

Abstract
Animal miRNAs silence the expression of mRNA targets through translational repression, deadenylation and subsequent mRNA degradation. Silencing requires association of miRNAs with an Argonaute protein and a GW182 family protein. In turn, GW182 proteins interact with poly(A)-binding protein (PABP) and the PAN2–PAN3 and CCR4–NOT deadenylase complexes. These interactions are required for the deadenylation and decay of miRNA targets. Recent studies have indicated that miRNAs repress translation before inducing target deadenylation and decay; however, whether translational repression and deadenylation are coupled or represent independent repressive mechanisms is unclear. Another remaining question is whether translational repression also requires GW182 proteins to interact with both PABP and deadenylases. To address these questions, we characterized the interaction of Drosophila melanogaster GW182 with deadenylases and defined the minimal requirements for a functional GW182 protein. Functional assays in D. melanogaster and human cells indicate that miRNA-mediated translational repression and degradation are mechanistically linked and are triggered through the interactions of GW182 proteins with PABP and deadenylases.
INTRODUCTION
miRNAs belong to a large family of non-coding RNAs that post-transcriptionally silence the expression of mRNAs containing fully or partially complementary binding sites. To exert their regulatory functions, miRNAs assemble into miRNA-induced silencing complexes (miRISCs), minimally comprising an Argonaute protein (AGO) and a protein of the GW182 family (1,2). GW182 proteins function downstream of AGOs and play an essential role in miRNA-mediated gene silencing in animal cells (1,2).
Three GW182 paralog proteins (termed TNRC6A, B and C) exist in vertebrates and various invertebrate species; however, only one family member exists in Drosophila melanogaster [GW182 (1,2)]. These proteins typically contain an N-terminal (N-term) Argonaute-binding domain (ABD) and a C-terminal (C-term) silencing domain (SD) [Figure 1 (1,2)]. The SDs of the human proteins are required for silencing and serve as binding platforms for the cytoplasmic poly(A)-binding protein (PABP), as well as PAN3 and NOT1, which are subunits of the PAN2–PAN3 and CCR4–NOT deadenylase complexes, respectively (3–10).
Figure 1.

Domain organization of Drosophila melanogaster GW182, Hs TNRC6C and the corresponding chimeric proteins. ABD, AGO-binding domain; ABD2, AGO-binding domain from Caenorhabditis elegans AIN-2; NED, N-terminal effector domain; UBA, ubiquitin associated-like domain; QQQ, region rich in glutamine; Mid, middle region containing the PAM2 motif (dark blue), which divides the Mid region into the M1 and M2 regions; RRM, RNA recognition motif; C-term, C-terminal region; SD, silencing domain. The position of the conserved CIM-1, CIM-2 and P-GL motifs are indicated. Amino acid positions at domain boundaries are indicated below the protein outlines. Vertical red lines indicate the positions of GW repeats. Vertical green lines indicate the positions of tryptophan residues in the M2 region that are involved in NOT1-binding (9). Sequence alignments of the PAM2, CIM-1, CIM-2 and P-GL motifs and the amino acids mutated in this study are shown in Supplementary Figure S7.
The SD is bipartite and comprises the middle (Mid) and C-term regions of the GW182 proteins that flank an RNA-recognition motif (RRM). The Mid region is further divided into the M1 and M2 regions (Figure 1), which, together with the C-term, contribute to the interactions with deadenylases in an additive manner (8–10). For example, the interaction between human TNRC6 SDs and PAN3 requires both the M2 and C-term regions of the SD (8,9). NOT1 binding is mediated through tryptophan-containing sequences in the M1, M2 and C-term regions of the SD [Figure 1 (9,10)]. The motifs in the M1 and C-term regions were termed CCR4–NOT-interacting motifs 1 and 2 (CIM-1 and CIM-2), respectively [Figure 1 (10)]. However, in addition to the CIM-1 and CIM-2 motifs, tryptophan residues in the M2 region of the SD contribute to interactions with NOT1 and PAN3 (9). Finally, PABP binds directly to a conserved PAM2 motif (PABP-interacting motif 2) located between the M1 and M2 regions of the SD (Figure 1 (3–7)).
Remarkably, although the interactions between GW182 proteins and PABP and deadenylase complexes are conserved in D. melanogaster, the mode of interaction differs (5,8). For example, the CIM-2 motif is absent in D. melanogaster GW182 (9,10). Moreover, in contrast to the human SDs, which are necessary and sufficient for NOT1 and PAN3 binding, the deletion of the SD from D. melanogaster GW182 reduces but does not abolish binding to deadenylases (8), indicating that sequences upstream of the SD contribute to these interactions (8,9). Finally, in contrast to the human proteins, D. melanogaster GW182 also indirectly interacts with PABP through the M2 and C-term regions in cultured cells (4,5). Consequently, the D. melanogaster GW182 PAM2 motif is dispensable for PABP binding and silencing in Drosophila cells (5,9,11,12).
The interactions between GW182 proteins and deadenylase complexes are required for miRNA target deadenylation and degradation (8–10). Whether these interactions are also required for miRNA-mediated translational repression remains unclear. Three lines of evidence support a role for the CCR4–NOT deadenylase complex in translational repression of miRNA targets. First, the direct tethering of subunits of the CCR4–NOT complex to mRNA reporters lacking poly(A) tails represses translation in the absence of deadenylation (9,13). Second, depletion of subunits of the CCR4–NOT complex partially suppresses the silencing of mRNA reporters that lack a poly(A) tail; these reporters are silenced at the translational level without undergoing deadenylation (8,9). Third, mutations or deletions in GW182 proteins that disrupt the interactions with the CCR4–NOT complex suppress silencing, that is, translational repression and degradation of miRNA targets (8,9). However, other studies reported that although the depletion of CCR4–NOT complex subunits abolished the deadenylation and degradation of miRNA reporters, some reporters remained translationally repressed, suggesting that an additional mechanism could contribute to the repression (8,14,15). In addition, several studies have indicated that translational repression precedes deadenylation (3,15–19), although it remains unclear whether these two modes of regulation are linked or whether they represent independent mechanisms that are used by miRNAs to silence their mRNA targets.
In this study, we investigated whether the interactions of GW182 proteins with PABP and deadenylase complexes are also required for the translational repression of miRNA targets and thus, for target silencing, independently of the extent of target mRNA degradation. Accordingly, we first identified the regions in D. melanogaster GW182 that are required for deadenylase complex binding. We subsequently generated mutants that are unable to interact with PABP and deadenylases and demonstrated that these mutants cannot rescue silencing in cells depleted of endogenous D. melanogaster GW182. These observations, when combined with the study of engineered minimal functional GW182 proteins, indicate that translational repression and target degradation are mechanistically linked and depend on the interaction of GW182 proteins with PABP and deadenylase complexes in both D. melanogaster and human cells.
MATERIALS AND METHODS
Plasmids
Luciferase reporters and plasmids for the expression of miRNAs, AGO1, GW182, PABP and subunits of the two deadenylase complexes studied in this article have been described previously (8,14,20).
Co-immunoprecipitation analyses and western blotting in S2 cells
S2 cells were transfected in six-well plates using Effectene transfection reagent (Qiagen). For co-immunoprecipitation assays, the transfection mixtures contained 1 µg of plasmid-expressing Green fluorescent Protein (GFP)-tagged GW182 or the corresponding mutants and 0.5 µg of human influenza hemagglutinin (HA)-tagged deadenylation subunits. Co-immunoprecipitations and western blots were performed as described by Braun et al. (8), except that cell lysates were supplemented with CaCl2 and treated with micrococcal nuclease before immunoprecipitation. HA- and GFP-tagged proteins were detected using horseradish peroxidase–conjugated monoclonal anti-HA (Roche 3F10; 1:5000) and anti-GFP antibodies (Roche 11814460001; 1:2000), respectively. V5-tagged proteins were detected using anti-V5 antibodies (Invitrogen, 1:5000). Endogenous AGO1 and α-tubulin were detected using commercial antibodies at the following dilutions: D. melanogaster AGO1 (Abcam ab5070; 1:1000) and α-tubulin (Sigma T6199; 1:2000). Endogenous D. melanogaster GW182 was detected with a rat polyclonal antibody prepared in our laboratory (14). All western blots were developed using the ECL western blotting detection system (GE Healthcare) as recommended by the manufacturer.
