


Abstract
Phosphorylation of the NF-κB subunit, p50, is necessary for cytotoxicity in response to DNA methylation damage. Here, we demonstrate that serine 329 phosphorylation regulates the interaction of p50 with specific NF-κB binding elements based on the identity of a single κB-site nucleotide. Specifically, S329 phosphorylation reduces the affinity of p50 for κB-sites that have a cytosine (C) at the −1 position without affecting binding to sequences with a −1 adenine. The differential interaction between phospho-p50 and the −1 base regulates the downstream transcriptional response and underlies the inhibition of anti-apoptotic gene expression following DNA damage. In genes with multiple κB-sites, the presence of a single −1C κB-site enables inhibition of NF-κB-dependent activity. The data suggest that interaction between phospho-p50 and the −1 κB nucleotide facilitates cytotoxicity in response to DNA damage. Moreover, although conservation of the entire κB-site sequence is not seen across species, the identity of the −1 nt in critical anti-apoptotic genes is conserved such that the overall response to DNA damage is maintained.
INTRODUCTION
DNA damage induces cytotoxicity through differential regulation of specific sets of downstream genes. This expressional profile is mediated by the interaction between transcription factors and their sequence-specific binding sites (1). Variations in the sequence of cis-regulatory elements and in the binding affinity or activity of trans-factors both contribute to the gene expression patterns that mediate the downstream response.
The nuclear factor-κB (NF-κB) family of proteins regulates the expression of >200 genes involved in pathways that mediate immunomodulation, cell survival and the response to DNA damage (www.bu.edu/nf-kb/). NF-κB is a dimeric transcription factor composed of five subunits unified by a conserved N-terminal Rel homology domain (RHD). While the prototypical NF-κB dimer is composed of p50 (NF-κB1, p105) and p65 (RelA), p52 (NF-κB2, p100), RelB, and cRel also contribute to the mature transcription factor. Under basal conditions, NF-κB dimers are retained in the cytoplasm through interaction with the inhibitor-κB (IκB) proteins. Stimulus-induced degradation of IκB proteins facilitates translocation of NF-κB dimers to the nucleus (2). While IκB degradation represents the primary control point for NF-κB activation, numerous post-translational modifications also modulate NF-κB activity (3).
In the nucleus, NF-κB dimers mediate their effect by binding to specific consensus sequences dispersed throughout the genome. The κB-site consists of a loosely conserved decameric series of nucleotides bearing the generic sequence 5′-G−5G−4G−3R−2N−1 W0 Y+1Y+2C+3C+4-3′ (where R represents a purine, N represents any nucleic acid, W represents an A or T and Y represents a pyrimidine) (4). Structural analyses of NF-κB dimers bound to a series of different κB-sites indicate that p50 and p52 bind to the 5′ nucleotides (5′-GGGRN), whereas p65, RelB and cRel bind preferentially to the 3′ nucleotides (YYCC-3′) (5). The importance of this cis-regulatory element is emphasized by the evolutionary conservation of the κB-site sequence (6). While differential NF-κB dimer binding to various κB-sites has been extensively documented (7,8), the impact of post-translational subunit modification on NF-κB recruitment to κB-sites is less well known.
We previously reported that SN1-type methylating agents such as temozolomide (TMZ) induce cytotoxicity through a signaling pathway involving p50 (9). Specifically, O6-methylguanine (O6-MeG):thymine (T) mismatches, formed in response to SN1-methylation, induce the checkpoint kinase, Chk1, to phosphorylate p50 at serine 329 (S329). This serine phosphorylation blocks DNA binding of p50-containing NF-κB dimers and attenuates transcription of NF-κB-regulated anti-apoptotic genes. Although p50 S329 phosphorylation inhibits NF-κB activity and gene expression, not all NF-κB-regulated genes are down-regulated in response to O6-MeG. Here, we show that phosphorylated p50 differentially interacts with κB-sites that vary only at the −1 nt. This cis-trans interaction regulates NF-κB DNA binding and transcriptional activity following DNA damage. Our results demonstrate that the relationship between phospho-p50 and the κB-site is evolutionarily conserved and that this interaction facilitates the cytotoxic response to DNA damage.
MATERIALS AND METHODS
Reagents, cells and recombinant protein expression
TMZ was obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute. U87 cells and 3T3 immortalized wt and p105−/− mouse embryonic fibroblasts (MEFs) were cultured and stable re-expression of p50 isoforms was performed as previously described (9). 6-Benzylguanine was obtained from Sigma Chemical Co and used in all experiments with MEFs (20 µM, 2 h pretreatment). Sh-control and sh-p105 U87 cells were also constructed as previously described (9). Bacterially expressed p50wt, p50S329A and p50S329D were purified as previously described (9).
