

Abstract
MepA is a multidrug and toxin extrusion (MATE) family protein and the only MATE protein encoded within the Staphylococcus aureus genome. Structural data for MATE proteins are limited to a single high-resolution example, NorM of Vibrio cholerae. Substitution mutations were created in MepA using gradient plates containing both a substrate and reserpine as an efflux pump inhibitor. Site-directed mutagenesis of plasmid-based mepA was used to reproduce these mutations, as well as unique or low-frequency mutations identified in mepA-overexpressing clinical strains, and to mutagenize conserved acidic residues. The effect of these changes on protein function was quantitated in a norA-disrupted host strain by susceptibility testing with and without inhibitors and by determining the proficiency of ethidium efflux. Up-function substitutions clustered in the carboxy half of MepA, near the cytoplasmic face of the protein. Repeated application of the same gradient plate conditions frequently reproduced identical substitution mutations, suggesting that individual residues are required for interaction with specific substrates. Acidic residues critical to protein function were identified in helices 4 and 5. In silico modeling revealed an outward-facing molecule, with helices 1, 2, 4, 7, 8, and 10 having contact with a central cavity that may represent a substrate translocation pathway. Functionally important residues within this cavity included S81, A161, M291, and A302. These data provide a critical starting point for understanding how MATE multidrug efflux proteins function and will be useful in refining crystallographic data when they are available.
INTRODUCTION
Efflux of antimicrobial agents and biocides is recognized as a significant mechanism of resistance in bacteria (1). The ability of selected efflux proteins to transport multiple structurally diverse substrates amplifies the problem, resulting in a multidrug resistance (MDR) phenotype. MDR-conferring efflux proteins are found in all bacteria, are membrane based, and belong to one of five protein families distinguished by secondary structure and the energy source utilized for substrate translocation (2). These families consist of the ATP-binding cassette (ABC), major facilitator superfamily (MFS), resistance-nodulation-division (RND), small multidrug resistance (SMR), and multidrug and toxin extrusion (MATE) proteins. All except ABC proteins, which cleave ATP to provide energy for their activity, utilize ion gradients as the energy source for substrate transport. Most commonly, this is the H+ gradient, but MATE family proteins also can utilize the Na+ gradient (3). Bacterial MATE transporters are the least numerous and least studied of the MDR efflux proteins, with only one (MepA) encoded within the genome of Staphylococcus aureus N315. A single homologue is also found within the genomes of coagulase-negative staphylococci, such as Staphylococcus carnosus (89% homology), Staphylococcus saprophyticus (81%), Staphylococcus haemolyticus (77%), and Staphylococcus lugdunensis (74%) (http://www.membranetransport.org and http://blast.ncbi.nlm.nih.gov). Their low numbers in most bacterial genera correlate with the relative paucity of biochemical and structural data for them.
Expression of mepA is regulated by MepR, a MarR family repressor encoded immediately upstream of mepA (4). Functional and structural analyses have been performed for MepR and have established that it is substrate responsive and binds to both the mepR and mepA promoter regions. The characteristics of its interaction with each operator site are profoundly different. MepR binds as a single dimer to the mepR operator but as a dimer of dimers to that of mepA. The first dimer binds to a primary, high-affinity site, followed by recruitment of a second dimer, perhaps in a cooperative fashion (5). In the absence of substrates, the expression of mepA is essentially abrogated. However, in their presence, the affinity of MepR for the mepA operator site is markedly reduced compared to a much more attenuated effect at the mepR operator (6).
Details regarding the functional characteristics of MepA are limited to identification of its substrate profile, which includes selected fluoroquinolones and other hydrophobic cations, such as monovalent and divalent biocides and dyes. Reserpine, a commonly employed efflux pump inhibitor (EPI), reverses MepA-mediated MIC increases to all substrates and blocks ethidium efflux (4). Other EPIs, such as paroxetine and selected phenothiazines and thioxanthenes, also inhibit its activity (7, 8). Further functional specifics are lacking.
Structural details of membrane-based proteins are limited due to the difficulty in obtaining diffraction quality crystals. Optimal conditions for crystallization may be different for different proteins, and as such, only a small number of structures have been solved. The most intensively studied MDR efflux protein structure is that of AcrB, a member of the RND family (9–12). This effort has identified substrate binding and translocation pathways in some detail, as well as providing information about the interaction between AcrB and its cognate membrane fusion and outer membrane proteins, AcrA and TolC, respectively.
Low-resolution structural data (6.5 Å) for the NorM MATE MDR efflux protein of Neisseria gonorrhoeae provided some very general structural parameters, but recent high-resolution data (3.65 Å) for the NorM MATE MDR protein of Vibrio cholerae offered real insight (13, 14). The solved structure, which is in the outwardly facing conformation, identified a large internal cavity within the lipid bilayer that is involved in substrate binding. Amino acid residues facing the cavity are mainly hydrophobic and/or aromatic, but a few are polar or charged. The binding of Rb+ and Cs+ ions, alkali-metal sodium analogues utilized due to the better resolution they provide on X-ray crystallography, identified residues from transmembrane segments (TMSs) 7, 8, and 10 to 12 as being involved in cation binding, with residues E255 and D371 critical, as mutation to either alanine or asparagine abolished binding. Earlier work done using the NorM homologue of Vibrio parahaemolyticus (76% sequence identity with the V. cholerae protein) identified residues homologous with E255 and D371 as being critical for transport function (15). MepA has limited homology with either Vibrio protein (∼17%), and neither E255 nor D371 is conserved (data not shown). While these data identify a binding site for the monovalent cation with which substrates are exchanged by NorM, no structural data are available regarding how or where substrates interact with any MATE protein. It is reasonable to presume that MepA and NorM share functional and structural characteristics, but sequence differences are likely to result in variations in particular residues involved in substrate and Na+ or H+ translocation.
In lieu of crystallographic data, this study was designed to define functionally critical regions and residues of MepA using microbiologic, genetic, and in silico modeling methods. The study of gradient plate-selected and naturally occurring MepA mutants, as well as mutants of conserved acidic residues, identified a number of positions important for protein function and inhibitor susceptibility. A model of MepA was generated using the Phyre2 algorithm, and structural correlations for the functional effects of selected mutations are proposed.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and reagents.
The working strains and plasmids used are described in Table 1. SA-K3731 was constructed by transducing the norA::erm mutation of SA-K1758 into SA-K2068, resulting in a norA null mepA overexpression phenotype (16). SA-K3486 was constructed by transduction of pK436 into SA-K2124. The pALC2073 backbone provides inducible expression of genes by way of the xyl-tetO promoter, which is unaffected by MepR.
Table 1.
