

Abstract
Vesicular stomatitis virus (VSV) has shown considerable promise both as an immunization vector and as an oncolytic virus. In both applications, an important concern is the safety profile of the virus. To generate a highly attenuated virus, we added two reporter genes to the 3′ end of the VSV genome, thereby shifting the NPMGL genes from positions 1 to 5 to positions 3 to 7. The resulting virus (VSV-12′GFP) was highly attenuated, generating smaller plaques than four other attenuated VSVs. In one-step growth curves, VSV-12′GFP displayed the slowest growth kinetics. The mechanism of attenuation appears to be due to reduced expression of VSV genes downstream of the reporter genes, as suggested by a 10.4-fold reduction in L-protein RNA transcript. Although attenuated, VSV-12′GFP was highly effective at generating an immune response, indicated by a high-titer antibody response against the green fluorescent protein (GFP) expressed by the virus. Although VSV-12′GFP was more attenuated than other VSVs on both normal and cancer cells, it nonetheless showed a greater level of infection of human cancer cells (glioma and melanoma) than of normal cells, and this effect was magnified in glioma by interferon application, indicating selective oncolysis. Intravenous VSV-12′GFP selectively infected human gliomas implanted into SCID mice subcutaneously or intracranially. All postnatal day 16 mice given intranasal VSV-12′GFP survived, whereas only 10% of those given VSV-G/GFP survived, indicating reduced neurotoxicity. Intratumoral injection of tumors with VSV-12′GFP dramatically suppressed tumor growth and enhanced survival. Together these data suggest this recombinant virus merits further study for its oncolytic and vaccine potential.
INTRODUCTION
Vesicular stomatitis virus (VSV) is an enveloped nonsegmented negative-strand RNA virus of the Rhabdoviridae family with a simply organized genome of 11.2 kb that encodes just five genes (N, P, M, G, and L) (1, 2). The ability to recover fully replication-competent VSV from suitably engineered plasmid DNA (3, 4) has enabled the generation of modified recombinant versions of VSV (rVSV), some of which are currently under active investigation for their therapeutic potential as replicating or nonreplicating vaccine vectors (5–8) and as oncolytic agents for the treatment of a number of different types of human cancer (9–12).
In nature, VSV is a pathogen of livestock, such as horses, cattle, and swine, with infection of humans being relatively rare and resulting typically in subclinical or mild flu-like symptoms (13, 14). Although encephalitis is not a characteristic of natural VSV infection (13), experimental infection of brain cells has been found in animal models (15–18). We have previously shown that the use of recombinant attenuated VSV and peripheral immunization reduced or blocked the ability of VSV to infect central nervous system (CNS) cells (12, 19). Further refinement of recombinant VSVs for therapeutic application, particularly within the brain, may benefit from additional viral attenuation to improve their margin of safety and curtail the undesired infection of normal tissue.
To date, a variety of strategies have been employed to attenuate the propagation of VSV (20). One strategy has been to incorporate mutations into the M protein (M33A, M51A, and deltaM51) (12, 21–25). Another strategy has been to delete small portions of the G protein (12, 17, 22, 23, 26, 27) or to delete the G protein entirely (12, 26, 28). Shuffling the normal order of VSV genes (22, 23, 29, 30) or insertion of a nonviral gene into the viral genome also attenuates the virus (23, 31, 32). Insertion of a gene into the first gene position yields a greater attenuation than in other genomic positions (12, 23, 33).
Here we report the construction of a recombinant VSV (VSV-12′GFP) that adds two (nonviral fluorescent reporter) genes, one in the first (1′) and another in the second (2′) genomic positions, thus shifting the viral genes NPMGL from positions 1′ through 5′ to positions 3′ through 7′. The level of gene expression is dependent on gene position, with the highest-expression gene located at the 3′ end of the genome (1′ gene position). The insertion of two genes on the 3′ end (1′ and 2′ gene positions) results in a highly attenuated viral phenotype with respect to growth kinetics and plaque size in vitro and improved tolerance in vivo. The virus, VSV-12′GFP, evokes a strong antibody response to the reporter gene and to the VSV G protein and displays selective targeting in vitro and in vivo against several human cancer cell types.
MATERIALS AND METHODS
VSV-12′GFP plasmid construction, virus recovery, and verification.
The 15.8-kb pVSV-12′GFP plasmid used to recover VSV-12′GFP (shown schematically in Fig. 1B) was constructed as follows. The plasmid pVSV1XN-dsRed (28, 34) was digested using XhoI and NotI to remove the 0.75-kb insert coding for dsRed in the first position, replacing it with a 1.5-kb insert containing two genes, one encoding a green fluorescent protein (GFP) derived from Aequorea victoria (avGFP) and the other humanized Renilla GFP (rrGFP) derived from Renilla reniformis. This 1,503-bp insert was commercially synthesized (Genscript, Piscataway, NJ) using the sequence shown in Fig. 1B and the sequences for avGFP (accession number U55761; nucleotides [nt] 97 to 816) and rrGFP (accession number GQ380658; nt 2895 to 3614). AscI and NheI restriction sites were included to more easily allow future manipulation of first- and second-position genes. The gene junction sequence between the newly created first and second genomic positions contains a transcription termination and polyadenylation signal (TATGAAAAAAA) for the first gene and a transcription initiation signal (AACAGATATC) for the second gene (33, 35).
Fig 1.

Attenuated VSV genomes and VSV-12′GFP construction. The six recombinant VSVs used in the present study (rVSV, VSV-G/GFP, VSV-M51, VSV-M51/CT9, VSV-1′GFP, and VSV-12′GFP) all derive from engineered versions of the original recombinant “wild-type” rVSV (3). (A) Diagrams depicting the VSV genomes of the present study. Each is a single negative strand of RNA, and the “wild-type” rVSV (top) encodes five genes, N, P, M, G, and L. GFP reporter genes (V and R) are shown in green and designate avGFP and rrGFP, respectively. RT-PCR products described in part C are indicated in purple. See Materials and Methods for further description. (B) Diagram of the plasmid that was used to recover VSV-12′GFP, showing the 1,503-bp DNA insert sequence that was synthesized and then ligated into the first position using XhoI and NotI restriction sites. The AscI restriction site used in panel C is shown in purple. (C) Agarose gel of purified RT-PCR products generated using a primer pair flanking the first-position insert site and genomic RNA from VSV-G/GFP, VSV-1′GFP, and VSV-12′GFP. Lanes 2 to 4 show the expected increasing product sizes of 1,059 bp (G/GFP), 1,817 bp (1′GFP), and 2,603 bp (12′GFP), respectively. Lane 5 shows the restriction digestion of 12′GFP using AscI and yields the two expected bands, of 1,798 and 805 bp. Lanes 1 and 6 are 1-kb molecular size markers (mw). (D) Photomicrographs taken 24 h post-inoculation of BHK-21 cells, showing the fluorescent plaque sizes of VSV-12′GFP compared to those of VSV-G/GFP and VSV-1′GFP. Scale bar, 1 mm. (E) BHK-21 cells (1.5 × 106) were infected (MOI = 10) with VSV-G/GFP (circles), VSV-1′GFP (triangles), and VSV-12′GFP (squares). Medium samples were collected at the indicated times postinfection, and titers were determined by plaque assay on BHK-21 cells. The graph displays the increase in VSV titer (PFU/ml) with respect to time after infection. Each point represents the mean value obtained from two separate growth experiments performed in parallel.
Recombinant VSV was recovered from pVSV-12′GFP plasmid DNA through transfection of BHK-21 cells and infection with vaccinia virus-T7, as described in detail elsewhere (3, 4). Of the 20 10-cm dishes of BHK-21 cells transfected with the pVSV-12′GFP plasmid and infected with vaccinia-T7, only 1 dish yielded a successful recovery of VSV-12′GFP. Seed stocks of the additional plasmids used in VSV recovery (pBS-N, pBS-P, and pBS-L), along with pVSV1XN-dsRed, were obtained from J. Rose (Yale University).