Plasmids and luciferase assay
The luciferase reporter plasmid bearing two sequential copies of the κB-enhancer site was based off of the Igκ κB sequence, a kind gift from Dr Joseph Anrather (10). The KpnI site of pGL4.20 (Promega) was used to generate luciferase reporter constructs bearing the human NOD2 promoter (sense CGGGTACCGTGACAGTTTCACTGGAGC, antisense GGCTTTTGGCGTTCTGGTACCGC) and a region of the mouse Cox2 gene spanning both the promoter and intron 1 (sense AAGGTACCGGCGAGTGCCAGGGG, antisense TTGGTACCAAGCAGCCACTCTTGTTCAGTTGC). To create the chimeric construct used for clonogenic assays, the luciferase ORF was first removed from the pGL4.20-NOD2 promoter construct by restriction digest with HindIII and XbaI. Bcl-xL cDNA, including a 5′ Kozack sequence, was amplified (sense AAGCTTGGGCCACCATGTCTCAGAGCAACCGG, antisense TTGCGAATTCTTAAGCGACTGAAGAGTGAGCC) and then inserted into the modified pGL4.20 vector using the HindIII and XbaI sites. Dr Miguel Iñiguez kindly provided the human Cox2 luciferase promoter reporter construct (11) and the mouse Cox2 promoter reporter (−963/+70) was a generous gift from Dr Susan Fischer (12). The p105wt plasmid was purchased from Addgene (plasmid no. 23 288). For all mutations, the QuikChange Lightning Site Directed Mutagenesis Kit (Stratagene) was used. Luciferase transfection was normalized using Renilla reniformis as previously described (9) and data are representative of more than two independent experiments.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) was performed essentially as described following treatment and nuclear fraction isolation (9). All double-stranded 30-mer oligos contain a single central κB-site (Supplementary Table S1) with 10 flanking base pairs on either side or the Oct-1 consensus sequence for control EMSA (TGTCGAATGCAAATCACTAGAA). Briefly, reactions contain 10 mM Tris–HCl (pH 7.5), 1 mM MgCl2, 50 mM NaCl, 0.5 mM EDTA, 0.5 mM DTT, 4% glycerol, 50 ng/μl poly(dI•dC), 1–3 μl of protein fraction and 2–4 fmol of 32P-labeled probe. Reaction mixtures were incubated at room temperature for 20 min prior to resolving on a 5% polyacrylamide gel (0.5× TBE) at ∼10 V/cm for 2–4 h at 4°C. Supershift assays were performed using antibody cocktails specific to the indicated NF-κB subunits.
Competition binding analysis and Scatchard analysis
For competition binding experiments, serially diluted non-radioactive oligonucleotides were mixed with the 32P-labeled oligonucleotide probe prior to adding purified recombinant p50 for the binding reaction. Protein–DNA complexes were resolved by native gel electrophoresis and the amount of bound 32P-labeled oligonucleotide probe was quantified by PhosphorImager (Molecular Dynamics).
The Kd of purified p50 isoforms was determined by Scatchard analysis in a manner similar to that described previously (13,14). Briefly, a constant amount (1 fmol) of purified recombinant p50 was incubated with serially diluted 32P-labeled oligonucleotide probe (<2 fmol) in a 20-μl reaction volume. The bound and free probes were separated by native gel electrophoresis and were quantified by PhosphorImager (Molecular Dynamics). The reactions were performed in the absence of competitor DNA. All reactions were performed in triplicate.
Immunoblotting
The concentration of cell lysates was determined using the Bradford protein assay (Bio-Rad) and equal quantities of protein were loaded. Following separation by SDS–PAGE and transfer to PVDF membrane, immunoblots were performed with the indicated antibodies (Santa Cruz Biotechnology, Inc.). Blots were counterstained with Alexa Fluor 680 or Alexa Fluor 800 fluorescent dye-conjugated secondary antibodies (Invitrogen) and visualized with Odyssey Infrared system (LICOR Biosciences).
Quantitative real-time polymerase chain reaction and chromatin immunoprecipitation
U87 and MEF samples were processed using the RNeasy Mini Kit with DNase treatment (Qiagen). The ProtoScript M-MuLV Taq RT-PCR Kit (New England Biolabs) was used for reverse transcriptase reaction with poly-T primers. Transcripts were quantified using SYBR Green PCR (Bio-Rad) and normalized to GAPDH expression (mouse and human: sense CTTCACCACCATGGAGAAGGC, antisense GGCATGGACTGTGGTCATGAG). Primers included NOD2 (human: sense GCACGTGGCCTGAATGTTGG, antisense CCGCGGCAGTGATGTAGTTATTC; mouse: sense CGACATCTCCCACAGAGTTGTAATCC, antisense GGCACCTGAAGTTGACATTTTGC), CSF1 (human: sense GTCATATGTTGAGCCTGTGG, antisense GGCTACGGAGATGACAGAAT; mouse: sense CTCATGAGCAGGAGTATTGCCA, antisense TTTGACTGTCGATCAACTGCTG), Bax (human: sense CCAGGATGCGTCCACCAAGAAG, antisense GGAGTCCGTGTCCACGTCAGC), IFN-β (human: sense CCCAGTGCTGGAGAAATTGT, antisense CCCTATGGAGATGACGGAGA), Fas (human: sense GTCCAAAAGTGTTAATGCCCAAGT antisense ATGGGCTTTGTCTGTGTACTCC), TAP1 (human: sense TGGTCTGTTGACTCCCTTACAC, antisense AAATACCTGTGGCTCTTGTCC) and LMP2 (human: sense ATGCTGACTCGACAGCCTTT, antisense GCAATAGCGTCTGTGGTGAA). Data are representative of greater than or equal to three experiments, each in triplicate.