Complementation assays in S2 cells
Complementation assays were performed as described previously (5). For miRNA-mediated silencing assays, the transfection mixtures contained 0.1 µg of firefly luciferase reporter plasmid, 0.4 µg of a Renilla transfection control and 0.1 µg of plasmids expressing miRNA primary transcripts or the corresponding vector without insert. Unless otherwise indicated, 20 ng (Figure 3) or 100 ng (all other Figures) of plasmids expressing recombinant proteins were cotransfected. Firefly and Renilla luciferase activities were measured 3 days after transfection using the Dual-Luciferase Reporter Assay System (Promega). Total RNA was isolated using TriFast (Peqlab Biotechnologies) and analyzed as described previously (21).
Figure 3.

The Drosophila melanogaster GW182 SD is generally required for miRNA-mediated translational repression and target degradation. (A–I) Control S2 cells (treated with glutathion S-transferase (GST) dsRNA) or cells depleted of endogenous GW182 were transfected with a mixture of three plasmids: one expressing the indicated F-Luc reporters; a second expressing miRNA primary transcripts or the corresponding empty vector (−) and a third expressing Renilla luciferase (R-Luc). Plasmids encoding HA-GW182 (wild-type or deletion mutants) or HA-MBP (negative control) were included in the transfection mixtures as indicated. For each condition, firefly luciferase activities and mRNA levels were normalized to those of the Renilla luciferase transfection control and set at 100% in cells transfected with the empty vector (i.e. in the absence of the miRNAs). (A and E) Normalized firefly luciferase activities and mRNA levels in the absence or presence of miRNAs in control cells (i.e. cells treated with GFP dsRNA and transfected with a plasmid expressing MBP). (B and F) Northern blot analysis of representative RNA samples. Numbers in italics below the panels indicate the levels of the F-Luc reporters normalized to that of R-Luc mRNA and set at 100 in the absence of the miRNAs. (C and G) Relative derepression of F-Luc activity for each condition. (D and H) Relative F-Luc mRNA levels. Throughout this study, error bars represent standard deviations from at least three independent experiments. Upper and lower dashed lines indicate maximal derepression and repression, respectively, observed in depleted cells. (I) A western blot showing that GW182 mutants were expressed at levels equivalent to that of the wild-type protein.
Immunoprecipitation analyses and luciferase assays in human cells
The R-Luc-3xlet-7 reporter and the corresponding R-Luc-Mut have been described previously (22). The R-Luc-Hmga2 wild-type and the R-Luc-Hmga2 m7 mutant described by Mayr et al. (23) were subcloned into the pCI-neo (Promega) expression vector. Immunoprecipitation assays were performed in human cells as described by Braun et al. (8). GFP- and HA-tagged proteins and endogenous α-tubulin were detected as described above. Endogenous PABP and TNRC6A were detected using commercial antibodies at the following dilutions: PABP (Abcam ab21060; 1:10 000) and human TNRC6A (Bethyl A302-329A; 1:2000).
For luciferase assays, human HeLa cells were seeded in six-well plates and transfected using Lipofectamine 2000 (Life technologies). The transfection mixtures contained 0.05 μg of R-Luc-3xlet-7 or 0.2 µg R-Luc-HMG2a reporter plasmids, or the corresponding reporters carrying mutations in the let-7 binding sites (R-Luc-Mut and R-Luc-Hmga2-mut7), and 0.3 μg of the pEGFP-N3-F-Luc transfection control. In the overexpression experiment described in Figure 9, the transfection mixtures contained in addition 1.5 μg of plasmids expressing GFP-CNOT1 fragments or the GFP-CNOT7 catalytically inactive mutant. The CNOT7 mutant carries alanine substitutions of the catalytic residues D40 and E42. A plasmid expressing GFP-tagged maltose binding protein (GFP-MBP) served as a negative control. R-Luc and F-Luc activities were measured 48 h after transfection using the Dual-Luciferase Reporter Assay System (Promega). Complementation assays were performed as described previously (5). The following siRNAs were used: TNRC6A 5′-GCCUAAUCUCCGUGCUCAATT-3′; TNRC6B 5′-GGCCUUGUAUUGCCAGCAATT-3′ and β-Gal 5′-CUACACAAAUCAGCGAUUUUU-3′ (Dharmacon).
Figure 9.

Overexpression of the CNOT1-M domain inhibits silencing in a dominant negative manner. (A) Domain organization of human CNOT1. CNOT1-N, N-terminal domain; CNOT1-M, middle domain containing the MIF4G domain (31); CNOT1-C, C-terminal domain containing the NOT homology domain. Amino acid positions at domain boundaries are indicated below the protein outlines and correspond to the NCBI protein sequence NP_057368.3. (B) Interaction of GFP-tagged human CNOT1 (wild-type or fragments) and human HA-TNRC6A. Inputs (1%) and immunoprecipitates (5% for CNOT1 or 10% for TNRC6A) were analyzed by western blotting. F-Luc-GFP served as a negative control. (C and D) Human cells were transfected with the indicated Let-7 reporters or the corresponding controls (Mut) as described in Figure 8. The transfection mixtures also contained plasmids encoding GFP-CNOT1-M and GFP-CNOT7 (catalytically inactive mutant, D40A+E42A), as indicated. A plasmid encoding GFP-MBP served as a negative control. For each condition, R-Luc activities were normalized to that of a F-Luc transfection control and set at 100% for the Mut reporters (gray bars, shown only for control cells). Error bars represent standard deviations from three independent experiments.
Reverse transcription and quantitative real-time polymerase chain reaction were performed as described by Zekri et al. (4) using the following oligos: R-Luc forward (5′-ACTTCGAAAGTTTATGATCC-3′), R-Luc reverse (5′-TGTTCATTTTTGAGAACTCG-3′), F-Luc forward (5′-GGTGAGCAAGGGCGAGGAGC-3′) and F-Luc reverse (5′-CGCCGGACACGCTGAACTTG-3′).
GST pull-down assays
A cDNA encoding the SD of TNRC6A was cloned into the BamH1–Not1 restriction sites of plasmid pGEX6P (GE Healthcare) and expressed in Escherichia coli as an N-term GST-fusion. Mutations were introduced using the QuikChange mutagenesis kit (Stratagene) and the appropriate oligonucleotides. For the GST pull-down assays shown in Figure 8, lysates from E. coli cells expressing GST, GST-TNRC6A-SD or the indicated mutants were incubated with 40 μl of Protino Glutathione Agarose 4B beads (Macherey Nagel; 50% slurry) in lysis buffer (10 mM Hepes [pH 7.5], 300 mM NaCl and 1 mM DTT) for 1 h at 4°C. The beads were washed three times using 1 ml of lysis buffer each time. The pre-coated beads were then incubated with lysates from HEK293T cells (ca. 2 × 106 cells/pull down) expressing CNOT1 in a total volume of 1 ml of NET buffer (10 mM Hepes [pH 7.5], 150 mM NaCl, 1 mM EDTA and 1% [v/v] Triton-X100) for 1 h at 4°C. The beads were washed three times using 1 ml of NET buffer each time. Proteins were eluted with 40 µl of sample buffer and separated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Figure 8.