Molecular verification of the successful recovery of VSV-12′GFP was performed using reverse transcriptase PCR (RT-PCR). Briefly, medium harvested from VSV-infected cultures was used to isolate VSV genomic RNA using the QIAamp viral RNA minikit (Qiagen, Valencia, CA). Reverse transcription of genomic RNA was performed using the SuperScript III reverse transcriptase kit (Invitrogen, Carlsbad, CA) and the RT oligonucleotide VSV1F, with the sequence 5′-ACG AAG ACA AAC AAA CCA TTA TTA TC-3′, designed to anneal to the initial 26 bases of the 3′ end of the VSV genome. VSV cDNA (2 μl) was then used as the template in PCRs using the Expand high-fidelity PCR kit (Roche Diagnostics, Indianapolis IN). The primer pair VSV1F (sequence above) and VSV1059R, with the sequence 5′-CAA ACC TGC TGT AGT AAG AG-3′, was used and was designed to flank the first genomic position insertion site and anneal to nt 1 to 26 and 1040 to 1059, respectively, of the VSV-G/GFP sequence (Fig. 1A) (accession number FJ478454). Three different PCR annealing temperatures and elongation times, corresponding to the type of VSV cDNA being amplified and the product length, were used, as follows: VSV-G/GFP (1,059 bp), 51.4°C, 60 s; VSV-1′GFP (1,817 bp), 53.8°C, 80 s; and VSV-12′GFP (2,603 bp), 54.9°C, 110 s. PCR products were then purified using the QIAquick PCR purification kit (Qiagen) and spectrophotometrically quantified, and 300 ng of each was run on a 1% agarose gel. One microgram of purified VSV-12′GFP PCR product was also restriction digested using AscI (New England BioLabs, Beverly, MA), and 300 ng was run on the agarose gel described above.
Other VSVs.
The six recombinant VSVs used for purposes of comparison in the present study (rVSV, VSV-G/GFP, VSV-M51, VSV-M51/CT9, VSV-1′GFP, and VSV-12′GFP) are all derived from the original “wild-type” recombinant rVSV (3), a chimera assembled from the San Juan and Mudd-Summers strains of the Indiana serotype (36–39). VSV-G/GFP incorporates an enhanced GFP (EGFP) reporter gene, derived from Aequorea victoria, fused to the C terminus of an additional copy of the G-protein gene, with this G/GFP fusion gene inserted into the fifth genomic position (18, 40). VSV-M51 contains an M gene that has been mutated to delete the methionine codon at position 51 of the M protein and includes an EGFP reporter gene in the fifth position (25). VSV-M51/CT9 incorporates the M51 mutation along with a truncation of the G gene that removes all but 9 of the 27 amino acids of the C-terminal cytoplasmic tail. Both of these mutations result in attenuated viral propagation (22, 25, 41). VSV-1′GFP contains an EGFP reporter gene inserted into the first genomic position, which results in a decreased level of transcription of the remaining five downstream genes, which in turn attenuates viral propagation (22, 29, 33). Seed stocks of rVSV, VSV-G/GFP, VSV-M51, VSV-M51/CT9, and VSV-1′GFP were obtained from J. Rose (Yale University) and were propagated along with determination of titers on BHK-21 cells.
Cell lines.
The baby hamster kidney cell line BHK-21, human glioblastoma (GBM) tumor cell line U-87MG, and human fibrosarcoma cell line HT1080 were obtained from the ATCC (Manassas, VA). The human glioma line rU-87, expressing a gene coding for a red fluorescent reporter, has been described previously (42). Normal human fibroblasts were purchased from Cambrex (Walkersville, MD). The human glioblastoma tumor cell lines U-373MG and U-118 were kindly provided by R. Matthews (Syracuse, NY). The primary human melanocyte cultures and YUMAC melanoma cancer cells were obtained from the Yale University Skin Diseases Research Center (SPORE Core) and were kindly provided by R. Halaban. Primary cultures of normal human astrocytes were established from surgery specimens of patients undergoing surgery for epilepsy (12). Human cell preparation and use were approved by the Yale University Human Investigation Committee. All cell lines were propagated using MEM growth medium, consisting of minimum essential medium (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin solution (Gibco) and housed in a humidified incubator at 37°C, supplied with 5% CO2. Phosphate-buffered saline solution (PBS) and trypsin solution were purchased from Gibco.
One-step growth curves.
One-step growth curves (43) were performed for VSV in a manner similar to that previously described (33, 44). Briefly, approximately 1.5 × 106 BHK-21 cells grown in a 35-mm well of a 6-well dish were infected (multiplicity of infection [MOI] = 10) with each of the VSVs to be tested and allowed to incubate for 30 min at 37°C in 5% CO2. After incubation, the inoculum was removed, and each well was washed 3× with PBS to remove unadsorbed virus and then replenished with 3 ml MEM and returned to the incubator. At the indicated time points (Fig. 1E), 125-μl samples of medium were removed and stored at −80°C for later determination of the VSV titer by fluorescent plaque count assays on BHK-21 cells. Growth curves for each virus tested (VSV-G/GFP, VSV-1′GFP, and VSV-12′GFP) were performed in duplicate, and the resulting sample titers were averaged.
Plaque assays.
Since five of the six VSVs of the present study express GFP as a reporter protein, fluorescent imaging could be utilized for both plaque counting and plaque size determination. Briefly, fluorescent plaque counting assays consisted of growing confluent monolayers of BHK-21 cells in six-well dishes and infecting them with 1-ml volumes of serially diluted samples of VSV-containing medium. After infection, plates were returned to the incubator for 1 h to allow time for the virus to adsorb into the cell monolayer. The infection medium was then aspirated, and the cell monolayers were overlaid with 2 ml of 0.5% (wt/vol) agarose (Ultrapure GPG/LE; American Bioanalytical, Natick, MA) in MEM growth medium. After solidification of the agarose, plates were incubated at 37°C in 5% CO2 for 18 to 24 h to allow time for plaque development. Plaque counting of nonfluorescent rVSV stocks was performed in a similar manner, and plaques were visualized by counterstaining using a solution of neutral red (N2889; Sigma, St. Louis, MO). Fluorescent plaque visualization was performed using a fluorescent imaging system that consisted of an Olympus IX71 inverted microscope (Tokyo, Japan) fitted with a GFP filter set and Olympus UplanFl 2×, 4×, 10× and 20× objectives. Fluorescent plaque counts were tallied by eye under fluorescent illumination, and images of fluorescent plaques were captured using a Spot digital camera (Diagnostic Instruments, Sterling Heights, MI) and further processed using the Adobe Photoshop 7.0 software program (Adobe Systems, San Jose, CA). Fluorescent-plaque size measurements were performed in a similar manner to that of plaque counting. Briefly, plaques were grown as above using the VSV and cell type to be tested. For each VSV, 60 plaques were randomly selected under fluorescent visualization and measured across their diameters to the nearest 0.05 mm. In Fig. 4D, the mean VSV-G/GFP plaque diameter in each cell line was used as the normalization factor for all plaque diameter measurements (normalized mean VSV-G/GFP plaque diameter = 100%). All uncertainties represent the standard error of the mean (SE). Statistical calculations were performed using the InStat 3.0 software program (GraphPad Software, La Jolla, CA). One-way analysis of variance (ANOVA) with post hoc Tukey-Kramer multiple-comparison tests was used to determine statistical significance, as were Student's t tests, where appropriate. Comparisons between groups yielding a P value of <0.05 were considered significantly different.
Fig 4.

VSV plaque size on normal brain and glioblastoma cells. A series of representative photos show the sizes of the fluorescent plaques that developed from each of the VSVs after 18 h postinfection on monolayers of normal human brain astrocytes (A) or U-373 human glioblastoma cells (B). Scale bar, 500 μm. The histograms display the distribution of plaque diameter measurements (n = 60) acquired from the same population from which the corresponding photos were taken. The triangles indicate the mean diameter of each plaque population, and the dotted line marks the mean for the VSV-G/GFP population for comparison. Circles depict the mean plaque size for each population, with the small vertical line above each circle representing the SE. Mean virus plaque sizes were all significantly different from that of VSV-G/GFP (ANOVA; P < 0.01) for each cell line except for the M51 plaques. (C) Bar graph summarizing the mean individual plaque areas (in square micrometers) measured in panels A and B, with error bars representing the SE. The ratio of glioblastoma to normal brain plaque area for each virus is indicated below the axis. Mean plaque size between normal brain and glioblastoma cells were all significantly different from one another for each virus tested (t test; P < 0.001). (D) Comparison of mean plaque sizes (n = 60) using normal human brain astrocytes, normal human fibroblasts, and hamster BHK-21 cells after infection with each of the VSVs. Plaque sizes for each cell type have been normalized with respect to the corresponding VSV-G/GFP mean plaque diameters. All mean plaque sizes were significantly different (ANOVA; P < 0.01) from those of corresponding G/GFP plaques, except the M51 plaques in human brain and BHK-21 cells and M51/CT9 plaques in BHK-21 cells. All VSV-12′GFP plaques were significantly different at the P < 0.001 confidence level.
qRT-PCR measurements of VSV genomes, N- and L-protein transcripts, and interferon-stimulated gene expression.
