

Abstract
In 2004, a previously undiscovered mycobacterium resembling Mycobacterium ulcerans (the agent of Buruli ulcer) was reported in an outbreak of a lethal mycobacteriosis in a laboratory colony of the African clawed frog Xenopus tropicalis. This mycobacterium makes mycolactone and is one of several strains of M. ulcerans-like mycolactone-producing mycobacteria recovered from ectotherms around the world. Here, we describe the complete 6,399,543-bp genome of this frog pathogen (previously unofficially named “Mycobacterium liflandii”), and we show that it has undergone an intermediate degree of reductive evolution between the M. ulcerans Agy99 strain and the fish pathogen Mycobacterium marinum M strain. Like M. ulcerans Agy99, it has the pMUM mycolactone plasmid, over 200 chromosomal copies of the insertion sequence IS2404, and a high proportion of pseudogenes. However, M. liflandii has a larger genome that is closer in length, sequence, and architecture to M. marinum M than to M. ulcerans Agy99, suggesting that the M. ulcerans Agy99 strain has undergone accelerated evolution. Scrutiny of the genes specifically lost suggests that M. liflandii is a tryptophan, tyrosine, and phenylalanine auxotroph. A once-extensive M. marinum-like secondary metabolome has also been diminished through reductive evolution. Our analysis shows that M. liflandii, like M. ulcerans Agy99, has the characteristics of a niche-adapted mycobacterium but also has several distinctive features in important metabolic pathways that suggest that it is responding to different environmental pressures, supporting earlier proposals that it could be considered an M. ulcerans ecotype, hence the name M. ulcerans ecovar Liflandii.
INTRODUCTION
An interesting member of the Mycobacterium marinum and Mycobacterium ulcerans complex was discovered in the summer of 2001, when an outbreak of generalized cutaneous lesions developed in a colony of Xenopus tropicalis at the University of Berkeley in California (1). Infected frogs developed granulomatous skin lesions along with coelomic distention, generalized edema, and septicemia (1). Cytological examinations confirmed the presence of acid-fast bacilli in smears from the liver, spleen, kidney, and skin. Based on histopathology and some molecular testing, it was concluded that these frogs were suffering from a mycobacteriosis caused by a Mycobacterium ulcerans-like bacterium (1). There have been ongoing reports and high lethality of this disease in captive frogs across the United States and those imported from the United States to Europe (2–4), but this mycobacterium has not been reported to cause illness in humans. Further characterization of the frog pathogen revealed that it harbored the M. ulcerans pMUM megaplasmid and produced mycolactone E, a unique structural variant of the polyketide toxin that is key for pathogenesis in M. ulcerans (5, 6). Limited genotype analysis suggests that a single clone of this pathogen is circulating worldwide in institutions housing and breeding anurans. The species name “Mycobacterium liflandii” was proposed, although as the authors admitted at that time, the data were lacking to conclude that this mycobacterium constituted a separate species (6).
A recent genomic study of 35 M. marinum-M. ulcerans complex isolates confirmed earlier indications that all mycolactone-producing mycobacteria, including those strains classically considered M. ulcerans, evolved by a process of lateral gene transfer and reductive evolution from a common M. marinum-like progenitor (7–9). These comparisons also show at least three discrete deep-branching lineages of mycolactone producers, which include the strains predominantly infecting ectotherms and those strains commonly causing Buruli ulcers in humans (7–9). All lineages showed evidence of strong selective pressures acting on the same cell wall-associated genes, but the lineages differed in the extent of genome reduction, suggesting that each lineage might be adapting to slightly different niche environments (8). The frog pathogen belongs to lineage 1, while M. ulcerans isolates from Africa and Australia belong to lineage 3. Here we will refer to this frog pathogen as M. ulcerans ecovar Liflandii, in line with our proposition that it and other lineage-specific isolates should be considered ecotypes of M. ulcerans (8, 10).
Like other M. marinum-M. ulcerans complex members, M. ulcerans ecovar Liflandii grows preferentially around 32°C. However, it exhibits several distinguishing microbiological characteristics. M. ulcerans ecovar Liflandii forms rough light-orange nonphotochromogenic colonies on Middlebrook 7H11 agar supplemented with oleic acid, albumin, dextrose, and catalase (1, 11). It grows better on charcoal medium than on Lowenstein-Jensen (LJ) medium, whereas classical (lineage 3) M. ulcerans isolates do not grow on charcoal. Like M. ulcerans, antibiograms of M. ulcerans ecovar Liflandii indicate resistance to isoniazid, ethambutol, and ethionamide, but M. ulcerans ecovar Liflandii is reportedly also resistant to rifampin and clarithromycin (11).
To date, the M. marinum-M. ulcerans complex is represented by only two fully assembled genome sequences, an M. ulcerans isolate from a Buruli ulcer patient isolated in Ghana in 1999 and an M. marinum clinical isolate obtained from a patient in the United States in 1994 (12, 13). The high complexity of M. ulcerans genomes, with the repeat-rich nature of the mycolactone polyketide synthase genes (harbored on the pMUM plasmid) and >200 chromosomal copies of IS2404, has been a barrier to completing assemblies of genome sequences from other members of this complex. Here, we describe and compare the sequenced and fully assembled chromosome of M. ulcerans ecovar Liflandii, which, together with our previous description of the pMUM002 mycolactone plasmid, represents the first complete genome for the strains of M. ulcerans that are increasingly associated with epizoonotics in fish, frogs, and other ectotherms around the world.
MATERIALS AND METHODS
Strain and culture conditions.
M. ulcerans ecovar Liflandii 128FXT was originally isolated from infected Xenopus tropicalis at the University of California, Berkeley (1). The isolate was cultured on Brown and Buckle egg yolk agar slopes at 30°C. Rifampin MIC testing was performed using mycobacterial growth indicator tubes (MGITs) as described previously (14). Briefly, M. ulcerans ecovar Liflandii at a McFarland standard of 0.5 was prepared as recommended by the manufacturer (Becton Dickinson) and diluted 1:5 (vol/vol) in sterile saline. MGITs were enhanced with 800 μl of MGIT streptomycin, isoniazid, rifampin, and ethambutol (SIRE) supplement (Becton Dickinson) and inoculated with 0.5 ml of the diluted bacterial suspension. MGITs contained 2-fold dilutions of rifampin from 0.0625 μg/ml to 16 μg/ml. Experiments were established in triplicate for each dilution. MGITs were incubated at 30°C and examined daily for 4 weeks at a wavelength of 365 nm while noting the times to fluorescence.
