harvested by centrifugation, and the cell pellets were stored at −80°C. The expression of uniformly 13C/15N-labeled and 15N-labeled protein was carried out by growing freshly transformed cells in M9 minimal medium containing 4 g/L [13C]-glucose and/or 1 g/L 15NH4Cl (Cambridge Isotope Laboratory) as the sole source of carbon and nitrogen, respectively. The cell pellets were resuspended in lysis buffer (50 mM Tris/HCl at pH 8.0, 5 mM imidazole, 500 mM NaCl, 0.1% Triton-X 100), supplemented with a Complete EDTA-free protease inhibitor tablet (Roche). Cells were lysed by high pressure homogenization (Avestin C-3 Emulsiflex), and cell debris was removed by centrifugation (50 000g, 40 min, 4°C). The clarified lysate was loaded onto a His-Trap HP Column (GE Healthcare), and the protein was eluted with an imidazole gradient of 5–500 mM. Fractions containing FHA domain were pooled, incubated with TEV protease to remove the N-terminal his6-MBP-tag and dialyzed against 20 mM Tris/HCl pH 7.5, 200 mM NaCl. The dialyzed sample, containing the cleaved FHA domain, was loaded onto a Ni-NTA column (Invitrogen), from which it eluted in the flow-through. For the final purification step, the protein was loaded onto a size-exclusion column (Superdex75 26/60; GE Healthcare), equilibrated in NMR buffer (20 mM BisTris at pH 6.3, 50 mM NaCl, 5 mM DTT). Fractions containing the pure NIPP1 FHA domain, as confirmed by SDS-PAGE, were pooled and concentrated.
Chemical shift perturbation calculation
Chemical shift perturbation (CSP) calculations were quantified to equation: 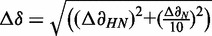 . Peptide binding affinity was determined by fitting the CSP as a function of peptide concentration using equation:
. Peptide binding affinity was determined by fitting the CSP as a function of peptide concentration using equation:
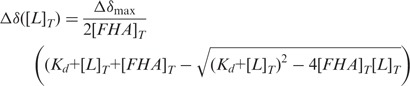 |
where Δδmax, [L]T, [FHA]T and Kd are the maximum chemical shift at saturation, total ligand concentration, total FHA domain concentration and dissociation constant, respectively. Data were simultaneously fit to Δδmax and Kd using the non-linear regression suite in SigmaPlot.
NMR spectroscopy
The binding affinity between EZH2 and NIPP1 was determined with NMR spectroscopy, by titrating increasing EZH2 peptide concentrations to 15N-labeled NIPP1 FHA domain. All NMR experiments were acquired at 298 K on a Bruker Avance 500 MHz spectrometer, using either 15N- or 15N/13C-labeled NIPP1 FHA domain at a final concentration of 1 mM (sequence-specific backbone assignment) or 0.1 mM (peptide interactions) in NMR buffer (20 mM Bis/Tris at pH 6.3, 50 mM NaCl, 5 mM DTT). The following spectra were used for the sequence-specific backbone assignments of the FHA domain: 2D [1H,15N] HSQC, 3D HNCA, 3D HNCACB, 3D CBCACONH and 3D CCCONH. TopSpin 2.1 (Bruker) was used for data acquisition and processing. NMR spectra were analyzed using the CARA software package (http://cara.nmr.ch). For NMR titration studies, all peptides were solubilized in NMR buffer, and 2D [1H,15N] HSQC spectra were used to monitor CSPs that occur owing to peptide binding. More details on CSP calculation can be found in the supplemental information section. Chemical shift assignments of the NIPP1 FHA domain were deposited in the Biological Magnetic Resonance Data Bank under accession number 18 448.
Mass spectrometry
The band corresponding to EGFP-EZH2 was excised from gel and subjected to an overnight digestion with trypsin (Roche) at 37°C. The resulting peptide mixture was analyzed by nanoLC-MS/MS, consisting of a precursor 79 (−) ion scan to signal the presence of putative phosphopeptides and a product (+) ion scan to determine the phosphorylated residue. Nano LC-MS/MS was performed on an Ultimate capillary liquid chromatography system (Dionex) coupled to a 4000 QTRAP mass spectrometer (Applied Biosystems). Peptides were separated on a PepMap C18 column developed with a 30-min linear gradient (0.1% formic acid/6% acetonitrile/water–0.1% formic acid/40% acetonitrile/water). Tandem MS spectra were interpreted manually.
DNA-adenine-methyltransferase-identification analysis
DNA-adenine-methyltransferase-identification (DamID) samples of EZH2-Dam, EZH2-mTP5-Dam and the Dam control were obtained as described before (10). All DamID data are available at Gene Expression Omnibus (GEO) under the accession number GSE39593. Analysis was performed by the model-based analysis of tiling arrays (MAT) algorithm. The derived bar files were converted to bigwig files allowing for downstream analysis by tools offered by the Galaxy/Cistrome website (22). Derived genes lists were functionally analyzed using the Ingenuity Pathway Analysis (IPA) tool (Ingenuity® Systems, www.ingenuity.com).
Hela cell histone modification data extraction
Values for histone modifications in HeLa cells were extracted from the data sets deposited at the University of California Santa Cruz (UCSC) genome site (23–25). Briefly, peaks overlapping with the ±4 kb region surrounding the transcription start sites (TSS) of the selected genes were extracted, and the percentage of TSS regions with the respective histone modification was calculated.