Quantitative RT-PCR (qRT-PCR) was carried out with total RNA isolated from cell lysates using the RNeasy kit (Qiagen, Valencia, CA) and reverse transcribed using random hexamers and the SuperScript III reverse transcriptase kit (Invitrogen, Carlsbad, CA). TaqMan gene expression assays for the VSV N- and L-protein genes along with the human β-actin and interferon-stimulated genes EIF2AK2, IFIT1, ISG-15, OAS-1, and Mx1 were purchased from Applied Biosystems (Foster City, CA). Measurement of VSV genomes was performed using a TaqMan assay designed to recognize the intergenic sequence between the N and P genes, thus attenuating any contribution to the VSV genomic RNA signal that might arise from mRNA transcripts (45). Genomic VSV values were found to represent less than 1% and 10% of the corresponding N and L transcript values, respectively, and were subtracted. Dilutions of the pVSV-12′GFP plasmid, described earlier, were used to establish a standard curve to determine transcript copy number per microliter of total RNA. Samples were collected in triplicate, and each sample was measured using triplicate PCRs.
Animal procedures.
For intranasal administration of VSVs, young postnatal day 16 Swiss/Webster mice (Taconic Farms, Germantown, NY) were lightly anesthetized using ketamine-xylazine and then inoculated with 15 μl of virus solution in each nostril (30 μl total per animal). Mouse health and weight were monitored daily, and animals displaying any neurological symptoms (abnormal walking, paralysis, etc.) or a body weight drop below 85% of the starting weight were euthanized according to institutional guidelines and were recorded as showing a lethal response.
To investigate the efficacy and safety of VSV-12′GFP in adult tumor-bearing mice, immunodeficient homozygous CB17-SCID mice (Taconic Farms) 6 to 8 weeks of age were used. Subcutaneous bilateral flank tumors were established by injection of approximately 1.25 × 105 rU-87 cells (42) in 50 μl sterile PBS. Tumors were allowed to develop to approximately 2 to 3 mm in diameter before the viral administration phase of the experiment was begun. VSV-12′GFP (150,000 PFU) or control medium was injected directly into each tumor. Tumor dimensions were measured using a caliper, and tumor volume (V) was estimated using the formula for the volume of a prolate spheroid of length a and uniform width b, i.e., V = 4/3π(a/2)(b/2)(b/2). When tumors reached 1 cm in diameter, animals were euthanized by mandate of institutional guidelines and were recorded as showing a lethal response.
In another set of experiments, 24 h after VSV-12′GFP injection, tumors were harvested in sterile PBS and then transferred to 500 μl of MEM and triturated. The triturated tissue sample was then microcentrifuged, and the cleared virus-bearing supernatant was used in a plaque assay to determine plaque size. Additional mice were perfused transcardially at 24 or 48 h post-tumor inoculation to determine virus spread. For histology, animals were given a pentobarbital overdose and perfused transcardially with 4% paraformaldehyde.
To determine the ability of the virus to selectively infect human cancer cells, U-87 and U-118 human glioma cells were injected subcutaneously (100,000 cells in sterile saline) or intracerebrally (50,000 cells in 0.5 μl sterile saline) into adult SCID mice. After establishment of the tumors, VSV-12′GFP (107 PFU in 100 μl sterile saline) was administered by intravenous injection into the tail vein. At indicated time points, mice were given an anesthetic overdose and perfused transcardially with saline followed by 4% paraformaldehyde. Brains and areas containing subcutaneous tumors were sectioned on a cryostat and mounted on glass slides. All animal experiments and postoperative care were performed in accordance with the institutional guidelines of the Yale University Animal Care and Use Committee.
Immunocytochemistry.
To demonstrate that VSV-12′GFP expressed both rrGFP and avGFP, we used antibodies against each of the different GFPs. Mice receiving intracranial injections of VSV-12′GFP were given an anesthetic overdose and perfused transcardially with 4% paraformaldehyde. After sucrose infiltration, frozen brain sections were cut. Separate sections were immunostained with either the rrGFP antibody (from Agilent Technologies, Santa Clara, CA) or avGFP antibody (Chemicon International, Billerica, MA) at a dilution of 1:2,500, followed by a secondary antiserum of goat anti-mouse conjugated to red fluorescent Alexa 594. As an additional control to demonstrate that the antisera used were selective for only one or the other of the two GFPs used, we tested each antibody on sections from transgenic mice in which either rrGFP was selectively expressed by cells containing neuropeptide Y (NPY) (46) or avGFP was selectively expressed by cells containing melanin-concentrating hormone (MCH) (47).
Antisera were raised against VSV-12′GFP by intranasal or subcutaneous inoculation of mice, followed by a second inoculation 5 weeks later. Antisera were used to detect both the GFP reporter and the VSV G protein, using dilutions from 1:1,000 to 1:250,000. Photomicrographs were taken with a Spot digital camera (Diagnostic Imaging), and contrast and brightness were corrected using Adobe Photoshop.
RESULTS
Addition of reporter genes in the first and second genomic positions substantially attenuates VSV virulence.
Starting with a genomic VSV plasmid carrying a single fluorescent reporter gene inserted in the first position of the VSV genome (28, 34), we replaced this insert with a 1.5-kb synthetically manufactured insert encoding two fluorescent reporter genes separated by a gene junction containing the appropriate gene transcript termination and initiation sequences (33, 35) (Fig. 1B). The resulting plasmid (pVSV-12′GFP) encodes a GFP derived from the jellyfish Aequorea victoria (avGFP) and a GFP derived from the sea pansy Renilla reniformis (rrGFP) in the first (1′) and second (2′) genomic positions, respectively, thereby shifting the remaining VSV genes (N, P, M, G, and L) two positions further down the genome (Fig. 1A). The avGFP and rrGFP genes share less than 20% nucleotide identity, with the resulting proteins sharing less than 30% amino acid identity and displaying similar fluorescent characteristics (48). In order to verify the identity of the recovered VSV, we isolated genomic RNA and performed RT-PCR using primers flanking the insertion site (Fig. 1A and C). Purified RT-PCR products from VSV-12′GFP (2,603 bp) were compared with those from VSV-G/GFP (1,059 bp) and VSV-1′GFP (1,817 bp), and all were found to have the expected molecular weights. Additionally, the identity of the VSV-12′GFP product was further confirmed through restriction digestion using AscI (Fig. 1B and C). This digestion of the 12′GFP product was complete and yielded bands of 1,798 and 805 bp, as expected (Fig. 1C, lane 5).
Next, the plaque sizes of different VSVs growing on BHK-21 cells were compared as an indicator of virus attenuation. The different VSVs displayed apparent differences in plaque size, with VSV-12′GFP plaques being smallest, VSV-G/GFP largest, and VSV-1′GFP plaques of intermediate size (Fig. 1D). To further assess the growth of VSV-12′GFP relative to that of other recombinant VSVs, we performed a one-step growth curve experiment (33, 43, 44) comparing VSV-12′GFP to VSV-G/GFP and VSV-1′GFP. These curves (Fig. 1E) display a clear progression of attenuated growth kinetics, with VSV-12′GFP being the most attenuated (5.5 × 107 PFU/ml at 8 h postinoculation [p.i.]), followed by VSV-1′GFP (3.4 × 108 PFU/ml at 8 h p.i.) and VSV-G/GFP (1.2 × 109 PFU/ml at 8 h p.i.) or a ratio between these titers of approximately 1 to 6 to 21, respectively. Additionally, the length of the eclipse period or time between viral inoculation and the entry into rapid growth phase (characterized by a logarithmic increase in the detected release of viral progeny) was longer for VSV-12′GFP (3.0 to 3.5 h) than for VSV-1′GFP (2.5 to 3.0 h) or VSV-G/GFP (2.0 to 2.5 h). The results of the one-step growth curve parallel those found with plaque size, as described above.
Transgene expression.
To ensure that the newly generated virus expressed both reporter genes, we used immunocytochemical staining with antisera selective for one or the other of the two fluorescent reporter gene products. We confirmed that the antisera were selective for each reporter by immunocytochemical testing of brain sections from transgenic mice that expressed either avGFP in neurons that synthesize melanin-concentrating hormone (MCH) (47) or rrGFP in neurons that synthesize neuropeptide Y (NPY) (46). Antiserum against avGFP stained GFP-fluorescent MCH cells of the lateral hypothalamus from MCH-avGFP mice (Fig. 2K and L) but did not stain GFP-fluorescent NPY cells of the arcuate nucleus from NPY-rrGFP mice (Fig. 2G and H). Conversely, antiserum against rrGFP stained GFP-fluorescent NPY cells from NPY-rrGFP mice (Fig. 2E and F) but did not stain GFP-fluorescent MCH cells from MCH-avGFP mice (Fig. 2I and J), thus demonstrating the immunocytochemical selectivity of each antiserum. Next, VSV-12′GFP (2 μl of 107 PFU/ml) was injected into the brains of normal mice, and the animals were euthanized 1 day later. The antiserum against avGFP stained GFP fluorescent cells infected with VSV-12′GFP (Fig. 2C and D), and the antiserum against rrGFP also stained GFP-fluorescent VSV-12′GFP-infected cells (Fig. 2A and B). Together, these data indicate that VSV-12′GFP expresses both the avGFP and rrGFP reporter genes and that the two antisera used display selective staining of the homotypic antigen. VSV-12′GFP was also given to mice subcutaneously and intranasally, and mice were boosted 5 weeks later. When used to immunostain brain sections from transgenic mice expressing the avGFP reporter gene selectively in neurons that synthesize melanin-concentrating hormone, the antiserum was positive for immunofluorescence when used at a very high dilution of 1:250,000 (Fig. 3A and B), indicating a robust ability of the virus to generate a humoral response to the transgene engineered into VSV-12′GFP. In addition, we infected BHK-21 cells in culture with a VSV that did not express a reporter gene (rVSV), and using the same antibody as above, we found strong immunostaining in infected cultures but not in noninfected control cultures (Fig. 3C and D). This indicates that the same antiserum bound not only to the GFP but also to VSV viral proteins.