Complementation assay in human cells. (A–C) Control HeLa cells (transfected with β-Gal siRNA) or cells codepleted of TNRC6A and TNRC6B were transfected with a mixture of three plasmids: the R-Luc-3xlet-7 or the corresponding reporter carrying mutations in let-7–binding sites (R-Luc-Mut), a plasmid expressing F-Luc as a transfection control, and a plasmid expressing GFP or siRNA-resistant versions of HA-TNRC6A (wild-type or mutant). For each condition, Renilla luciferase activity was measured, normalized to that of the F-Luc transfection control and set at 100% in cells expressing R-Luc-Mut. (A) Normalized Renilla luciferase activities in control cells (i.e. cells treated with β-Gal siRNA and expressing GFP). (B) Relative fold derepression for each condition. Mean values ± standard deviations are shown. (C) Western blot showing the efficiency of the TNRC6A knockdown. Dilutions of control cell lysates were loaded in lanes 1–4 to estimate the efficacy of the depletion. α-tubulin served as a loading control. (D) Interaction of GST-tagged TNRC6A-SD (wild-type or mutants) with CNOT1 and endogenous PABP. Inputs (1%) and bound fractions (40%) were analyzed by western blotting. (E) Western blot analysis showing that all proteins were expressed at comparable levels.
RESULTS
Drosophila melanogaster GW182 interacts with NOT1 through an extended silencing domain
The conserved core of the D. melanogaster CCR4–NOT complex consists of NOT1, NOT2, NOT3 and two catalytic subunits, CCR4 and POP2 (24). Of these, NOT1 and NOT2 are efficiently co-immunoprecipitate with D. melanogaster GW182 from Schneider cell (S2 cells) lysates (8,9). In addition, similar to human TNRC6 proteins, D. melanogaster GW182 interacts with PAN3 (8,9). In contrast to the human proteins, however, the deletion of the SD from D. melanogaster GW182 does not prevent interactions with deadenylation factors, indicating that sequences upstream of the SD also contribute to binding (8,9). In agreement with these observations, an N-term region of D. melanogaster GW182 (containing the N-term effector domain [NED]; Figure 1) has been shown to bind NOT1 and possess silencing activity (9,11,25).
To precisely define the regions of D. melanogaster GW182 involved in deadenylase binding, we performed co-immunoprecipitation experiments in S2 cells using a series of GW182 deletion mutants. First, we confirmed that deletion of the GW182 SD (ΔSD) reduced but did not abolish D. melanogaster GW182 binding to NOT1, whereas the binding to NOT2 and PAN3 was unaffected (Figure 2A–C, lanes 8). As expected, deletion of the SD did not prevent binding to AGO1 (which is mediated through the ABD); however, PABP binding was abrogated [Figure 2D and E, lanes 8 (4,5,12)].
Figure 2.

Interaction of Drosophila melanogaster GW182 with NOT1, NOT2 and PAN3. (A–G) S2 cells were cotransfected with plasmids expressing GFP-tagged D. melanogaster GW182 (wild-type or mutants) and HA-tagged deadenylase subunits or V5-tagged PABP as indicated. Cell lysates were immunoprecipitated using a polyclonal anti-GFP antibody. GFP-tagged firefly luciferase served as a negative control. Inputs (1%) and immunoprecipitates (5% for GFP-tagged proteins or 40% for HA- or V5-tagged proteins) were analyzed by western blotting using the corresponding antibodies. In all panels, cell lysates were treated with micrococcal nuclease before immunoprecipitation. The presence of endogenous AGO1 in the immunoprecipitates was determined using a specific anti-AGO1 antibody (E).
Notably, although the SD is sufficient for PABP binding (Figure 2D, lane 9 (4,5)), in isolation, this domain interacted with NOT1 much less efficiently than full-length D. melanogaster GW182 and did not interact with NOT2 and PAN3 (Figures 2A–C, lanes 9). These results indicate that sequences upstream of the SD contribute to the interaction between GW182 and deadenylases. Furthermore, these results also indicate that deadenylases and PABP can independently interact with D. melanogaster GW182. The interactions with deadenylases are also independent of AGO1, because a D. melanogaster GW182 mutant that does not interact with AGO1, that is, in which all 12 N-term GW-repeats are mutated to alanines (12xGW mutant; Figure 2E, lane 10), interacted with NOT1, NOT2 and PAN3 as efficiently as wild-type D. melanogaster GW182 (Figure 2A–C, lanes 10).
To further delineate the regions of D. melanogaster GW182 required for NOT1 deadenylase binding, we extended the SD to include additional N-term sequences of increasing length (Figure 2F). We observed that full binding activity required fragment 713–1384 or the combination of the entire Q-rich region and the SD (Q+SD; i.e. 635–1384) (Figure 2F and G). Conversely, a D. melanogaster GW182 protein lacking both the Q-rich region and the SD (ΔQ+SD) did not interact with NOT1 (Figure 2G, lane 7). Surprisingly, PAN3 interacted with these two non-overlapping protein fragments, whereas NOT2 interacted primarily with the Q+SD region (Supplementary Figure S1A and B, lanes 7 and 8).
We conclude that the interaction of D. melanogaster GW182 with NOT1 is mediated by the Q-rich region together with the SD. Another important conclusion from these results is that PAN3 can interact with the D. melanogaster GW182 fragment 1–634 (i.e. ΔQ+SD) independently of NOT1, although the binding efficiency in this case is reduced relative to that observed with full-length D. melanogaster GW182. In addition, PAN3 and NOT2 also interact with the Q+SD region.
A complex network of interactions recruits deadenylases to D. melanogaster GW182
To identify the sequences sufficient for NOT2 and PAN3 binding, we performed co-immunoprecipitation assays using a series of D. melanogaster GW182 N-term fragments of increasing length (Supplementary Figure S1). These fragments were chosen based on the published activity of these fragments in tethering assays (11,25,26). Collectively, these experiments revealed the following observations.
First, GW182 residues 1–830 and 1–1115 are sufficient for NOT2 and PAN3 binding, respectively (Supplementary Figure S1C–E), whereas, as mentioned earlier, NOT1 binding requires residues 635–1384 (i.e. Q+SD).
Second, the GW182 ABD (residues 1–539) does not interact with deadenylase subunits (Supplementary Figure S1C–E, lanes 11) despite the fact that this fragment contains the NED. In accordance with these results, deletion of the NED from D. melanogaster GW182 did not affect NOT1, NOT2 or PAN3 binding (Supplementary Figure S1C–E, lanes 16). All fragments interacted with endogenous AGO1 as expected (Supplementary Figure S1C).
Third, the contribution of GW182 N-term sequences (residues 1–539) to deadenylase binding becomes apparent only when the C-term region is deleted (Supplementary Figure S1F and G; compare fragments 1–1115 versus 539–1115 for PAN3, and fragments 1–830 versus 539–830 for NOT2).
We conclude that D. melanogaster GW182 interacts with deadenylases through multiple binding sites that appear to contribute additively to the affinity of the interaction. Moreover, because deadenylase subunits interact with each other (24), these subunits could bind D. melanogaster GW182 either directly or indirectly. For example, although NOT2 interacts with GW182 N-term sequences, it also interacts with the Q+SD region (most likely via NOT1). This connectivity and redundancy of the GW182 interaction network helps to explain why the contribution of the NED to deadenylase binding becomes apparent only when SD regions are deleted.
GW182 generally requires the SD to silence miRNA targets
Our previous results demonstrated that deleting the SD from D. melanogaster GW182 abrogates its silencing activity in complementation assays, wherein GW182 protein mutants are tested for their ability to restore silencing in cells that are depleted of endogenous GW182 (5,8,12). In contrast, other studies have shown that N-term fragments of D. melanogaster GW182 (e.g. 1–605 and 1–830) can rescue the silencing of at least one miRNA reporter in S2 cells (11,25). Furthermore, D. melanogaster GW182 N-term fragments confer strong repression in vivo when artificially tethered to reporter mRNAs, independently of whether the reporter contains a poly(A) tail [Supplementary Figure S2A and B (11,25–27)]. Because D. melanogaster GW182 N-term fragments do not interact with PABP, it was important to determine whether these fragments complement silencing in cells that are depleted of endogenous D. melanogaster GW182.
