

Abstract
We present a detailed characterization of a single-cycle infection of the bocavirus minute virus of canines (MVC) in canine WRD cells. This has allowed identification of an additional smaller NS protein that derives from an mRNA spliced within the NS gene that had not been previously reported. In addition, we have identified a role for the viral NP1 protein during infection. NP1 is required for read-through of the MVC internal polyadenylation site and, thus, access of the capsid gene by MVC mRNAs. Although the mechanism of NP1's action has not yet been fully elucidated, it represents the first parvovirus protein to be implicated directly in viral RNA processing.
INTRODUCTION
The Bocavirus genus of the Parvovirus family has gained renewed interest recently because of the identification of a bocavirus of humans (HBoV) that has been associated with lower respiratory tract infections in children (1, 2). Although an infectious molecular clone of the HBoV genome has recently been reported, characterization of this virus has previously been limited by the lack of a robust tissue culture system that supports efficient replication (3).
We have chosen to more fully characterize the bocavirus minute virus of canines (MVC) as a prototype of the bocavirus genus. An infectious molecular clone of the MVC genome has been generated, and the virus grows well in tissue culture. In addition, we have developed accurate traditional plaque assays and plaque lift assays for titering virus.
The general transcription maps for MVC, HBoV, and a third bocavirus, bovine parvovirus (BPV), have been delineated and have been found to be similar (3–5). They feature a single promoter at the left-hand end of the genome which generates multiple mRNAs that are derived from a single pre-mRNA by alternative splicing and alternative polyadenylation at either a proximal (pA)p or distal (pA)d site. The proximal polyadenylation signal must be suppressed for viral RNAs to extend into the right-hand end of the genome and access the capsid-coding region. This signal must be retained during export to the cytoplasm and must remain in these mRNAs throughout their existence. Another important novel feature of the bocaviruses is the presence of the viral NP1 protein (5, 6). This abundantly produced protein has no homologue in parvoviruses of other genera (7). The BPV life cycle has been examined in a general way previously (5, 6), and more recently, an examination of the cellular DNA damage response to MVC infection has been reported (8, 9).
In this article, we present the results of a detailed examination of MVC infection of canine cells. We describe basic features of MVC single-cycle infection, including a kinetic analysis of viral products and analysis of viral protein stability and localization. We have identified an additional smaller NS protein that derives from an mRNA spliced within the NS gene that had not been previously reported. In addition, we have identified a role for the viral NP1 protein in RNA processing. This is the first parvovirus protein to be associated with such a role.
MATERIALS AND METHODS
Viruses and cells.
The minute virus of canine (MVC) used in this study was the original strain (GA3) isolated from lung tissue (10) and was kindly provided by Colin Parrish at Cornell University. It was grown in the WRD cell line (11) through two passages (MVC GA3 P2). An infectious molecular clone of wild-type MVC (pIMVC) was described previously (5). The GenBank accession number for that clone is FJ214110.1. Plaque assays and plaque lift assays were developed to titer virus. All cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal calf serum in 5% CO2 at 37°C.
Infection and transfection.
For single-cycle infections, WRD cells were parasynchronized by isoleucine deprivation as described previously (12), followed by MVC infection (plaque assay-derived multiplicity of infection [MOI] = 10); reinfection was blocked by the addition of 20 μl of neutralizing anti-MVC antibody 2 h after the initial MVC infection. The neutralizing MVC antibodies from naturally infected canines were provided by Colin Parrish. Transfections were performed with WRD or 293T cells using either Lipofectamine Plus (Invitrogen, CA) or LipoD293 transfection reagent (SignaGen Laboratories, MD) according to the manufacturer's instructions.
Antibodies.
Polyclonal anti-NS1 or anti-NP1 antibodies were produced by immunizing rabbits (New Zealand Whites; SPF) on day 0, followed by two boosts with either NS1- or NP1-specific peptides coupled to keyhole limpet hemocyanin (KLH). Two distinct anti-NS1 antibodies were generated against NS1 epitopes targeting either the N-terminal amino acids 30 to 40 (anti-NS1 NH2) or the C-terminal amino acids 687 to 700 (anti-NS1 COOH) of the NS1 protein. A peptide consisting of amino acids 1 to 13 of the NP1 protein was used as the immunogen to produce anti-NP1 antibodies. Anti-capsid antibodies produced in rabbits were kindly provided by Colin Parrish. Additional antibodies used in this study were purchased from commercial sources.
Cycloheximide experiments.
The stability of NS1 and NP1 proteins was assessed by cycloheximide inhibition of protein synthesis, as described previously (13). Briefly, WRD cells were plated in six-well dishes to 60 to 70% confluence and then infected with MVC at a plaque assay-derived MOI of 10. At 30 h postinfection, cycloheximide (CHX) (Sigma Chemicals, MO) was added to the cells at 100 μg/ml. Control wells received dimethyl sulfoxide (DMSO) vehicle instead of CHX. Cells were then collected at the time points described in the text and subjected to immunoblot analysis as described.