DNA methods.
Genomic DNA was extracted from M. ulcerans ecovar Liflandii as described previously (9). Standard methods were used for PCR and Sanger sequencing.
Whole-genome sequencing and assembly.
Roche 454 GS-FLX sequencing was employed to obtain 260,654 single-end reads (106.3 Mbp) and assembled using gsAssembler v2.5.3 into 91 scaffolds containing a total of 622 contigs. The scaffolds were ordered by reference to an optical map (see below) and BLAT searches in Projector 2 against the M. marinum M and M. ulcerans Agy99 reference genomes (15). Genome finishing was managed with Gap4 (16). Specific oligonucleotides were designed to contig-flanking sequences, and PCR with Sanger sequencing was used to close gaps. Wherever it was established that a gap contained a single copy of IS2404, the gap was filled with a copy of IS2404 from the previously sequenced M. ulcerans Agy99. It is important to note, therefore, that the exact sequence of each copy of IS2404 in M. ulcerans ecovar Liflandii has not been established. A 3-kbp Roche 454 GS-FLX mate pair library was also constructed and yielded 218,423 reads (63.6 Mbp). Finally, 13,778,118 Illumina paired-end sequencing reads (487.61 Mbp) obtained from a previous project (8) were mapped to the final assembly using an in-house Python utility called Nesoni (Victorian Bioinformatics Consortium) (P. Harrison and T. Seemann, unpublished data) with SHRiMP 2.2 (51). Nesoni was used to do a global variant analysis, generate a list of differences, and correct 454 sequencing errors.
Optical mapping.
An NheI optical map of M. ulcerans ecovar Liflandii was prepared by OpGen (Madison, WI, USA) to guide assembly. Contigs were aligned to the optical map using MapSolver software (v3.2.0).
Annotation and comparative genome analysis.
Genome annotation was performed by Prokka (Victorian Bioinformatics Consortium) (T. Seemann, unpublished data) utilizing the previously annotated M. marinum M and M. ulcerans Agy99 genomes as referenced (12, 13). Manual curation of the annotation was then performed using Wasabi, a Web-based annotation editor and database, as described previously (12). Regions of difference (RDs) were identified by mapping M. ulcerans Agy99 and M. marinum de novo contigs to the assembled M. ulcerans ecovar Liflandii genome. This mapping and other comparisons were visualized with Circos (17), Mauve (18), and the Artemis Comparison Tool (ACT) (19). Secondary metabolite biosynthesis gene clusters were identified using antiSMASH (20). Additional pseudogene clusters that were too degraded for detection by antiSMASH were identified by MultiGeneBlast (http://multigeneblast.sourceforge.net/) (M. H. Medema, E. Takano, and R. Breitling, unpublished data). The final manually determined cutoff used to define gene cluster orthology was based on the premise that at least 40% of the genes in gene clusters should have at least 60% amino acid sequence identity. All gene clusters connected by orthology were grouped together into a single gene cluster family. Chromosomal maps of the biosynthetic gene clusters were generated with in-house Python scripts using pySVG (http://codeboje.de/pysvg/).
Nucleotide sequence accession number.
The chromosome sequence of M. ulcerans ecovar Liflandii 128FXT was submitted to GenBank under BioProject number PRNJA128960.
RESULTS AND DISCUSSION
General features of the M. ulcerans ecovar Liflandii genome.
M. ulcerans ecovar Liflandii has a 6,399,542-bp genome comprising a single circular 6,208,954-bp chromosome and the previously described 190,588-bp pMUM002 mycolactone megaplasmid, with G+C contents of 65.61% and 62.9%, respectively (21). The chromosome is predicted to contain 4,994 protein-coding DNA sequences (CDSs) and 436 pseudogenes (Fig. 1). One rRNA operon and 50 tRNA genes were identified (Table 1). Sequence assembly was complicated by the presence of 231 chromosomal copies of the previously described M. ulcerans insertion sequence IS2404 (208 complete copies) (Fig. 1). The integrity of the finished sequence was verified by reference to a high-resolution NheI whole-chromosome restriction map (Fig. 2A).
Fig 1.

Genome map of M. ulcerans ecovar Liflandii. DNA sequence mapping of the three mycobacterial genomes using M. ulcerans ecovar Liflandii as a reference displayed in Circos. The tracks from inside to outside represent the GC skew, GC content, M. marinum M, M. ulcerans Agy99, ancestral copies of IS2404, all M. ulcerans ecovar Liflandii IS2404 copies, pseudogenes present in both mycolactone-producing strains but absent in M. marinum M, all M. ulcerans ecovar Liflandii pseudogenes, reverse-strand CDSs, forward-strand CDSs, the assembled M. ulcerans ecovar Liflandii genome, and major regions of difference in MULRD3 and MULRD7. Mapping to pMUM002 is also presented, with sequences from M. marinum M excluded from the plot. The sequences for M. ulcerans Agy99 and M. marinum M were obtained from Stinear et al. (12, 13).
Table 1.
Comparison of key genomic features between the three fully assembled genomes of the M. marinum-M. ulcerans complex
| Feature | M. ulcerans ecovar Liflandii | M. ulcerans Agy99 | M. marinum M |
|---|---|---|---|
| Chromosome size (bp) | 6,208,955 | 5,631,606 | 6,636,827 |
| Size of pMUM plasmid (bp) | 190,588 | 174,155 | Not present |
| G+C content (%) | 65.62 | 65.47 | 65.73 |
| No. of CDSs | 4,994 | 4,160 | 5,424 |
| No. of unique CDSs | 268 | 30 | 395 |
| No. of pseudogenes | 436 | 771 | 65 |
| No. of IS2404 copies | 239 | 213 | Not present |
| No. of IS2606 copies | 4 | 91 | Not present |
Fig 2.