Study strains and plasmids
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| NCTC 8325-4 | NCTC 8325 cured of prophages | 39 |
| SH1000 | NCTC 8325-4 with rsbU mutation corrected | 40 |
| SA-K1758 | NCTC 8325-4 norA::erm | 41 |
| SA-K2068 | NCTC 8325-4 with increased mepA expression | 4, 42 |
| SA-K3731 | SA-K2068 norA::erm | This study |
| SA-K2124 | SH1000 norA::lacZ | 43 |
| SA-K3486 | SA-K2124 + pK436 | This study |
| pALC2073 | xyl-tetO-inducible promoter controlling expression of cloned genes; Cmr | 44 |
| pK436 | pALC2073-mepA (wild-type gene from NCTC 8325-4) | 4 |
Cmr, chloramphenicol resistance selection.
Bloodstream isolates of S. aureus (one per patient) collected from various locations in 2005 (n = 232; Detroit area) and again in 2009 (n = 563; Boston, Detroit, Houston, Omaha, and Freiburg, Germany) were analyzed using quantitative reverse transcription-PCR (qRT-PCR) for increased expression of several MDR efflux pump genes, including mepA (17–19). An additional 126 unique nonbloodstream isolates collected in 2009 from San Francisco were also evaluated in the same manner. Sequence analysis of mepRA was performed on all strains demonstrating increased mepA expression (≥4 times that of SH1000) using an automated method (described below) and identified several unique or low-frequency MepA substitutions in them. Based on our observations that single amino acid substitutions in MepA can affect efflux efficiency (see below), these substitutions were selected for further analysis. Unless otherwise noted, all media and reagents were the highest quality available and were obtained from Sigma Chemical Co. (St. Louis, MO) or BD Biosciences (Sparks, MD).
Microbiologic procedures.
MICs were determined in quadruplicate by broth microdilution according to CLSI guidelines, with the exception that tetracycline (0.05 μg/ml) was included to induce expression of mepA derivatives from pALC2073 (20). In some instances, the effect of reserpine (final concentration, 20 μg/ml) on MICs was also determined. EtBr efflux was quantitated at least 3 times for each test strain using a fluorometric approach, as described previously, modified by including tetracycline during growth to induce mepA expression, as described above (21). Efflux inhibition dose-response profiles for reserpine, and in some cases paroxetine, were constructed by determining ethidium bromide (EtBr) efflux in the presence of increasing concentrations of inhibitor. The 50% inhibitory concentration (IC50; in μM) for efflux was determined by inspection of dose-response plots and then rounding to the nearest 0.1 μM.
The residues involved in efflux and susceptibility to efflux pump inhibitors were sought by passing SA-K3731 on gradient plates, which were constructed using Mueller-Hinton agar containing a variety of MepA substrates plus a steady reserpine concentration (20 μg/ml) (22). Inclusion of the inhibitor was anticipated to increase the likelihood of recovery of mutants with changes in both parameters and, if successful, would support the conclusion that binding sites for the substrate and inhibitor may overlap. The gradient concentrations of substrates were increased until growth progression of the test strain across plates ceased. The compounds and final MIC gradients used included benzalkonium chloride (BAC) (0 to 5×), chlorhexidine (CHX) (0 to 25×), 4′,6-diamidino-2-phenylindole (DAPI) (0 to 32×), dequalinium (DQ) (0 to 32×), EtBr (0 to 10×), Hoechst 33342 (H33342) (0 to 8×), pentamidine (0 to 4×), rhodamine (RD) (0 to 4×), and tetraphenylphosphonium bromide (TPP) (0 to 3×). Each selection condition was employed twice to determine if the resultant mutants could be reproduced and thus be deemed drug specific. All derivatives of SA-K3731 produced by gradient plate passage were single-colony purified, passed three times on drug-free Mueller-Hinton agar, and then evaluated by MIC testing in the presence and absence of reserpine (20 μg/ml). Those demonstrating differences from SA-K3731 were selected for further analyses.
Genetic procedures.
Nucleotide sequencing of the mepRA region of gradient plate-passaged and clinical mepA-overexpressing strains was performed using an automated dideoxy chain termination method by the Applied Genomics Technology Center, Wayne State University (23). Identified mepA mutations resulting in a residue substitution in clinical mepA-overexpressing isolates, and for gradient plate mutants a phenotype change, were replicated in pK436 using the QuikChange Lightning site-directed mutagenesis kit following directions provided by the manufacturer (Agilent Technologies, Inc., Santa Clara CA). Following sequence confirmation, these constructs were transduced into SA-K2124.
Negatively charged (acidic) residues within MDR efflux pump proteins of the MATE, MFS, and SMR families, especially those predicted to lie in TMSs, have been found to be of significant functional importance (14, 15, 24–26). Such residues may participate in substrate binding and translocation, as the preferred substrates for these pumps are hydrophobic cations. Additionally, negatively charged residues may be involved in H+ or Na+ antiport. Conserved negatively charged residues of MepA were identified and changed to alanine, as well as aspartic acid and glutamine for glutamate, and glutamic acid and asparagine for aspartate, by site-directed mutagenesis of pK436. The mutated plasmids were sequence confirmed and then transduced into SA-K2124 for further study.
The efficiency of expression of all plasmid-based mepA derivatives was verified using qRT-PCR, which was performed using a multiplex approach exactly as described previously, using the Quantitect multiplex RT-PCR kit (Qiagen, Inc., Valencia, CA) and an ABI 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA) (27). All strains were grown under the same conditions used for EtBr efflux assays, including tetracycline induction. Beacon Designer 7.80 (Premier Biosoft International, Palo Alto, CA) was used to design TaqMan probes and primers, which were purchased commercially (Eurofins MWG/Operon, Huntsville, AL). The comparative threshold cycle method was used to determine the relative expression of mepA compared to that of SA-K3486, in which mepA expression was considered to be 1.0.
Quantitation of MepA.
The relative amount of MepA present in the membrane of SA-K2124 containing pALC2073, into which various mepA derivatives were cloned, was determined by Western blotting analyses. The derivatives studied included the wild type (WT), as well as selected versions conferring large gain- or loss-of-function phenotypes, including two that were gradient plate generated (S81L and F375I), one that was naturally occurring (A397V), and three with mutations in conserved acidic residues (E156A, D183A, and E423Q) (Tables 2 and 3). A hexahistidine tag was inserted at the carboxy terminus by site-directed mutagenesis, which had no adverse effect on EtBr efflux (data not shown). Strains were grown as for the EtBr efflux assay, except that brain heart infusion broth was used as the growth medium. The cells were recovered by centrifugation and frozen at −80°C until use. The thawed cells were resuspended in 0.9% NaCl containing lysostaphin, RNase, and DNase (100, 50, and 50 μg/ml, respectively) and incubated for 10 min at 37°C. Cellular debris, including cytoplasmic membranes, was recovered by centrifugation and resuspended in water, and the protein content was determined (Bio-Rad Life Science Research, Hercules, CA). After separation of a 7-μg sample of each derivative on a 12% polyacrylamide gel, the proteins were transferred to a nitrocellulose membrane, and a Western blot was made using the SuperSignal West HisProbe Kit following the directions provided by the manufacturer (Pierce Biotechnology, Inc., Rockford, IL). MepA bands were quantitated by densitometry using Phoretix 1D software, and all mutants were normalized to the wild type (Nonlinear Dynamics Ltd., Newcastle upon Tyne, United Kingdom).