SDS-PAGE, immunoblot, and immunofluorescence assays.
SDS-PAGE, Western blotting, and immunofluorescence assays were performed as previously described (14). For immunofluorescence, parasynchronized WRD cells were grown on glass coverslips in 24-well plates and infected with MVC at a plaque assay-derived MOI of 10. After 18 h, cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 15 min, and extracted with 0.5% Triton X-100 in PBS for 10 min. Nuclei were visualized by staining with 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were mounted in Fluoromount-G (Southern Biotech), and light microscopy images were taken at a magnification of ×200 or ×400 (objective lens) with an Olympus IX-70 inverted fluorescence microscope. Anti-NS1 or anti-NP polyclonal rabbit antibodies raised against specific epitopes as described above were used at a 1/1,000 dilution followed by incubation with a goat anti-rabbit IgG conjugated with fluorescein isothiocyanate (FITC) (Sigma). DAPI was utilized as a nuclear counterstain.
Southern blotting.
WRD cells were transfected with the MVC constructs as described in the text and figure legends. At 48 h posttransfection, DNA was extracted from transfected cells and DNA replication was accessed by Southern blotting as described previously (15) using whole-MVC-genome probes derived by NotI digestion of the full-length MVC wild-type (WT) infectious clone (IC). A one-kilobase fragment from the MVM capsid region was inserted into the plasmid backbone to clearly separate input plasmid from the monomer replicative form (RF). Loading of samples was standardized using a NanoDrop spectrophotometer and normalized to 0.1% SDS lysis buffer.
Plasmid constructs.
Multiple constructs were used in this study as described below.
MVC pIMVC.
The MVC wild-type infectious clone was generated as previously described (5).
NS1 (full length)/pcDNA 3.1.
MVC NS1 was constructed by inserting the MVC NS1 open reading frame (ORF) with additional flanking sequences (nucleotides [nt] 340 to 2750 of the MVC pIMVC) into cytomegalovirus (CMV)-driven pcDNA 3.1.
NS1 mutant infectious clone.
The NS1 ORF was terminated by introducing a TAG stop codon at nt 416 with a single T insertion in the MVC IC.
Spliced NS1/pcDNA 3.1.
The NS1 spliced cDNA was generated by reverse transcription and cloned into CMV-driven pcDNA 3.1 (Invitrogen, CA).
NP1 (full length)/pcDNA 3.1.
MVC NP1 was constructed by inserting the MVC NS1 ORF (nt 2537 to 3096) into CMV-driven pcDNA 3.1.
NP1 premature termination mutants.
The NP1 ORF was terminated by introducing a TAG stop codon at nt 2735 with a single T mutation in the MVC IC using overlapping PCR techniques. Additional NP1 premature termination mutants were made by converting either nt 2738 to 2740 or nt 2837 to 2839 to TAA using site-directed mutagenesis.
NP1-ATGm.
The NP1 initiating codon was mutated at nt 2537 from AUG to GCG.
NP1-5XPro.
This mutant was made by introducing 5 prolines in a row starting at nt 2780 in the MVC IC.
NP1 capsid fusion mutant.
An NP1 mutant was made by a single nucleotide deletion at nt 3089 resulting in a fusion of the NP1 and capsid ORFs.
3A splice acceptor mutant.
A 3Am mutant was constructed by an in-frame G-to-A substitution at nt 3037.
Capsid-lacking mutant.
A VP1/VP2-negative mutant was generated by a 4-nt deletion (3700 to 3703) in the capsid gene which shifted the reading frame and truncated the translation of VP1 and VP2.
RT-PCR.
Reverse transcriptase PCR (RT-PCR) was performed with total RNA isolated from WRD cells infected with MVC GA3 virus or transfected with either the MCV infectious clone (pIMVC) or CMV NS1 (spliced) cDNA. The RT-PCR was performed with the TaKaRa Blueprint RT-PCR kit (Clontech, CA). The 5′ primer CCTGTATGGATCCAGATAAAGCCA and the 3′ primer GTCTCTCTCCCCATTCGGACGGCGTG were used for the RT-PCR.
DNA sequencing.
Sequence analysis of DNA fragments from RT-PCR experiments was done by the University of Missouri DNA Core Facility.
RPAs.