Fig 2.

Both avGFP and rrGFP are expressed by VSV-12′GFP. To demonstrate antibody selectivity, we used sections from transgenic mice that express either avGFP in neurons that synthesize melanin-concentrating hormone (MCH) or rrGFP in neuropeptide Y neurons (NPY). Panel A shows VSV-12′GFP infection of normal mouse brain under GFP illumination, and panel B shows the same field after staining with an rrGFP antibody. Panel C shows VSV-12′GFP infection, and panel D shows the same field immunostained with a second primary antiserum against avGFP. In NPY-rrGFP transgenic mouse sections (E), the rrGFP antiserum stains the NPY neurons (F), whereas NPY neurons (G) are not stained with the avGFP antibody (H). In the MCH-avGFP mouse sections, MCH neurons (I) express avGFP and are not stained with the rrGFP antibody (J), whereas MCH neurons (K) express avGFP and are labeled with the avGFP antibody (L). Scale bar, 25 um.
Fig 3.

VSV-12′GFP generates an antibody response against transgene and viral proteins. (A) Antisera were raised against VSV-12′GFP. At a substantial dilution of 1:250,000, the antibody was used to visualize MCH neurons in the brain that expressed avGFP. (B) The GFP expression of the MCH cells immunolabeled in panel A is shown. (C) A VSV that did not express a fluorescent reporter was used to infect BHK cells. The antibody raised against VSV-12′GFP was used to stain infected cells with red immunofluorescence. On the right is the phase image of the same field as shown in the fluorescence micrograph on the left. (D) Control BHK cells show no immunolabeling when stained with the VSV-12′GFP antibody, corroborating the selectivity of the antiserum for the virus protein.
Attenuated viral plaque size on normal and transformed human cells.
To quantitatively compare differences in plaque size between VSV-12′GFP and a number of additional attenuated recombinant VSVs expressing GFP, a fluorescence microscopic imaging-based plaque size assay was used. Monolayer cultures of normal human astrocytes and U-373 glioblastoma cells were assayed using five separate VSVs of various degrees of attenuation (VSV-G/GFP, VSV-M51, VSV-M51/CT9, VSV-1′GFP, and VSV-12′GFP). Fluorescent-plaque size measurements were made 18 h postinfection (Fig. 4). In normal brain cells, the mean plaque diameter was smallest for VSV-12′GFP plaques and largest for VSV-G/GFP plaques, with the sizes arranged from smallest to largest being in the order 12′GFP, 1′GFP, M51/CT9, M51, and G/GFP (Fig. 4A and B). VSV-12′GFP plaques in both cell types were dramatically smaller than those of all other VSVs tested (ANOVA; n = 60/group; P < 0.001). Comparison of plaque sizes shows a highly significant difference (P < 0.001; t test; n = 60/group), with much larger plaques generated on human GBM cells (U-373) than on normal human astrocytes in all VSVs tested (Fig. 4C). The plaques formed by VSV-12′GFP on glioblastoma were approximately 9.5-fold larger than the plaques formed on normal astrocytes, suggesting a more enhanced propagation in glioblastoma cells relative to normal cells.
Plaque sizes were also assayed using normal human fibroblasts and BHK-21 cells (Fig. 4D). Similar to the previous results, VSV-12′GFP yielded smaller plaques (ANOVA; n = 60; P < 0.001) than all the other VSVs tested. Taken together, these results suggest that the highly attenuated VSV-12′GFP phenotype is maintained across a number of different cell types, both normal and cancer, and that, although attenuated in both cell types, the virus continues to propagate in an enhanced fashion in cancer cells compared to that in normal cells.
Attenuated production of VSV N and L transcripts.
Here we tested the hypothesis that the insertion of two genes at the 3′ end of the VSV genome reduces expression of viral genes to a degree even greater than that with insertion of a single gene in gene position 1. Measurement of the amount of VSV N-protein and L-protein mRNA transcript generated by VSV-12′GFP and three other recombinant VSVs (rVSV, VSV-G/GFP, and VSV-1′GFP) was performed using quantitative RT-PCR (qRT-PCR) on total RNA isolated from infected cultures (MOI = 0.1) of human glioblastoma U-87 cells 12 h postinfection (Fig. 5). The N-protein mRNA transcript copy number per μl of total RNA harvested from infected cells was lowest in VSV-12′GFP and highest in rVSV with ratios (from lowest to highest) of approximately 1.0, 2.9, 11.2, and 17.8 (for 12′GFP, 1′GFP, G/GFP, and rVSV). A qualitatively similar set of values was also found for the L-protein transcript copy number, with VSV-12′GFP again showing the lowest and rVSV the highest with ratios, of approximately 1.0, 2.6, 4.8, and 10.6 (for 12′GFP, 1′GFP, G/GFP, and rVSV). The magnitude of the L transcript values (107 copies/μl) is considerably less than that of the N transcript values (108 copies/μl), with each L transcript being approximately 4.9, 4.4, 2.1, and 2.9% of the corresponding N transcript for VSV-12′GFP, VSV-1′GFP, VSV-G/GFP, and rVSV, respectively. These N/L transcript ratios are consistent with previous reports of recombinant VSV N/L protein ratios based on radiolabel experiments using [35S]methionine (49, 50). Taken together, these results demonstrate a progressive reduction in both N and L transcript levels that corresponds with increasing VSV attenuation, with VSV-12′GFP displaying the lowest transcript levels and highest degree of attenuation.
Fig 5.
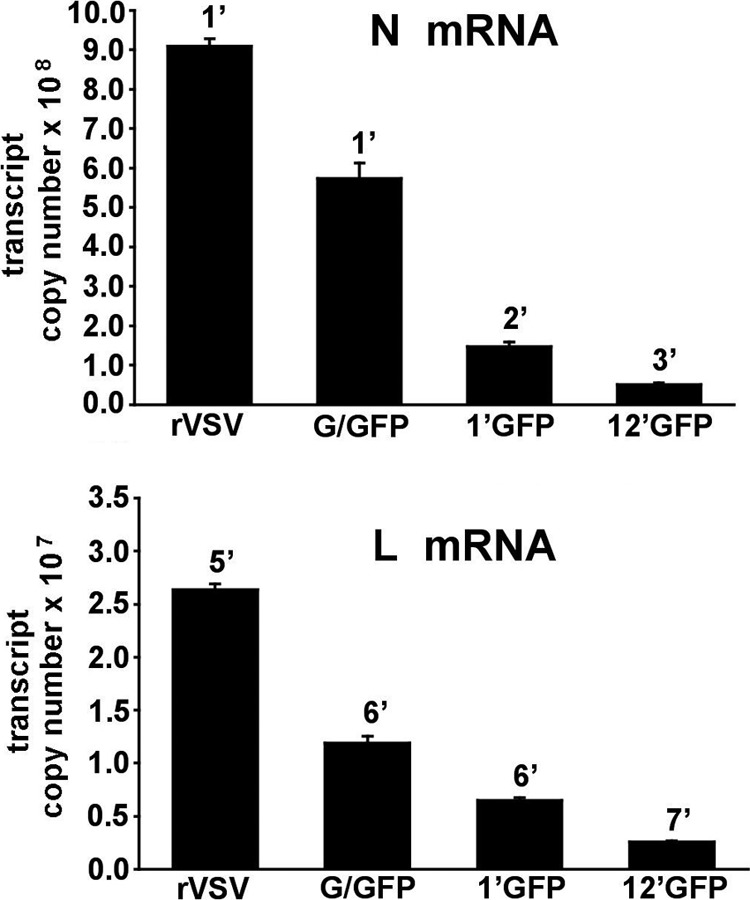
N and L RNA transcript levels of recombinant VSVs. The amounts of N-protein mRNA (A) or L-protein mRNA (B) generated by VSV-12′GFP and three other recombinant VSVs (rVSV, VSV-G/GFP, and VSV-1′GFP) were compared using quantitative RT-PCR and are expressed as transcript copy number per μl of total RNA. Genomic RNA was distinguished from mRNA by utilizing a probe specific to an intergenic region of the VSV genome; genomic values were subtracted from total RNA to yield mRNA values. Cultures of U-87 cells were inoculated (0.1 MOI) in triplicate for each condition, and total RNA was harvested 12 h postinfection. The number above each bar indicates the genomic position from which the transcript arises, and the error bars represent the standard errors of the means. All mean transcript values were significantly different (ANOVA; P < 0.01) from each other except those for 1′GFP and 12′GFP in the N mRNA panel.