RESULTS
Phosphorylated EZH2 binds to the FHA domain of NIPP1
To define the role of NIPP1 in EZH2 signaling, we first mapped the EZH2-NIPP1 interaction sites. EZH2 is known to bind to the N-terminal 142 residues of NIPP1 (12), which mainly consist of a pTP interacting FHA domain [(26); Figure 1A]. In accordance with a phosphorylation-dependent interaction, endogenous EZH2 immunoprecipitated from HEK293T cell lysates was found to be phosphorylated at TP-dipeptide motifs (Figure 1B). To investigate the contribution of the FHA domain of NIPP1 to EZH2 binding, we performed pull-down assays with GST-tagged NIPP11−142 (GST-FHA) or the same fusion with a non-functional FHA domain (27), generated by alanine substitution of residues 68-71 (Figure 1A) in the essential FHA phosphate-binding loop (GST-FHAm). EZH2 from HEK293T cell lysates co-sedimented with GST-FHA, but not with GST-FHAm, suggesting that the FHA domain of NIPP1 mediates the binding to EZH2 (Figure 1C). Consistently, the binding of EZH2 to GST-FHA was abolished by prior dephosphorylation of EZH2 with lambda phosphatase (Figure 1B and C), but the binding could be restored by in vitro phosphorylation with cyclinA/CDK2 (Figure 1D). An FHA and phosphorylation-dependent interaction could also be demonstrated with purified baculovirus-expressed His-EZH2 (Supplementary Figure S1A and B), supporting a direct interaction between the two proteins. These results demonstrate that the interaction between NIPP1 and EZH2 is phosphorylation-dependent and involves the FHA domain.
Figure 1.

EZH2 phosphorylation on TP-dipeptide motifs mediates binding to the FHA domain of NIPP1. (A) Domain structures of NIPP1 and EZH2. NIPP1 contains an N-terminal FHA-domain. The N-terminal fragment of 142 residues that was used for pull-down experiments, and the residues of the FHA-domain that were alanine mutated (FHAm) are indicated. EZH2 contains two SANT-domains, a catalytic SET-domain and a cysteine-rich (CXC) domain. The six TP-dipeptide motifs of mouse EZH2 are numbered TP1-6 from the N to C-terminus. Also indicated are the corresponding threonine residue numbers. (B) Endogenous EZH2 was immunoprecipitated from HEK293T cell lysates that were pre-incubated with phosphatase inhibitors (NT) or with lambda phosphatase (λPPase). The (de)phosphorylation of EZH2 was verified by immunoblotting with pan-pTP antibodies. (C) GST pull-down assays were performed with HEK293T lysates and GST, GST-FHA or GST-FHAm. Assays were performed after incubation of the lysate as such (NT) or with lambda phosphatase (λPPase). Equal loading of GST-fusions was verified by Coommassie. (D) EGFP-EZH2 was immunoprecipitated, dephosphorylated with lambda phosphatase and extracted from the beads with 0.2 M glycine at pH 2.5. After neutralization of the pH, the fusions were incubated for 60 min at 30°C, as such (Ctrl), with MgATP, cyclinA/CDK2 or MgATP plus cyclinA/CDK2, and then subjected to GST pull-downs. Phosphorylation was validated by immunoblotting with pan-pTP antibodies. (E) EGFP (Ctrl) or EGFP-NIPP1 (WT or FHAm) was immunoprecipitated from HEK293T cell lysates using EGFP traps. The co-precipitation of core PRC2 components was quantified by immunoblotting with antibodies against EZH2, EED and SUZ12.
As our previous (12,13) and present (Figure 1C and D) data identified distinct NIPP1 binding sites for the PRC2 components EZH2 and EED, we have re-examined whether these proteins can be recruited independently by full-length NIPP1. Both EZH2 and EED co-precipitated with ectopically expressed EGFP-NIPP1, but only EED co-sedimented with the corresponding FHA mutant. In contrast, the PRC2 component SUZ12, which interacts directly with EZH2 but not with EED (19), only co-precipitated with the WT NIPP1 fusion (Figure 1E). These data confirm that full-length NIPP1 has distinct interaction sites for EZH2 and EED, and that its interaction with EZH2 depends on a functional FHA domain. Taken together, these data imply that EED can be complexed to NIPP1 in absence of EZH2.
EZH2 phosphorylation at Thr416 creates a docking site for NIPP1
A comparative analysis of the binding sequences of established FHA ligands of NIPP1 delineated pTP as the only common binding determinant (Supplementary Figure S2A). In total, EZH2 contains six TP-dipeptide motifs, all grouped in the central region and further denoted as TP1-6 (Figure 1A). These TP-dipeptide motifs are highly conserved from fish to man (Supplementary Figure S2B), and TP3, TP4 and TP6 are already known to be phosphorylated in vivo (5,7–9). In addition, TP5 was identified as an in vitro CDK1 target site (8). To identify the TP-dipeptide motif of EZH2 that mediates the binding to NIPP1, we made Thr to Ala substitution mutants for all six TP motifs. Pull-down experiments with GST-FHA revealed a decreased binding of EZH2 after mutation of TP3 and TP6, but a complete loss of binding after mutation of TP5 (Figure 2A). Additional pull-downs with combined TP mutants of EZH2 confirmed an essential role for TP5 in the binding to NIPP1. Indeed, any combination of mutations that lacked TP5 resulted in undetectable binding to GST-FHA, whereas EZH2 with all TPs mutated except for TP5 still co-sedimented with GST-FHA (Figure 2B). Consistent with these data, competition studies with synthetic dodecapeptides comprising (non-) phosphorylated TP3, TP5 or TP6, and their flanking sequences only showed a decreased EZH2-FHA interaction after addition of the pTP5 peptide (Figure 2C). Finally, after mutation of TP5 (EZH2-mTP5), EZH2 no longer co-immunoprecipitated with endogenous NIPP1 from HEK293T cell lysates (Figure 2D). Collectively, these data disclosed a key role for pTP5 (Thr416) of EZH2 in the binding to the FHA domain of NIPP1.