Total and cytoplasmic RNAs were isolated using TRIzol reagent (Invitrogen), and RNase protection assays (RPAs) were performed as previously described (16). Two distinct MVC homologous RNase protection probes were used in this study. A proximal polyadenylation probe [the (pA)p probe] (17), which spanned MVC nt 3107 to 3333, distinguished RNAs that were polyadenylated either proximally at (pA)p or at the distal site (pA)d, and a capsid probe (the cap gene probe), which spanned nt 2941 to 3333, distinguished individual RNA transcripts that were polyadenylated at either site as well as RNAs resulting from splicing at the 3A acceptor. These probes were generated from linearized templates by in vitro transcription using SP6 polymerase, as previously described (16). RNA hybridizations for RPAs were done in substantial probe excess, and RPA signals were analyzed with the Typhoon FLA9000 and quantified with Multi Gauge software (GE). Relative molar ratios of individual RNA species were calculated after adjustment for the number of 32P-labeled uridines in each protected fragment as previously described (16).
RESULTS
Single-cycle infections.
We initially performed a detailed characterization of a low-multiplicity, single-cycle MVC infection of parasynchronized canine WRD cells. Single-cycle infections were carried out in the presence of an anti-MVC neutralizing antibody that was previously optimized to block reinfection (data not shown). Figure 1A shows the coordinate expression over 36 h of replicating viral DNA, the various major mRNAs, as previously described, and the major nonstructural NS1 protein, the NP1 protein, and the capsid proteins VP1 and VP2. Viral RNA was analyzed using an RNase protection probe across the internal polyadenylation site (pA)p [probe (pA)p]. This probe distinguished RNAs that read through (pA)p from RNAs polyadenylated internally at (pA)p. Internal polyadenylation at (pA)p actually occurs at 4 sites within a 120-nt region in that area (O. Fasina and D. J. Pintel, unpublished data), similar to what has been seen for B19 (18). Under the parasynchronous conditions used, no differences in the ratios of the relative macromolecular forms, and only a continual increase in all forms, were detected.
Fig 1.

Characterization of the kinetics of single-cycle MVC infection in parasynchronized permissive WRD cells. (A, left) DNA replication. Southern blot analysis of samples taken from MVC-infected WRD cells in the presence of a neutralizing antibody as described in the text. Representative samples were taken at 12, 18, 24, or 36 h following infection, as shown in lanes 1, 2, 3, and 4, respectively. Viral DNA replication is evidenced by the presence of monomer and dimer replication intermediates. Identification of single-stranded DNA (SS) is also indicated but is underrepresented in this experiment. (Middle) RNA expression. RNase protection assay (RPA) showing RNA expression from the same single-cycle MVC infection shown in panel A. The (pA)p probe used to detect RNA expression is depicted in Fig. 3A. The relative size of the probe is shown on the left, and RNA transcripts are seen in lanes 1 to 4. Read-through transcripts that polyadenylate at the distal polyadenylation site (pA)d are indicated by RT. Transcripts that are polyadenylated at the proximal site are indicated as (pA)p. A full transcription map generated previously (5), updated as described below, is shown in Fig. 3A. (Right) Expression of NS1, NP1, and capsid proteins. Immunoblots of cell extracts from a time course of single-cycle MVC infection probed with anti-NS1, anti-NP1, or anti-capsid antibodies as described in Materials and Methods. Cellular tubulin was used as a loading control. (B) Localization of NS1 and NP1 proteins. Cellular localization of NS1 (top) and NP1 (lower) proteins using specific antibodies as described in Materials and Methods. DAPI was used as a nuclear counterstain. (C) Stability of NS1 and NP1 proteins. Immunoblots of extracts harvested at 0, 2, 4, 6, 8, and 10 h following cycloheximide treatment of MVC-infected WRD cells. The relative levels of the two forms of the NS1 protein, approximately 84 kDa and 66 kDa, are shown in the top panel. The expression of NP is shown in the lower panel, and cellular tubulin is shown as a control for the amount of protein loaded per lane.
Viral infection generated two nonstructural proteins of approximately 66 and 84 kDa in size, respectively, that reacted with an antibody to an epitope from amino acids (aa) 687 to 700 in the COOH terminus of these proteins. This was unexpected, as the large ORF that comprises the purported NS1 gene would be predicted to encode only the larger protein. The derivation of these two proteins will be discussed further below. Immunofluorescence assays demonstrated that MVC NS1 proteins were primarily nuclear throughout infection, as seen for the NS1 proteins of other autonomous parvoviruses (Fig. 1B). While NP1 expression is primarily nuclear, cytoplasmic expression is consistently seen in a small population (approximately 10%) of the total NP1-positive cells.
Immunoblots assaying remaining protein after cycloheximide inhibition indicated that both NS1 species were very stable while the NP1 protein had an intermediate stability (Fig. 1C). Quantification of multiple experiments showed that both NS1 proteins had a half-life of at least 10 to 12 h while the NP1 protein had a half-life of approximately 6 to 8 h.
MVC generates two NS proteins.
We were surprised to see that viral infection generated two abundant proteins that reacted with an antibody directed against the COOH terminus of NS1. The smaller protein was seen to be present in greater abundance. As mentioned, the NS1 gene ORF was predicted to encode the larger protein, and the single, unspliced mRNA species identified as expressed from this gene would generate such a protein.