Enhanced survival in vivo.
A common method of testing the virulence of VSV is with intranasal inoculation of young postnatal day 16 mice (19, 51). Mice at this age are more susceptible to VSV than adults are. We compared VSV-12′GFP with VSV-G/GFP. Mice received 30 μl intranasal VSV (50,000 PFU total). Any mice developing obvious neurological dysfunction or severe weight loss (greater than 15% of initial body weight) were euthanized and considered to show a lethal effect (Fig. 6). As shown in Fig. 6, 9 of 10 mice administered VSV-G/GFP showed a lethal response, and the surviving mouse showed reduced body weight. In contrast, all 10 mice treated with an equal dose of VSV-12′GFP survived.
Fig 6.
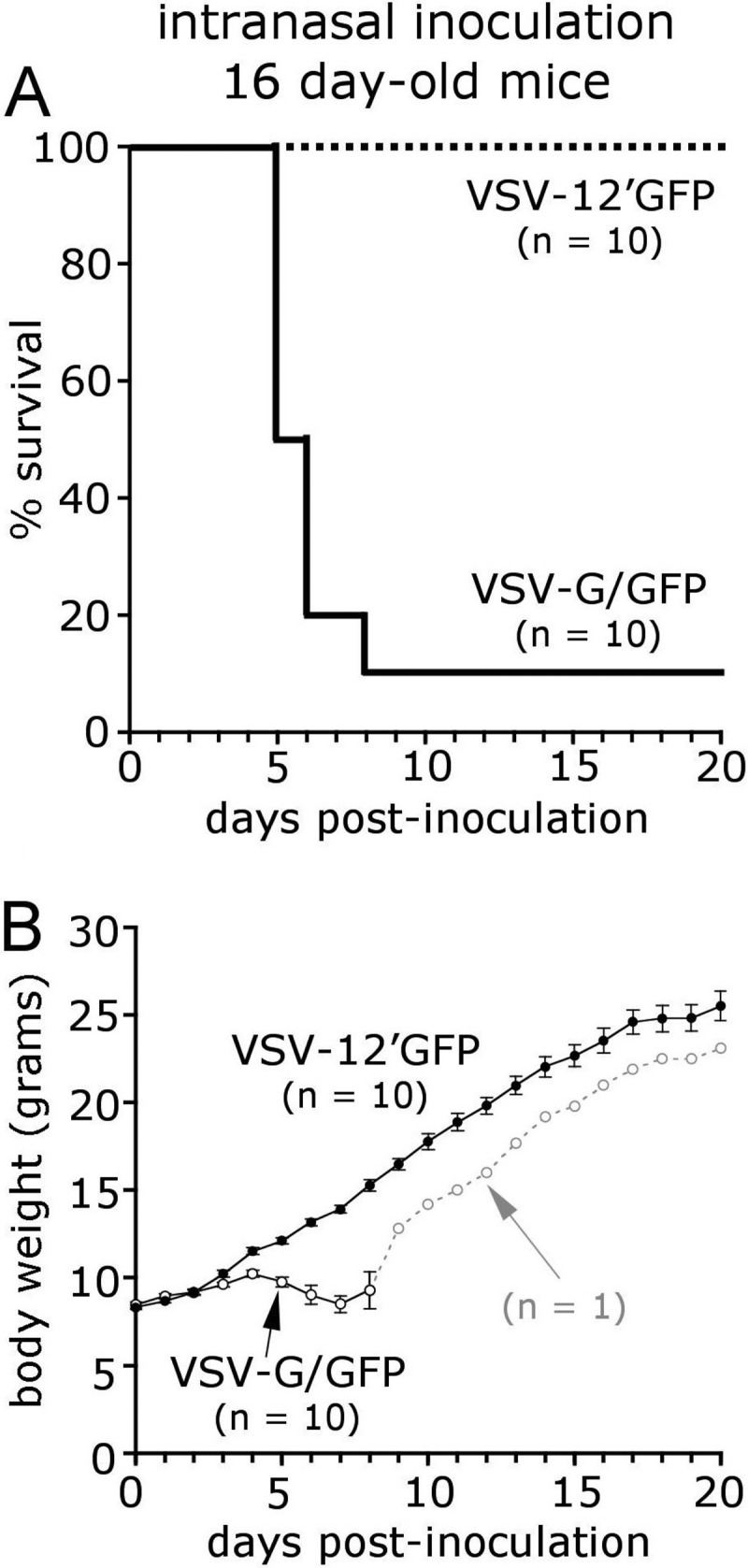
Intranasal inoculation of mice with VSV-12′GFP. VSV is often lethal in young mice when delivered through an olfactory entry route. A total of 50,000 PFU of either VSV-G/GFP or attenuated VSV-12′GFP was administered intranasally to young 16-day-old mice. Of the mice inoculated with VSV-G/GFP, 9 of 10 failed to gain body weight (B) and ultimately succumbed (A). In contrast, littermates receiving an equal dose of VSV-12′GFP steadily gained weight, and no mortality was observed. The number of mice inoculated with virus is indicated (n = 10), and error bars represent the standard errors of the means.
Fig 9.

Tail vein injection of VSV-12′GFP targets glioma in the brain. Human glioblastoma U-87 cells that had been stably transfected with a gene coding for a red fluorescent reporter were transplanted into the brains of adult SCID mice (A through G). VSV-12′GFP was subsequently injected into the tail vein. (A) Red glioblastoma cells are seen in the brain. (B) VSV-12′GFP selectively infected the tumor cells and not nearby normal brain cells. (C) Overlay of panels A and B. Scale bar, 30 um. (D) A large red GBM is seen in the left brain. (E) Peripheral intravenous VSV-12′GFP infected the tumor, whereas in the contralateral brain no tumor cells (F) or infection (G) are found. Scale bar, 70 um. (H) Human glioblastoma U-118 cells were implanted subcutaneously in adult SCID mice, and VSV-12′GFP was administered by tail vein injection. Four days after injection, the GFP-fluorescent virus had selectively infected the tumor at multiple sites (arrows). The boundary of the tumor is shown by the white line. Infection was not found in normal tissue surrounding the tumor, indicating that intravenous application of VSV-12′GFP successfully targets multiple derivations of glioma.
Enhanced infection of human glioma and melanoma cancer cells in vitro.
A variety of recombinant VSVs have been used to target cancer cells (12, 52–57). The plaque size experiments described above indicated a preference for human glioma/glioblastoma U-373 cells over normal human glia. Here we tested the hypothesis that despite its attenuated nature, VSV-12′GFP will still preferentially infect a variety of other cancer cells compared with normal cells. We compared the infection of human melanoma cells with infection of normal melanocytes. One day after inoculation (0.01 MOI), about 1% of the melanoma cells were infected, and fewer melanocytes (<0.1%) showed signs of infection. By 2 days p.i. (dpi), we found a dramatic difference in the level of infection: all melanoma cells were infected and dead or dying. Upon infection, the cells showed a striking pathological shift in morphology (Fig. 7B) and expressed the viral GFP reporter genes (Fig. 7B and C). In contrast, even after 4 dpi, less than 1% of the normal melanocytes had been infected (Fig. 7A and C); melanocytes from inoculated cultures looked normal and showed little pathology.
Fig 7.

Oncolytic activity of VSV-12′GFP against melanoma cells. Representative GFP fluorescent (left) and phase-contrast (right) photos of normal melanocyte (A) or YUMAC melanoma (B) cell culture 2 days after infection using a low MOI (0.01) of VSV-12′GFP are shown. (C) Bar graphs showing the mean percentages of infected cells in the cultures shown in panels A and B over a period of 4 days postinfection; linear presentation of values on the left and logarithmic on the right. Each bar represents the mean for six randomly selected microscopic fields, and the error bars represent the SE.
Next, we examined plaque size in two other human cancer cell types together with the normal noncancer control cells. The plaque size of VSV-12′GFP in U-118 glioblastoma was about 3-fold greater than that in normal human astrocytes (Fig. 8A and B). Similarly, the plaque size on fibrosarcoma was about 5-fold greater than that on normal human fibroblasts (Fig. 8B). We then used sister cultures from the same cancer and control cells to determine if the presence of alpha interferon (IFN) would alter the ratio of infection of cancer cells to that of normal cells. IFN added 12 h prior to viral inoculation completely blocked infection of normal astrocytes but did not block infection of glioblastoma cells (Fig. 8A and B). In contrast, IFN blocked infection of both the normal fibroblasts and the fibrosarcoma (Fig. 8B).