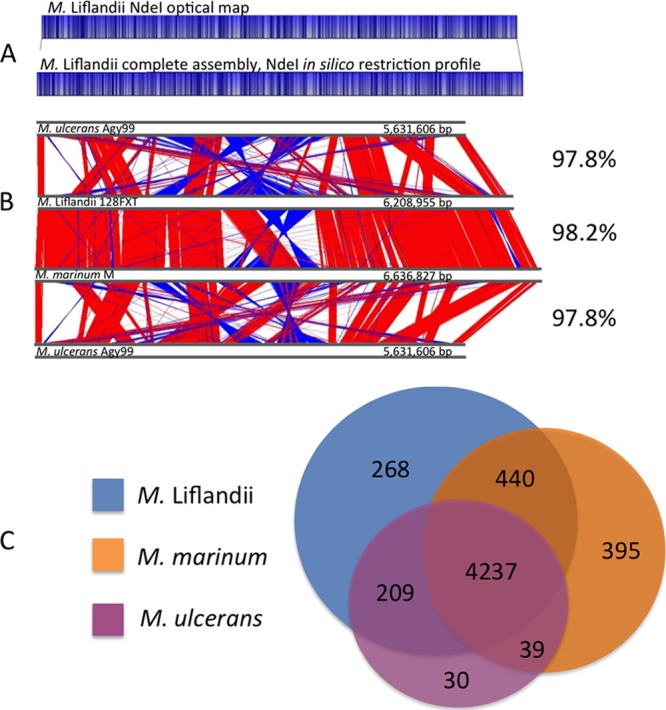
Comparisons of M. ulcerans ecovar Liflandii. (A) Alignment of assembled M. ulcerans ecovar Liflandii chromosomes against an NheI optical map. (B) ACT comparisons of M. ulcerans Agy99 to M. ulcerans ecovar Liflandii, M. marinum M, and M. ulcerans Agy99. The regions in red indicate identical sequences and orientation, and those in blue indicate identical sequences in the reverse orientation. Indicated are the percent nucleotide identities among core orthologs. (C) Venn diagram highlighting the number of orthologs between M. marinum M, M. ulcerans Agy99, and M. ulcerans ecovar Liflandii.
Comparisons of the complete chromosomal sequences of M. marinum M and M. ulcerans Agy99 against M. ulcerans ecovar Liflandii identified a core genome of 4,237 CDSs, which is 85% of the total number of M. ulcerans ecovar Liflandii coding sequences (Fig. 2B and C). The genetic functional group distributions among the three sequenced genomes did not differ significantly across the classes “intermediary metabolism and respiration,” “cell wall and cell processes,” “conserved hypothetical,” and “lipid metabolism” (Fig. 3A). Nucleotide identities among the three-way genome comparison of a subset of 3,391 strict core orthologs (excludes paralogs and pseudogenes) were greater than 97% and showed that the highest sequence similarities are between M. ulcerans ecovar Liflandii and M. marinum (Fig. 2B). Similarly, the chromosome architecture of M. ulcerans ecovar Liflandii is more closely related to M. marinum M (Fig. 2B), with few genome rearrangements and few large DNA deletions, such as those seen in the reduced M. ulcerans Agy99 genome. These data do not contradict previous genomic comparisons that indicated a common ancestor for the three lineages of mycolactone-producing mycobacteria, including M. ulcerans ecovar Liflandii (lineage 1) and M. ulcerans Agy99 (lineage 3), and the data also suggest that lineage 3 isolates have undergone accelerated evolution through additional niche adaptation, while M. ulcerans ecovar Liflandii has retained a more pleomorphic state.
Fig 3.
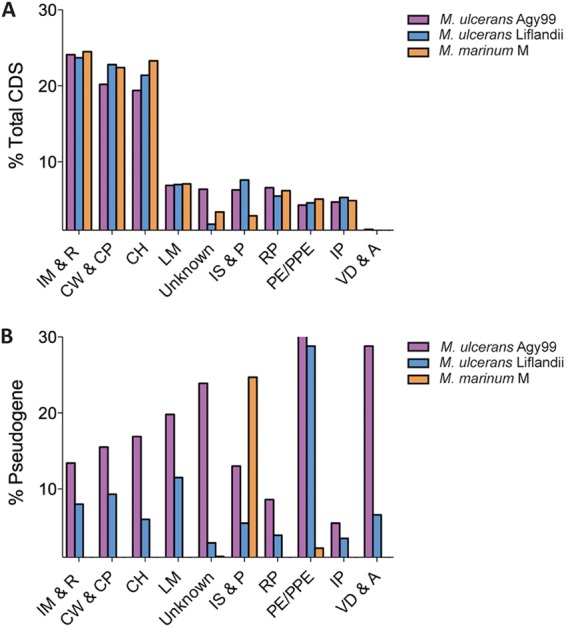
Functional group distribution. Percent proportions of total coding sequences (A) and pseudogenes (B) by functional group for the sequenced M. ulcerans-M. marinum isolates. CDSs were classified into one of ten classes: intermediary metabolism and respiration (IM & R), cell wall and cell processes (CW & CP), conserved hypotheticals (CH), lipid metabolism (LM), unknown, insertion sequences and phages (IS & P), regulatory proteins (RP), PE/PPE proteins, information pathways (IP), and virulence, detoxification, and adaptation (VD & A).
Insertion sequences and phage elements.
Isolates from all lineages of M. ulcerans have more than 200 chromosomal copies of IS2404 (9, 10, 22). This element is known to have promoted, at least in part, significant genomic rearrangements in M. ulcerans Agy99, including many large DNA deletions and gene disruptions by insertion (23). The M. ulcerans ecovar Liflandii chromosome is 430 kb smaller than that of M. marinum, but considering that IS2404 sequences contribute another 305 kb, it appears that approximately 780 kb has been lost since the divergence from an M. marinum-like ancestor. Arguably, this difference is largely due to IS2404-mediated deletions. Also, 89 of the 224 chromosomal copies of IS2404 (40%) have become inserted within CDSs. This is comparable to the situation in M. ulcerans Agy99, where 97 CDSs have been disrupted by IS2404 (45% of the 213 copies), although only six copies of IS2404 appear in the same location in both isolates, indicating independent expansion of IS2404 in each lineage. Most startling, however, is that despite the obvious activity of IS2404 in M. ulcerans ecovar Liflandii, the 224 copies of this element have had an unexpectedly modest impact on overall chromosome architecture. There are only four major DNA rearrangements in M. ulcerans ecovar Liflandii relative to M. marinum M (all flanked by copies of IS2404), such as a 440-kb inversion between nucleotide positions 2891467 and 3144394. These four regions have been unchanged in M. ulcerans Agy99 relative to M. marinum M. The conserved chromosome structure of M. ulcerans ecovar Liflandii suggests positive selection for this chromosome arrangement, despite the expansion of IS2404. One conclusion from these observations is that both strains are undergoing independent processes of reductive evolution, where expansion of IS2404 has been equally tolerated but there has been strong selection for the maintenance of chromosome structure in M. ulcerans ecovar Liflandii.