To begin to determine the origin of the smaller NS1 protein, we repeated our virally infected cell immunoblots using an antibody developed against the amino terminus of NS1. This antibody also detected both the 84-kDa and 66-kDa proteins (Fig. 2A, left panel). Suspecting at first that the larger protein might be either processed or alternatively initiated to generate the smaller protein, we tagged the NS1 ORF in a CMV-driven expression vector with hemagglutinin (HA) at either the amino or carboxyl terminus. The small protein generated from both of these constructs reacted with anti-HA antibody, suggesting that the smaller protein and larger protein contained the same termini (data not shown). This led us to suspect that these constructs generated a previously undetected spliced mRNA that encoded the 66-kDa protein. RT-PCR using primers at the ends of the NS1 gene identified a spliced product in the expression constructs (data not shown), and additional RT-PCR assays showed that similar-sized spliced RNA was generated by both the wild-type infectious MVC clone (Fig. 2A, center panel, lane 3) and the original Cornell strain GA3 virus (Fig. 2A, center panel, lane 1) as well. Sequencing of the cDNA product demonstrated both a splice that joined a previously unidentified donor at nt 1272 with the previously identified A1 acceptor (Fig. 2B) and another splice that joined the previously identified 2D-2A. This mRNA is expected to be approximately 3.2 kb in size and was predicted to encode an NS protein of approximately 66 kDa. When the NS1 gene representing this cDNA was cloned into a CMV expression vector, a similar 66-kDa protein was produced (Fig. 2A, right panel). As expected, this clone also generated a same-sized spliced RNA (Fig. 2A, center panel, lane 5). A revised transcription map showing the additional spliced species (R1′ and R2′) is shown in Fig. 3A.
Fig 2.
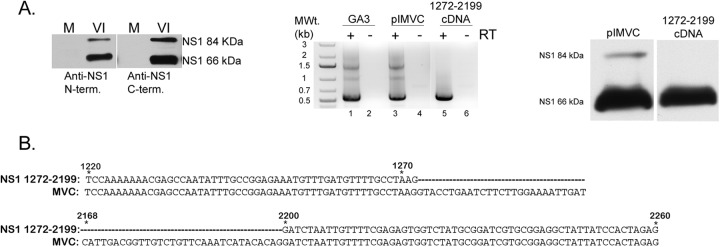
MVC generates two NS1 proteins during viral replication. (A, left) NS1 expression. Immunoblot of mock (M) or virus-infected (VI) WRD cell extracts probed with either an N or C terminus-specific anti-NS1 antibody showing the expression of two distinct NS1 proteins. (Middle) RT-PCR using 5′ and 3′ primers as described in Materials and Methods was done in the presence or absence of reverse transcriptase (RT). cDNA products from the RT-PCR are shown for GA3 virus infection (lane 1), WT pIMVC (lane 2), and the 1272-2199 cDNA (lane 5). (Right) Expression of spliced NS1/pcDNA. Immunoblot of 293T cell extracts transfected with either WT pIMVC or NS1 1272-2199/cDNA probed with an anti-NS1 (C terminus-specific) antibody. (B) NS1 DNA sequence analysis. Alignment of the nucleotide sequences of MVC and NS1 1272-2199/cDNA is shown. The regions depicted are from nt 1220 to 1272 and nt 2168 to 2260. The dashed lines in the NS1 1272-2199/cDNA sequence represent the spliced region from the newly identified 1D′-1A intron.
Fig 3.

Revised genetic map of MVC. (A) The genetic map of MVC drawn to scale, highlighting the P6 promoter, splice donors (D) and acceptors (A), and the proximal (pA)p and distal (pA)d polyadenylation sites. The numbers denote the nucleotide positions within the MVC genome. The newly identified splice site (1D′1A), as used in RNAs R1′ and R2′, is incorporated in the map. The locations of the cap gene and (pA)p probes relative to the MVC genome are shown. (B) Diagram (not to scale) indicating the relative locations of the NP1 termination mutants NP1-2735TAG, NP1-2740TAA, and NP1-2839TAA and the NP1-5XPro mutant.
NP1 mutants have a significant effect on RNA processing independent of genome replication.
As previously reported, termination mutations that prevented production of either the NS1 proteins or NP1 greatly inhibited replication of the MVC infectious clone (5). In addition to its anticipated role in viral genome replication, we detected no evidence that NS1 played a role in transactivation of the viral P6 promoter or in RNA processing (data not shown). In contrast, however, we observed that NP1 mutants displayed a striking phenotype in which the profiles of accumulated RNA forms were altered in a manner that was mainly independent of replication. As mentioned, NP1 is unique to the bocaviruses. It currently has no apparent homologue in the database.