Table 2.
Phenotypic effects of gradient plate-generated mepA mutations replicated in pK436 and expressed in SA-K2124
| Parameter | Value in strain (mutation)a: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K3486 | K3809 (N369Y) | K3811 (M312T) | K4538 (F375V) | K4670 (A161V) | K4842 (S81L) | K4898 (K242E) | K4900 (N308)b | K4902 (F375I) | K4904 (A161T) | K4908 (M391T) | K4984 (M291I) | |
| Selecting conditions applied to SA-K3731 | H33342, RD | DQ | EtBr, Pentc | CHX | BAC | TPP | TPP, CHX | EtBr | Pent | DAPI | DAPI | |
| Location of substitutiond | TMS 10 | Loop 8-9 | TMS 10 | TMS 4e | Loop 2-3 | TMS 7e | Loop 8-9 | TMS 10 | TMS 4 | TMS 11 | TMS 8 | |
| Relative susceptibility | ||||||||||||
| BAC | 1 (1) | 2 (0.5) | 2 (0.5) | 2 (0.5) | 1 (0.25) | 2 (0.13) | 2 (0.06) | 2 (0.06) | 2 (0.06) | 2 (0.06) | 2 (0.06) | 2 (0.25) |
| CHX | 1 (1) | 1 (0.13) | 1 (0.5) | 1 (0.25) | 1 (0.25) | 1 (0.25) | 1 (0.25) | 1 (0.13) | 0.5 (0.13) | 1 (0.25) | 1 (0.25) | 1 (0.25) |
| DAPI | 1 (1) | 4 (1) | 2 (1) | 1 (0.5) | 0.5 (0.5) | 0.5 (1) | 1 (1) | 2 (0.5) | 4 (1) | 1 (1) | 2 (1) | 2 (1) |
| DQ | 1 (1) | 2 (0.5) | 2 (0.5) | 1 (0.25) | 1 (0.25) | 1 (0.25) | 1 (0.25) | 2 (0.5) | 1 (0.25) | 1 (0.25) | 2 (0.5) | 1 (0.5) |
| EtBr | 1 (1) | 2 (0.5) | 2 (0.5) | 2 (0.5) | 1 (0.5) | 2 (0.5) | 1 (0.5) | 2 (0.5) | 2 (0.5) | 2 (1) | 2 (1) | 2 (0.5) |
| H33342 | 1 (1) | 8 (2) | 8 (2) | 1 (1) | 1 (1) | 8 (8) | 1 (1) | 4 (2) | 2 (1) | 2 (1) | 2 (0.5) | 4 (1) |
| Norfloxacin | 1 (1) | 4 (2) | 4 (1) | 0.5 (0.5) | 0.5 (0.5) | 2 (2) | 1 (0.25) | 1 (0.5) | 1 (0.5) | 1 (0.5) | 1 (0.5) | 1 (0.5) |
| Pent | 1 (1) | 4 (2) | 4 (2) | 2 (1) | 1 (0.5) | 2 (0.5) | 2 (0.5) | 2 (0.25) | 2 (0.25) | 1 (1) | 2 (0.25) | 2 (0.25) |
| RD | 1(1) | 2(2) | 2(2) | 1(1) | 1(1) | 1(2) | 0.5(0.5) | 0.5(0.5) | 0.5(0.5) | 1(0.5) | 1(0.5) | 1(1) |
| 0 | ||||||||||||
| TPP | 1(1) | 2(0.25) | 2(0.25) | 2(0.5) | 2(0.5) | 2(0.13) | 2(0.25) | 1(0.25) | 1(0.25) | 1(0.25) | 2(0.25) | 2(0.5) |
| 1 (1)EtBr efflux (%) ± SD | 27.2 ± 4.6 | 82.2 ± 0.4 | 83.1 ± 0.6 | 56.0 ± 5.8 | 40.4 ± 3.0 | 80.4 ± 2.2 | 52.9 ± 2.8 | 63.0 ± 2.0 | 66.4 ± 4.7 | 38.4 ± 5.7 | 60.3 ± 13.0 | 36.2 ± 6.6 |
| % Changef | 202 | 206 | 106 | 49 | 196 | 94 | 132 | 144 | 41 | 122 | 33 | |
| P value | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | 0.001 | |
| IC50 for EtBr efflux (reserpine/paroxetine) | 3/1.8 | 5.2/2.7 | 3/2.6 | 4.6/3.6 | 3.8/>4.5 | 2.4/1.5 | 2.4/2.2 | 1.8/2.5 | 3.2/1.8 | 1/0.8 | 0.6/0.5 | |
Susceptibility, fold reduction in MIC in the presence of 20 μg/ml reserpine (in parentheses), and IC50 data were normalized to K3486, in which wild-type mepA is expressed.
Mutation consisted of duplication of indicated residue.
Pent, pentamidine.
Predicted by SOSUI. Loop x-y, extramembrane loop connecting TMSs x and y (Fig. 1 shows details).
Alternate location predicted by TMHMM algorithm. See the text for details.
Compared to K3486.
Table 3.