For quantitative ChIP, IP was performed from treated U87 or wt MEF cells with anti-p50, anti-GFP, anti-Histone H1 and IgG to control for non-specific binding. Promoter specific primers included the following: TAP1/LMP2 (human: sense GCAGGGAGAGGCGAGAAGGGTGTGC, antisense GGTGGGGCCTGAAGCTCCGGGTACC), mouse Cox2 Promoter (sense ACCGGTAGCTGTGTGCGTGC, antisense CAGTCGCGCATCCAGTGGGG), mouse Cox2 Intron (sense GCATCCTGCCAGCTCCACCG, antisense ACAGCCAGGCCCACACTGCT) and NOD2 (human: sense TAGTTCTGGAAGGCTGGT, antisense CCCATCAAAGCCCATTAG; mouse: sense GAGTTCCTGCACATTACCTTCCA, antisense AGAGGCCACCGATGTGTCAG). Amplification was performed as described earlier and quantification of the change in DNA enrichment for each IP condition was determined relative to input DNA as previously described (9).
Clonogenic assay
Cells were plated and allowed to attach overnight. After treatment with TMZ or vehicle, colony formation assay was performed as previously described (9). The surviving fraction was calculated based on the plating efficiency of untreated cells.
Statistical analyses
Statistical significance was performed using a two-tailed Student’s t-test.
RESULTS
The κB-site −1 nt regulates NF-κB binding in response to DNA damage
Inhibition of NF-κB-dependent transcription following methylation damage requires p50-mediated attenuation of DNA binding. Therefore, the interaction between p50 and the κB-site may play a critical role in the inhibition of gene expression. As the heterogeneity of the p50 half-site is primarily restricted to the −1 and −2 positions (Figure 1A), we hypothesized that this region of the κB-site is involved in the response to methylation damage. Using a systematic approach, we assessed binding of NF-κB to probes that vary in this region of the p50 half-site (Figure 1B, upper). While inhibition of NF-κB following TMZ treatment is seen in the presence of −1C or G, binding to −1A or T κB-sites is minimally affected by TMZ, findings that are independent of the −2 purine. Notably, the nucleotides at the +1 and +2 positions of the p65 half-site also have significant sequence variability. However, consistent with the observation that p65 is dispensable for inhibition of NF-κB by TMZ (9), changes to these positions do not influence NF-κB binding in response to DNA damage (Figure 1B, lower). TMZ also inhibits NF-κB binding to −1C sequences when κB probes from a variety of known NF-κB-regulated genes are examined, again without affecting binding to −1A-bearing probes (Figure 1C). Importantly, TMZ does not alter the nuclear level of p50, p105, p65 or crel nor does TMZ affect nuclear extract binding to an Oct-1 probe (Supplementary Figure S1A). Furthermore, supershift studies demonstrate that p50 is present in NF-κB complexes bound to −1A sequences at baseline and after treatment (Supplementary Figure S1B and S1C), indicating that the inability of TMZ to block NF-κB binding to −1A sequences is not due to the absence of p50 in the transcription factor.
Figure 1.

The κB-site −1 nt regulates NF-κB binding in response to DNA damage. (A) Alignment of human κB-sites, where R represents a purine, N represents any nucleic acid, W represents an A or T and Y represents a pyrimidine. (B) EMSA using oligo probes containing κB-sites that vary at the −1 and −2 positions (upper) or +1 and +2 positions (lower) as indicated. U87 glioma cells were treated with vehicle (U) or 100 µM TMZ (T) for 16 h, nuclear extract isolated and EMSA performed. Non-specific (N) and specific (S) competitors were used as shown. (C) EMSA as in (B) using κB-site probes corresponding to NF-κB-regulated genes. Probe sequences are indicated in (A). (D) Luciferase assay in U87 cells following treatment with vehicle or TMZ (16 h). Reporter constructs contain two identical tandem κB-sites that either have −1C or −1A bases (upper). Data represent mean κB-dependent luciferase expression relative to renilla and are normalized to control treatment, ±SD of triplicate samples. *P < 0.05.
We next sought to determine whether the −1 nt also influences NF-κB-dependent transcriptional activity following TMZ treatment. Because of the well-described prevalence of A and C at the −1 κB-site position (14), we restricted our analysis to consensus sequences bearing these bases. NF-κB-dependent transcription was assessed following induction of DNA damage using luciferase reporter constructs bearing two sequential κB-sites that vary only at the −1 position. While TMZ inhibits the transcriptional activity of a −1C-bearing reporter, TMZ has no effect on a −1A-bearing reporter (Figure 1D). In sum, these findings indicate that the −1 nt of the NF-κB binding site influences both DNA binding and transcriptional activity of NF-κB in response to DNA alkylation.