There are 91 copies of IS2606 in M. ulcerans Agy99 (12, 24). M. ulcerans ecovar Liflandii has only 4 copies of IS2606, three of which are on pMUM002 (21). The presence of a copy of IS2606 in the same location on both pMUM001 and pMUM002 suggests that this insertion sequence (IS) originated on pMUM and expanded in lineage 3 (21). The single IS2606 chromosomal copy (Mlif_03910) is in stark contrast to the 83 copies in M. ulcerans Agy99 and also highlights the likely role that IS2606 has played in promoting the extensive chromosome remodeling seen in the lineage 3 isolates, particularly the preference for IS2606 to insert in close proximity to IS2404. Of the 83 chromosome copies of IS2606, there are 39 instances of IS2606 inserting within 100 bp of IS2404 (59 copies within 500 bp), and the combination of IS2606 and IS2404 is associated with at least 30 instances of inversions and/or deletions (>5,000 bp) in M. ulcerans Agy99. It is not clear why IS2606 has not similarly expanded in M. ulcerans ecovar Liflandii.
Two mycobacteriophages, phiMU01 (18 kb) and phiMU02 (24 kb), are variously present in all M. ulcerans but not in M. marinum M (8). Both phages are present in M. ulcerans ecovar Liflandii, yet phiMU02 is significantly smaller (13 kb), and phiMU02 CDSs disrupted by IS2606 in M. ulcerans Agy99 are mostly deleted or have acquired frameshift mutations in M. ulcerans ecovar Liflandii.
M. ulcerans ecovar Liflandii regions of difference.
To further explore genetic features that might help explain the specific phenotypes of M. ulcerans ecovar Liflandii, we examined the regions of DNA present only in M. ulcerans ecovar Liflandii compared to the other two genomes. Stretches of DNA were present in M. marinum M but absent from M. ulcerans Agy99 (12) in the original description of the M. ulcerans Agy99 genome regions of difference (MURDs). Here, we have defined M. ulcerans ecovar Liflandii regions of difference (MULiRDs) as regions present only in M. ulcerans ecovar Liflandii compared with M. ulcerans and M. marinum. Eleven MULiRDs, spanning 290 CDSs, were identified in the genome (see Tables S1 and S2 in the supplemental material). The two largest regions, MULiRD3 (73.9 kb) and MULiRD7 (18.3 kb) (Fig. 1), harbor CDSs with possible roles in secondary metabolism. Both regions are flanked by IS2404 elements, and it appears that in M. ulcerans Agy99 these regions were lost by insertion sequence element (ISE)-mediated deletion, where a single copy of IS2404 remains in these locations. MULiRD7 harbors a seven-gene hyc operon (Mlif_03568 to Mlif_03574) that is duplicated on the chromosome (Mlif_01806 to Mlif_01813), although hycQ (Mlif_01811) is a predicted pseudogene. M. ulcerans Agy99 has only one copy of this operon, with the associated transcriptional regulator (MUL_1889) disrupted by a copy of IS2404 and hycE (MUL_1896) containing a 317-amino-acid C-terminal truncation. The orthologous hyc locus in Mycobacterium tuberculosis is thought to encode a formate hydrogenlyase complex that is part of a dormancy regulatory network involving MprA and DevR, for which in M. tuberculosis the ortholog of the transcriptional regulator in this locus (Mlif_01806) is upregulated in response to increased concentrations of nitric oxide (25). The preservation and duplication of this system in M. ulcerans ecovar Liflandii suggest that a similar dormancy response may be important for the lifestyle of the frog pathogen.
Pseudogene composition of M. ulcerans ecovar Liflandii.
Approximately 8% of all CDSs annotated in M. ulcerans ecovar Liflandii were predicted to be pseudogenes. Pseudogenes in prokaryote genomes generally occupy 1 to 5% of all CDSs (26). However, obligate pathogens are thought to be an exception, with higher levels of gene inactivation (26). CDSs associated with lipid metabolism (11.5%), cell wall and cell processes (9.3%), and intermediary metabolism-associated proteins (8.0%) were overrepresented with pseudogenes (Fig. 3B). In comparison, M. ulcerans Agy99 has 19.8%, 15.5%, and 13.4% pseudogenes in the same classes, respectively. The percentages of total coding sequences in M. ulcerans ecovar Liflandii that are pseudogenes are intermediate between M. marinum and M. ulcerans in all families of proteins (Fig. 3B), with 169 pseudogenes conserved in both M. ulcerans Agy99 and M. ulcerans ecovar Liflandii.
Unique to mycobacteria, the cell envelope-associated PE/PPE proteins have been suggested to modulate host immune responses, among other potential functions (27). High DNA identity in the 3′ region of these genes is a substrate for recombination and is thought to provide a source of antigenic variation among the mycobacteria (28, 29). The highly reduced genome of Mycobacterium leprae contains very few intact CDSs of this family (30). In M. ulcerans Agy99, 48.7% of all PE/PPE CDSs are pseudogenes. A significant proportion of these CDSs are also pseudogenes in the M. ulcerans ecovar Liflandii genome (28.8%), compared with only 2.2% in M. marinum M.
The mutation and inactivation of certain CDSs (pseudogene formation) in M. ulcerans Agy99 are predicted to have caused some significant phenotypic changes. A frameshift mutation in M. ulcerans Agy99 has resulted in a disruption of cydA, a component of the cytochrome bd oxidase transporter. However, cydA is intact in M. ulcerans ecovar Liflandii. This system is involved in responses to anaerobic and hypoxic conditions in vitro in M. tuberculosis (31). Conservation of this locus in M. ulcerans ecovar Liflandii may indicate an increased ability of the frog mycobacterium to survive under low-oxygen conditions, although, like M. ulcerans Agy99, the selenocysteine-containing formate dehydrogenase complex, with a predicted role in anaerobiosis, is likely to be inactive in M. ulcerans ecovar Liflandii, suggesting that it too has an impaired anerobic respiration capacity.