Phenotypic effects of mepA mutations found in clinical strains and those produced in conserved negatively charged residues replicated in pK436 and expressed in SA-K2124
| Parameter | Value in strain (mutation)a/location of substitutionb: |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical origin |
Conserved negatively charged residues |
||||||||||||||
| K4916 (A302S)/loop 8–9c | K4920 (A392V)/TMS 11 | K4950 (A397V)/TMS 11 | K5084 (D48A)/loop 1–2 | K5260 (D48E) | K5262 (D48N) | K5086 (E156A)/TMS 4c | K5264 (E156D) | K5266 (E156Q) | K5088 (D183A)/TMS 5 | K5268 (D183E) | K5270 (D183N) | K5090 (E423A)/TMS 12 | K5272 (E423D) | K5274 (E423Q) | |
| Relative susceptibility | |||||||||||||||
| BAC | 4 (0.25) | 2 (2) | 2 (1) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 |
| CHX | 0.5 (0.5) | 0.5 (0.5) | 0.5 (0.5) | 0.5 | 0.5 | 1 | 1 | 0.25 | 0.25 | 0.5 | 0.5 | 0.5 | 1 | 0.25 | 1 |
| DQ | 1 (0.5) | 1 (4) | 1 (1) | 0.5 | 0.5 | 1 | 0.5 | 1 | 0.25 | 1 | 2 | 1 | 1 | 1 | 2 |
| EtBr | 2 (1) | 2 (2) | 1 (0.5) | 1 (1) | 1 (0.5) | 1 (0.5) | 1 (0.5) | 0.5 (0.5) | 0.5 (0.5) | 1 (0.5) | 0.5 (1) | 0.5 (1) | 1 (0.5) | 1 (0.5) | 2 (0.5) |
| Hoechst 33342 | 2 (1) | 2 (4) | 1 (2) | 1 | 0.5 | 0.5 | 1 | 0.5 | 0.25 | 1 | 0.5 | 0.25 | 0.5 | 0.25 | 0.5 |
| Norfloxacin | 1 (1) | 0.5 (1) | 0.5 (1) | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 0.5 |
| Pentd | 2 (1) | 2 (8) | 4 (8) | 2 | 2 | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 1 | 2 |
| TPP | 1 (0.5) | 1 (1) | 1 (0.5) | 1 | 0.5 | 0.5 | 1 | 0.5 | 0.25 | 0.5 | 0.5 | 0.25 | 0.5 | 1 | 1 |
| EtBr efflux (%) ± SD | 52.6 ± 14.9 | 60.1 ± 6.0 | 62.6 ± 2.9 | 48.9 ± 3.0 | 41.4 ± 3.5 | 28.2 ± 7.0 | 5.4 ± 0.8 | 12.5 ± 5.0 | 6.5 ± 3.2 | 9.3 ± 2.4 | 12.9 ± 3.4 | 8.0 ± 1.8 | 18.0 ± 2.0 | 42.1 ± 7.1 | 65.1 ± 6.2 |
| % Changee | 93 | 121 | 130 | 80 | 52 | 4 | −80 | −54 | −76 | −66 | −53 | −71 | −34 | 55 | 139 |
| P value | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | NS | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| IC50 of EtBr efflux (reserpine/paroxetine) | 0.6/0.5 | 0.4/0.7 | 0.6/1.2 | 1.6/ND | 0.6/ND | 0.4/ND | ND/ND | ND/ND | ND/ND | ND/ND | ND/ND | ND/ND | 1/ND | 0.8/ND | 1/ND |
Susceptibility, fold reduction in MIC in the presence of 20 μg/ml reserpine (in parentheses), and IC50 data were normalized to K3486, in which wild-type mepA is expressed. ND, not done.
Predicted by SOSUI.
Alternate location predicted by the TMHMM algorithm. See the text for details.
Pent, pentamidine.
Compared to K3486.
Bioinformatics and statistics.
CLC Main Workbench 6.6.2 (CLC Bio, Cambridge, MA) was used for DNA sequence analyses. The MepA input sequence was that of S. aureus NCTC 8325, which was obtained from available genome sequence data and is identical to that of S. aureus 8325-4 and SH1000. Conserved acidic residues were identified by BLAST analysis using resources available from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). Determination of transmembrane helices and extramembrane loops was predicted using SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui) (28). This algorithm takes into account hydropathy, the presence of amphiphilic residues and their positions, charge, and overall protein length in making its prediction. For comparative purposes, the TMHMM (Trans-Membrane Prediction Using Hidden Markov Models) transmembrane segment prediction algorithm was also used (http://www.cbs.dtu.dk/services/TMHMM/). TMHMM makes its prediction based on hydrophobicity, charge, parameters for typical helical lengths, and the constraint that predicted cytoplasmic and extracytoplasmic loops must alternate. The volumes of substrate molecules were determined using online resources available at http://www.molinspiration.com/services/volume.html. Phyre2 (Protein Homology/Analogy Recognition Engine) was used to predict the three-dimensional structure of MepA (http://www.sbg.bio.ic.ac.uk/phyre2), and I-TASSER (Iterative Threading Assembly Refinement) was used to confirm the Phyre2 prediction (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) (29–31). These algorithms utilize different approaches to arrive at structural predictions. The residues studied were indicated on the MepA structural model using UCSF Chimera 1.6 (32). Statistical analyses were performed employing the t test or Mann-Whitney rank-sum test using modules embedded within SigmaPlot 12.0 (Systat Software, Inc., Chicago, IL).
RESULTS AND DISCUSSION
Gradient plate mutants.
Twenty mutants demonstrating a phenotypic change in the MIC profile were produced using gradient plates containing different MepA substrates plus reserpine. Sequence analyses of the mepRA region identified 9 affected residues, among which were 10 substitution mutations and 1 residue duplication, which were replicated in pK436 and studied in the SA-K2124 background (Table 2). Of significant interest was the fact that on several occasions an identical genetic change was produced upon repeated application of the same selecting conditions. An ATT→TAT (N→Y) mutation at codon 369 was produced four times, twice each with exposure to H33342 or RD. These substrates have similar molecular volumes (451 and 425 Å3, respectively), suggesting they may fit into a common substrate binding site that includes, or is dependent on, residue 369. The beneficial effect of tyrosine at this position with respect to EtBr transport (see below) may be related to improved hydrophobic interactions with that substrate. Alternatively, the tyrosine substitution may improve protein function by altering substrate binding site geometry without actually being part of the site.
In a similar manner, repeated selection of the same mutation occurred with DQ, CHX, BAC, and TPP exposure at codons 312 (ATG→ACG; M→T), 161 (GCA→GTA; A→V), 81 (TCA→TTA; S→L), and 242 (AAA→GAA; K→E), respectively. Mutations within the same codon resulting in a different substitution were observed with EtBr exposure (TTT→GTT or ATT in codon 375 and F→V or I, respectively). Both substitutions replace a large hydrophobic residue with a smaller one, with isoleucine somewhat more beneficial functionally than valine with respect to EtBr transport (66% versus 56% [see below]). Improved protein-EtBr interaction may be achieved by the same mechanisms discussed above with respect to the N369Y substitution.
Pentamidine exposure reproduced the EtBr-induced TTT→GTT (F→V) mutation at codon 375, as well as a mutation similar to that selected by CHX at codon 161 (GCA→ACA; A→T). The molecular volumes of pentamidine and EtBr are similar (325 and 299 Å3, respectively) but are very different from that of CHX (446 Å3). These data are consistent with a large and highly promiscuous binding pocket similar to those described for the multidrug binding proteins QacR and AcrB (33, 34). Finally, DAPI exposure resulted in mutations at two different codons, but the native residue at both positions was methionine. This observation suggests that the native methionine at both positions compromises DAPI recognition within a binding pocket.
All of the aforementioned observations imply that specific residues are involved in the interaction between MepA and different substrates and that more than one substrate can interact with the same residue. This conclusion is supported by pentamidine and EtBr exposure resulting in the F375V substitution and pentamidine and CHX exposure resulting in a substitution at position 161.
The expression of gradient plate-selected mepA derivatives, when replicated in pK436, were high level and similar to that observed for SA-K3486 (range, 0.6 to 2.3×) (data not shown). The contribution of the single chromosomal copy of mepA to data for these or any derivative examined in this study was negligible, as mean plasmid-based mepA expression was 1.4 × 104 (range, 3.8 × 103 to 4.4 × 104) times that of native mepA.