Phosphorylation of S329 attenuates p50 affinity for −1C-containing κB-sites
Consistent with the observation that inhibition of NF-κB binding is mediated by phosphorylation of p50 S329, a p50 phosphomimetic mutant, p50S329D, binds Igκ DNA less well than wild-type (wt) p50 (p50wt) (9). To determine if phospho-S329 p50 displays preferential binding for a specific sequence, competition studies were performed. Purified p50S329D or p50wt was incubated with radiolabeled −1A probe in the presence of increasing unlabeled −1A, −1C or non-specific competitor DNA. Binding of p50wt to the −1A probe is competed equally by −1A and −1C sequences, suggesting that unphosphorylated p50 does not preferentially bind one sequence over the other (Figure 2A, upper; Supplementary Figure S2). By contrast, binding of p50S329D to the −1A probe is only competed by −1A oligonucleotide, while competition by −1C DNA is only marginally better than non-specific DNA (Figure 2A, lower; Supplementary Figure S2).
Figure 2.

p50S329D has reduced binding affinity for −1C-bearing κB-sites. (A) Competition binding analysis of p50wt (upper) and p50S329D (lower). Quantitative EMSA was performed with 32P-labeled oligonucleotide probe containing the −1A site (GGGAATTTCC) and a constant amount of recombinant p50. Increasing concentrations of non-labeled −1A, −1C (GGGACTTTCC) or non-specific DNA was added as indicated (see ‘Materials and Methods’ section). The amount of bound probe is plotted as a percent of control samples without competitor DNA. Data represent mean value ± SD of three separate experiments. (B) Scatchard plot analysis of p50wt (upper) and p50S329D (lower) binding affinity for −1C and −1A sequences. Quantitative binding analysis was performed using various probe concentrations and a constant amount of recombinant p50 (see ‘Materials and Methods’ section). Samples were analyzed by EMSA.
Scatchard plots were next generated to examine the binding affinity of phosphomimetic p50. By this method, the dissociation constant of p50wt is similar for both −1A and −1C sites (Kd −1A = 4.4 × 10−12 M; Kd −1C = 5.8 × 10−12 M) (Figure 2B, upper). Notably, these affinities are comparable to previously reported values for NF-κB binding to other κB-sites (13). On the other hand, while p50S329D binds to −1A sites with a similar affinity to that of p50wt, the binding affinity for −1C sequence is almost 10-fold lower (Kd −1A = 4.2 × 10−12 M; Kd −1C = 38.4 × 10−12 M) (Figure 2B, lower). Together, these data suggest that phosphorylation at S329 alters p50 affinity for −1C but not −1A sites. Furthermore, as p50wt and p50S329D have similar affinity for −1A DNA, the data suggest that S329 phosphorylation has an inhibitory affect on NF-κB binding to −1C-containing binding sites.
S329 phosphorylation reduces NF-κB binding and transcription from promoters with −1C κB-sites
To assess NF-κB binding and transcriptional activity from endogenous promoters, we began by identifying genes with solitary κB-sites that vary only at the −1 nt. The bidirectional promoter for the neighbouring TAP1 and LMP2 genes has a single −1C-containing site identical to the Igκ κB-site (15). Conversely, the NOD2 κB-site differs from the TAP1/LMP2 site only by the presence of an A at the −1 position (Figure 1A) (16). Consistent with EMSA studies, quantitative ChIP assays demonstrate that TMZ attenuates recruitment of p50 to the TAP1/LMP2 promoter, but has no effect on p50 binding to the NOD2 promoter (Figure 3A and B).
Figure 3.

S329 phosphorylation reduces NF-κB binding to promoters with −1C-bearing κB-sites. (A–D) Quantitative ChIP assays using promoter specific primers for the TAP1/LMP2 or NOD2 promoters as indicated. IP was performed with anti-p50, anti-GFP or anti-histone H1 (positive control) as shown. Data represent promoter enrichment of p50, GFP or histone H1 relative to IgG control ± SEM of three separate experiments. (A and B) U87 cells were treated with 100 µM TMZ or vehicle for 16 h. (C and D) U87 cells were treated as in (A) following expression of the GFP-tagged p50 isoforms. *P < 0.05 relative to untreated p50wt samples. Inset: immunoblot with anti-p50 demonstrates equal expression of GFP-p50 mutants in stable clones. (E and F) Luciferase assays using a human NOD2 promoter/reporter construct with the indicated −1 nt mutation following treatment with TMZ for 16 h. (E) U87 cells were treated with the indicated TMZ concentrations. (F) p105−/− MEFs were co-transfected with either p50wt (wt) or p50S329A (S329A) as well as the indicated reporter construct and then treated 24 h later with vehicle or 100 µM TMZ. Data show mean relative luciferase, normalized to control conditions, ±SD of triplicate samples. *P < 0.05 relative to untreated. (G) Left: stable expression of Bcl-xL cDNA under the control of the wt (−1A) or mutant (−1C) NOD2 promoter in U87 glioma cells. Cells were treated with vehicle (U) or 100 µM TMZ (T) and IB performed with the indicated antibody. Right: colony formation assay in stable transfectants treated with TMZ. Cell lines include U87 cells expressing empty vector (U87) or Bcl-xL under the control of the wt (−1A) or the mutated (−1C) NOD2 promoter. Data show mean value from three independent experiments, each with three separate clones, ±SEM. *P<0.05.