Figure 2.

Thr416 (TP5) of EZH2 is the key NIPP1-binding site. (A) Left: EGFP-EZH2 was mutated at the indicated TP-motifs (Thr to Ala mutations) and used for GST pull-down assays with the NIPP1-FHA domain. Right: Quantification and statistical analysis of the immunoblots. Data are shown as means ± SEM (n = 4). Significant differences compared with WT are indicated with *(P < 0.05) or **(P < 0.01) (paired Student’s t-test). (B) Same as in (A) with combined Thr to Ala mutation of the indicated TP-motifs. (C) GST pull-downs of EGFP-EZH2 were performed in the presence of 1 mM of the indicated EZH2 dodecapeptides. (D) Endogenous NIPP1 was immunoprecipitated from HEK293T cell lysates that were transfected with expression vectors for EGFP-tagged EZH2-WT or EZH2-mTP5. The co-immunoprecipitation of the EZH2 fusions was analyzed by immunoblotting with EGFP-antibodies.
Molecular basis of the EZH2–NIPP1 interaction
CSP NMR experiments using the 15N-labeled FHA domain of NIPP1 and the aforementioned dodecapeptides confirmed that only the pTP5 peptide caused significant CSPs for residues in pThr-binding loops of the FHA domain (Figure 3A and B, Supplementary Figure S3A and B). pTP5 mainly interacted with the canonical binding site of the FHA domain on the β3/β4 (R53,N54), β4/β5 (S66, S68, R69,V70) and β6/β7 loops (H92) (Figure 3B and C). The observed CSPs were significant (0.09 < Δδ < 0.21 ppm) and in the fast exchange regime on the chemical shift time scale. Saturation was not achieved even after a 10-fold molar excess of the peptide, indicating a rather weak interaction. An estimation of the dissociation constant for six residues that exhibited CSPs when the pTP5 peptide was titrated into the 15N-labeled FHA domain yielded an average Kd of ∼1.9 ± 0.8 mM (Supplementary Figure S3C).
Figure 3.

The EZH2-derived pTP5 peptide binds to the FHA domain of NIPP1. (A) 2D [1H,15N] HSQC spectrum of 15N-labeled NIPP1-FHA (red) and different ratios of NIPP1-FHA:pEZH2410-421; 1:1 (orange), 1:2 (yellow), 1:5 (green) and 1:10 (blue). CSPs are readily followed; some peaks have significantly reduced intensity in the 1:10 spectrum most likely due to intermediate conformation exchange; few residues are annotated. (B) Histogram showing the combined 1H/15N CSPs versus residue. Experimentally determined secondary structure elements of NIPP1-FHA are shown. CSP deviations larger than 0.1 ppm (pink) and 0.2 ppm (blue) are indicated. The 11 β-strands that constitute the FHA domain are shown as arrow representations on top of the graph, and the two anti-parallel β-sheets are colored green and blue, respectively. (C) Left: ribbon representation of NIPP1-FHA. Right: surface representation of NIPP1-FHA. Residues that exhibit CSPs in the presence of pEZH2410-421 are shown as stick representation and are colored.
EZH2 is phosphorylated at Thr416 in cultured cells
To explore the phosphorylation of pTP5 in intact cells, we generated a phospho-TP5 specific antibody. The affinity-purified antibody clearly recognized purified EGFP-EZH2 that was phosphorylated in vitro by cyclinA/CDK2 (Figure 4A). However, no signal was detected in these conditions after mutation of TP5, although this mutation only moderately reduced the phosphorylation of EZH2 by CDK2, as determined by autoradiography. Conversely, the mutation of other TP-motifs did not have an effect on the immunodetection of pTP5, providing additional evidence for the specificity of the antibody.
Figure 4.

EZH2 is phosphorylated in vivo on TP5. (A) Validation of pTP5 antibody specificity. The indicated EGFP-EZH2 variants were affinity-purified, dephosphorylated with lambda phosphatase, in vitro phosphorylated using γ32P-ATP or cold ATP and cyclinA/CDK2. Phosphorylation was detected by autoradiography or immunoblotting with the pTP5-specific antibody. (B) Lysates from HEK293T cells were used for immunoprecipitation with the pTP5-specific antibody, and the immmunoprecipitate was subjected to SDS-PAGE. The band corresponding to EZH2 was excised, subjected to in-gel trypsin digestion and analyzed by nano LC–MS/MS mass spectrometry. The MS/MS spectrum of the doubly charged ion (m/z 385) shows that Thr416 (TP5) is phosphorylated in the 414CQTPIK419 peptide. (C) Endogenous EZH2 was immunoprecipitated from HEK293T cell lysates and analyzed by immunoblotting for phosphorylation on TP5 (pTP5) and TP in general (pTP). Immunoprecipitation with immunoglobulins was used as a control (Ctrl). (D) GST pull-downs were performed using GST or GST-FHA in lysates from HEK293T cells overexpressing EGFP (Ctrl) or EGFP-EZH2. Cells were pre-incubated with dimethylsulfoxide (DMSO) (−) or 100 µM of the CDK-inhibitor roscovitine (+) for 2 h. Equal loading of the GST fusions was verified by Coomassie staining.