As can be seen by the RNase protection assays using a probe that spanned the (pA)p site, following either viral infection (Fig. 1, middle panel) or transfection of the replicating infectious clone (Fig. 4A, lane 2), the majority of wild-type MVC RNAs read through the internal polyadenylation site (pA)p extend through the capsid gene and polyadenylate at the (pA)d site at the right-hand end. However, three individual mutants of the infectious clone containing premature translation termination mutations within the NP1 coding region all show a very different phenotype: the majority of the steady-state RNAs in total RNA preparations generated from these mutants do not extend beyond the internal polyadenylation site (Fig. 4A, NP1-2735TAG, NP1-2740TAA, and NP1-2839TAA in lanes 3 to 5, respectively; see the mutant diagram in Fig. 3B).
Fig 4.

MVC NP1 mutants have a significant effect on RNA processing independent of genome replication. (A) RNA expression determined by an RNase protection assay (RPA). WRD cells were transfected with the wild-type infectious clone pIMVC (lane 1) or three distinct NP1 termination mutants, NP1-2735TAG, NP1-2740TAA, or NP1-2839TAA (lanes 3, 4, and 5). At 2 days posttransfection, total RNAs were isolated and probed for MVC-specific mRNAs using the (pA)p probe that is depicted in Fig. 3A. The sizes of protected bands are shown on the left, and the designated (pA)d or (pA)d RNA species are shown on the right. The RNA probe is shown in lane 1. (B) RNase protection assays using the (pA)p probe showing RNA expression of WRD cells transfected with WT pIMVC (lane 2) or the replication-deficient mutants RC WT, NP1-2735TAG, pIMVC-NS1−, and pIMVC-FD, as described in the text (lanes 3, 4, 5, and 6, respectively). The (pA)p probe is shown in lane 1. RNA species are designated (pA)d or (pA)p. (C) RNA expression assayed by RPAs using the (pA)p probe of WRD cells transfected with WT pIMVC (lane 2) or the NP1-lacking mutants ATGm (lane 4), 5XPro (lane 5), or NP1-2735TAG (lane 6) as described in the text. The (pA)p probe is shown in lane 1. RNA species are designated (pA)d or (pA)p. (D) RPA utilizing the cap gene probe as depicted in Fig. 3A of either virus-infected WRD cells (VI; lane 3) or WRD cells transfected with WT pIMVC (lane 4) or the third intron splice mutant 3Am (lane 5), as described in the text. The WT cap gene probe (lane 1) and the homologous 3A cap gene probe (lane 2) are shown. Individual RNA species and their relative sizes are designated on the right and left of the image, respectively. (E) Quantification of RPAs showing the average polyadenylation ratios [(pA)p/(pA)d] of RNA transcripts from WT pIMVC, 3Am, NP1-5XPro, or NP1-2735TAG in either a replicating infectious clone (TR+) or replicating-deficient (TR−) background. Error bars indicate the standard deviation from 3 independent experiments. (F) RPA utilizing the cap gene probe (lane 1) showing RNA expression and distinguishing individual transcripts from viral infection (lane 2), transfection of WT pIMVC (lane 3), or transfection of the NP1 mutants 2735TAG, 2740TAA, and 2839TAA (lanes 4, 5, and 6, respectively). The individual RNA species R1 to R6 are labeled on the right, and their respective sizes are shown on the left. (G) Expression of NP1, NS1, and capsid proteins. Immunoblots of 293T cell extracts either mock transfected or transfected with WT pIMVC, the NP1 termination mutant NP1-2735TAG, or the NP1 proline mutant NP1-5XPro, probed with anti-NS1, anti-NP1, or anti-capsid antibodies.
It has been suggested that the processing of B19 RNA is governed by replication of the viral genome (19). Although NP1 mutants are deficient for viral replication, the RNA phenotype we observed is not governed by genome replication: a nonreplicating MVC rep/cap-like construct lacking the viral hairpins (RC WT), a nonreplicating hairpin-containing NS1-lacking mutant of the MVC infectious clone (pIMVC-NS1−), and a mutant severely impaired for replication due to deletion mutations in the right-hand-end hairpin acquired during passage of the infectious clone in recombination-proficient Escherichia coli (pIMVC-FD) all displayed an RNA phenotype indistinguishable from viral infection (Fig. 4B, compare lanes 3 [RC WT], 5 [pIMVC-NS1−], and 6 [pIMVC-FD] to lane 2 [the wild type] and lane 4 [NP1-2735TAG]). Results with pIMVC-NS1− also indicated that the phenotype that was observed was independent of NS1.
To rule out nonspecific cis-acting effects of the introduction of termination mutations in the NP1 coding region, we also generated and characterized a single nucleotide mutation destroying NP1 AUG (NP1-ATGm) and another that introduced a series of 5 proline mutations in the center of the NP1 coding region (NP1-5XPro) in the nonreplicating rep/cap construct. These mutations had the same phenotype as the termination mutations (Fig. 4C, NP1-ATGm and NP1-5XPro; compare lanes 4 and 5 to lane 6).