For these complementation assays, we used two previously characterized firefly luciferase reporters: the F-Luc-Nerfin-1 and F-Luc-Par-6 reporters silenced by miR-279 and miR-1, respectively (20,21). At steady state, the F-Luc-Nerfin-1 reporter is predominantly repressed at the level of translation, with a small reduction in mRNA abundance (Figure 3A and B, lane 2 versus 1); however, the F-Luc-Par-6 mRNA is degraded in a miR-1-dependent manner (Figure 3E and F, lane 2 versus 1). The depletion of endogenous D. melanogaster GW182 suppressed the silencing of both reporters, leading to a 4- to 5-fold increase in firefly luciferase expression (Figure 3C and G). For the F-Luc-Par-6 reporter, a corresponding increase in mRNA levels was also observed (Figure 3F and H). These results confirm that D. melanogaster GW182 is required for both miRNA-mediated translational repression and mRNA degradation, as previously reported (14,28).
In depleted cells, a dsRNA-resistant version of D. melanogaster GW182 fully rescued the silencing of both reporters (Figure 3B–D and F–H). In contrast, D. melanogaster GW182 fragments lacking the SD (1–539, 1–605 and ΔSD) did not restore silencing, whereas a protein lacking the NED was fully active (Figure 3B–D and F–H). All proteins were expressed at comparable levels (Figure 3I). Notably, the inactive GW182 protein fragments and fragment 1–830 remained inactive, even when expressed at higher levels (Supplementary Figure S3A–C). Finally, a D. melanogaster GW182 mutant lacking the RRM and the C-term region (fragment 1–1115) complemented silencing, although less efficiently than full-length GW182 (Figure 3B–D and F–H and Supplementary Figure S3A and B). A western blot analysis indicated that the levels of D. melanogaster GW182 in the depleted cells were reduced below 10% of the control levels (Supplementary Figure S3D). We concluded that the ability of GW182 protein fragments to complement silencing is independent of whether silencing occurs at the level of translation (the F-Luc-Nerfin-1 reporter) or mRNA stability (the F-Luc-Par-6 reporter), suggesting that these two modes of regulation are not mediated through different GW182 protein domains; thus, these effects might be mechanistically linked.
Previous studies have reported that GW182 N-term fragments can rescue the silencing of the F-Luc-Nerfin-1 reporter when silenced by miR-9b (11,25). In line with those studies, we observed that GW182 protein fragments 1–605 and 1–830 (but not 1–539) restored the silencing of the F-Luc-Nerfin-1 reporter by miR-9b (Supplementary Figure S4A–C). Paradoxically, as shown above, these N-term protein fragments did not rescue the silencing of the F-Luc-Par-6 and F-Luc-Nerfin-1 reporters when silencing was mediated via miR-1 and miR-279, respectively.
To resolve this apparent discrepancy, we further examined the silencing of additional reporters, including the F-Luc-CG3548, F-Luc-CG5281 and F-Luc-CG7709 reporters silenced by miR-12; the F-Luc-Vha-68-1 reporter silenced by miR-9b and the F-Luc-CG11206 reporter silenced by both miR-9b and miR-12 (20,21). We observed that D. melanogaster GW182 N-term fragments (1–605, 1–830 and ΔSD) did not rescue the silencing of these reporters (Figure 4A–F). Furthermore, these N-term fragments inhibited silencing in a dominant-negative manner in control cells (Supplementary Figure S5A–G).
Figure 4.

The Drosophila melanogaster GW182 SD is generally required for silencing. (A–F) Complementation assays using the indicated miRNA reporters were carried out as described in Figure 3. The graphs on the left of each panel show normalized firefly luciferase activities in the absence or presence of miRNAs in control cells (i.e. cells treated with GFP dsRNA and expressing MBP). The graphs on the right of each panel show the relative derepression of the F-Luc reporters for each condition. Mean values ± standard deviations from three independent experiments are shown. Labels are as described in Figure 3.
We conclude that although the N-term region of D. melanogaster GW182 is sufficient for the silencing of the F-Luc-Nerfin-1 reporter via miR-9b, this region was not sufficient to rescue the silencing of the additional miRNA reporters tested. Furthermore, in the context of the full-length protein, the SD is required for the silencing of a majority of the reporters tested, whereas the NED is dispensable. Finally, an N-term fragment containing the NED (1–539) was not sufficient to rescue silencing, although this fragment binds AGO1. On the basis of these results and our previous observations (12), we conclude that the SD is required for the silencing of most miRNA targets (5,8,12). Nevertheless, because one of nine reporters was silenced independently of the SD, it would be interesting to determine how many targets are SD independent (and thus independent of the GW182–PABP interaction) on a genome-wide level, and what features confer this independence.
Design of a minimal functional GW182 protein
Having established that the SD is generally required for silencing, we next examined whether this domain is also sufficient for silencing. Because the SD does not interact with AGO1, we therefore generated a chimeric protein containing a minimal AGO-binding domain fused to the D. melanogaster GW182 SD. We selected the AGO-binding domain (ABD) of the highly divergent Caenorhabditis elegans GW182 protein AIN-2, which binds to D. melanogaster AGO1 [Figure 5 (29)]. The AIN-2 ABD (herein referred to as ABD2) comprises 147 amino acids and contains only 3 GW repeats (Figure 1). Importantly, and in contrast to the D. melanogaster GW182 ABD, ABD2 does not interact with deadenylases and has no silencing activity in complementation assays (Figure 5).
Figure 5.

The Drosophila melanogaster GW182 Q+SD region is sufficient for silencing. (A and B) The silencing activity of chimeric proteins containing the Caenorhabditis elegans ABD2 fused to various GW182 fragments was tested in complementation assays as described in Figure 3 except that control cells were treated with GST dsRNA and transfected with a plasmid expressing GFP. (C–E) The interactions of the chimeric ABD2–GW182 proteins with AGO1, PABP and NOT1 were analyzed as described in Figure 2.
We constructed a chimeric protein containing ABD2 fused to the isolated D. melanogaster GW182 SD or the SD plus additional N-term sequences. We also generated chimeric proteins containing the NED, the Q+SD fragment (635–1384) or the complementary N-term fragment (1–634, i.e. ΔQ+SD). Remarkably, in cells depleted of endogenous D. melanogaster GW182, the chimeric protein containing the Q+SD region rescued the silencing of the F-Luc-Nerfin-1 and F-Luc-Par6 reporters although not as efficiently as wild-type D. melanogaster GW182 (Figure 5A and B), suggesting that sequences upstream of the Q-rich region although not essential, contribute to silencing. The chimeric protein containing the isolated SD or the SD and additional N-term sequences partially rescued silencing (Figure 5A and B, and Supplementary Figure S5H and I). In contrast, the chimeric proteins containing the NED or residues 1–634 did not rescue silencing, even when expressed at higher levels (Figure 5A and B, and Supplementary Figure S6A–C). Notably, a chimeric protein containing only the Q-rich region was also inactive in complementation assays (Supplementary Figure S6A and B), although this region is active in tethering assays (11,25,26). All proteins were expressed at comparable levels and interacted with D. melanogaster AGO1 (Figure 5C and Supplementary Figure S6C).
Consistent with the results shown in Figure 2, the chimeric protein containing the Q+SD region interacted with PABP and NOT1 as efficiently as the full-length D. melanogaster GW182 (Figure 5D and E, lanes 12) and also bound PAN3 and NOT2 (Supplementary Figure S7A and B). The chimera containing the SD interacted with PABP (Figure 5D, lane 13), whereas the chimera containing the NED did not exhibit any binding affinity towards PABP or NOT1 (Figure 5D and E, lanes 14). Finally, the chimeric protein that contained fragment 1–634 did not rescue silencing, despite the observation that this fragment interacts with PAN3 and NOT2 (Figure 5A and B and Supplementary Figure S1A and B). We concluded that the silencing activity of the chimeric GW182 protein correlates with binding to both PABP and deadenylases because only the Q + SD fragment efficiently rescues silencing.