Next, to examine the role of p50 S329 phosphorylation in the differential recruitment of NF-κB to −1C and −1A promoters, a series of stable cell lines expressing EGFP-tagged p50wt, p50S329D or a non-phosphorylateable S329A mutant (p50S329A), were constructed (Figure 3C, inset). While p50wt and p50S329A are recruited equally to the TAP1/LMP2 promoter at baseline, p50S329D is recruited substantially less to this promoter (Figure 3C). On the other hand, p50S329D is recruited to the same extent as the other p50 isoforms to the NOD2 promoter, which contains an A at the −1 position (Figure 3D). Furthermore, only recruitment of p50wt is inhibited by TMZ and specifically only to the TAP1/LMP2 promoter. These results support the hypothesis that inhibition of NF-κB binding in response to DNA damage is regulated by both the −1 nt and the phosphorylation status of p50 at S329.
To further examine the role of the −1 nt in the regulation of transcription from a promoter, luciferase constructs containing 1.4 kb of the human NOD2 promoter were fashioned (Figure 3E, upper). TMZ does not inhibit transcriptional activity from the wt NOD2 promoter (−1A), but does attenuate expression from a mutant construct containing a C at the −1 position (Figure 3E). Notably, the level of transcriptional activity from these two constructs is equivalent at baseline (data not shown). Next, the effect of TMZ on these reporter constructs was examined in cells stably expressing either p50wt or p50S329A. TMZ can only inhibit the transcriptional activity of the −1C-containing promoter reporter and only in cells expressing p50wt but not p50S329A (Figure 3F). These findings are consistent with the DNA binding data and suggest that the interaction between the −1 base and S329 regulates transcription in response to DNA damage.
The above data raise the question of whether the interaction between p50 and the −1 nt actually plays a functional role in the cytotoxic response to methylating agents. We previously demonstrated the importance of p50 to damage-induced cytotoxicity (9), determining the role of the −1 site ideally requires simultaneous ‘knock-in’ mutations at multiple promoters, an unfeasible undertaking. In order to examine the functional role of the −1 base, an exogenous system was chosen where the coding sequence of the anti-apoptotic protein, Bcl-xL, was expressed under the control of either a −1A or −1C promoter. As the promoter of Bcl-xL has more than one identified κB-site (17,18), the NOD2 promoter was used. Stable U87 cells expressing Bcl-xL constructs were made and clones with equal basal Bcl-xL expression selected. TMZ decreases Bcl-xL protein level in clones expressing the −1C-promoter construct or in control cells but does not affect Bcl-xL level in cells expressing the −1A-promoter construct (Figure 3G, left). Importantly, cells expressing the −1A-promoter construct have significantly more clonogenic survival following TMZ treatment than −1C-promoter expressing or control cell lines (Figure 3G, right). These results suggest that by controlling the expression of downstream genes, the κB-site −1 nt may play a functional role in regulating the cytotoxic response to DNA damage.
Phospho-p50 and the −1 nt facilitate differential gene expression in response to DNA damage
Binding and ChIP studies suggest that the interaction between p50 and the −1 base in the κB-site influences gene expression in response to DNA damage. In this regard, examination of endogenous mRNA demonstrates that TMZ inhibits expression of TAP1 and LMP2, genes with −1C-bearing κB-sites, but not NOD2, a −1A-bearing gene (Figure 4A). Similarly, expression of CSF1, another gene with a solitary reported −1C κB-site identical to that of TAP1 (19), is inhibited by TMZ. Furthermore, examination of genes with a range of κB-sites demonstrates that while TMZ inhibits anti-apoptotic genes that have −1C-bearing κB-sites, including Bcl-xL, Cox2 and Gadd45β (9), TMZ does not inhibit NF-κB-dependent genes that have −1A-bearing κB-sites, including Bax, IFNβ, IκBα and Fas (Figure 4A). Moreover, depletion of p105 attenuates inhibition of the genes containing −1C-bearing κB-sites, but does not do so for −1A-containing genes (Figure 4A).
Figure 4.
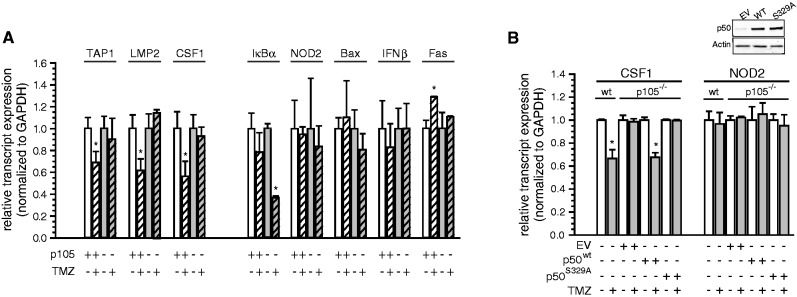
Differential expression of genes with −1C and −1A κB-sites following DNA damage. (A) qPCR of endogenous mRNA in sh-control and sh-p105 expressing U87 cells treated with 100 μM TMZ or vehicle (16 h). Data show relative mRNA expression normalized to control ± SEM of triplicate samples from three separate experiments. *P < 0.05. (B) qPCR analysis of CSF1 and NOD2 mRNA expression in wt MEFs and p105−/− MEF stable clones expressing empty vector (EV), p50wt or p50S329A following treatment with 100 μM TMZ or vehicle (16 h). Data show mean values normalized to control ± SEM of triplicate samples from three experiments. *P < 0.05. Inset: immunoblot with anti-p50 and anti-actin.