Fig 8.
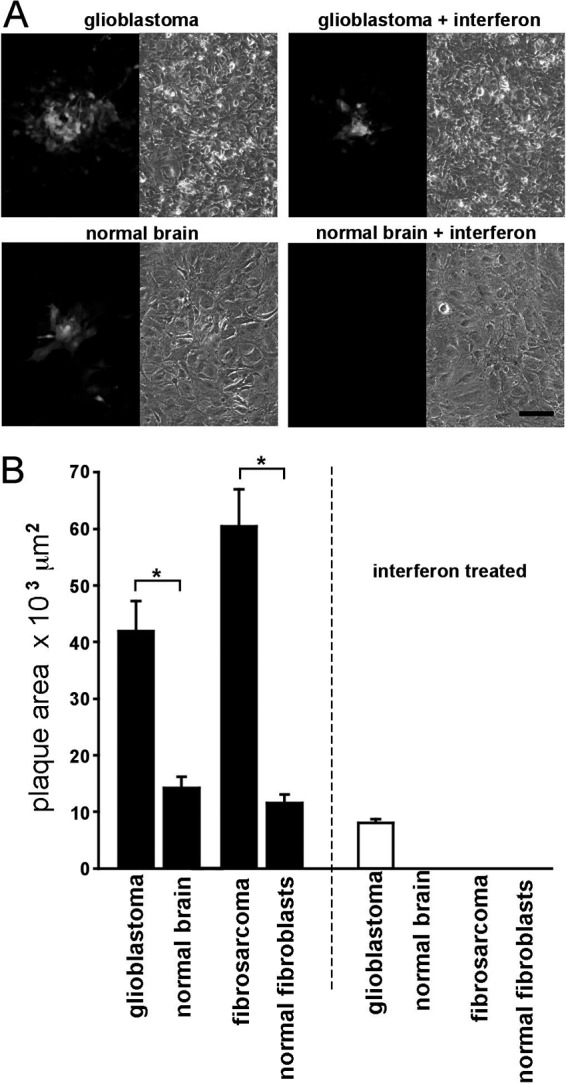
VSV-12′GFP shows enhanced infection of cancer cells. (A) Representative photos showing plaques under fluorescent (left photo) and phase-contrast (right photo) illumination. Cultured human glioblastoma cells (U-118) are shown in the top row, and primary normal human brain astrocytes are shown below, with parallel interferon-treated cultures (100 U/ml) shown on the right. No plaques were found in any of the interferon-treated normal brain cultures up to the highest concentration of virus tested (200,000 PFU/well), in contrast to the plaques observed in the interferon-treated glioblastoma cultures. Scale bar,100 μm. (B) Bar graph showing the sizes of fluorescent plaques in square micrometers (μm2) grown on human cancer cell cultures (glioblastoma U-118 and fibrosarcoma HT1080) and comparable primary normal human cells (brain astrocytes and fibroblasts, respectively). Each bar represents the mean for 60 plaques; error bars are SE. Plaque sizes in both pairs of cancer and normal cells were significantly different from one another (*; t test; P < 0.001). Interferon treatment (100 U/ml) of parallel cultures (white bars on right) blocked plaque formation in all cultures except glioblastoma up to the highest concentration of virus tested (200,00 PFU/well).
VSV-12′GFP targets brain tumors in vivo.
We have previously found that some less-attenuated recombinant VSVs target brain tumors in vivo, infecting experimental subcutaneous and intracranial tumors (12, 19, 42). To test the hypothesis that, despite its highly attenuated nature, VSV-12′GFP can still target brain tumors, we generated human glioblastoma tumors both in peripheral subcutaneous locations and within the brains of SCID mice.
The human glioma line rU-87, expressing a gene coding for a red fluorescent reporter (42), and U-118 glioma cells were used peripherally, and rU-87 cells were used intracranially. After establishment of the in vivo tumors, VSV-12′GFP (107 PFU in 100 μl sterile saline) was injected into the tail vein of the mice. Two to three days after virus inoculation, mice were harvested. In two mice with two U-118 tumors each and in 3 mice with two U-87 subcutaneous tumors each, intravascular inoculation 12 to 19 days after tumor establishment resulted in infection in all tumors studied at 3 days postinoculation; an example of an infected U-118 glioma is seen in Fig. 9H. No infection was found in normal tissue surrounding the tumors.
After intravenous inoculation, histological examination of red U-87 GBM tumors (Fig. 9A and D) in the brain displayed selective virus infection, shown by the green fluorescence of the viral GFP reporter (Fig. 9B and E). VSV-12′GFP was found in glioma cells expressing red fluorescence (Fig. 9C), indicating selective targeting of the tumor cells in 4 of 5 mice. VSV-12′GFP was found in the tumor but not in the normal contralateral side of the brain (Fig. 9G). One mouse showed no sign of virus in the tumor 2 days after intravenous virus inoculation. An additional mouse received a needle penetration in the brain without any cells. Twelve days after injury and three days after virus inoculation, that glioma-free mouse showed no sign of virus in the brain.
VSV-12′GFP intratumoral injection suppresses tumor growth and enhances survival.
To further assess the ability of VSV-12′GFP to safely target and suppress the growth of tumors in vivo, we used SCID mice (n = 12) bearing bilateral subcutaneous human glioma rU-87 tumors. Twelve days after the injection of 1.3 × 105 rU-87 cells, tumors had developed in both flanks of all mice and reached a size of 2 to 3 mm. Half of the mice were then injected intratumorally with 1.5 × 105 PFU of VSV-12′GFP (n = 6), and the remaining half were injected with buffer as controls (n = 6). Eleven days later, the mean volume of uninfected control tumors had grown 6.4-fold ± 1.7 SE (n = 12). In contrast, VSV-12′GFP-injected tumors (n = 12) displayed only a marginal increase, of 1.2-fold ± 0.3 SE compared to their size on the day of virus injection (day 0), a significant difference (Mann-Whitney test; P < 0.01) between populations (Fig. 10A). By day 28, control tumors had grown an average of 13.9-fold (±3.8 SE) their original size, whereas VSV-treated tumor growth appeared arrested, being only 1.2-fold ± 3.8 SE greater in volume than that on day 0.
Fig 10.

Intratumoral injection of VSV-12′GFP suppresses the growth of subcutaneous tumors and enhances survival. Twelve SCID mice with human rU-87 tumors 2 to 3 mm in size were established through bilateral subcutaneous implantation (2 tumors per mouse) and then intratumorally injected with VSV-12′GFP (n = 6 mice) or medium control (n = 6 mice) at day 0 and monitored for 52 days. (A) Tumor size was measured externally using calipers and is displayed as mean tumor volume in VSV-12′GFP-treated tumors (squares) and untreated control tumors (circles). Numbers in parentheses indicate the numbers of tumors measured in the controls, and this number decreased as mice were euthanized. Error bars (SE) (Mann-Whitney test; **, P < 0.01; ***, P < 0.001). (B) Survival curve showing the deaths of animals in the untreated tumor population over time. Mice that developed tumors reaching 1 cm in diameter were euthanized in accordance with institutional guidelines. All control tumor mice were dead by day 45. (C) Graph showing mean body weight in VSV-12′GFP-treated (squares) and untreated control (circles) mice. Numbers in parentheses indicate the numbers of mice measured in the control population and decreased as animals died. Error bars, SE.
Tumors in control mice not treated with virus grew to 1 cm in diameter, and mice were euthanized in accordance with institutional guidelines; tumors of some controls reached 1 cm by day 29, and all of the control mice were dead by day 45 (Fig. 10B). All VSV-12′GFP-treated mice survived until the end of the study at day 52 with no detectable adverse effects; the majority of tumors had regressed to a point of not being detectable. At the conclusion of the study, all VSV-12′GFP-treated mice were euthanized, dissected, and examined histologically for tumor masses. The tumors dissected from control mice not treated with virus were dramatically larger than the residual tumor masses remaining in VSV-12′GFP-treated mice (Fig. 11A to C). Importantly, during histological analysis of VSV-12′GFP mice, none of the 12 tumors injected with virus showed any remaining dsRed-fluorescent tumor cells, suggesting that the virus had successfully eliminated all tumor cells. The small “tumor” masses appeared to be composed of scar tissue and other nontumor cells. All tumors in the control mice not treated with virus showed dsRed-expressing tumor cells.
Fig 11.