A minimal GW182 protein reveals interactions that are required for silencing
Recent studies have identified W-containing motifs in the M1, M2 and C-term regions of human SDs that are required for the interaction with NOT1 and PAN3 and described mutations in these motifs that abolish binding and silencing (9,10). The motifs in the M1 and C-term regions were termed CIM-1 and CIM-2, respectively (10). The CIM-2 motif is absent in D. melanogaster GW182; however, D. melanogaster GW182 contains a CIM-1 motif and six tryptophan residues in the M2 region (9,10). The contribution of these motifs to deadenylase binding in the context of the full-length D. melanogaster GW182 protein has not been analyzed.
The finding that a minimal chimeric protein consisting of ABD2 and the D. melanogaster GW182 Q+SD region could complement silencing in S2 cells provided an opportunity to test how the specific disruption of PABP or deadenylase binding interferes with silencing in a cellular context in the absence of the contribution of the GW182 N-term sequences. Therefore, we introduced amino acid substitutions into the PAM2 and CIM-1 motifs and in the M2 region (individually or in combination) and assessed the interaction of the mutant proteins with PABP and deadenylases using immunoprecipitation assays; the silencing activity was also tested using complementation assays. We again used a reporter silenced at the translational level (F-Luc-Nerfin-1) and a reporter that is degraded under steady-state conditions (F-Luc-Par-6). These studies revealed the following observations.
First, a single amino acid substitution in the PAM2 motif (F961A) or mutations in the CIM-1 motif did not affect PABP or NOT1 binding (Figure 6A and B, lanes 10 and 11) and had no significant effect on silencing activity (Figure 6C and D). The alanine substitution of all six tryptophan residues in the M2 region (6xW) reduced both PABP and NOT1 binding and, consequently, silencing activity (Figure 6A and B, lanes 12 and Figure 6C and D), as previously reported (9). For all mutants tested, PAN3 and NOT2 binding mirrored NOT1 binding (Supplementary Figure S7A and B).
Figure 6.

PABP and deadenylase binding are required for silencing. Mutations in the PAM2 and CIM-1 motifs and the M2 region were introduced in a minimal GW182 protein consisting of Caenorhabditis elegans ABD2 fused to the GW182 Q+SD region (ABD2-Q+SD). The PAM2 mutant carries a single amino acid substitution (F961A) in the PAM2 motif. Mutations in the CIM-1 motif are shown in Supplementary Figure S7. The 6xW mutant carries alanine substitutions of all six tryptophan residues in the M2 region of the SD. (A and B) The interactions of the ABD2-Q+SD protein (wild-type or mutant) with PABP and NOT1 were analyzed as described in Figure 2. (C–F) The silencing activity of the ABD2-Q+SD protein (wild-type or mutants) was tested in complementation assays as described in Figure 5.
Second, when combined, mutations in the CIM-1 motif and the M2 region strongly reduced PABP and NOT1 binding and silencing activity (Figure 6A and B, lanes 13 and Figure 6C and D). The silencing activity of the chimeric protein was abolished when the PAM2 motif was mutated in combination with the CIM-1 and the 6xW mutations (Figure 6C and D). Importantly, similar results were obtained when F-Luc-Par-6 mRNA levels were analyzed (Figure 6E and F), indicating that the mutations affect translational repression and mRNA degradation in a similar way. Together, these results indicate that the silencing activity of GW182 proteins correlates with both PABP and deadenylase binding.
A recent study reported the identification of another conserved motif, the P-GL motif, in the M2 region of GW182 proteins [Figure 1 and Supplementary Figure S7F (30)]. This motif was shown to contribute to the translational repression and deadenylation of polyadenylated targets in zebrafish embryos (30). We observed that the corresponding mutations in the D. melanogaster GW182 P-GL motif did not affect the silencing activity of the chimeric ABD2-Q+SD protein and did not exacerbate the effect of mutations in the CIM-1 or PAM2 motifs (Supplementary Figure S8A and B). Furthermore, mutations in the P-GL motif did not affect binding to PABP and deadenylases [Supplementary Figure S8C–E (30)]. These results confirm that the P-GL motif does not contribute to silencing in S2 cells (9).
The human TNRC6C SD is sufficient for silencing
Because the mechanism of silencing is conserved and we have previously shown that human TNRC6 proteins complement silencing in Drosophila cells (5), we next determined whether a minimal protein consisting of ABD2 fused to the human TNRC6C SD could rescue silencing in S2 cells depleted of endogenous D. melanogaster GW182. This question was particularly interesting because, unlike the D. melanogaster GW182 SD, human SDs are sufficient for the interaction with PABP and deadenylases (3,5,8–10). Quite remarkably, we observed that a chimeric protein containing ABD2 fused to the TNRC6C SD complemented the silencing of the F-Luc-Nerfin-1 and F-Luc-Par-6 reporters in GW182-depleted cells (Figure 7A and B).
Figure 7.
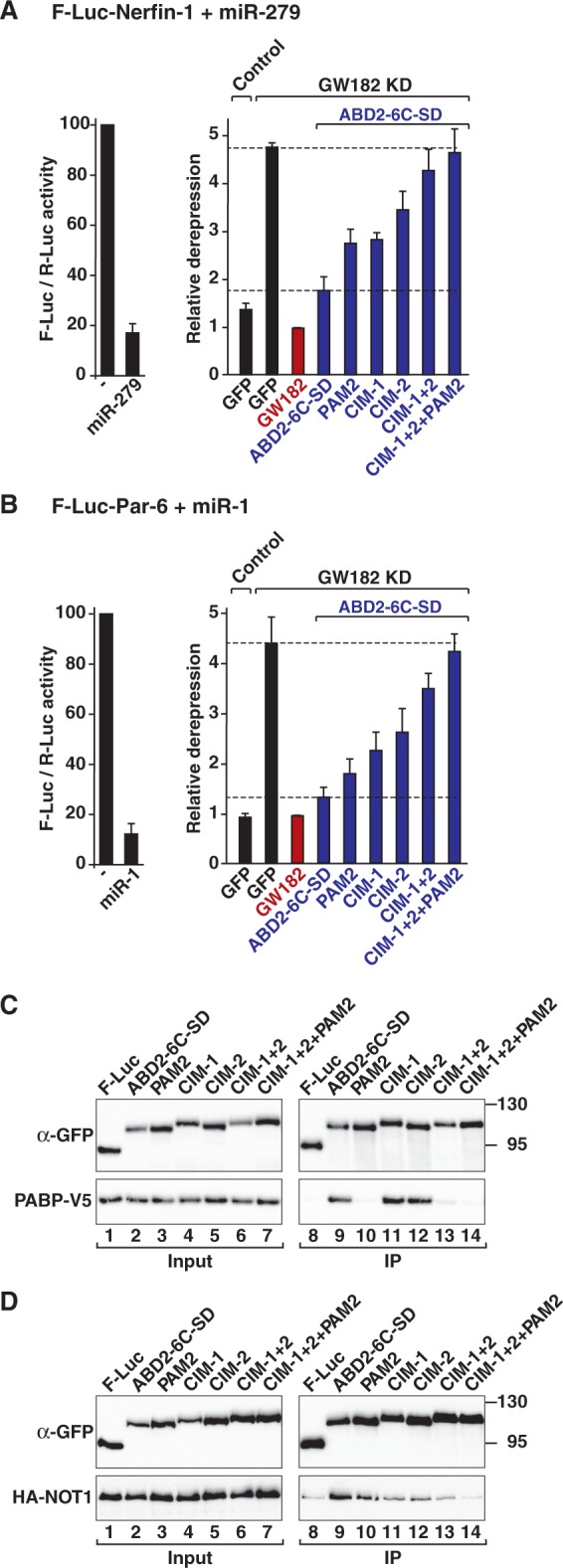
The TNRC6 SD is sufficient for silencing. Mutations in the PAM2, CIM-1 and CIM-2 motifs were introduced in a minimal GW182 protein consisting of Caenorhabditis elegans ABD2 fused to the human TNRC6C SD region (ABD2-6C-SD). The PAM2 mutant carries a single amino acid substitution (F1389A) in the PAM2 motif. Mutations in the CIM-1 and CIM-2 motifs are shown in Supplementary Figure S7. (A and B) The silencing activity of the chimeric ABD2-6C-SD protein (wild-type or mutant) was tested in complementation assays as described in Figure 5. (C and D) The interactions of the ABD2-6C-SD protein (wild-type or mutant) with PABP and NOT1 were analyzed as described in Figure 2.