The termination mutations so far described in NP1 exist within the third intron. It was therefore possible that the primary effect of these mutations was on splicing of this intron, which subsequently affected downstream polyadenylation. This was unlikely for the following reasons. First, an acceptor mutation in the MVC rep/cap background that prevented splicing of the third intron had no effect on the relative uses of the two polyadenylation sites (Fig. 4C, 3Am, compare lanes 3 and 2). An RPA utilizing the cap gene probe (diagrammed in Fig. 3A) that monitors splicing of the third intron confirmed that this mutant did not generate RNAs spliced at this acceptor (Fig. 4D, compare lane 5 to lanes 3 and 4). Second, a single nucleotide mutation in the rep/cap background downstream of the third intron, designed to inactivate NP1 by fusing it to the capsid ORF, had the same phenotype as that seen for the other NP1 mutations (data not shown).
Figure 4E shows a quantification of multiple experiments with these mutants, both in a replication-deficient rep/cap background (TR−) and within the replicating infectious clone (TR+). The NP1-5XPro and NP1-2735TAG constructs showed a >10-fold increase in relative internal polyadenylation, while the 3Am mutant performed like the wild type. It is not yet clear why there is a difference (albeit statistically insignificant) in the polyadenylation ratios of the NP1-2735TAG construct in replicating versus nonreplicating backgrounds.
The previous analysis monitored levels of RNA that were alternatively polyadenylated at either (pA)p or (pA)d. To determine which MVC RNAs had been specifically affected by the lack of NP1 function, we utilized an RNase protection assay using the cap gene probe that specifically distinguished relative levels of (i) R1 and R3, which utilize (pA)p and encode NS1 and NP1, respectively, (ii) read-through R2 and R4, which encode NS1 and NP1, respectively, and (iii) the read-through R5 and R6 mRNAs, which together encode capsid proteins. As described above, the wild-type virus and infectious clone encode primarily read-through products, and these can here be seen to include both the R2 and R4 mRNAs as well as predominantly the capsid-encoding R5 and R6 mRNAs (Fig. 4F, lanes 2 and 3). RNase protections of RNA generated from the NP1 mutants using this probe showed a deficiency in the accumulation of both the read-through R2 and R4 mRNA and a drastic reduction in the capsid-encoding mRNAs R5 and R6 (Fig. 4F, lanes 4 to 6). Both the NP1-2735TAG mutant and the NP1-5XPro mutant generated dramatically reduced levels of VP1 and VP2, although they both expressed NS1 and the NP1-5XPro mutant generated high levels of NP1 (Fig. 4G, lanes 3 and 4).
Taken together, our results demonstrate that NP1 has a significant effect on viral RNA processing and that in the absence of wild-type NP1, read-through of the internal (pA)p site, which is necessary to access the capsid gene ORF, is deficient.
If the sole defect of the NP1 mutants was a deficiency in capsid protein production, NP1 mutants in a replicating background should replicate similarly to a capsid-lacking mutant, i.e., they should generate the double-stranded 5-kb monomer RFs but no single-stranded DNA (ssDNA). However, in multiple experiments (in which reinfection was blocked), the NP1 mutants were found to be more deficient in generating the monomeric replicative forms (mRFs) than a capsid-lacking mutant (Fig. 5A, compare lanes 3 [NP1-2735TAG] and 4 [NP1-5XPro] to lane 5 [cap-negative]) and to lane 1 [viral infection] and lane 2 [pIMVC]). This suggested that NP1 had at least one additional function during viral replication.
Fig 5.
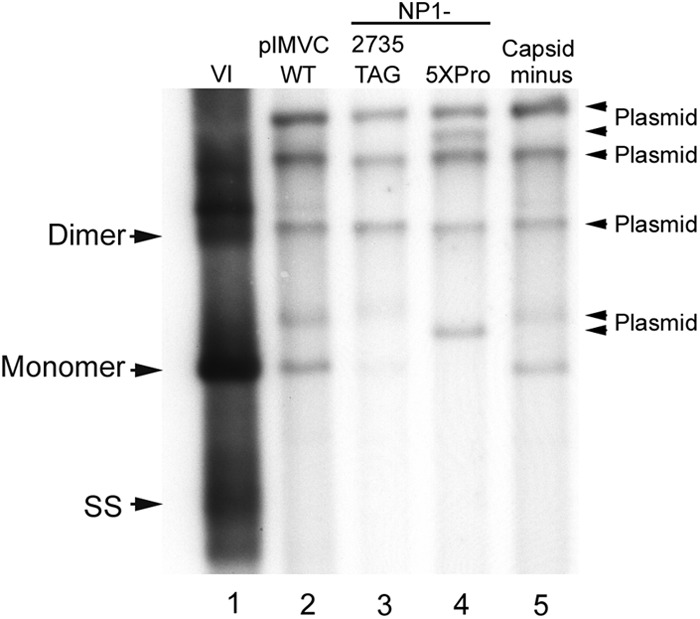
NP1 mutants exhibit a greater defect in viral replication than a capsid-lacking mutant. Southern blot analysis of samples taken from WRD cells infected with MVC (lane 1) or transfected with one of the following: WT pIMVC (lane 2), the NP1 mutant (NP1-2735TAG) (lane 3), NP1-5XPro (lane 4), or a capsid-lacking mutant described in Materials and Methods (lane 5). The infection or transfections were done in the presence of a neutralizing antibody, and Southern blot analyses were done as described in the text. Total DNA content was measured, and equivalent amounts of total DNA were loaded in each lane. DNA replication forms are indicated by monomer, dimer, and single-stranded (SS) DNA.