Next, we performed a mass-spectrometric analysis of EGFP-EZH2 that was immunoprecipitated from HEK293T cell lysates with the pTP5-specific antibody. In addition to the previously characterized phosphorylation sites Thr345 (TP3) and Thr487 (TP6), this analysis also identified the phosphopeptide 414CQpTPIK419, with Thr416 (TP5) as the phosphorylated residue (Figure 4B). This shows that EZH2 is phosphorylated at TP5 in intact cells and that TP5-phosphorylated EZH2 is, at least partially, also phosphorylated at TP3 and/or TP6.
The pTP5 antibody recognized endogenous EZH2 that was immunoprecipitated from total HEK293T cell lysates (Figure 4C) and from the chromatin-enriched fraction (Supplementary Figure S4A). In addition, the pTP5 antibody detected ectopically expressed EGFP-EZH2, but the latter fusion was not detected after mutation of TP5 (Supplementary Figure S4B) or after a preincubation of HEK293T cells with the CDK inhibitor roscovitine (Figure 4D). A preincubation of the cells with roscovitine also abolished the binding of EGFP-EZH2 to GST-FHA. These data suggest that TP5, like TP3 and TP6 (6–9), is phosphorylated by CDKs.
NIPP1 opposes the dephosphorylation of EZH2 by PP1
To investigate the interdependency of the phosphorylation of EZH2 on distinct TP-dipeptide motifs, we examined the phosphorylation status of TP mutants of EZH2. The mutation of all TPs but TP5 strongly reduced TP-dipeptide phosphorylation, as detected with a pan-pTP antibody, but only mildly decreased the pTP5 signal (Figure 5A). This demonstrates that EZH2 is phosphorylated on multiple TP-sites in intact cells and that pTP5 only contributes marginally to the pan-pTP signal (Figure 5A). Yet, the mutation of TP5 nearly abolished the pan-pTP signal (Figure 5A and Supplementary Figure S4B). A similar massive loss of TP-phosphorylation was not seen after the individual mutation of any of the other TP-motifs (Supplementary Figure S4B). Collectively, these data represent sound evidence that the phosphorylation of TP5 is necessary to facilitate or maintain the phosphorylation of other TP-dipeptide motifs of EZH2.
Figure 5.

NIPP1 inhibits the dephosphorylation of EZH2 by PP1. (A) EGFP (Ctrl) or the indicated EGFP-EZH2 variants were affinity purified by EGFP trap and examined for pTP and TP5 phosphorylation by immunoblotting. (B) Dephosphorylation of EZH2 by PP1. EGFP-EZH2 was immunoprecipitated from HEK293T cell lysates and in vitro dephosphorylated using PP1 (1.5 µM, 500 nM and 166 nM) that was purified from rabbit skeletal muscle. Remaining phosphorylation levels were analyzed by immunoblotting with pan-pTP antibodies. (C) Same as in (B, 500 nM PP1) with the addition of decreasing amounts of recombinant His-NIPP1 (4.5 µM–1.5 µM–500 nM–166 nM–55 nM). (D) EGFP-EZH2 was purified by EGFP trap and incubated as such (−), with 500-nM skeletal muscle PP1 or with the immunoprecipitated EGFP-NIPP1/PP1 complex, before and after trypsinization (Tryp). As a control, an EGFP-trap of EGFP-βGal was used (Ctrl IP). Dephosphorylation of EZH2 was examined by immunoblotting with pan-pTP antibodies. The presence of NIPP1 and PP1 was also verified by immunoblotting. The PP1 antibody does not recognize trypsin-pretreated PP1 because of the removal of the C-terminal epitope. Trypsinolysis during the phosphatase assay was prevented by the addition of soybean trypsin inhibitor. (E) EGFP-βgal (Ctrl) or EGFP-EZH2 (WT or mTP5) were immunoprecipitated out of HEK293T cell lysates and investigated for associated PP1 by immunoblotting. (F) Phosphorylation of EZH2 on TP-dipeptide motifs was visualized by immunoblotting after immunoprecipitation of endogenous EZH2 from nocodazole-arrested HeLa cells treated with RNAi for NIPP1, PP1 (α + γ isoform) or a scrambled Ctrl.
As phosphorylated TP5 mediates the interaction of EZH2 with NIPP1, this prompted us to ask whether the recruitment of NIPP1 serves to inhibit the dephosphorylation of EZH2 by PP1. Consistent with this notion, ectopically expressed EGFP-EZH2 was dephosphorylated at TP-motifs by purified PP1 (Figure 5B), but this dephosphorylation was abolished by the addition of purified NIPP1 (Figure 5C). Also, EGFP-EZH2 was not dephosphorylated by immunoprecipitated NIPP1, in spite of the presence of co-precipitated PP1 (Figure 5D). However, after a pretreatment with trypsin, which destroys NIPP1 and releases a fully active PP1 catalytic subunit (28), the immunoprecipitate dephosphorylated EGFP-EZH2. Together, these data demonstrate that NIPP1 inhibits the PP1-mediated dephosphorylation of EZH2 at TP-motifs. Importantly, immunoprecipitates of EZH2-WT and EZH2-mTP5 contained the same amount of PP1 (Figure 5E), indicating that the NIPP1-EZH2 interaction is not required to recruit PP1 to the EZH2 complex.