Next, to specifically investigate a link between S329 phosphorylation and differential gene expression, p105−/− MEFs stably expressing equal levels of either p50wt or p50S329A were constructed (Figure 4B, inset). As has been noted for other genes (6), κB-site sequences of CSF1 and NOD2 are conserved between the human and murine genomes (Supplementary Figure S1D). Consistent with this conservation, TMZ inhibits the expression of CSF1 but not NOD2 in wt-MEFs and loss of p105 blocks the down-regulation of CSF1, but not that of NOD2 (Figure 4B). Re-expression of p50wt but not p50S329A (or p105wt but not p105S329A—Supplementary Figure S3) in p105−/− MEFs restores inhibition of CSF1 transcription by TMZ but does not change expression of NOD2. Taken together, the above data indicate that DNA damage regulates the expression of endogenous NF-κB-dependent genes in a manner involving S329 phosphorylation and the −1 nt, and further, suggest that this pathway is conserved across species.
The −1 nt and κB-dependent transcription in the presence of >1 κB-site
Although some genes have a single reported κB-site, many NF-κB targets contain more than one NF-κB binding site. The human Cox2 promoter is reported to have two functional κB-sites, one of which contains a −1A base (−443) and the other has a −1C (−227) (Figure 5A, upper) (20). To examine a gene with multiple κB-sites, a luciferase reporter containing the human Cox2 promoter was obtained and transcriptional activity from this reporter assessed following mutation of the κB-sites (Figure 5A, upper). TMZ inhibits the activity of both the wt construct and a construct carrying an A to C mutation in the −443 κB-site (Figure 5A, lower). However, a C to A mutation in the −227 κB-site, which creates a construct with two −1A κB-sites, blocks the inhibitory effect of TMZ. These findings, coupled with our prior observation that TMZ down-regulates human Cox2 gene expression in a p50-dependent manner (9), suggest that the presence of a single −1C κB-site in a gene with multiple κB-sites is sufficient to mediate inhibition of gene expression following DNA damage.
Figure 5.

The role of the −1C base in the presence of >1 κB-site. (A, B and E) Luciferase reporter assays in U87 cells using the indicated Cox2 promoter/reporter after 16 h of treatment with TMZ at 0, 50, 100 and 250 μM. (A) Human Cox2 promoter reporter with the indicated −1 position mutation. (B) Murine Cox2 promoter (Pr) reporter. (C) Upper: sequences of the murine Cox2 promoter and intronic κB-sites. Lower: EMSA using the indicated κB probe following treatment with vehicle (U) or 100 μM TMZ (T). N and S: non-specific and specific competitors, respectively. *Non-specific bands. (D) Quantitative ChIP in wt-MEFs transfected with the indicated GFP-tagged p50 isoforms (wt, S329A and S329D) following 16-h treatment with 100 μM TMZ or vehicle. IP was performed with anti-GFP, anti-histone H1 or anti-IgG and qPCR performed using primers flanking the mouse Cox2 promoter (left) or intronic (right) κB-sites. Data represent promoter enrichment of GFP-p50 relative to IgG control and H1 ± SEM of three separate experiments. *P < 0.05 relative to vehicle treatment in p50wt expressing cells. (Inset: immunoblot of transfectants with anti-p50 antibody.) (E) Upper: murine Cox2 promoter/intron 1 (−730/+301) reporter with the indicated (C–A) mutation at the −1 position. Lower left: luciferase assay using either the murine Cox2 promoter (Pr) or promoter/intronic (Pr+In) reporter with the indicated mutation or κB-site ablation (ΔκB). Cells were either untreated or stimulated with 10 ng/ml TNFα. Lower right: Luciferase assay using the murine Pr+In reporter with the indicated −1 position mutation in the intronic (+77) κB-site. Luciferase data show mean value, normalized to value without TMZ, in all except (E) which is normalized to the untreated Pr value, ±SD of triplicate samples. *P < 0.05 relative to untreated.
Next, we examined murine Cox2 and noted that there is only a single reported κB-site in the promoter at −525 relative to the translational start site (21). Interestingly, this is a −1A-containing sequence (Figure 5C, upper) and consistent with our data, TMZ neither inhibits the activity of a reporter construct containing the murine Cox2 promoter (Figure 5B) nor blocks NF-κB binding to this sequence on EMSA (Figure 5C, lower left). Nevertheless, we previously noted that TMZ inhibits murine Cox2 mRNA expression (9). This inconsistency led us to search the mouse Cox2 sequence for other potential κB-sites and a putative consensus sequence was identified at +77 within the first intron (Figure 5C). EMSA demonstrates that NF-κB specifically binds to this putative intronic sequence and that binding is inhibited by TMZ (Figure 5C, lower right). Next, to examine p50 recruitment to this region of Cox2, quantitative ChIP was performed using primers specific to both the promoter and intronic region (Figure 5D). Consistent with the binding data, TMZ specifically inhibits recruitment of p50wt, but not p50S329A, to the intronic κB-site but not the −1A-bearing promoter site. Moreover, p50S329D has significantly reduced recruitment, compared to p50wt, to the intronic site (Figure 5D). Together these findings suggest that p50 binding to the intronic κB-site is blocked by TMZ in an S329-dependent manner.