We next examined the contribution of the CIM-1, CIM-2 and PAM2 motifs to PABP and NOT1 binding and silencing activities. We observed that mutations in the CIM-1 or CIM-2 motifs reduced silencing activity, and the effect of mutations in CIM-2 was stronger (Figure 7A and B); these findings are in agreement with the observation that the CIM-1 and CIM-2 motifs are not functionally equivalent (10). The CIM-1 and CIM-2 mutations reduced NOT1 binding without affecting the interaction with PABP (Figure 7C and D, lanes 11 and 12). When combined, mutations in the CIM-1 and CIM-2 motifs strongly reduced NOT1 binding and silencing activity (Figure 7A–D). The double mutations also abrogated PABP binding (Figure 7C, lane 13), consistent with the observation that the GW182-PABP interaction in S2 cells is predominantly indirect. The effect of the CIM-1+2 double mutation was exacerbated when combined with the F1389A substitution in the PAM2 motif (Figure 7A–D). The F1389A mutation alone abolished PABP binding (Figure 7C, lane 10) and impaired silencing activity, particularly for the F-Luc-Nerfin-1 reporter [Figure 7A and B (5,8)], although NOT1 binding was only slightly reduced (Figure 7D, lane 10). Collectively, the results obtained with the chimeric proteins demonstrate that full silencing activity requires interactions between GW182 proteins and both PABP and deadenylases.
The interaction of TNRC6s with PABP and deadenylases is required for silencing in human cells
Having established that the interaction with NOT1 and PABP is important for silencing in the context of the chimeric ABD2-TNRC6C SD, we subsequently investigated the contribution of these interactions to the silencing activity of TNRC6A protein in human cells. Accordingly, we used a previously characterized F-Luc reporter containing three Let-7 miRNA binding sites in the 3′ UTR, which is primarily regulated at the translational level [Figure 8A (22)]. The knockdown of TNRC6 proteins was achieved using siRNAs targeting TNRC6A and TNRC6B. This double depletion inhibited the silencing of the reporter, leading to a 2.8-fold increase in F-Luc activity (Figure 8A and B). A western blot analysis indicated that the levels of TNRC6A in the depleted cells were reduced to 10% of the control levels (Figure 8C). The silencing of F-Luc-3xlet-7 was rescued in cells expressing a siRNA-resistant form of wild-type TNRC6A (Figure 8B).
As shown above, mutations in either the CIM-1 or the CIM-2 motifs or alanine substitutions of the six tryptophan residues in the M2 region (6xW) reduced human NOT1 (CNOT1), but not PABP binding (Figure 8D). These mutations impaired silencing activity in complementation assays; a stronger effect was observed for the mutations in the M2 region and CIM-2 motif (Figure 8B). The CIM-1+2 double mutant was strongly impaired with respect to CNOT1 binding and silencing activity (Figure 8B and D). However, in contrast to the results observed in S2 cells, this mutant retained the ability to bind PABP in human cells (Figure 8D). Thus, the interaction of TNRC6A with PABP is not sufficient for silencing. Conversely, CNOT1 binding is also insufficient for full silencing activity because a TNRC6A mutant carrying a F1359A substitution in the PAM2 motif interacted with CNOT1, but not with PABP, and was impaired in complementation assays (Figure 8B and D). The silencing activity was reduced further when the mutation in the PAM2 motif was combined with the mutations in the CIM-1 and CIM-2 motifs or in the M2 region (Figure 8B). A protein carrying mutations in the M2 region and the CIM-1, CIM-2 and PAM2 motifs was inactive as observed using a protein lacking the entire SD (Figure 8B). All proteins were expressed at comparable levels (Figure 8E) and have no dominant negative effects in control cells (Supplementary Figure S9). These results demonstrated that the interactions of human TNRC6 proteins with PABP and CNOT1 are also required for full silencing activity in human cells.
Overexpression of the CNOT1 Mid domain suppresses silencing
The experiments described above suggest that silencing (i.e. translational repression and target degradation) requires an interaction between GW182 proteins and the CCR4–NOT complex. We next sought to further validate the contribution of this complex to silencing using an overexpression approach. These experiments were conducted in human cells, wherein the interaction of TNRC6s with CNOT1 is direct and mediated through the SD, without any contribution from N-term sequences (8–10).
CNOT1 is a large protein containing 2376 amino acids and is predicted to be mainly α-helical. Sequence alignments and secondary structure predictions suggest that CNOT1 consists of N-term (CNOT1-N), Mid (CNOT1-M) and C-term (CNOT1-C) domains (Figure 9A). In immunoprecipitation assays, we observed that the CNOT1-M domain was sufficient for the interaction with the TNRC6A SD (Figure 9B, lane 9). CNOT1-N showed no detectable binding, whereas CNOT1-C exhibited some residual binding affinity (Figure 9B, lane 10). The CNOT1-M domain also interacts with CNOT7 [also known as CAF1 (31)]. Therefore, we tested whether overexpression of the CNOT1-M domain, either alone or together with a catalytically inactive CNOT7 mutant, could inhibit silencing in a dominant negative manner.
Previous studies have demonstrated that the overexpression of a catalytically inactive CNOT7 mutant or the depletion of the subunits of the CCR4–NOT complex suppressed miRNA-mediated mRNA deadenylation and degradation, although translation of the reporters was not fully restored (14,15,21,31–33). Consistent with these studies, we observed that overexpression of the CNOT7 catalytically inactive mutant partially suppressed silencing of the R-Luc-3xlet-7 reporter (Figure 9C). Interestingly, overexpression of the CNOT1-M domain also partially suppressed the silencing of the R-Luc-3xlet-7 reporter (Figure 9C). Silencing was abrogated in cells coexpressing the NOT1-M domain together with the CNOT7 catalytically inactive mutant (Figure 9C).
To further confirm these results, we analyzed the effects of coexpressing the CNOT1-M domain together with the CNOT7 mutant on silencing of the R-Luc-Hmga2 reporter, which is also silenced by Let-7 (23). Silencing of this reporter was also suppressed in cells that coexpressed both proteins (Figure 9D). Taken together with results of recent reports showing that translational repression precedes target degradation and decay (15–17), these results further support the conclusion that the interaction of GW182 proteins with the CCR4–NOT complex is required for both miRNA-mediated translational repression and target degradation. Thus, the CCR4–NOT complex is a major effector complex of silencing.
DISCUSSION
Recent studies indicate that translational repression of miRNA targets precedes deadenylation and decay (3,15–19). Here, we show that these two functional outcomes of miRNA regulation are linked and both require the interaction of GW182 proteins with PABP and deadenylases.