For SA-K3486, in which WT mepA was expressed from pK436 in the SA-K2124 background, MICs (fold reduction in the presence of 20 μg/ml reserpine) in μg/ml were as follows: BAC, 1.25 (16-fold); CHX, 1.25 (16-fold); DAPI, 12.5 (4-fold); DQ, 2.5 (8-fold); EtBr, 25 (8-fold); H33342, 1.25 (4-fold); norfloxacin, 1.25 (4-fold); pentamidine, 25 (4-fold); RD, 0.63 (2-fold); and TPP, 50 (8-fold). Replication of gradient plate-selected mutations in pK436 and expression in SA-K2124 provided the results shown in Table 2, in which all data were normalized to SA-K3486. Increases in MICs to the substrate(s) used in the selection process were observed in all but three instances, i.e., CHX for strains K4670 and K4900 and pentamidine for strain K4904 (Table 2). It is clear that exposure to one substrate may result in the appearance of substitution mutations having interesting pleiotropic effects, including not stably affecting susceptibility to the selecting agent.
The majority of gradient plate substitutions were associated with 2- to 8-fold MIC increases for three or more substrates, augmenting the preexisting MDR phenotype conferred by wild-type MepA. Strain K4670 (A161V) provided an intriguing exception to this, demonstrating a raised MIC to only a single substrate (TPP). A different substitution at the same position (A161T; K4904) was associated with raised MICs to three substrates, not including TPP (BAC, EtBr, and H33342). Different substitutions at F375 (V or I) also conferred different MIC phenotypes. The fact that MIC increases were confined to selected MepA substrates for each studied mutation, and with the exception of N369Y and M312T differed for each mutation, including those at the same position, is consistent with binding site alterations uniquely affecting affinity to specific substrates. The absence of an effect on all substrates by any substitution mutation also eliminates the possibility of improved stability of mutant proteins affecting our data, as does the relative equivalence of MepA protein observed in SA-K2124 expressing wild-type, S81L, and F375I versions of MepA (Fig. 1). Structural analyses will provide insight into the pleiotropic behavior of these mutations, especially those at the same residue.
Fig 1.
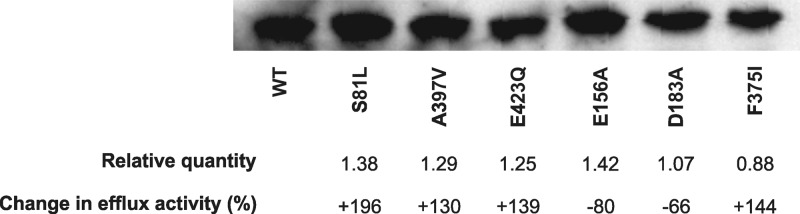
Western blot analyses of MepA derivatives. The amount of MepA and change in efflux activity observed for each mutant, relative to the wild type, are provided.
With the exception of DAPI, H33342, and possibly RD, the effect of reserpine on reversing the MICs of tested compounds against mutant MepA proteins was generally reduced. This was not surprising, as reserpine was included in the gradient plates. This observation suggests that the interaction of reserpine with MepA may involve binding to specific residues, which may overlap those for some, but not all, substrates. Alternatively, reserpine may bind to MepA remote from the substrate binding site(s), resulting in a shift in protein structure so that affinity for selected substrates is reduced. The identified substitution mutations may reduce this binding.
All gradient plate substitution mutants demonstrated improved EtBr efflux, with increases ranging from 33 to more than 200%, with increased efflux accompanied by a concomitant increase in the EtBr MIC in 9 of 11 instances. These data indicate that these mutations augment the affinity for EtBr capture and/or the efficiency of its translocation, possibly as a result of a favorable modification of the geometry of the EtBr binding site(s). This modification may include a salutary reorientation of critical charged, polar, or even hydrophobic residues that change affinity for EtBr. These changes are not dependent on EtBr itself, as exposure to it did not result in production of all mutations augmenting its efflux. Specific changes induced by substrates other than EtBr may only be possible with that substrate but may have the same result of improved EtBr efflux.
Discordance between the EtBr MIC and augmented efflux occurred in strains K4670 and K4898. The unique nature of the A161V substitution has been discussed previously. The conditions under which MIC testing and efflux are determined are not the same, a fact likely contributing to the observed discordances. Differences include their time courses, preconditioning strains with carbonyl cyanide m-chlorophenyl hydrazone and EtBr loading for the efflux assay and the absence of this in MIC testing, and the presence of an inducing concentration of tetracycline throughout MIC testing and its absence subsequent to organism harvest for the efflux assay. In addition, an MIC increase to a level greater than 25 (the MIC of SA-K3486) but less than 50 μg/ml would be missed in our MIC testing scheme. Thus, it is not surprising that these parameters may occasionally be dissociated.
EtBr MICs increased no more than 2-fold regardless of the magnitude of increased EtBr efflux observed. This likely is related to the already high EtBr MIC conferred by WT MepA (25 μg/ml), and maximally functioning MepA may not be capable of increasing this to values higher than 50 μg/ml. Our recent evaluation of clinical MDR-efflux pump-overexpressing strains, in which we identified 41 strains overexpressing mepA with or without increased expression of other pump genes, supports this hypothesis. None of these strains had an EtBr MIC above 50 μg/ml, including two in which EtBr efflux was nearly 80% (17).
IC50 determinations demonstrated that augmented EtBr efflux was associated with nearly 2-fold or greater resistance of the efflux process to inhibition by reserpine and/or paroxetine. The effect of reserpine on EtBr MICs correlated with IC50 data in the majority of mutants. Exceptions included one strain (K4904) with an unchanged reserpine effect on the MIC but a 3.2-fold increase in the IC50 and a second strain (K4984) showing a reduction in the reserpine effect on the EtBr MIC in the absence of a change in the IC50. The hypotheses advanced above regarding the occasional lack of correlation between the EtBr MIC and augmented EtBr efflux also apply here, as the approaches employed for their determinations are vastly different. It is also possible that EtBr efflux and inhibitor sensitivity are not strictly linked. Mutations affecting only inhibitor susceptibility are likely possible, as has been described for the Bmr MDR efflux protein of Bacillus subtilis (35).
The SOSUI- and TMHMM-predicted secondary structures of MepA are provided in Fig. 2. The two predictions are very similar, with the only discrepancies among gradient plate mutants being the locations of A161 and K242. Phyre2 modeling of MepA supports the TMHMM prediction for A161 (loop 4-5) and the SOSUI prediction for K242 (TMS 7) (Fig. 3); all three models agree on the positions of the remaining gradient plate mutants. It is noteworthy that all but S81 and A161 are located in the carboxy-terminal half of MepA (TMSs 7, 8, 10, and 11 and selected intervening cytoplasmic loops). Additionally, those residues that are within TMSs cluster near the cytoplasmic face of the protein. This positioning suggests that this region of MepA is instrumental in substrate recognition, capture, and transport, either directly or indirectly by influencing the geometry of a substrate binding pocket. It may be that substrate is captured from within the inner lipid bilayer of the cytoplasmic membrane, as well as at the bilayer-cytoplasm interface. A similar mechanism of substrate capture has been proposed for the AcrB MDR pump of Escherichia coli, differing from the proposed MepA mechanism in that substrate capture likely occurs at the periplasm-cytoplasmic membrane interface (12). It is noteworthy that this region of V. cholerae NorM has been shown to be involved in cation binding, which raises the possibility that the mechanism of increased EtBr transport by MepA mutants may be the effect of combined improvements in substrate and antiported cation translocation (14).