To further test the functional relevance of this intronic κB-site, we constructed a luciferase reporter containing a contiguous DNA sequence that includes the proximal promoter and first intron of murine Cox2 (Figure 5E, upper). Luciferase expression from this reporter is greater than that of a construct carrying only the Cox2 promoter and the expression is enhanced by TNFα (Figure 5E, lower left). Furthermore, deletion of the intronic κB-site abolishes the response to TNFα. These findings suggest that the intronic NF-κB consensus sequence is a functional κB-site. Most importantly, while TMZ inhibits transcription from the promoter/intronic reporter, a C to A mutation at the −1 position in the intronic κB-site abrogates the inhibitory effect of TMZ (Figure 5E, lower right). These data indicate that the −1C base in the intronic κB-site is important for inhibition of murine Cox2 expression by TMZ and highlight the functional conservation of the κB-dependent response following DNA damage.
DISCUSSION
It has been proposed that κB-sites enable differential gene regulation by acting as allosteric regulators of NF-κB and restricting the conformation of bound dimers (22,23). Consistent with this hypothesis, structural studies indicate that NF-κB dimers adopt unique conformations when bound to κB-sites that minimally differ in their nucleotide sequence (13,24,25). While studies of unmodified NF-κB dimers indicate that binding affinities are similar across a range of κB-sites (13,25), we demonstrate that S329 phosphorylation reduces the affinity of p50 for κB-sites with −1C nt. In considering potential mechanisms for this attenuated affinity, it is notable that G:C-rich DNA is less flexible than A:T-containing stretches. The rigidity that potentially results from an additional G:C base pair at the −1 position may contribute to differential NF-κB binding in response to DNA damage. Although S329 is located on the outer surface of p50 far from both the dimerization and DNA contact regions (5,26), the addition of a negative charge following phosphorylation may induce a conformation change that reduces dimer affinity for −1C κB-sites. Phosphorylation may also affect the interaction of p50 with cofactors that can alter binding affinity and refine the downstream expression profile. In this regard, single nucleotide changes in the κB-site have previously been reported to be critical in regulating co-activator recruitment and gene expression (6). Interestingly, while the presence of −1C enables inhibition of κB-dependent transcription, −1G but not −1T, also allows inhibition of binding (Figure 1B). Although our work focuses on −1C and −1A because of their predominant expression in κB-sites (14), it is likely that the presence of a −1G or −1T also contributes to the overall transcriptional response.
Many NF-κB-dependent genes contain >1 κB-site and the cooperative interaction between two κB-sites has been previously reported (6). One hypothesis for the regulation of transcription factor binding by multiple κB-sites is that one site imposes a dominant effect over others. This observation is consistent with our data in that a −1C-bearing κB-site enables inhibition of NF-κB regardless of the identity of the −1 nt in the other κB-site. In examining genes with two κB-sites, we focused on the Cox2 locus and identified a novel κB-site in the first intron of the mouse gene. In general, NF-κB binding in the first intron is relatively common (27). Despite the evolutionary conservation of κB-site sequences, it is apparent from the cox2 gene that this is not a universal finding. While sequence conservation between human and mouse genomes is seen at the first Cox2 κB-site, the murine intronic κB-site differs at 2 nt from the human analog. Nevertheless, the intronic κB-site retains the −1C base that enables the inhibition of NF-κB-dependent transcription following DNA damage. Similarly, although human and murine Bcl-xL κB-sites are not 100% identical (17,18,28) (Figure 1A and Supplementary Figure S1D), a −1C base is retained in both species, a finding consistent with the observation that Bcl-xL expression is inhibited in both human and mouse cells following DNA damage (9). Taken together, these data suggest that although the entire κB-site sequence is not strictly conserved, in specific anti-apoptotic genes, the critical −1 nt is maintained and enables p50-dependent inhibition in response to DNA damage. Interestingly, it is notable that κB-sites found in several pro-apoptotic genes such as Fas, Bax and IκBα lack −1C bases and preferentially contain −1A nt. These genes are not inhibited in response to damage (Figure 4A). While our studies emphasize −1C and its role in enabling inhibition of anti-apoptotic genes, cytotoxicity following DNA damage likely results from a concerted downstream response that also includes up-regulation or lack of inhibition, of pro-apoptotic factors, an observation that further supports the critical role of the −1 base in the damage response.
Although the interaction between post-translationally modified NF-κB subunits and κB-elements has not been robustly investigated, it was noted in one study that p65 RHD mutants interact in a variable way with different κB-sites (10). However, a specific functional role for the differential interaction of these mutants was not noted. Also, while p65 phosphorylation can induce differential gene expression (29), a specific interaction with individual κB-sites is not apparent. Our work focuses on p50 because of its central role in methylation-induced cytotoxicity (9). While p50 S329 phosphorylation is not sufficient to induce cell death, this modification facilitates killing by ‘priming’ cells for DNA strand break-induced cytotoxicity. We propose that the cis-trans interaction between the −1 nt of the κB-site and phospho-p50 is functional in that it enables regulation of specific NF-κB-dependent genes that facilitates cytotoxicity. This response not only mediates chemotherapy-induced killing but also is important following intrinsic and environmental damage. By enabling removal of damaged cells, this cytotoxic pathway maintains genomic stability, an observation that underlines the importance of the evolutionary conservation of the −1 nt.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–3.