The GW182–PABP interaction is required for maximal silencing activity
The interaction of GW182 proteins with PABP has been well documented using biochemical and structural studies, and the PAM2 motif is highly conserved among vertebrate and insect GW182 proteins (3–7). Despite conservation, the study of the role of PABP in silencing in different systems has led to conflicting conclusions. For example, several studies have reported that the PABP–GW182 interaction is important for silencing in D. melanogaster and human cells and in cell-free systems that recapitulate silencing (3,5,6,8,10,34,35). Furthermore, PABP depletion prevented miRNA-mediated deadenylation in cell-free extracts from mouse Krebs-2 ascites cells (3), and mutations in the PAM2 motif of TNRC6C reduced the rate of deadenylation in tethering assays (6). In addition, a study in D. melanogaster cell-free extracts wherein silencing is mediated through endogenous preloaded miRISCs indicated that PABP stimulates silencing by facilitating the association of miRISC complexes with mRNA targets (35). It was also shown that on miRISC binding, PABP progressively dissociated from the mRNA target, in the absence of deadenylation (35).
In contrast to the studies mentioned above, studies in zebrafish embryos and in a D. melanogaster cell-free assay wherein miRISCs are loaded with exogenously supplemented miRNA duplexes indicate that PABP is dispensable for miRNA-mediated silencing (26,30). Intriguingly, efficient silencing in zebrafish embryos required the GW182 PAM2 motif (30). Moreover, the observation that multiple and non-overlapping fragments of D. melanogaster GW182 (including N-term fragments that do not interact with PABP) silenced mRNA reporters in tethering assays was interpreted as evidence that the interaction of GW182 proteins with PABP is not required for silencing (26). In this study, we show that unlike in tethering assays, N-term fragments of GW182 fail to restore the silencing of a majority of the reporters tested in complementation assays. Thus, tethering assays bypass the requirement for PABP binding, and may not faithfully recapitulate silencing. Furthermore, the observation that PABP dissociates from the poly(A) tail of miRNA targets in the absence of deadenylation (35) provides one explanation for the occurrence of silencing in extracts in which PABP has been depleted or displaced from the poly(A) tail using an excess of Paip2 (26,30).
In summary, our results confirm and further extend previous observations that a single amino acid substitution in the PAM2 motif of human TNRC6 proteins abolishes PABP binding and impairs silencing activity, despite the interaction of this mutant with deadenylases (3–8). Furthermore, D. melanogaster GW182 N-term protein fragments that bind deadenylases, but not PABP, failed to complement the silencing of eight of the nine reporters tested, although they are active in tethering assays. These results provide evidence for a role of PABP in silencing in human and Drosophila cells. However, it is possible that PABP becomes dispensable for silencing depending on cellular conditions or the nature of the specific mRNA target, as shown, for example, for the F-Luc-Nerfin-1 reporter when silencing is mediated by miR-9b [this study (11,25,26,30)].
Drosophila melanogaster GW182 establishes an intricate network of interactions with deadenylases
The SDs of human TNRC6 proteins directly interact with CNOT1 through tryptophan-containing motifs in the M1, M2 and C-term regions of the SD (9,10). Here, we show that these motifs contribute additively to CNOT1 binding and silencing activity in human cells. Indeed, when at least two motifs are simultaneously mutated, CNOT1 binding is strongly reduced and silencing activity impaired.
The interaction between GW182 and deadenylases is conserved in D. melanogaster; however, in contrast to human SDs, the D. melanogaster SD is not sufficient for NOT1 binding. Here, we show that in addition to the SD, the Q-rich region is required for full NOT1 binding activity. Thus, although D. melanogaster GW182 has lost the CIM-2 motif, this protein has acquired additional motifs that can interact with NOT1. We also show that in contrast to the human proteins, D. melanogaster GW182 can interact with NOT2 and PAN3 via N-term sequences. Consequently, D. melanogaster GW182 can recruit deadenylases in multiple ways. Considering that (i) NOT1 interacts with NOT2 [reviewed in (36)], (b) the PAN2–PAN3 complex interacts with PABP (37) and (c) the CCR4–NOT and PAN2–PAN3 complexes form a larger multiprotein complex in vivo (38), our observations indicate a high degree of connectivity and redundancy within the GW182 interaction network, which could explain why mutations in individual motifs do not abolish partner binding or silencing activity, but a combination of two or more mutations is required to abrogate binding and silencing activity.
In addition, the ability of D. melanogaster GW182 N-term fragments to bind deadenylases also explains why these fragments are potent triggers of translational repression and mRNA degradation in tethering assays (9,11,25–27), whereas the corresponding fragments of the human proteins exhibit only residual activity (11,25,39,40). As discussed previously, despite their activity in tethering assays, D. melanogaster GW182 N-term fragments failed to complement the silencing of several of the reporters tested. The reason for the different activities of these fragments in tethering and complementation assays remains unknown.
Definition of a minimal protein interaction network required for silencing
In this study, we demonstrated that silencing (i.e. translational repression and target degradation) requires the interaction between GW182 proteins and both PABP and deadenylases. Several lines of evidence support this conclusion. First, the TNRC6C SD, which is sufficient for PABP and deadenylase binding, rescues silencing when fused to a minimal ABD. Similarly, the minimal fragment of D. melanogaster GW182 that rescues silencing comprises the Q+SD region, which also binds both deadenylases and PABP. Second, the D. melanogaster GW182 N-term fragments that bind deadenylases but not PABP are generally inactive in complementation assays. Third, mutations that specifically disrupt TNRC6 binding to PABP or deadenylase impair silencing, and mutations that disrupt deadenylase binding exhibit a stronger deleterious effect. Silencing activity is abolished when these mutations are combined. Finally, silencing is inhibited in human cells overexpressing the CNOT1 Mid domain together with a catalytically inactive CNOT7 mutant. In combination with the previously published data (1,2), our results indicate that silencing minimally requires an AGO, a GW182 protein, PABP and deadenylases, thus defining the minimal interaction network required for silencing. Our findings do not rule out that additional interactions are potentially required to achieve maximal repression, depending on the cellular context or the mRNA target. For example, the P-GL motif is highly conserved and important for silencing in zebrafish embryos (30). This motif may mediate interactions with additional partners.
The finding that deadenylase complexes, in particular, are required for miRNA-mediated translational repression has broad implications regarding post-transcriptional mRNA regulation. Indeed, in addition to the GW182 proteins, various sequence-specific mRNA-binding proteins, such as Nanos, Bicaudal-C and Pumilio, recruit the CCR4–NOT complex to their mRNA targets [reviewed in (36)]. Furthermore, the direct tethering of the subunits of the CCR4–NOT complex represses the translation of mRNA reporters lacking a poly(A) tail, suggesting that the CCR4–NOT complex promotes translational repression in the absence of deadenylation (9,13). Therefore, elucidating the mechanism by which the CCR4–NOT complex regulates the fates of mRNA targets promises to increase our understanding of the mechanism underlying repression by miRNAs and diverse sequence-specific RNA-binding proteins.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–9.
FUNDING
Max Planck Society; the Deutsche Forschungsgemeinschaft (DFG, FOR855 and the Gottfried Wilhelm Leibniz Program awarded to E.I.). Funding for open access charge: Max Planck Society.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank S. Heimstädt for providing the plasmid used for the expression of GW182 protein fragments and performing preliminary assays, and we also thank M. Fauser and S. Helms for excellent technical assistance.