To determine whether the phospho-regulation of EZH2 by PP1/NIPP1 also applies to intact cells, we investigated the effect of depleting either PP1 or NIPP1 on the phosphorylation of EZH2, using previously validated siRNAs (10,28). The combined knockdown of PP1α and PP1γ resulted in a hyperphosphorylation of EZH2, as detected with pan-pTP antibodies (Figure 5F). In contrast, the knockdown of NIPP1 caused EZH2 to be hypophosphorylated. These data are consistent with EZH2 being a substrate for PP1 and suggest that a net phosphorylation of EZH2 at TP-dipeptide motifs requires the inhibition of PP1 by TP5-bound NIPP1.
EZH2-mTP5 shows a deficient association with proliferation-related target loci
TP-dipeptide phosphorylation has been implicated in both the assembly of the PRC2 complex and the association of EZH2 with subsets of Polycomb target loci (5,7–9). Immunoblot analysis of the immunoprecipitated EGFP-tagged EZH2-WT, EZH2-mTP3, EZH2-mTP5 or EZH2-mTP6 showed that the PRC2 components EED, SUZ12 and RbAp48 were equally abundant in all studied complexes (Supplementary Figure S5). This indicates that TP-dipeptide phosphorylation of EZH2 is not needed for the formation of the PRC2 core complex. Next, we compared the distribution of EZH2-WT and EZH2-mTP5 at promoters, using the DamID approach [(29); Supplementary Figure S6A]. For this purpose, stable HeLa cell lines were generated that express trace amounts of Dam or Dam that was C-terminally fused to either EZH2-WT or EZH2-mTP5. Two distinct polyclonal cell lines were generated and analyzed for each transgene. From each cell line, three independent samples were prepared, pooled and hybridized to GeneChip Human Promoter 1.0 R Arrays (Affymetrix), covering the −7.5/+2.5 kb region of ∼25 000 TSS. Heatmaps were generated to identify sets of TSS with robust-binding patterns for Dam-fused EZH2-WT and EZH2-mTP5. When clustered by best fitting of either the EZH2-WT or EZH2-mTP5 binding patterns, the clusters were mirrored to some extent by the binding sites of the respective other factor, indicating an overlapping set of binding sites (Supplementary Figure S6B, center). However, the NIPP1-binding mutant clearly formed smaller clusters, hinting at fewer specific target locations.
To compose EZH2 target gene lists, we subsequently extracted the TSS corresponding to the distinct clusters and assigned the respective genes. When limited to the genes that were common to four independent clustering experiments with random k-means seed setting, this resulted in 2826 target genes for EZH2-WT and 1885 target genes for EZH2-mTP5 (Supplementary Figure S6B) which, however, overlapped for 94% with the EZH2-WT targets (Figure 6A and Supplementary Table S1). The identified EZH2-WT and EZH2-mTP5 bound genes fulfilled the expectations for PRC2 targets: (i) IPA revealed that they were highly enriched for developmental genes (Supplementary Figure S6B), which are among the key targets of Polycomb signaling; (ii) the identified EZH2 targets were enriched for the H3K27me3 mark, but were underrepresented for the active gene markers H3K4me2 and H3K36me3 (Supplementary Figure S6C); (iii) Gene Set Enrichment Analysis (30,31) showed the most significant overlaps with gene sets that identified target genes of EED and SUZ12 or localization of H3K27 trimethylation (32); and (iv) the EZH2 targets identified by DamID showed a clear overlap with previously published PRC2 target lists (32,33) from other cell types and using different mapping strategies (Supplementary Figure S6D).
Figure 6.

A NIPP1-binding mutant of EZH2 shows aberrant association with proliferation-related target genes. (A) Target genes of EZH2-WT (green) and EZH2-mTP5 (red) are widely overlapping (1776 TP5-independent targets), but an important subset is EZH2-WT specific (1050 TP5-dependent target genes). (B) Clustering in relation to the TSS of the derived target genes. Bigwig files were clustered (k-means 10) in relation to the TSS of the TP5-independent target genes (left) or the TP5-dependent target genes (right) for the −7.5 kb to +2.5 kb region, using the heatmap tool of the cistrome online resource. Calculation of the clusters was based on the combined WT and mTP5 data sets. Although a high degree of similarity of the two data sets is observed for the set of TP5-independent target genes, the mTP5 signal is clearly reduced in comparison with WT for the TP5-dependent target genes. Moreover, EZH2 chromatin binding at TP5-dependent target genes is generally located more upstream of the TSS. (C) DamID-binding profile of EZH2 on a selected TP5-independent and TP5-dependent target gene across the promoter region. IFNA5 is found in cluster 3 of the TP5-independent gene list and UBE2E2 belongs to cluster 10 of the TP5-dependent gene list. (D) The relative expression levels of the indicated genes were determined by qRT-PCR in HeLa cells that were treated with control (Ctr), NIPP1 or EZH2 siRNAs. ACTIN was used as control for normalization. (E) Pie-charts showing the top five classes of a comparative analysis for molecular and cellular functions using IPA. Genes can be present in more than one functional category.