FUNDING
National Institutes of Health (NIH) [1R01CA136937 to B.Y.]; and the Ludwig Center for Metastasis Research. Funding for open access charge: NIH [1R01CA136937 to B.Y.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Miguel A Iñiguez, Susan Fischer and Josef Anrather for plasmid constructs.
REFERENCES
- 1.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 5.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 6.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Kunsch C, Ruben SM, Rosen CA. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell. Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitt AM, Crawley CD, Kang S, Raleigh DR, Yu X, Wahlstrom JS, Voce DJ, Darga TE, Weichselbaum RR, Yamini B. p50 (NF-kappaB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol. Cell. 44:785–796. doi: 10.1016/j.molcel.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anrather J, Racchumi G, Iadecola C. cis-acting, element-specific transcriptional activity of differentially phosphorylated nuclear factor-kappa B. J. Biol. Chem. 2005;280:244–252. doi: 10.1074/jbc.M409344200. [DOI] [PubMed] [Google Scholar]
- 11.Iniguez MA, Martinez-Martinez S, Punzon C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J. Biol. Chem. 2000;275:23627–23635. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Fischer SM. Transcriptional regulation of cyclooxygenase-2 in mouse skin carcinoma cells. Regulatory role of CCAAT/enhancer-binding proteins in the differential expression of cyclooxygenase-2 in normal and neoplastic tissues. J. Biol. Chem. 1998;273:27686–27694. doi: 10.1074/jbc.273.42.27686. [DOI] [PubMed] [Google Scholar]
- 13.Fujita T, Nolan GP, Ghosh S, Baltimore D. Independent modes of transcriptional activation by the p50 and p65 subunits of NF-kappa B. Genes Dev. 1992;6:775–787. doi: 10.1101/gad.6.5.775. [DOI] [PubMed] [Google Scholar]
- 14.Zabel U, Schreck R, Baeuerle PA. DNA binding of purified transcription factor NF-kappa B. Affinity, specificity, Zn2+ dependence, and differential half-site recognition. J. Biol. Chem. 1991;266:252–260. [PubMed] [Google Scholar]
- 15.Wright KL, White LC, Kelly A, Beck S, Trowsdale J, Ting JP. Coordinate regulation of the human TAP1 and LMP2 genes from a shared bidirectional promoter. J. Exp. Med. 1995;181:1459–1471. doi: 10.1084/jem.181.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, Nunez G, Fernandez-Luna JL. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J. Biol. Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol. Cell. Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong QG, Sclabas GM, Fujioka S, Schmidt C, Peng B, Wu T, Tsao MS, Evans DB, Abbruzzese JL, McDonnell TJ, et al. The function of multiple IkappaB: NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene. 2002;21:6510–6519. doi: 10.1038/sj.onc.1205848. [DOI] [PubMed] [Google Scholar]
- 19.Harrington MA, Edenberg HJ, Saxman S, Pedigo LM, Daub R, Broxmeyer HE. Cloning and characterization of the murine promoter for the colony-stimulating factor-1-encoding gene. Gene. 1991;102:165–170. doi: 10.1016/0378-1119(91)90074-l. [DOI] [PubMed] [Google Scholar]
- 20.Tazawa R, Xu XM, Wu KK, Wang LH. Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem. Biophys. Res. Commun. 1994;203:190–199. doi: 10.1006/bbrc.1994.2167. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J. Biol. Chem. 1995;270:31315–31320. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]
- 22.Lefstin JA, Yamamoto KR. Allosteric effects of DNA on transcriptional regulators. Nature. 1998;392:885–888. doi: 10.1038/31860. [DOI] [PubMed] [Google Scholar]
- 23.Natoli G. Little things that count in transcriptional regulation. Cell. 2004;118:406–408. doi: 10.1016/j.cell.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Berkowitz B, Huang DB, Chen-Park FE, Sigler PB, Ghosh G. The x-ray crystal structure of the NF-kappa B p50.p65 heterodimer bound to the interferon beta-kappa B site. J. Biol. Chem. 2002;277:24694–24700. doi: 10.1074/jbc.M200006200. [DOI] [PubMed] [Google Scholar]
- 25.Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G. The kappa B DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J. Biol. Chem. 2002;277:24701–24708. doi: 10.1074/jbc.M200007200. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature. 1995;373:303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- 27.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl Acad. Sci. USA. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsukahara T, Kannagi M, Ohashi T, Kato H, Arai M, Nunez G, Iwanaga Y, Yamamoto N, Ohtani K, Nakamura M, et al. Induction of Bcl-x(L) expression by human T-cell leukemia virus type 1 Tax through NF-kappaB in apoptosis-resistant T-cell transfectants with Tax. J. Virol. 1999;73:7981–7987. doi: 10.1128/jvi.73.10.7981-7987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell. Biol. 2008;28:3623–3638. doi: 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.