Fig 2.

Secondary structure of MepA, predicted using SOSUI (A) and TMHMM (B). Positions at which substitution mutations resulted in ≥50% increase in efflux activity are indicated in green, those resulting in increases of less than 50% in yellow, and those resulting in loss of efflux activity in red. Substitutions at positions 48 and 423 gave variable results depending on the particular amino acid present. This characteristic is illustrated by split colors for these residues.
Fig 3.

Residues at which substitution mutations affect efflux function, mapped onto the Phyre2 predicted structure of MepA. Wild-type side chains are depicted. The colors are the same as in Fig. 2, with the exception of residues 48 and 423, which are indicated as fully up function here (green). (A) Front. (B) Rear, rotated 180o from panel A. (C) Top. (D) Bottom. The cytoplasmic face of the protein is toward and away from the reader in panels C and D, respectively, as indicated by the arrows.
Naturally occurring mutants found in mepA-overexpressing clinical strains.
Unique or low-frequency MepA substitution mutations discovered among mepA-overexpressing clinical strains, when replicated in pK436, were expressed at similarly high levels (range, 0.8 to 1.7 times that of SA-K3486 [data not shown]). The majority of these had no effect on MepA function, including S12L, A123M, K140E, M166I, I227F, L229F, Y268H, V328I, V329L, I398V, L428F, and D439N. However, three substitutions did affect the EtBr efflux phenotype (A302S, A392V, and A397V) (Table 3). Compared to wild-type MepA, all were up function in nature with respect to EtBr efflux (93 to 130% increase), as well as conferring a 2-fold increase in the MICs of two to four MepA substrates. It is highly unlikely that changes in MepA quantity played a role in the observed up-function phenotypes observed. This was confirmed for the A397V derivative, in which the quantity of MepA protein was similar to that in the wild type (Fig. 1).
In contrast to gradient plate mutants, both increases and decreases in the effect of reserpine on substrate MICs were observed. The most pronounced difference was seen for pentamidine, where, despite a 2- to 4-fold increase in the MIC, a significantly greater reserpine effect was noted for the A392 and A397 mutants. These phenotypic differences from gradient plate mutants are most likely related to the fact that these clinical mutants arose in the absence of reserpine pressure. Nevertheless, similar to gradient plate mutants, the combination of efflux augmentation with changes in reserpine phenotype suggests these naturally occurring mutations also change the dynamics of the reserpine-MepA interaction.
SOSUI predicts A302 to be the first residue at the cytosol-membrane interface of TMS 8 (Fig. 2), while TMHMM predicts it to be in TMS 8. The latter is supported by Phyre2 modeling (Fig. 3). It is not surprising that discrepancies in positioning of residues near a membrane surface of MepA occur with different prediction algorithms. Consistent with our findings that the majority of gradient plate substitution mutants affecting MepA function lie within the carboxy half of MepA and near the cytoplasmic face of the protein, residues A302, A392, and A397 also lie in this region. These data provide additional support for the hypothesis that this region of MepA is important with respect to substrate and/or antiported cation capture and transport.
Substitution mutations at conserved negatively charged residues.
As discussed previously, negatively charged residues are important in the coordination and ultimate translocation of hydrophobic cation substrates, as well as in antiported cation exchange (i.e., Na+ or H+), in MATE, MFS, and SMR family proteins (14, 24–26). The possibility of an important functional role is increased if such residues are highly conserved, such as those we chose for study. The expression levels of all mepA mutants we constructed encoding substitutions at negatively charged residues were similar to that of SA-K3486 (range, 0.8 to 1.8×) (data not shown). Consistent with expression data, protein quantitation for derivatives containing E156A, D183A, and E423Q substitutions was similar to that of the wild type (Fig. 1). These results support the conclusion that any functional changes observed are related to MepA itself and not a secondary effect of altered transcription or translation.
Substitution of alanine for glutamate at position 156 or aspartate at position 183 resulted in a profound reduction in EtBr efflux (Table 3). Alternative substitutions designed to maintain charge (E↔D) or residue size (E→Q or D→N) had no appreciable salutary effect, in that EtBr efflux remained severely compromised. Clearly, the specific wild-type negatively charged residues at both positions are critical to MepA function. SOSUI predicts that these residues are located in TMSs 4 (E156) and 5 (D183); TMHMM differs by locating E156 in loop 4-5. Phyre2 supports the SOSUI model for E156. These residues may be involved in sequential transfer of a cation, either a substrate or an antiported ion, such as Na+ or H+. Alternatively, substitution at either position may disrupt the positioning of helices in such a manner as to inactivate the protein.
Substitutions for D48 had variable effects on MepA function. SOSUI, TMHMM, and Phyre2 locate D48 in loop 1-2 and E423 in TMS 12. Replacement of the native aspartate with either alanine (+80%) or glutamate (+52%) at position 48 resulted in an up-function phenotype, whereas maintenance of the residue size by a D→N change had no effect on function (Table 3). These data indicate that neither size nor charge at this position is essential. The up-function effect of alanine or glutamate may result from a beneficial repositioning of neighboring helices, resulting in improved affinity of EtBr within the substrate binding site.
Substitutions for E423 also had variable effects, but they differed from those seen at position 48. A loss of efflux function was observed when glutamate was replaced with the much smaller alanine (−34%) (Table 3). Replacement of glutamate with the slightly smaller aspartate provided a modest gain-of-function phenotype (+55%), and substitution of the similarly sized glutamine provided a larger gain of function (+139%). Changes in both size and charge had a significant negative functional impact (E→A), whereas charge removal with size preservation (E→Q) actually improved efflux function. These data support the conclusion that size at position 423 has greater importance than charge. The unexpected observation of augmented efflux activity associated with a modest size reduction (E→D) accompanied by maintenance of charge is difficult to explain with the data in hand. Perhaps the small size difference between glutamate and aspartate (∼10%) allows a beneficial alteration in binding site geometry, as hypothesized for position 48.
For both naturally occurring and site-directed mutations at conserved negatively charged residues, several strains demonstrated an apparent disconnect between EtBr efflux and the MIC. Hypotheses to explain this phenomenon have been provided previously.
Functional considerations based on in silico modeling.
Figure 3 provides the Phyre2-predicted model of MepA, with the positions of substitutions of functional significance indicated. Modeling included 98% of MepA residues, and the calculated confidence of the model was 100%. I-TASSER generated a highly similar model using 99% of MepA residues, with a predicted accuracy of 95% (data not shown). Because of the similarity in models, the Phyre2 prediction was chosen for further analyses. Generation of the model was limited by the paucity of solved MATE family structures, of which there is only NorM of V. cholerae (14). Superimposition of the NorM structure on the Phyre2 model for MepA revealed the two to be highly similar, but not identical (data not shown). The Phyre2 model is also presented in Fig. 4, except that helices are depicted as cylinders to better visualize their association with each other, as well as with the presumptive central-cavity region of the protein.