Phenolic glycolipids (PGLs) are potent antigens and virulence factors produced by mycobacterial pathogens. PGLs are composed of a polyketide backbone, decorated with species-specific combinations of sugar(s) via a phenolic head group. Genome analysis suggested that M. ulcerans ecovar Liflandii produces the same PGL as M. marinum M (sometimes called mycoside G). This is distinct to M. ulcerans Agy99, where two genes (MUL_1998 and MUL_2001) have been inactivated by mutation, resulting in the synthesis of an aglycosylated molecule with a modified polyketide backbone. These two genes are intact in M. ulcerans ecovar Liflandii (Mlif_01910 and Mlif_01913). The predicted presence of intact PGLs in M. ulcerans ecovar Liflandii might have implications for the interactions between host and bacteria and again points to likely differences between the lifestyle of M. ulcerans Agy99 and that of M. ulcerans ecovar Liflandii (12).
ESX loci.
Mycobacterial intracellular pathogens such as M. tuberculosis and M. marinum have at least five ESX (or type VII) ATP-dependent protein secretory systems named ESX-1, -3, -4, -5, and -6. The best defined of these systems, ESX-1, has been implicated in virulence via secretion of certain effectors, including the antigens ESAT-6 and CFP-10 (32). M. ulcerans ecovar Liflandii has six predicted ESX loci. ESX-1 (M. ulcerans ecovar Liflandii nucleotide positions 6138025 to 6171960) appears intact in M. ulcerans ecovar Liflandii, although there are two mutations that might impact function. A copy of IS2404 has inserted within the intergenic region between the divergently transcribed eccCb1 and the PE35 ortholog (Mlif_05720), and this may impact the expression of either gene. Also of note, Mlif_05724, the gene immediately downstream of esxA, is a pseudogene, although the M. tuberculosis ortholog (Rv3876) is thought to be nonessential for ESX function (33). ESX-2, immediately downstream of ESX-1, shares the same arrangement as that of M. marinum M and might be inactive, whereas Mlif_05735 (the ortholog of MMAR_5460) also appears truncated. The 10-kb region immediately downstream of ESX-2 is disrupted by four copies of IS2404 and, thus, is distinct to M. marinum. ESX-3 (M. ulcerans ecovar Liflandii nucleotide positions 218100 to 230400), ESX-4 (M. ulcerans ecovar Liflandii nucleotide positions 1319227 to 1332538), and ESX-5 (M. ulcerans ecovar Liflandii nucleotide positions 2592830 to 2614870) all appear intact, although ESX-5 has a single copy of IS2404 between Mlif_02416 and Mlif_02418. ESX-6 (M. ulcerans ecovar Liflandii nucleotide positions 182073 to 187394) is incomplete, with a 7-kb deletion compared to the 12-kb version in M. marinum M, although esxB_2 and esxA_2 in this locus remain intact. In M. marinum M, but not in other pathogenic mycobacteria, the region immediately upstream of ESX-1 contains 11 paralogous predicted membrane proteins of unknown function. This 15.7-kb region is deleted in M. ulcerans ecovar Liflandii and has been replaced with a single copy of IS2404. This region is also deleted in M. ulcerans Agy99. Eleven ESX-1 secretion-associated proteins (Esp) are present in M. marinum, and only four of these proteins are intact in the M. ulcerans Agy99 genome. M. ulcerans ecovar Liflandii has mostly conserved these proteins, with 10 intact Esp paralogs and 1 (Mlif_04556) pseudogenized.
Lipoproteins.
Lipoproteins in mycobacteria have been implicated in signal transduction (34) and evasion of mammalian cells (35), and some have a direct role in virulence as a part of transport systems (36). These proteins can be surface exposed and anchored by hydrophobic interactions, potentially to mycolic acids within the cell wall (37, 52). Serine proteases like SppA are responsible for hydrolyzing signal peptides prior to export across the cytoplasmic membrane (38). Prelipoproteins are then acylated after export from the cytoplasmic membrane by Lgt prior to cleavage by Lsp and Lnt (39). Lipoprotein synthesis appears to be considerably disrupted in M. ulcerans ecovar Liflandii, as sppA is a pseudogene. In Escherichia coli, the sppA homolog specifically cleaves the signal peptide of a major lipoprotein (40). The predicted inability of M. ulcerans ecovar Liflandii to cleave N-terminal signal peptides from lipoproteins is likely to significantly hamper preprocessing of lipoproteins. The lgt gene is also likely to be inactivated in M. ulcerans ecovar Liflandii (8). While not well studied in mycobacteria, prolipoprotein acylation by Lgt is not essential for cleavage by Lsp in different Gram-positive bacteria (41). Despite this, the absence of SppA and Lgt in these M. ulcerans strains may provide a mechanism for reducing the lipoprotein-induced Toll-like receptor 2 (TLR2) response, as described for other Gram-positive pathogens (41, 42).
Unique metabolic features of M. ulcerans ecovar Liflandii.
Genome inspection and metabolic pathway analyses suggest that M. ulcerans ecovar Liflandii may have some distinctive phenotypic characteristics. Genes encoding 3-deoxy-d-arabinoheptulosonic acid 7-phosphate (DAHP) synthases, which are important in the first step of the shikimate enzyme pathway, are pseudogenes. DAHP synthases are responsible for converting erythrose-4-phosphate into 3-deoxy-d-arabino-heptulosonate 7-phosphate during chorismate synthesis (43). The paralogs aroG (Mlif_02016) and aroG_1 (Mlif_03449) are both disrupted, while in M. marinum M, there are intact copies of both aroG (MMAR_3222) and aroG_1 (MMAR_1854). In M. ulcerans, aroG (MUL_2100) is a pseudogene, but aroG_1 (MUL_3533) is intact. Chorismate is an essential precursor for the synthesis of the aromatic amino acids tyrosine, phenylalanine, and tryptophan (43), and in M. tuberculosis, this pathway is essential for survival (44, 45). The lack of intact aroG suggests that M. ulcerans ecovar Liflandii may be a tryptophan, tyrosine, and phenylalanine auxotroph and may inhabit an environment where these amino acids are available. Experiments to confirm this predicted auxotrophy using Sauton's medium supplemented with aromatic amino acids have so far been unsuccessful.
Antibiotic susceptibility of M. ulcerans ecovar Liflandii.
In M. tuberculosis, there is a correlation between isoniazid and ethambutol resistance and the presence of an intact iniA gene, as iniA is part of an operon proposed to encode an efflux pump involved in resistance to a wide range of antibiotics that target cell wall biosynthesis (46, 47). Upon deletion of iniA, M. tuberculosis shows increased susceptibility to isoniazid (47). In M. ulcerans ecovar Liflandii, the three genes iniA, iniB, and iniC are pseudogenes, whereas these genes are intact in both M. marinum and M. ulcerans Agy99, suggesting that other genetic differences in M. ulcerans ecovar Liflandii could explain its reportedly increased resistance to isoniazid and ethambutol compared to that of M. ulcerans Agy99 (2).