Human rU-87 subcutaneous tumor suppression after VSV-12′GFP infection. (A to C) From tumor-bearing SCID mice shown in Fig. 10. (A) Photo of control human rU-87 tumor and comparable region from a VSV-12′GFP-treated tumor mouse 52 days after intratumoral viral injection. The arrow indicates residual tumor mass. (B) Section of an rU-87 control tumor showing robust expression of red fluorescent protein throughout the tumor mass. The dotted line indicates the nonfluorescent surface layer of skin. (C) Histological sections of a residual tumor mass from VSV-12′GFP-treated mice 52 days after intratumoral injection. The scale is same as that for panel B. (D) Comparison of plaque sizes of VSV-12′GFP harvested from rU-87 tumors 24 h after injection and in vitro-maintained stocks of VSV-12′GFP, VSV-1′GFP, and VSV-G/GFP. No statistically significant difference in size was found between VSV-12′GFP harvested from tumors and the in vitro-maintained stock. (E to H) Subcutaneous rU-87 tumor established in a SCID mouse 48 h after injection with VSV-12′GFP. Red fluorescence (E) is from tumor cells, and green fluorescence (F) is from VSV-12′GFP, with panel G showing an overlay of the two images. (H) Nonfluorescent image of the same field showing the border between the tumor mass and normal tissue. The virus fairly selectively infects the tumor cells and not surrounding normal tissue.
Given the possibility of mutations in the VSV genome (34, 58), one concern is the stability of VSV-12′GFP over time. To study the phenotype of VSV-12′GFP, we injected tumors in vivo with the virus and harvested virus-infected tumors 24 h later for comparison in a plaque size assay. Measurement of plaque sizes from tumor-harvested VSV-12′GFP showed only a slight increase that was not statistically significant compared to sizes of plaques from the viral stock used for tumor injection (Fig. 11D). VSV-1′GFP and VSV-G/GFP plaques were also compared for control purposes and were found to be dramatically larger than VSV-12′GFP, suggesting no significant loss of attenuation in the VSV-12′GFP viral population over 24 h, a period that would allow about 4 to 6 cycles of replication (1, 2). Although we found no obvious change in the attenuated phenotype of VSV-12′GFP harvested from tumors as determined by a characteristically small plaque size, the possibility remains that over many generations the virus could nonetheless evolve a more rapidly replicating genotype. We also examined subcutaneous tumors 48 h after inoculation with VSV-12′GFP. Virus was found selectively in the tumor tissue and not in surrounding normal tissue (Fig. 11E to H).
Interferon and ISG expression.
To determine if VSV-12′GFP evoked an interferon and interferon-stimulated gene (ISG) response similar to that of other VSVs, we infected normal human astrocytes with VSV-G/GFP and VSV-12′GFP with a low MOI of 0.3 and a high MOI of 3. Since we found no readily measurable IFN in noninfected controls, we compared IFN transcript stimulation in cells infected with either VSV-12′GFP or with VSV-G/GFP (Fig. 12A). VSV-G/GFP induced a greater IFN response than VSV-12′GFP; when the data were reanalyzed on the basis of virus genome copy number, VSV-12′GFP induced a slightly greater upregulation than VSV-G/GFP at an MOI of 0.3 but similar levels at an MOI of 3. Both viruses showed a much greater activation of ISGs. IFIT1, ISG15, MX1, OAS, and EIF2AK2 mRNA transcript expression was measured using qRT-PCR. In both high and low MOIs, VSV-12′GFP generated a level of ISG expression similar to or slightly greater than that with VSV-G/GFP (Fig. 12B and D). To determine the level of ISG stimulation per virus genome, we normalized to the genome copy number as measured with qRT-PCR; this ratio represents the strength of the innate immune response relative to the replicative efficiency of the virus. VSV-12′GFP generated a greater level of ISG stimulation per genome than VSV-G/GFP (Fig. 12 C and E). These data suggest that despite a reduced proliferation of virions, VSV-12′GFP generated a strong ISG response per virion.
Fig 12.

Interferon and interferon-stimulated gene (ISG) expression are upregulated by VSV-12′GFP. qRT-PCR was used to measure mRNA levels of interferon (A) and five interferon-stimulated genes (B to E) in cultures of human astrocytes 10 h after infection with either VSV-G/GFP (white bars) or VSV-12′GFP (black bars). Two concentrations of virus were tested; low MOI (0.3) (A, B, and C) and high MOI (3.0) (A, D, and E), and mRNA levels were computed as multiples of the levels found in uninfected control cultures, except in panel A, where the level of interferon gene expression in uninfected cultures was too low to be detected and VSV-G/GFP-infected levels were used instead. Since VSV-12′GFP propagates more slowly than VSV-G/GFP, we also normalized the results with respect to levels of genomic VSV measured using a VSV genome-specific probe (A, C, and E). The bars represent the standard deviation (SD), and *, **, and *** represent a statistically significant difference between viruses (Student's t test) at the P < 0.05, P < 0.01, and P < 0.001 levels, respectively.
DISCUSSION
In the present article, we show that a recombinant VSV (VSV-12′GFP) with two reporter genes added in the first and second gene positions displays a high degree of attenuation, yielding the smallest plaques and generating the fewest progeny of any of the VSVs tested. VSV-12′GFP evoked a strong immune response in vivo against inserted transgenes. Despite its attenuated nature, VSV-12′GFP also displayed an enhanced infection of cancer cells compared with that of noncancer control cells. The relative selectivity of the virus for gliomas was further enhanced in the presence of interferon. In vivo, intravenous inoculation of VSV-12′GFP resulted in selective targeting and killing of both subcutaneous and intracranial glioma cells. In an in vivo test, VSV-12′GFP was much less neurotoxic than VSV-G/GFP. Finally, direct intratumoral injection of the virus appeared to eliminate the tumor and prolong life for tumor-bearing mice.
Attenuation compared with other VSVs.
We recently showed that of 11 recombinant VSVs, one of the most attenuated contained a single reporter gene in the first position (12). We reasoned that if one gene in the first position reduced virulence, then inserting two nonviral genes at gene positions 1 and 2 would further attenuate the virus. Comparisons of VSV-12′GFP with VSV-1′GFP support this view; in all tests, VSV-12′GFP was more attenuated than VSV-1′GFP and showed a substantially greater degree of attenuation than all other attenuated VSVs tested. VSV-12′GFP was significantly more attenuated than VSV-M51, VSV-CT9/M51, VSV-G/GFP, and VSV-1′GFP. Of the attenuating gene insertions compared, the VSV-12′GFP double gene insertion had the greatest attenuating effect on plaque size and progeny generation as measured with a one-step growth curve.
To address the hypothesis that the mechanism of attenuation was reduced expression of viral genes, we examined the virus mRNA transcripts of what would normally be the first and fifth viral genes, N and L, in 4 recombinant VSVs. Expression of both the N and L transcripts was reduced by an order of magnitude in VSV-12′GFP compared with that in unattenuated recombinant VSV devoid of nonviral genes or other mutations. In VSV-12′GFP, N and L expression was about half that found in the most closely related virus, VSV-1′GFP, which contains only a single nonviral reporter gene in position 1. During transcription, the VSV polymerase progresses sequentially in the 3′-to-5′ direction along the viral genome, transcribing genes from position 1 to position 5, or 1 to 7 in the case of VSV-12′GFP. Upon reaching the transcription termination signal at the end of each gene, the polymerase may continue on to the transcription initiation signal of the next gene; alternately, the polymerase can dissociate from the genome at a rate of 20 to 30% for each gene junction and thereby not transcribe genes that are further along the VSV genome (59, 60). Thus, adding more transgenes to the 3′ end of the genome, as we did with VSV-12′GFP, reduces the probability that the polymerase will continue through the genome to express viral genes, particularly a gene such as L which is last in order and the gene showing the greatest reduction of expression in VSV-12′GFP.
VSV-12′GFP appeared to infect selectively and eliminate tumors in SCID mice. It is remarkable that even after more than 7 weeks of infection, immunocompromised SCID mice remained healthy, gained weight, and showed no obvious signs of poor health despite the absence of a fully functional T- and B-cell systemic immune response. Some human viruses, such as poliovirus and herpesvirus, have oncolytic potential and are sometimes used in mouse models but can infect only cells from humans. In contrast, that mouse cells and mice are susceptible to VSV is shown by the fatal response of most postnatal day 16 mice to VSV-G/GFP but not to VSV-12′GFP.
Targeting cancer cells.