Fig 4.
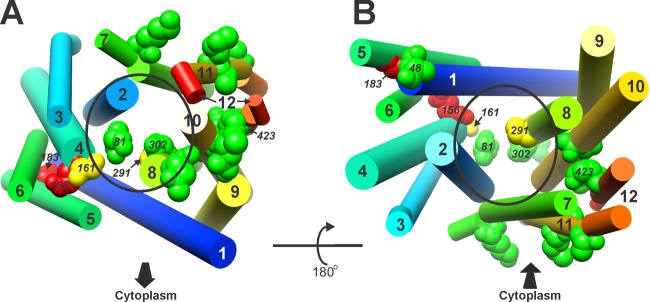
Phyre2 model of MepA, cylindrical view. Residues and colors are the same as in Fig. 2. (A and B) The cytoplasmic face of the protein is toward (A) and away from (B) the reader, as indicated and correlating with Fig. 3C and D. Helix numbers and specific residues are indicated, as is the presumptive central cavity (oval).
The solved structure of V. cholerae NorM is in the outward-facing orientation, and thus, the Phyre2 model of MepA has the same conformation. Helix positioning and residue orientation are likely to change as the molecule shifts to the inward-facing structure. It is highly likely that substrate affinity will be greatest when MepA is inwardly facing and that affinity will decrease upon a shift to the outward-facing conformation, favoring substrate release. The affinity for a cotransported cation, either Na+ or H+, would be expected to be opposite to that of the substrate. This mechanism of substrate translocation has been referred to as the “alternating-access model” and has been established for the MFS pump LacY and the ABC multidrug resistance pump MsbA (36, 37). While it is reasonable to presume that MepA functions in a similar manner, evidence supporting this will require the determination of structure in the absence, as well as the presence, of substrate. These conditions will favor outward- and inward-facing conformations, respectively.
The MepA model identified helices 1, 2, 4, 7, 8, and 10 as having contact with the central region of the protein. These data are consistent with the known role of helices 7, 8, and 10 of V. cholerae NorM in cation binding and provide support for the accuracy of the model. Also similar to NorM, hydrophobic or aromatic residues are well represented among residues facing this region (18 of 27) (data not shown). Of the residues investigated in this study, S81, A161, M291, and A302 appear to lie within the presumptive central cavity, with D48 positioned near the external opening (Fig. 4). A shift to the inward-facing orientation will alter these relationships. What is clear is that the highly functionally significant negatively charged residues (E156 and D183) are not proximate to this region in the outward-facing orientation, suggesting that in this conformation, they are not available for electrostatic interactions with cationic ions or molecules. They may be available for such interactions in the inward orientation, rotating to a more favorable location as the protein transitions. As noted previously, membrane-embedded negatively charged residues have been shown to have a role in Na+-substrate exchange in the NorM MATE protein of V. parahaemolyticus, and these MepA residues may have a similar function (15). If these residues play a role in substrate transport, their positioning in the outward-facing orientation is consistent with a low-affinity state in which their negative charges are unavailable for coordination with cationic compounds.
Residues with an intimate relationship with the central cavity may have a close association with the substrate or antiported cation. The salutary effect of the S81L substitution may be related to the changes in both residue size and polarity, with leucine being larger and nonpolar. Substitution of isoleucine for methionine at position 291 reduces the size of the side chain projecting into the central cavity, perhaps providing better hydrophobic interactions with substrates, including EtBr. Substitution of either valine or threonine for alanine at position 161 results in a modest improvement in efflux efficiency (+40 to 50%); these substitutions provide nearly identical increases in bulk at this position, with molecular weights of 117 and 119, respectively. Bulk appears to be most important, as the polarity provided by threonine has no significant positive or negative impact compared to valine. Finally, replacement of A302 with serine provides increased polarity in this region of the central cavity, which may provide a more favorable interaction with polar/charged regions of substrates.
Examination of the model with respect to residues facing the putative central cavity revealed that the side chain of a glutamic acid at position 295 in TMS 8 projected into this region. This is not a conserved residue but rather is unique to S. aureus MepA and homologues found among other staphylococci, such as S. saprophyticus and S. haemolyticus. In order to test the possible functional importance of this residue, we used site-directed mutagenesis as described for the other negatively charged residues examined here. A significant loss of efflux proficiency was observed for E295A and E295D mutants (−67 and −83% compared to wild-type MepA, respectively) (data not shown). However, conservation of residue size by an E295Q substitution resulted in relative maintenance of efflux activity (−19%) and reserpine susceptibility (IC50, 1.6; normalized to wild-type MepA). Clearly, residue size, but not charge, at position 295 is of the greatest functional importance. Smaller residues here clearly affect central-cavity geometry in a negative way.
Excluding the negatively charged residues already discussed, other functionally significant residues outside the predicted central-cavity region include K242, N308, M312, N369, F375, M391, A392, and A397. These residues lie entirely within TMSs 7 to 11 and the intervening cytoplasmic loops. The substitutions at these positions that we studied were all up function in nature and likely conferred this effect by a salutary repositioning of critical residues in a manner productive for EtBr transport. Crystal structure analyses will be required to substantiate or refute this hypothesis.
Concluding remarks.
The model of MepA presented here provides a framework upon which mechanistic considerations can be proposed and further tested. The observations in hand, while mainly descriptive, will be of great use in refining MepA structural data. Such data in the presence of substrates would provide great detail with respect to which residues of MepA interact with different substrates. It is likely that substrates bind to MepA in the same general region but through interactions with different residues lining an expansive drug binding cavity. This has been shown to be true for other multidrug binding proteins, such as QacR and AcrB (11, 38). While it is possible that our data may be limited by the lack of protein quantitation in all test strains, complete gene expression data and Western blots of selected strains demonstrating a large phenotypic variance from wild-type MepA suggest that this is not likely to be a significant concern.
We have established the critical nature of E156 and D183 in the substrate translocation process, but the exact manner of their involvement in this process remains uncertain. Our findings also support the conclusion that substrate and inhibitor can interact with MepA in similar locations and that certain residues are crucial to interaction with selected substrates. The latter point is supported by the production of mutations at the same positions with repeated exposure to the same substrate. An alternating-access mechanism for substrate translocation by MepA is feasible but awaits confirmation by structural determinations in the presence and absence of substrate.
ACKNOWLEDGMENTS
This work was supported by Veterans Affairs Biomedical Laboratory Research and Development funds (grant no. 1IO1BX000465).
We thank Muthiah Kumaraswami for critical review of the manuscript.