M. ulcerans lineage 3 strains are sensitive to rifampin, which is used in combination with streptomycin to treat Buruli ulcer. In contrast, M. ulcerans ecovar Liflandii is reported to be resistant to rifampin (2). Examination of the genome revealed a single amino acid substitution in M. ulcerans ecovar Liflandii rpoB (T713M), whereas neither M. marinum nor M. ulcerans has this substitution. To our knowledge, the mutation has not been previously reported, but it may represent a novel rifampin resistance mutation. We therefore conducted MIC testing and found that M. ulcerans ecovar Liflandii 128FXT was fully susceptible to rifampin (MIC < 0.0625 μg/ml), indicating that the T713M mutation does not cause rifampin resistance. This discrepancy with the previous report remains to be explained.
Secondary metabolism in M. ulcerans ecovar Liflandii.
Mycobacteria have diverse secondary metabolite repertoires that include toxins, siderophores, and complex cell wall lipids (48). M. marinum M in particular has one of the largest arrays of secondary metabolite gene clusters yet described among bacteria (13), although the metabolites produced by most of these clusters are unknown. We explored the secondary metabolome of M. ulcerans ecovar Liflandii using the biosynthetic gene cluster search tool antiSMASH (20), and we detected 28 distinct secondary biosynthesis clusters on the chromosome. This compares with 33 in M. marinum M, 11 in M. ulcerans Agy99, and 15 in M. tuberculosis F11. To try to compare the secondary biosynthetic potentials between these mycobacteria, we classified each of the 88 antiSMASH-identified gene clusters as 1 of 37 families based on their sequence homology (see Table S3 in the supplemental material). M. ulcerans ecovar Liflandii has 10 more gene clusters than M. ulcerans Agy99, while the numbers of pseudogenized gene clusters are similar in both strains (Fig. 4). This analysis reflected the general trend already observed, that is, that lineage 3 M. ulcerans has proceeded further along a reductive evolutionary trajectory than have lineages 1 and 2, given that in the genomes of both species numerous gene clusters appear to have been deactivated by pseudogene formation or have been lost by deletion (Fig. 4A).
Fig 4.

Reductive evolution of secondary metabolite gene clusters in M. ulcerans ecovar Liflandii. (A) Numbers of pseudogene clusters and intact gene clusters in four mycobacterial genomes. Note here that M. marinum and M. tuberculosis may also contain some pseudogene clusters that may have escaped detection due to the lack of a reference strain in which these gene clusters are still intact. (B) Phylogram with maximum-parsimony inferred evolutionary events (+, gene cluster gain; -, gene cluster loss; P, pseudogene cluster) in the four mycobacteria, assuming that the 10 gene clusters shared by all four mycobacteria represented the ancestral gene cluster repertoire. The probabilities of the three event types were regarded as equal. The numbers after the species names represent the numbers of intact and pseudogene clusters in their genomes. Note that reality does not necessarily adhere to maximum parsimony; for example, deletions that have occurred in the line toward M. ulcerans may in fact have been preceded by pseudogenes that formed before the divergence from M. ulcerans ecovar Liflandii. (C) Venn diagrams showing which intact and pseudogene clusters are shared between the four mycobacterial genomes.
Neither M. ulcerans ecovar Liflandii nor M. ulcerans Agy99 has any gene cluster that is not observed in the M. marinum M genome, and all gene clusters of M. ulcerans Agy99 (whether intact or pseudogenized) are also present in M. ulcerans ecovar Liflandii (Fig. 4C). This is the expected state for strains that have evolved from a common M. marinum-like ancestor. However, there is one intact M. ulcerans nonribosomal peptide synthetase (NRPS) gene cluster that is pseudogenized in M. ulcerans ecovar Liflandii (Mlif_01390 to Mlif_01430), and there are eight intact M. ulcerans ecovar Liflandii gene clusters that have been pseudogenized in M. ulcerans, indicating that a significant part of reductive biosynthetic evolution has taken place independently in both strains and that the products of these loci are not required by the respective specialized bacteria. Mapping the chromosomal positions of the intact and the pseudogene clusters from M. ulcerans ecovar Liflandii and M. ulcerans, we observed that gene cluster inactivation has primarily occurred on the leading strand of the right-hand replichore, closer to the origin of replication than the terminus (Fig. 5). In both M. ulcerans ecovar Liflandii and M. ulcerans, seven of the eight gene clusters closest to the origin of replication on the leading strand have been inactivated by pseudogene formation (71% of all pseudogene clusters), even though only two gene clusters are shared in this region between the two genomes. This suggests a general phenomenon and might be explained by the fact that in other bacterial species the same chromosomal region contains the most highly expressed genes (49).
Fig 5.
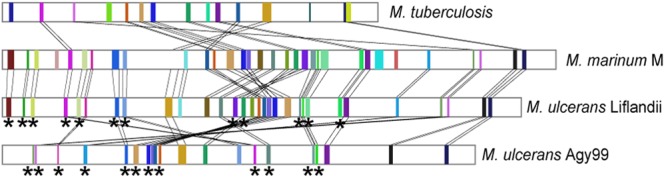
Inactivation of secondary metabolite gene clusters. The circular chromosomes are displayed in a linearized way, with the origin of replication shown on the left. Depicted is the predominance of pseudogenes on the leading strand of mycobacterial chromosomes near the origin of replication. The colored rectangles indicate biosynthetic gene clusters. The colors and connecting lines represent homology between gene clusters in the different genomes. Stars mark the clusters that have been inactivated through pseudogene formation.
M. ulcerans ecovar Liflandii.
There is no consensus on what defines an ecotype, but it has been suggested that an ecotype is a bacterial strain that conserves the genetic potential of a species with genetic differences, allowing it to exploit a slightly different ecological niche (50). As discussed previously, M. ulcerans ecovar Liflandii fulfills the criteria required for classification within the species M. ulcerans, and this is also true for the other mycolactone-producing mycobacteria (9, 10). The comparative genome data we have presented here show that M. ulcerans ecovar Liflandii, like all M. ulcerans strains, is undergoing reductive evolution and is also likely to be adapting to a niche environment. However, the pattern of mutations, the conserved arrangement of the chromosome, and other features, such as the distinctive structure and activity of mycolactone E, suggest that M. ulcerans ecovar Liflandii is responding to a different set of environmental pressures from those of M. ulcerans Agy99. These lines of evidence lead us to propose that M. ulcerans ecovar Liflandii is an ecotype within the species M. ulcerans.