To identify NIPP1-regulated EZH2 target genes, we generated new heatmaps with the obtained EZH2-WT and EZH2-mTP5 targets (Figure 6B). When clustered using the best fit for the overlapping (TP5-independent) target genes, the two heatmaps were nearly identical. However, when clustered for the EZH2-WT specific target genes (TP5-dependent targets), the binding of EZH2-mTP5 was nearly absent (Figures 6B and C and Supplementary Figure S7). In addition, EZH2 binding at the TP5-dependent target genes clustered in more sharply defined regions, only upstream and generally more remote from the TSS when compared with the TP5-independent target genes (Figure 6B and Supplementary Figure S7A).
To validate by an independent approach that NIPP1 specifically regulates TP5-dependent target genes, we first examined the effect of a knockdown of either NIPP1 or EZH2 on the expression of randomly selected TP5-dependent and TP5-independent target loci (Figure 6D). TP5-independent target genes were upregulated by the knockdown of EZH2, whereas the loss of NIPP1 did not affect their expression level. Conversely, TP5-dependent target genes were upregulated by a knockdown of either NIPP1 or EZH2, validating a role of NIPP1 in the pTP-dependent repression by EZH2. Next, we performed ChIP assays to study the recruitment of EZH2 at TP5-specific genes before and after the knockdown of NIPP1. These data confirmed that the UBE2E2, BDNF, BTBD7 and SEC24D genes are EZH2 targets and that the knockdown of NIPP1 resulted in a reduced association of EZH2 with these loci (Supplementary Figure S8). Finally, a comparative IPA analysis for molecular and cellular functions revealed that the TP5-dependent target genes in HeLa cells were enriched for genes involved in various aspects of proliferation (Figure 6E). In contrast, TP5-independent targets were enriched for developmental genes. Thus, our data clearly define TP5-dependent and TP5-independent EZH2 target loci in HeLa cells and show that NIPP1 is implicated in the pTP-regulated association of EZH2 with proliferation-related target loci.
DISCUSSION
Here, we identify Thr416 (TP5) of EZH2 as a novel CDK phosphorylation site in proliferating cells and demonstrate that pTP5 serves as a docking site for the FHA domain of NIPP1. Although it can form a complex with TP5-phosphorylated EZH2, the FHA domain of NIPP1 interacts rather poorly with an EZH2-derived dodecapeptide comprising pTP5. This is similar to the FHA domain of the proliferation marker Ki67, which also interacts weakly with short pTP-peptides but shows a dramatic increase in binding affinity for larger peptides (34). This is due to the extension of a β-sheet of the FHA domain by the addition of a β-strand from the longer ligand peptide. Thus, an interaction between the FHA domain of NIPP1 or Ki67 and a pTP-motif of their ligand appears to be essential, but not sufficient to form a stable complex. We speculate that for the assembly of these complexes, the pTP-FHA interaction only represents a first binding step that, by increasing the local concentration of both components, promotes secondary interactions needed to create a high-affinity binding. Our EZH2 mutagenesis studies showed that, in addition to TP5, TP3 and TP6 may also contribute to the recruitment of NIPP1. However, mutation of TP3 or TP6 did not affect the phosphorylation of TP5, suggesting that their role in the binding of NIPP1 is indirect. Intriguingly, TP5 is also conserved in EZH1, a paralog of EZH2 that is more abundant in adult, non-proliferative cells (35) and also associates with actively transcribed genes (36). It will be interesting to examine whether the phospho-regulation by NIPP1 also applies to EZH1.
Our biochemical data demonstrate that PP1 acts as an EZH2 phosphatase that is inhibited by the CDK-induced recruitment of NIPP1. This conclusion is supported by observations in intact cells showing that the loss of PP1 results in the hyperphosphorylation of EZH2 while the knockdown of NIPP1 has the opposite effect. Furthermore, an ectopically expressed NIPP1-binding mutant of EZH2 is hypophosphorylated, and NIPP1-associated EZH2 is phosphorylated at TP5 as well as TP3 and/or TP6. Thus, CDKs might preserve the phosphorylation of EZH2 at TP3, TP6 and possibly other sites, through phosphorylation of TP5, which functions as a docking site for NIPP1 and thereby inhibits the dephosphorylation of EZH2 by PP1 (Figure 7). This regulation is reminiscent of the induction of mitosis by CDK1, which is also critically dependent on the CDK-mediated inhibition of counteracting phosphatases (37). The constitutive dephosphorylation of EZH2 by PRC2-associated PP1 in the absence of NIPP1 provides a possible explanation for the observation that EZH2 is only phosphorylated at substoichiometric levels at any time during the cell cycle (8), and suggests that a net phosphorylation of EZH2 is only achieved after the recruitment of NIPP1. However, the regulation of the PRC2 complex by NIPP1 is likely to be more complex, as it not only involves the phosphorylation-dependent recruitment of NIPP1 by EZH2 (this work) and the RNA-stimulated interaction of NIPP1 with EED (13) but also the modulation of the PP1-inhibitory potency of NIPP1 by reversible phosphorylation (38,39). A further complexity stems from our observation that PP1 appears to have a dual function, in that it not only dephosphorylates EZH2 but is also required for the recruitment of NIPP1, which inhibits the dephosphorylation of EZH2 (10). We have recently found that a large portion of NIPP1 is intrinsically disordered, but folds on binding of PP1 (40). Hence, the contribution of PP1 to the recruitment of NIPP1 may well reflect a structural rather than a catalytic role. It will be important to explore how all these regulatory mechanisms are integrated to modulate PRC2 signaling.
Figure 7.