Footnotes
Published ahead of print 23 November 2012
REFERENCES
- 1.Li XZ, Nikaido H. 2009. Efflux-mediated drug resistance in bacteria: an update. Drugs 69:1555–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borges-Walmsley MI, McKeegan KS, Walmsley AR. 2003. Structure and function of efflux pumps that confer resistance to drugs. Biochem. J. 376:313–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morita Y, Kataoka A, Shiota S, Mizushima T, Tsuchiya T. 2000. NorM of Vibrio parahaemolyticus is an Na(+)-driven multidrug efflux pump. J. Bacteriol. 182:6694–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaatz GW, McAleese F, Seo SM. 2005. Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob. Agents Chemother. 49:1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumaraswami M, Schuman JT, Seo SM, Kaatz GW, Brennan RG. 2009. Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res. 37:1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaatz GW, DeMarco CE, Seo SM. 2006. MepR, a repressor of the Staphylococcus aureus MATE family multidrug efflux pump MepA, is a substrate-responsive regulatory protein. Antimicrob. Agents Chemother. 50:1276–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaatz GW, Moudgal VV, Seo SM, Hansen JB, Kristiansen JE. 2003. Phenylpiperidine selective serotonin reuptake inhibitors interfere with multidrug efflux pump activity in Staphylococcus aureus. Int. J. Antimicrob. Agents 22:254–261 [DOI] [PubMed] [Google Scholar]
- 8.Kaatz GW, Moudgal VV, Seo SM, Kristiansen JE. 2003. Phenothiazines and thioxanthenes inhibit multidrug efflux pump activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 47:719–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587–593 [DOI] [PubMed] [Google Scholar]
- 10.Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. 2004. Structure of the periplasmic component of a bacterial drug efflux pump. Proc. Natl. Acad. Sci. U. S. A. 101:9994–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu EW, Aires JR, McDermott G, Nikaido H. 2005. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J. Bacteriol. 187:6804–6815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Husain F, Nikaido H. 2010. Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol. Microbiol. 78:320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su CC, Long F, McDermott G, Shafer WM, Yu EW. 2008. Crystallization and preliminary X-ray diffraction analysis of the multidrug efflux transporter NorM from Neisseria gonorrhoeae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64:289–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X, Szewczyk P, Karyakin A, Evin M, Hong WX, Zhang Q, Chang G. 2010. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467:991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otsuka M, Yasuda M, Morita Y, Otsuka C, Tsuchiya T, Omote H, Moriyama Y. 2005. Identification of essential amino acid residues of the NorM Na+/multidrug antiporter in Vibrio parahaemolyticus. J. Bacteriol. 187:1552–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster TJ. 1998. Molecular genetic analysis of staphylococcal virulence. Methods Microbiol. 27:433–454 [Google Scholar]
- 17.Kosmidis C, Schindler BD, Jacinto PL, Patel D, Bains K, Seo SM, Kaatz GW. 2012. Expression of multidrug resistance efflux pump genes in clinical and environmental isolates of Staphylococcus aureus. Int. J. Antimicrob. Agents 40:204–209 [DOI] [PubMed] [Google Scholar]
- 18.Frempong-Manso E, Raygada JL, DeMarco CE, Seo SM, Kaatz GW. 2009. Inability of a reserpine-based screen to identify strains overexpressing efflux pump genes in clinical isolates of Staphylococcus aureus. Int. J. Antimicrob. Agents 33:360–363 [DOI] [PubMed] [Google Scholar]
- 19.DeMarco CE, Cushing LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW. 2007. Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 51:3235–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.CLSI 2006. Approved standard M7-A7. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 7th ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 21.Kaatz GW, Seo SM, O'Brien L, Wahiduzzaman M, Foster TJ. 2000. Evidence for the existence of a multidrug efflux transporter distinct from NorA in Staphylococcus aureus. Antimicrob. Agents Chemother. 44:1404–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryson V, Szybalski W. 1952. Microbial selection. Science 115:45–51 [PubMed] [Google Scholar]
- 23.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74:5463–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yerushalmi H, Mordoch SS, Schuldiner S. 2001. A single carboxyl mutant of the multidrug transporter EmrE is fully functional. J. Biol. Chem. 276:12744–12748 [DOI] [PubMed] [Google Scholar]
- 25.Braibant M, Chevalier J, Chaslus-Dancla E, Pages JM, Cloeckaert A. 2005. Structural and functional study of the phenicol-specific efflux pump FloR belonging to the major facilitator superfamily. Antimicrob. Agents Chemother. 49:2965–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paulsen IT, Brown MH, Littlejohn TG, Mitchell BA, Skurray RA. 1996. Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: membrane topology and identification of residues involved in substrate specificity. Proc. Natl. Acad. Sci. U. S. A. 93:3630–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel D, Kosmidis C, Seo SM, Kaatz GW. 2010. Ethidium bromide MIC screening for enhanced efflux pump gene expression or efflux activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 54:5070–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirokawa T, Boon-Chieng S, Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379 [DOI] [PubMed] [Google Scholar]
- 29.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 33.Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. 2001. Structural mechanisms of QacR induction and multidrug recognition. Science 294:2158–2163 [DOI] [PubMed] [Google Scholar]
- 34.Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr 2003. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science 300:976–980 [DOI] [PubMed] [Google Scholar]
- 35.Ahmed M, Borsch CM, Neyfakh AA, Schuldiner S. 1993. Mutants of the Bacillus subtilis multidrug transporter Bmr with altered sensitivity to the antihypertensive alkaloid reserpine. J. Biol. Chem. 268:11086–11089 [PubMed] [Google Scholar]
- 36.Dong J, Yang G, McHaourab HS. 2005. Structural basis of energy transduction in the transport cycle of MsbA. Science 308:1023–1028 [DOI] [PubMed] [Google Scholar]
- 37.Jiang X, Nie Y, Kaback HR. 2011. Site-directed alkylation studies with LacY provide evidence for the alternating access model of transport. Biochemistry 50:1634–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters KM, Sharbeen G, Theis T, Skurray RA, Brown MH. 2009. Biochemical characterization of the multidrug regulator QacR distinguishes residues that are crucial to multidrug binding and induction of qacA transcription. Biochemistry 48:9794–9800 [DOI] [PubMed] [Google Scholar]
- 39.Novick R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155–166 [DOI] [PubMed] [Google Scholar]
- 40.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. 2002. sB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 184:5457–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price CT, Kaatz GW, Gustafson JE. 2002. The multidrug efflux pump NorA is not required for salicylate-induced reduction in drug accumulation by Staphylococcus aureus. Int. J. Antimicrob. Agents 20:206–213 [DOI] [PubMed] [Google Scholar]
- 42.Kaatz GW, Moudgal VV, Seo SM. 2002. Identification and characterization of a novel efflux-related multidrug resistance phenotype in Staphylococcus aureus. J. Antimicrob. Chemother. 50:833–838 [DOI] [PubMed] [Google Scholar]
- 43.Kaatz GW, Seo SM. 2004. Effect of substrate exposure and other growth condition manipulations on norA expression. J. Antimicrob. Chemother. 54:364–369 [DOI] [PubMed] [Google Scholar]
- 44.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 69:7851–7857 [DOI] [PMC free article] [PubMed] [Google Scholar]