Conclusions.
M. ulcerans ecovar Liflandii is a member of the M. marinum-M. ulcerans complex. The data presented here represent the third complete genome sequence for a mycobacterium from this complex. Like M. ulcerans, the frog mycobacterium has the signature of a niche-adapted organism and contains the pMUM virulence plasmid, several hundred copies of the M. ulcerans-specific insertion sequence IS2404, and many pseudogenes. However, it lacks the dramatic DNA rearrangements and deletions seen in M. ulcerans and has a chromosome architecture more closely aligned with M. marinum. M. ulcerans ecovar Liflandii shares a large gene repertoire with other members of the M. marinum-M. ulcerans complex. However, loss-of-function mutations in key metabolic genes, such as aroG and aroG_1, may have a profound impact regarding the environments in which M. ulcerans ecovar Liflandii can survive. As with M. marinum, ESX cell wall secretion systems appear to be largely intact in M. ulcerans ecovar Liflandii. However, other components of the cell wall are characteristically distinctive, with lipoprotein processing likely to be significantly hampered with nonfunctional lgt and sppA, dissimilar to both M. ulcerans Agy99 and M. marinum M. We propose that M. ulcerans ecovar Liflandii is an ecotype of M. ulcerans and is adapting to a niche that is related to but distinct from those of other M. ulcerans lineages. The genome sequence of M. ulcerans ecovar Liflandii provides an important snapshot of short-term reductive evolution as highlighted by the impact on the repertoires of secondary metabolite biosynthesis gene clusters. Comparisons with the genomes of M. ulcerans and M. marinum have shown that such repertoires can change very rapidly and that they can leave distinct genomic scars which remain visible for some time and can be uncovered when a closely related genome is available for comparison. Future research could be aimed at improving our understanding of the microbiology (e.g., physiology, biochemistry, and antibiotic susceptibility) of M. ulcerans ecovar Liflandii and ascribing the genomic features we have observed and the predictions we have made here to the confirmed phenotypes for this unusual pathogen.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Health and Medical Research Council of Australia (628640) and the Dutch Technology Foundation STW (STW10463).
We are grateful to Rainer Breitling and Eriko Takano for constructive comments that helped improve the manuscript.
Footnotes
Published ahead of print 30 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02132-12.
REFERENCES
- 1. Trott KA, Stacy BA, Lifland BD, Diggs HE, Harland RM, Khokha MK, Grammer TC, Parker JM. 2004. Characterization of a Mycobacterium ulcerans-like infection in a colony of African tropical clawed frogs (Xenopus tropicalis). Comp. Med. 54:309–317 [PubMed] [Google Scholar]
- 2. Nigou J, Gilleron M, Puzo G. 2003. Lipoarabinomannans: from structure to biosynthesis. Biochimie 85:153–166 [DOI] [PubMed] [Google Scholar]
- 3. Fremont-Rahl JJ, Ek C, Williamson HR, Small PL, Fox JG, Muthupalani S. 2011. Mycobacterium liflandii outbreak in a research colony of Xenopus (Silurana) tropicalis frogs. Vet. Pathol. 48:856–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chai N, Bronchain O, Panteix G, Godreuil S, de Medeiros C, Saunders R, Bouts T, de Luze A. 2012. Propagation method of saving valuable strains from a Mycobacterium liflandii infection in Western clawed frogs (Silurana tropicalis). J. Zoo Wildl. Med. 43:15–19 [DOI] [PubMed] [Google Scholar]
- 5. Hong H, Stinear T, Skelton P, Spencer JB, Leadlay PF. 2005. Structure elucidation of a novel family of mycolactone toxins from the frog pathogen Mycobacterium sp. MU128FXT by mass spectrometry. Chem. Commun. (Camb.) 34:4306–4308 [DOI] [PubMed] [Google Scholar]
- 6. Mve-Obiang A, Lee RE, Umstot ES, Trott KA, Grammer TC, Parker JM, Ranger BS, Grainger R, Mahrous EA, Small PL. 2005. A newly discovered mycobacterial pathogen isolated from laboratory colonies of Xenopus species with lethal infections produces a novel form of mycolactone, the Mycobacterium ulcerans macrolide toxin. Infect. Immun. 73:3307–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaser M, Rondini S, Naegeli M, Stinear T, Portaels F, Certa U, Pluschke G. 2007. Evolution of two distinct phylogenetic lineages of the emerging human pathogen Mycobacterium ulcerans. BMC Evol. Biol. 7:177 doi:10.1186/1471-2148-7-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doig KD, Holt KE, Fyfe JA, Lavender CJ, Eddyani M, Portaels F, Yeboah-Manu D, Pluschke G, Seemann T, Stinear TP. 2012. On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genomics 13:258 doi:10.1186/1471-2164-13-258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yip MJ, Porter JL, Fyfe JA, Lavender CJ, Portaels F, Rhodes M, Kator H, Colorni A, Jenkin GA, Stinear T. 2007. Evolution of Mycobacterium ulcerans and other mycolactone-producing mycobacteria from a common Mycobacterium marinum progenitor. J. Bacteriol. 189:2021–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pidot SJ, Asiedu K, Kaser M, Fyfe JA, Stinear TP. 2010. Mycobacterium ulcerans and other mycolactone-producing mycobacteria should be considered a single species. PLoS Negl. Trop. Dis. 4:e663 doi:10.1371/journal.pntd.0000663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suykerbuyk P, Vleminckx K, Pasmans F, Stragier P, Ablordey A, Tran HT, Hermans K, Fleetwood M, Meyers WM, Portaels F. 2007. Mycobacterium liflandii infection in European colony of Silurana tropicalis. Emerg. Infect. Dis. 13:743–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stinear TP, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T, Meurice G, Simon D, Bouchier C, Ma L, Tichit M, Porter JL, Ryan J, Johnson PD, Davies JK, Jenkin GA, Small PL, Jones LM, Tekaia F, Laval F, Daffe M, Parkhill J, Cole ST. 2007. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. 17:192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, Clarke K, Cronin A, Davis P, Goodhead I, Holroyd N, Jagels K, Lord A, Moule S, Mungall K, Norbertczak H, Quail MA, Rabbinowitsch E, Walker D, White B, Whitehead S, Small PL, Brosch R, Ramakrishnan L, Fischbach MA, Parkhill J, Cole ST. 2008. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18:729–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heysell SK, Mtabho C, Mpagama S, Mwaigwisya S, Pholwat S, Ndusilo N, Gratz J, Aarnoutse RE, Kibiki GS, Houpt ER. 2011. Plasma drug activity assay for treatment optimization in tuberculosis patients. Antimicrob. Agents Chemother. 55:5819–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Hijum SA, Zomer AL, Kuipers OP, Kok J. 2005. Projector 2: contig mapping for efficient gap-closure of prokaryotic genome sequence assemblies. Nucleic Acids Res. 33:W560–W566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bonfield JK, Smith K, Staden R. 1995. A new DNA sequence assembly program. Nucleic Acids Res. 23:4992–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19:1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147 doi:10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. 2005. ACT: the Artemis Comparison Tool. Bioinformatics 21:3422–3423 [DOI] [PubMed] [Google Scholar]