REFERENCES
- 1.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 2.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 3.Fabian MR, Mathonnet G, Sundermeier T, Mathys H, Zipprich JT, Svitkin YV, Rivas F, Jinek M, Wohlschlegel J, Doudna JA, et al. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol. Cell. 2009;35:868–880. doi: 10.1016/j.molcel.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zekri L, Huntzinger E, Heimstädt S, Izaurralde E. The silencing domain of GW182 interacts with PABP to promote translational repression and degradation of miRNA targets and is required for target release. Mol. Cell. Biol. 2009;29:6220–6231. doi: 10.1128/MCB.01081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huntzinger E, Braun JE, Heimstädt S, Zekri L, Izaurralde E. Two PABP-binding sites in GW182 proteins promote miRNA-mediated gene silencing. EMBO J. 2010;29:4146–4160. doi: 10.1038/emboj.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jínek M, Fabian MR, Coyle SM, Sonenberg N, Doudna JA. Structural insights into the human GW182-PABC interaction in microRNA-mediated deadenylation. Nat. Struct. Mol. Biol. 2010;17:238–240. doi: 10.1038/nsmb.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozlov G, Safaee N, Rosenauer A, Gehrin K. Structural basis of binding of P-body associated protein GW182 and Ataxin-2 by the MLLE domain of poly(A)-binding protein. J. Biol. Chem. 2010;285:13599–13606. doi: 10.1074/jbc.M109.089540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun JE, Huntzinger E, Fauser M, Izaurralde E. GW182 proteins recruit cytoplasmic deadenylase complexes to miRNA targets. Mol. Cell. 2011;44:120–133. doi: 10.1016/j.molcel.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Chekulaeva M, Mathys H, Zipprich JT, Attig J, Colic M, Parker R, Filipowicz W. miRNA repression involves GW182-mediated recruitment of CCR4-NOT through conserved W-containing motifs. Nat. Struct. Mol. Biol. 2011;18:1218–1226. doi: 10.1038/nsmb.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabian MR, Cieplak MK, Frank F, Morita M, Green J, Srikumar T, Nagar B, Yamamoto T, Raught B, Duchaine TF, et al. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4–NOT. Nat. Struct. Mol. Biol. 2011;18:1211–1217. doi: 10.1038/nsmb.2149. [DOI] [PubMed] [Google Scholar]
- 11.Chekulaeva M, Filipowicz W, Parker R. Multiple independent domains of dGW182 function in miRNA-mediated repression in Drosophila. RNA. 2009;15:794–803. doi: 10.1261/rna.1364909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eulalio A, Helms S, Fritzsch C, Fauser M, Izaurralde E. A C-terminal silencing domain in GW182 is essential for miRNA function. RNA. 2009;15:1067–1077. doi: 10.1261/rna.1605509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooke A, Prigge A, Wickens M. Translational repression by deadenylases. J. Biol. Chem. 2010;285:28506–28513. doi: 10.1074/jbc.M110.150763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006;20:1885–1898. doi: 10.1101/gad.1424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Béthune J, Artus-Revel CG, Filipowicz W. Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep. 2012;13:716–723. doi: 10.1038/embor.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Djuranovic S, Nahvi A, Green A. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012;336:237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, Biffo S, Merrick WC, Darzynkiewicz E, Pillai RS, et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science. 2007;17:1764–1767. doi: 10.1126/science.1146067. [DOI] [PubMed] [Google Scholar]
- 19.Zdanowicz A, Thermann R, Kowalska J, Jemielity J, Duncan K, Preiss T, Darzynkiewicz E, Hentze MW. Drosophila miR2 primarily targets the m7GpppN cap structure for translational repression. Mol. Cell. 2009;35:881–888. doi: 10.1016/j.molcel.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 20.Eulalio A, Rehwinkel J, Stricker M, Huntzinger E, Yang SF, Doerks T, Dorner S, Bork P, Boutros M, Izaurralde E. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev. 2007;21:2558–2570. doi: 10.1101/gad.443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E. Deadenylation is a widespread effect of miRNA regulation. RNA. 2009;15:21–32. doi: 10.1261/rna.1399509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 23.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Temme C, Zaessinger S, Meyer S, Simonelig M, Wahle E. A complex containing the CCR4 and CAF1 proteins is involved in mRNA deadenylation in Drosophila. EMBO J. 2004;23:2862–2871. doi: 10.1038/sj.emboj.7600273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chekulaeva M, Parker R, Filipowicz W. The GW/WG repeats of Drosophila GW182 function as effector motifs for miRNA-mediated repression. Nucleic Acids Res. 2010;38:6673–6683. doi: 10.1093/nar/gkq501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukaya T, Tomari Y. PABP is not essential for microRNA-mediated translational repression and deadenylation in vitro. EMBO J. 2011;30:4998–5009. doi: 10.1038/emboj.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao B, Li S, Jung HM, Lian SL, Abadal GX, Han F, Fritzler MJ, Chan EK. Divergent GW182 functional domains in the regulation of translational silencing. Nucleic Acids Res. 2011;39:2534–2547. doi: 10.1093/nar/gkq1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eulalio A, Huntzinger E, Izaurralde E. GW182 interaction with Argonaute is essential for miRNA-mediated translational repression and mRNA decay. Nat. Struct. Mol. Biol. 2008;15:346–353. doi: 10.1038/nsmb.1405. [DOI] [PubMed] [Google Scholar]
- 29.Kuzuoglu-Öztürk D, Huntzinger E, Schmidt S, Izaurralde E. The Caenorhabditis elegans GW182 protein AIN-1 interacts with PAB-1 and subunits of the PAN2-PAN3 and CCR4-NOT deadenylase complexes. Nucleic Acids Res. 2012;40:5651–5665. doi: 10.1093/nar/gks218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishima Y, Fukao A, Kishimoto T, Sakamoto H, Fujiwara T, Inoue K. Translational inhibition by deadenylation-independent mechanisms is central to microRNA-mediated silencing in zebrafish. Proc. Natl Acad. Sci. USA. 2012;109:1104–1109. doi: 10.1073/pnas.1113350109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petit AP, Wohlbold L, Bawankar P, Huntzinger E, Schmidt S, Izaurralde E, Weichenrieder O. The structural basis for the interaction between the CAF1 nuclease and the NOT1 scaffold of the human CCR4-NOT deadenylase complex. Nucleic Acids Res. 2012;40:11058–11072. doi: 10.1093/nar/gks883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CY, Zheng D, Xia Z, Shyu AB. Ago-TNRC6 triggers microRNA-mediated decay by promoting two deadenylation steps. Nat. Struct. Mol. Biol. 2009;16:1160–1166. doi: 10.1038/nsmb.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piao X, Zhang X, Wu L, Belasco JG. CCR4-NOT deadenylates mRNA associated with RNA-induced silencing complexes in human cells. Mol. Cell. Biol. 2010;30:1486–1494. doi: 10.1128/MCB.01481-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walters RW, Bradrick SS, Gromeier M. Poly(A)-binding protein modulates mRNA susceptibility to cap-dependent miRNA-mediated repression. RNA. 2010;16:239–250. doi: 10.1261/rna.1795410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moretti F, Kaiser C, Zdanowicz-Specht A, Hentze MW. PABP and the poly(A) tail augment microRNA repression by facilitated miRISC binding. Nat. Struct. Mol. Biol. 2012;19:603–608. doi: 10.1038/nsmb.2309. [DOI] [PubMed] [Google Scholar]
- 36.Collart MA, Panasenko OO. The Ccr4–not complex. Gene. 2012;492:42–53. doi: 10.1016/j.gene.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 37.Siddiqui N, Mangus DA, Chang TC, Palermino JM, Shyu AB, Gehring K. Poly(A) nuclease interacts with the C-terminal domain of polyadenylate-binding protein domain from poly(A)-binding protein. J. Biol. Chem. 2007;282:25067–25075. doi: 10.1074/jbc.M701256200. [DOI] [PubMed] [Google Scholar]
- 38.Zheng D, Ezzeddine N, Chen CY, Zhu W, He X, Shyu AB. Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J. Cell. Biol. 2008;182:89–101. doi: 10.1083/jcb.200801196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zipprich JT, Bhattacharyya S, Mathys H, Filipowicz W. Importance of the C-terminal domain of the human GW182 protein TNRC6C for translational repression. RNA. 2009;15:781–793. doi: 10.1261/rna.1448009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazzaretti D, Tournier I, Izaurralde E. The C-terminal domains of human TNRC6A, B and C silence bound transcripts independently of the Argonaute proteins. RNA. 2009;15:1059–1066. doi: 10.1261/rna.1606309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.