DISCUSSION
In this article, we first present a characterization of a single-cycle bocavirus infection. In the course of this analysis, we have identified a second nonstructural protein and determined its origin from a previously unidentified spliced mRNA. The second protein lacks the typical Walker-type helicase motifs (20) and so may fall into the category of parvovirus nonstructural proteins lacking helicase activity, like MVM NS2 (21). The smaller protein is present in greater abundance than the larger protein for reasons that are under investigation.
Perhaps most importantly, we have also shown that the MVC NP1 protein is required for read-through of the internal polyadenylation site and access into the capsid gene. This is the first parvovirus protein shown to have a direct effect on RNA processing.
It is not known how NP1 functions. It may be directly at the level of splicing, polyadenylation, or export. Its activity is independent of genome replication, independent of NS1 function, and not due to effects on splicing of the upstream 3D-3A intron. Attempts at complementing the NP1 mutants with NP1 in trans have as yet been only partially successful; however, multiple types of mutations, including missense, nonsense, frameshift, and initiating AUG mutations, at different places within NP1, all have similar phenotypes. Interestingly, it has been pointed out to us (Florence Baudin, personal communication) that NP1 has multiple arginine/serine direpeats characteristic of an SR protein, a class known to function in RNA processing (22).
Although parvoviruses have only subtle early to late expression shifts, they all mediate access to their capsid genes, and different parvoviruses do so in different ways. Those parvoviruses with separate capsid gene promoters, like the Parvovirus MVM (23) and the Dependovirus AAV (17, 24, 25), do so by transcriptional trans-activation of those promoters. The erythroviruses (26) have a single promoter at the left-hand end of the genome and a polyadenylation site within an intron in the center of the genome. Splicing of this intron to remove the polyadenylation signal helps govern access to the capsid gene for these viruses (27). The amdoviruses (28, 29) and the bocaviruses (3) have polyadenylation sites that lie within their capsid gene; however, these potent motifs are retained in the capsid-encoding mRNA and must be somehow suppressed to allow export and accumulation of mRNAs that encode capsid protein information. We have shown here that the bocavirus MVC has evolved yet another way to allow essential access to its capsid protein gene, namely, by the action of its novel NP1 protein, which is required to read through its internal (pA)p site.
ACKNOWLEDGMENTS
We thank members of our labs for helpful discussions. We thank Colin Parrish, Cornell, for antibody and virus and Florence Baudin, EMBL, Heidelberg, Germany, for sharing information concerning NP1 homology.
This work was supported by PHS grant AI 046458 from the NIH to D.J.P. and grants AI 085236 and AI070723 from the NIH to J.Q.