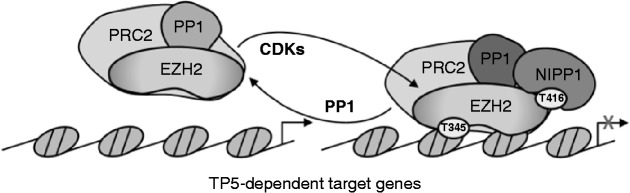
Proposed model for the NIPP1-mediated regulation of EZH2 chromatin association by reversible phosphorylation. PP1 dephosphorylates EZH2 on TP-motifs. The CDK-mediated phosphorylation of EZH2 on TP5 (T416) results in the recruitment of NIPP1, which inhibits PP1. This enables the net phosphorylation of EZH2 on other TP-dipeptide motifs, including TP3 (T345), which was previously shown to mediate EZH2 binding to chromatin. The NIPP1-regulated phosphorylation of EZH2 is required for its recruitment to target genes that are implicated in proliferation and contributes to their silencing.
Using genome-wide DamID profiling in stably transfected HeLa cells, we found that EZH2-WT and EZH2-mTP5 are both associated with a large set of genes that have the hallmarks of Polycomb targets and show a significant overlap with previously reported lists of PRC2-bound genes. However, although about two-thirds of the identified EZH2-WT targets were also occupied by EZH2-mTP5, the remaining targets were largely unique to EZH2-WT. This defines a set of ∼1000 TP5-dependent target genes in HeLa cells for which the binding of EZH2 to the promoter region depends on the recruitment of NIPP1, resulting in the inhibition of PP1 and the phosphorylation of EZH2 by CDKs. This conclusion is consistent with previous reports showing that the CDK-mediated phosphorylation of EZH2 at TP3 is required for its association with chromatin and the non-coding RNAs HOTAIR and RepA (7,8). Interestingly, TP5-dependent target genes are enriched for functions related to proliferation. This fits in nicely with observations that the CDK-mediated phosphorylation of EZH2 is important for cell proliferation, as blocking the phosphorylation of EZH2 on TP3 mitigates cell proliferation (7), whereas the expression of a TP3/TP6 double alanine substitution mutant confers a proliferative disadvantage when compared with cells expressing WT EZH2 (6). This suggests that NIPP1 may affect the expression of proliferation-related genes by regulating the phosphorylation of EZH2. Consistent with a role for NIPP1 in cell proliferation, the siRNA-mediated depletion of NIPP1 in PC-3 (11) and HEK293T cells (14) significantly reduces cell proliferation, and this correlates with a reduced trimethylation at H3K27. Moreover, NIPP1−/− embryos display an early embryonic lethality that is correlated with impaired cell proliferation (14).
In conclusion, our data indicate that NIPP1 is part of a feed-forward loop that preserves the CDK-catalyzed phosphorylation of EZH2 and thereby contributes to the targeting of EZH2 to proliferation-related targets. We propose that the phospho-regulation of EZH2 by NIPP1 is an essential component of the dynamic integration of cell proliferation and EZH2-mediated gene silencing.
ACCESSION NUMBERS
Chemical shift assignments of the NIPP1 FHA domain were deposited in the Biological Magnetic Resonance Data Bank under accession number 18448. All DamID data are available at GEO under the accession number GSE39593.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–8 and Supplementary Reference [41].
FUNDING
The Fund for Scientific Research-Flanders [G.0478.08]; the Flemish Concerted Research Action [GOA 10/16]; the Prime Minister's office (IAP/P6-28P) and de Foundation against cancer (to M.B.). Additional support came from the National Institute of Neurological Disorders and Stroke [R01NS056128 to W.P.]; the National Institute of General Medicine [R01GM098482]; American Cancer Society Research Scholar [RSG-08-067-01-LIB to R.P.]. Funding for open access charge: The Flemish Concerted Research Action [GOA 10/16].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Annemie Hoogmartens, Scott Nichols, Nicole Sente and Fabienne Withof, who provided expert technical assistance. They also thank Stein Aerts, Rudy Van Eijsden and Gert Hulselmans for their help with the DamID analysis. N.M. is a research fellow of the FWO-Flanders.