- 20. Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39:W339–W346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pidot SJ, Hong H, Seemann T, Porter JL, Yip MJ, Men A, Johnson M, Wilson P, Davies JK, Leadlay PF, Stinear TP. 2008. Deciphering the genetic basis for polyketide variation among mycobacteria producing mycolactones. BMC Genomics 9:462 doi:10.1186/1471-2164-9-462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaser M, Pluschke G. 2008. Differential gene repertoire in Mycobacterium ulcerans identifies candidate genes for patho-adaptation. PLoS Negl. Trop. Dis. 2:e353 doi:10.1371/journal.pntd.0000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahillon J, Chandler M. 1998. Insertion sequences. Microbiol. Mol. Biol. Rev. 62:725–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stinear T, Ross BC, Davies JK, Marino L, Robins-Browne RM, Oppedisano F, Sievers A, Johnson PD. 1999. Identification and characterization of IS2404 and IS2606: two distinct repeated sequences for detection of Mycobacterium ulcerans by PCR. J. Clin. Microbiol. 37:1018–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198:705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moran NA. 2002. Microbial minimalism: genome reduction in bacterial pathogens. Cell 108:583–586 [DOI] [PubMed] [Google Scholar]
- 27. Ramakrishnan L, Federspiel NA, Falkow S. 2000. Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science 288:1436–1439 [DOI] [PubMed] [Google Scholar]
- 28. Bottai D, Brosch R. 2009. Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol. Microbiol. 73:325–328 [DOI] [PubMed] [Google Scholar]
- 29. Delogu G, Brennan MJ. 2001. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect. Immun. 69:5606–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007–1011 [DOI] [PubMed] [Google Scholar]
- 31. Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. U. S. A. 102:15629–15634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U. S. A. 100:12420–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, Cole ST, Brosch R. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect. Immun. 74:88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steyn AJ, Joseph J, Bloom BR. 2003. Interaction of the sensor module of Mycobacterium tuberculosis H37Rv KdpD with members of the Lpr family. Mol. Microbiol. 47:1075–1089 [DOI] [PubMed] [Google Scholar]
- 35. Casali N, Riley LW. 2007. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics 8:60 doi:10.1186/1471-2164-8-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. 1999. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol. Microbiol. 34:257–267 [DOI] [PubMed] [Google Scholar]
- 37. Kovacs-Simon A, Titball RW, Michell SL. 2011. Lipoproteins of bacterial pathogens. Infect. Immun. 79:548–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hussain M, Ozawa Y, Ichihara S, Mizushima S. 1982. Signal peptide digestion in Escherichia coli. Effect of protease inhibitors on hydrolysis of the cleaved signal peptide of the major outer-membrane lipoprotein. Eur. J. Biochem. 129:233–239 [DOI] [PubMed] [Google Scholar]
- 39. Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu. Rev. Microbiol. 65:239–259 [DOI] [PubMed] [Google Scholar]
- 40. Ichihara S, Beppu N, Mizushima S. 1984. Protease IV, a cytoplasmic membrane protein of Escherichia coli, has signal peptide peptidase activity. J. Biol. Chem. 259:9853–9857 [PubMed] [Google Scholar]
- 41. Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, Hubner J, Santos-Sierra S, Teti G, Golenbock DT, Poyart C, Trieu-Cuot P. 2008. Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J. Immunol. 180:6149–6158 [DOI] [PubMed] [Google Scholar]
- 42. Machata S, Tchatalbachev S, Mohamed W, Jansch L, Hain T, Chakraborty T. 2008. Lipoproteins of Listeria monocytogenes are critical for virulence and TLR2-mediated immune activation. J. Immunol. 181:2028–2035 [DOI] [PubMed] [Google Scholar]
- 43. Pittard AJ. 1987. Biosynthesis of the aromatic amino acids, p 368–394 In Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology, Washington, DC [Google Scholar]
- 44. Barker C, Lewis D. 1974. Impaired regulation of aromatic amino acid synthesis in a mutant resistant to p-fluorophenylalanine. J. Gen. Microbiol. 82:337–343 [DOI] [PubMed] [Google Scholar]
- 45. Parish T, Stoker NG. 2002. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 148:3069–3077 [DOI] [PubMed] [Google Scholar]
- 46. Alland D, Steyn AJ, Weisbrod T, Aldrich K, Jacobs WR., Jr 2000. Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 182:1802–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Colangeli R, Helb D, Sridharan S, Sun J, Varma-Basil M, Hazbon MH, Harbacheuski R, Megjugorac NJ, Jacobs WR, Jr, Holzenburg A, Sacchettini JC, Alland D. 2005. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol. Microbiol. 55:1829–1840 [DOI] [PubMed] [Google Scholar]
- 48. Chopra T, Gokhale RS. 2009. Polyketide versatility in the biosynthesis of complex mycobacterial cell wall lipids. Methods Enzymol. 459:259–294 [DOI] [PubMed] [Google Scholar]
- 49. Rocha EP, Fralick J, Vediyappan G, Danchin A, Norris V. 2003. A strand-specific model for chromosome segregation in bacteria. Mol. Microbiol. 49:895–903 [DOI] [PubMed] [Google Scholar]
- 50. Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102:2567–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 5:e1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu CH, Tsai-Wu JJ, Huang YT, Lin CY, Lioua GG, Lee FJ. 1998. Identification and subcellular localization of a novel Cu, Zn superoxide dismutase of Mycobacterium tuberculosis. FEBS Lett 439:192–196 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.