Footnotes
Published ahead of print 7 November 2012
REFERENCES
- 1. Allander T. 2008. Human bocavirus. J. Clin. Virol. 41:29–33 [DOI] [PubMed] [Google Scholar]
- 2. Tattersall P. 2006. The evolution of parvovirus taxonomy, p 5–14 In Kerr JR, Cotmore SF, Bloom ME, Linden ME, Parish CR. (ed), Parvoviruses, 6th ed Hodder Arnold, London, United Kingdom [Google Scholar]
- 3. Chen AY, Cheng F, Lou S, Luo Y, Liu Z, Delwart E, Pintel D, Qiu J. 2010. Characterization of the gene expression profile of human bocavirus. Virology 403:145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qiu J, Cheng F, Johnson FB, Pintel D. 2007. The transcription profile of the bocavirus bovine parvovirus is unlike those of previously characterized parvoviruses. J. Virol. 81:12080–12085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun Y, Chen AY, Cheng F, Guan W, Johnson FB, Qiu J. 2009. Molecular characterization of infectious clones of the minute virus of canines reveals unique features of bocaviruses. J. Virol. 83:3956–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lederman M, Patton JT, Stout ER, Bates RC. 1984. Virally coded noncapsid protein associated with bovine parvovirus infection. J. Virol. 49:315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lederman M, Bates RC, Stout ER. 1983. In vitro and in vivo studies of bovine parvovirus proteins. J. Virol. 48:10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen AY, Luo Y, Cheng F, Sun Y, Qiu J. 2010. Bocavirus infection induces mitochondrion-mediated apoptosis and cell cycle arrest at G2/M phase. J. Virol. 84:5615–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo Y, Chen AY, Qiu J. 2011. Bocavirus infection induces a DNA damage response that facilitates viral DNA replication and mediates cell death. J. Virol. 85:133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carmichael LE, Schlafer DH, Hashimoto A. 1991. Pathogenicity of minute virus of canines (MVC) for the canine fetus. Cornell Vet. 81:151–171 [PubMed] [Google Scholar]
- 11. Binn LN, Lazar EC, Eddy GA, Kajima M. 1970. Recovery and characterization of a minute virus of canines. Infect. Immun. 1:503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schoborg RV, Pintel DJ. 1991. Accumulation of MVM gene products is differentially regulated by transcription initiation, RNA processing and protein stability. Virology 181:22–34 [DOI] [PubMed] [Google Scholar]
- 13. Nayak R, Farris KD, Pintel DJ. 2008. E4Orf6-E1B-55k-dependent degradation of de novo-generated adeno-associated virus type 5 Rep52 and capsid proteins employs a cullin 5-containing E3 ligase complex. J. Virol. 82:3803–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller CL, Pintel DJ. 2002. Interaction between parvovirus NS2 protein and nuclear export factor Crm1 is important for viral egress from the nucleus of murine cells. J. Virol. 76:3257–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi EY, Newman AE, Burger L, Pintel D. 2005. Replication of minute virus of mice DNA is critically dependent on accumulated levels of NS2. J. Virol. 79:12375–12381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venkatesh LK, Fasina O, Pintel DJ. 2012. RNAse mapping and quantitation of RNA isoforms. Methods Mol. Biol. 883:121–129 [DOI] [PubMed] [Google Scholar]
- 17. Qiu J, Pintel DJ. 2002. The adeno-associated virus type 2 Rep protein regulates RNA processing via interaction with the transcription template. Mol. Cell. Biol. 22:3639–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoto Y, Qiu J, Pintel DJ. 2006. Identification and characterization of two internal cleavage and polyadenylation sites of parvovirus B19 RNA. J. Virol. 80:1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guan W, Cheng F, Yoto Y, Kleiboeker S, Wong S, Zhi N, Pintel DJ, Qiu J. 2008. Block to the production of full-length B19 virus transcripts by internal polyadenylation is overcome by replication of the viral genome. J. Virol. 82:9951–9963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walker SL, Wonderling RS, Owens RA. 1997. Mutational analysis of the adeno-associated virus type 2 Rep68 protein helicase motifs. J. Virol. 71:6996–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kerr JR, Cotmore SF, Bloom ME, Linden ME, Parish CR. (ed). 2006. Parvoviruses, 6th ed Hodder Arnold, London, United Kingdom [Google Scholar]
- 22. Shepard PJ, Hertel KJ. 2009. The SR protein family. Genome Biol. 10:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pintel D, Dadachanji D, Astell CR, Ward DC. 1983. The genome of minute virus of mice, an autonomous parvovirus, encodes two overlapping transcription units. Nucleic Acids Res. 11:1019–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mouw MB, Pintel DJ. 2000. Adeno-associated virus RNAs appear in a temporal order and their splicing is stimulated during coinfection with adenovirus. J. Virol. 74:9878–9888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qiu J, Pintel D. 2008. Processing of adeno-associated virus RNA. Front. Biosci. 13:3101–3115 [DOI] [PubMed] [Google Scholar]
- 26. Liu Z, Qiu J, Cheng F, Chu Y, Yoto Y, O'Sullivan MG, Brown KE, Pintel DJ. 2004. Comparison of the transcription profile of simian parvovirus with that of the human erythrovirus B19 reveals a number of unique features. J. Virol. 78:12929–12939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guan W, Huang Q, Cheng F, Qiu J. 2011. Internal polyadenylation of the parvovirus B19 precursor mRNA is regulated by alternative splicing. J. Biol. Chem. 286:24793–24805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang Q, Deng X, Best SM, Bloom ME, Li Y, Qiu J. 2012. Internal polyadenylation of parvoviral precursor mRNA limits progeny virus production. Virology 426:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qiu J, Cheng F, Burger LR, Pintel D. 2006. The transcription profile of Aleutian mink disease virus in CRFK cells is generated by alternative processing of pre-mRNAs produced from a single promoter. J. Virol. 80:654–662 [DOI] [PMC free article] [PubMed] [Google Scholar]