REFERENCES
- 1.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer. 2009;9:773–784. doi: 10.1038/nrc2736. [DOI] [PubMed] [Google Scholar]
- 3.Sparmann A, van LM. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 4.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 5.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7:455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J. Biol. Chem. 2011;286:28511–28519. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell Biol. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Dessel N, Beke L, Gornemann J, Minnebo N, Beullens M, Tanuma N, Shima H, Van Eynde A, Bollen M. The phosphatase interactor NIPP1 regulates the occupancy of the histone methyltransferase EZH2 at Polycomb targets. Nucleic Acids Res. 2010;38:7500–7512. doi: 10.1093/nar/gkq643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nuytten M, Beke L, Van Eynde A, Ceulemans H, Beullens M, Van Humelen P, Fuks F, Bollen M. The transcriptional repressor NIPP1 is an essential player in EZH2-mediated gene silencing. Oncogene. 2008;27:1449–1460. doi: 10.1038/sj.onc.1210774. [DOI] [PubMed] [Google Scholar]
- 12.Roy N, Van Eynde A, Beke L, Nuytten M, Bollen M. The transcriptional repression by NIPP1 is mediated by Polycomb group proteins. Biochim. Biophys. Acta. 2007;1769:541–545. doi: 10.1016/j.bbaexp.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Jin Q, Van Eynde A, Beullens M, Roy N, Thiel G, Stalmans W, Bollen M. The protein phosphatase-1 (PP1) regulator, nuclear inhibitor of PP1 (NIPP1), interacts with the polycomb group protein, embryonic ectoderm development (EED), and functions as a transcriptional repressor. J. Biol. Chem. 2003;278:30677–30685. doi: 10.1074/jbc.M302273200. [DOI] [PubMed] [Google Scholar]
- 14.Van Eynde A, Nuytten M, Dewerchin M, Schoonjans L, Keppens S, Beullens M, Moons L, Carmeliet P, Stalmans W, Bollen M. The nuclear scaffold protein NIPP1 is essential for early embryonic development and cell proliferation. Mol. Cell Biol. 2004;24:5863–5874. doi: 10.1128/MCB.24.13.5863-5874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin-Granados C, Prescott AR, Van Dessel N, Van Eynde A, Arocena M, Klaska I, Goernemann J, Beullens M, Bollen M, et al. A role for PP1/NIPP1 in steering migration of human cancer cells. PLoS One. 2012;7:e40769. doi: 10.1371/journal.pone.0040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahimi A, Vande VG, Reumers V, Toelen J, Thiry I, Vandeputte C, Vets S, Deroose C, Bormans G, Baekelandt V, et al. Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Hum. Gene Ther. 2009;20:845–860. doi: 10.1089/hum.2008.188. [DOI] [PubMed] [Google Scholar]
- 17.Jagiello I, Van Eynde A, Vulsteke V, Beullens M, Boudrez A, Keppens S, Stalmans W, Bollen M. Nuclear and subnuclear targeting sequences of the protein phosphatase-1 regulator NIPP1. J. Cell Sci. 2000;113(Pt 21):3761–3768. doi: 10.1242/jcs.113.21.3761. [DOI] [PubMed] [Google Scholar]
- 18.Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, Cheung E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012;31:2810–2823. doi: 10.1038/emboj.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasini D, Bracken AP, Jensen MR, Lazzerini DE, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeGuzman A, Lee EY. Preparation of low-molecular-weight forms of rabbit muscle protein phosphatase. Methods Enzymol. 1988;159:356–368. doi: 10.1016/0076-6879(88)59036-5. [DOI] [PubMed] [Google Scholar]
- 21.Brown NR, Noble ME, Endicott JA, Johnson LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 22.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, III, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 25.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumeta H, Ogura K, Adachi S, Fujioka Y, Tanuma N, Kikuchi K, Inagaki F. The NMR structure of the NIPP1 FHA domain. J. Biomol. NMR. 2008;40:219–224. doi: 10.1007/s10858-008-9222-x. [DOI] [PubMed] [Google Scholar]
- 27.Boudrez A, Beullens M, Groenen P, Van Eynde A, Vulsteke V, Jagiello I, Murray M, Krainer AR, Stalmans W, Bollen M. NIPP1-mediated interaction of protein phosphatase-1 with CDC5L, a regulator of pre-mRNA splicing and mitotic entry. J. Biol. Chem. 2000;275:25411–25417. doi: 10.1074/jbc.M001676200. [DOI] [PubMed] [Google Scholar]
- 28.Qian J, Lesage B, Beullens M, Van Eynde A, Bollen M. PP1/Repo-man dephosphorylates mitotic histone H3 at T3 and regulates chromosomal aurora B targeting. Curr. Biol. 2011;21:766–773. doi: 10.1016/j.cub.2011.03.047. [DOI] [PubMed] [Google Scholar]
- 29.Vogel MJ, Peric-Hupkes D, van SB. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat. Protoc. 2007;2:1467–1478. doi: 10.1038/nprot.2007.148. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byeon IJ, Li H, Song H, Gronenborn AM, Tsai MD. Sequential phosphorylation and multisite interactions characterize specific target recognition by the FHA domain of Ki67. Nat. Struct. Mol. Biol. 2005;12:987–993. doi: 10.1038/nsmb1008. [DOI] [PubMed] [Google Scholar]
- 35.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell. 2008;32:503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mousavi K, Zare H, Wang AH, Sartorelli V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol. Cell. 2012;45:255–262. doi: 10.1016/j.molcel.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma HT, Poon RY. How protein kinases co-ordinate mitosis in animal cells. Biochem. J. 2011;435:17–31. doi: 10.1042/BJ20100284. [DOI] [PubMed] [Google Scholar]
- 38.Beullens M, Van Eynde A, Vulsteke V, Connor J, Shenolikar S, Stalmans W, Bollen M. Molecular determinants of nuclear protein phosphatase-1 regulation by NIPP-1. J. Biol. Chem. 1999;274:14053–14061. doi: 10.1074/jbc.274.20.14053. [DOI] [PubMed] [Google Scholar]
- 39.Beullens M, Vulsteke V, Van Eynde A, Jagiello I, Stalmans W, Bollen M. The C-terminus of NIPP1 (nuclear inhibitor of protein phosphatase-1) contains a novel binding site for protein phosphatase-1 that is controlled by tyrosine phosphorylation and RNA binding. Biochem. J. 2000;352(Pt 3):651–658. [PMC free article] [PubMed] [Google Scholar]
- 40.O'Connell N, Nichols SR, Heroes E, Beullens M, Bollen M, Peti W, Page R. The Molecular Basis for Substrate Specificity of the Nuclear NIPP1:PP1 Holoenzyme. Structure. 2012;20:1746–1756. doi: 10.1016/j.str.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.