A number of VSVs have shown promise in targeting and killing cancer cells (12, 42, 45, 57, 61–64). VSV-12′GFP showed a greater degree of infection of three different cancers than of their normal noncancerous control cells. Whereas we found almost no infection of normal melanocytes, cultures of melanoma cells were completely infected and ultimately killed by VSV-12′GFP. The virus showed an increased level of infection of glioma and fibrosarcoma cells compared with normal astrocytes or fibroblasts in vitro. When normal cells were treated with IFN, infection of astrocytes and fibroblasts was completely blocked. In contrast, even in the presence of IFN, VSV-12′GFP still infected and killed glioma cells, although at a lower rate than in the absence of IFN. These data are consistent with the view that one central mechanism underlying VSV's enhanced infection of cancer cells is an altered IFN response. Although many tumor types show IFN deficiencies (57), some do not, and the finding that fibrosarcomas were protected by IFN is consistent with the view that some tumor lines retain a sufficient IFN response to be protected against infection by VSV-12′GFP.
Due to the slow expansion of VSV-12′GFP, cells have more time to upregulate an IFN-based antiviral defense relative to the rate of viral replication, as shown by our qRT-PCR experiments. By protecting nearby uninfected cells, a relatively enhanced IFN response/virion would further retard the spread of VSV-12′GFP within normal tissue compared with that of faster-acting VSVs.
VSV is an arbovirus with cattle, pigs, and horses as primary mammalian hosts. VSV shows little toxicity in humans; in regions of central America where VSV is endemic, large portions of the human population are seropositive for the virus with no obvious associated serious illness (65). In humans, use of a highly attenuated VSV, such as VSV-12′GFP, as an immunization or oncolytic vector further reduces the probability of an unexpected adverse consequence.
For use as a potential oncolytic virus, VSV can either directly kill infected cells or can initiate an immune response to target infected cancer cells. The substantive attenuation found in VSV-12′GFP may make it less suitable for directly killing peripheral cancer cells, since its slow actions may reduce the probability that all cancer cells would be infected before upregulation of a systemic immune response that would eliminate the virus; a faster-acting virus, such as VSVrp30 (62), may be more effective to achieve a direct virus-mediated oncolysis outside the brain. However, in terms of its second mechanism of action, the initiation of an immune response against infected cancer cells, an attenuated virus, particularly one such as VSV-12′GFP that elicits a substantive IFN response, may be safer as well as effective. Because most cells of the brain are not replaced after loss, for tumors in the brain, such as glioma, to prevent neurotoxicity, a highly attenuated virus would provide the greatest level of safety.
In the present article, we added genes coding for proteins (GFPs) that would be unlikely to exert a direct effect on VSV or cell function. Alternate strategies that would further enhance the utility of this type of approach (i.e., adding two genes to the 3′ end of the virus genome) for cancer targeting may include substitution for GFP of genes such as those encoding interleukin 28 (IL-28), IL-2, FLT3L, or granulocyte-macrophage colony-stimulating factor (GM-CSF) that can stimulate an immune response against infected cancer cells (64, 66–68). An upregulated immune attack on infected cells that generalizes to similar cancer cells represents a second line of oncolysis if the immediate virus-mediated cell death does not involve all cancer cells. As an alternate approach, the GFP reporter genes could be exchanged for reporter genes that have utility as measurable markers of in vivo viral activity, including carcinoembryonic antigen, secreted alkaline phosphatase, and the beta subunit of chorionic gonadotropin; these reporters are released by infected cells into the blood and can be measured peripherally to determine viral activity (69–72). Because two gene positions are available, more than one immune response enhancer gene could be included, or one immune enhancer and one reporter gene. In addition to the substitution of genes coding for efficacy-enhancing proteins in positions 1 and 2, introduction of the M51 mutation, which disables the virus' counter-IFN strategy (and thereby would amplify the antiviral response from interferon-stimulated genes), or shuffling viral genes downstream of the first- and second-position transgenes (23) could further reduce the potential infection of normal cells (57, 73). Similarly, the VSV G protein could be truncated on the cytoplasmic side, as done with the CT9 mutation (27), to provide further attenuation of the virus.
ACKNOWLEDGMENTS
We thank Guido Wollmann and Justin Paglino for suggestions, Y. Yang and V. Rogulin for technical assistance, J. Rose for providing plasmids used in the generation and recovery of recombinant VSVs, and M. Robek and M. Cobleigh for experimental facilitation and helpful suggestions.
Grant support was provided by NIH CA124737 and CA161048.
Footnotes
Published ahead of print 7 November 2012
REFERENCES
- 1. Lyles DS, Rupprecht CE. 2007. Rhabdoviridae, p 1363–1408 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 2. Rose JK, Whitt MA. 2001. Rhabdoviridae: the viruses and their replication, p 1221–1244 In Knipe DM, Howley PM. (ed), Fields virology, 4th ed, vol 1 Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 3. Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. U. S. A. 92:4477–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whitt MA. 2010. Generation of VSV pseudotypes using recombinant deltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 169:365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brandsma JL, Shlyankevich M, Su Y, Zelterman D, Rose JK, Buonocore L. 2010. Reversal of papilloma growth in rabbits therapeutically vaccinated against E6 with naked DNA and/or vesicular stomatitis virus vectors. Vaccine 28:8345–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cobleigh MA, Buonocore L, Uprichard SL, Rose JK, Robek MD. 2010. A vesicular stomatitis virus-based hepatitis B virus vaccine vector provides protection against challenge in a single dose. J. Virol. 84:7513–7522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geisbert TW, Geisbert JB, Leung A, Daddario-DiCaprio KM, Hensley LE, Grolla A, Feldmann H. 2009. Single-injection vaccine protects nonhuman primates against infection with Marburg virus and three species of Ebola virus. J. Virol. 83:7296–7304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iyer AV, Pahar B, Boudreaux MJ, Wakamatsu N, Roy AF, Chouljenko VN, Baghian A, Apetrei C, Marx PA, Kousoulas KG. 2009. Recombinant vesicular stomatitis virus-based West Nile vaccine elicits strong humoral and cellular immune responses and protects mice against lethal challenge with the virulent West Nile virus strain LSU-AR01. Vaccine 27:893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gao Y, Whitaker-Dowling P, Griffin JA, Barmada MA, Bergman I. 2009. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 16:44–52 [DOI] [PubMed] [Google Scholar]
- 10. Jenks N, Meyers R, Greiner SM, Thompson J, Mader EK, Greenslade A, Griesmann GE, Federspiel MJ, Rakela J, Borad MJ, Vile RG, Barber GN, Meier TR, Blanco MC, Carlson SK, Russell SJ, Peng KW. 2010. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum. Gene Ther. 21:451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le Boeuf F, Diallo JS, McCart JA, Thorne S, Falls T, Stanford M, Kanji F, Auer R, Brown CW, Lichty BD, Parato K, Atkins H, Kirn D, Bell JC. 2010. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 18:888–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Letchworth GJ, Rodriguez LL, Del cbarrera J. 1999. Vesicular stomatitis. Vet. J. 157:239–260 [DOI] [PubMed] [Google Scholar]
- 14. Wagner RR, Rose JK. 1996. Rhabdoviridae: the viruses and their replication, p. 1121–1136 In Fields BN, Knipe DM. (ed), Fields virology. Lippincott-Raven, New York, NY [Google Scholar]
- 15. Bi Z, Barna M, Komatsu T, Reiss CS. 1995. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J. Virol. 69:6466–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huneycutt BS, Plakhov IV, Shusterman Z, Bartido SM, Huang A, Reiss CS, Aoki C. 1994. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 635:81–95 [DOI] [PubMed] [Google Scholar]
- 17. Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, Udem SA. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 360:36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van den Pol AN, Dalton KP, Rose JK. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 76:1309–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozduman K, Wollmann G, Ahmadi SA, van den Pol AN. 2009. Peripheral immunization blocks lethal actions of vesicular stomatitis virus within the brain. J. Virol. 83:11540–11549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clarke DK, Cooper D, Egan MA, Hendry RM, Parks CL, Udem SA. 2006. Recombinant vesicular stomatitis virus as an HIV-1 vaccine vector. Springer Semin. Immunopathol. 28:239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahmed M, Cramer SD, Lyles DS. 2004. Sensitivity of prostate tumors to wild-type and M protein mutant vesicular stomatitis viruses. Virology 330:34–49 [DOI] [PubMed] [Google Scholar]
- 22. Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J. Virol. 81:2056–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cooper D, Wright KJ, Calderon PC, Guo M, Nasar F, Johnson JE, Coleman JW, Lee M, Kotash C, Yurgelonis I, Natuk RJ, Hendry RM, Udem SA, Clarke DK. 2008. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and G gene truncation reduces neurovirulence and enhances immunogenicity in mice. J. Virol. 82:207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jayakar HR, Whitt MA. 2002. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J. Virol. 76:8011–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Publicover J, Ramsburg E, Robek M, Rose JK. 2006. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J. Virol. 80:7028–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roberts A, Buonocore L, Price R, Forman J, Rose JK. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J. Virol. 73:3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schnell MJ, Buonocore L, Boritz E, Ghosh HP, Chernish R, Rose JK. 1998. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 17:1289–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van den Pol AN, Ozduman K, Wollmann G, Ho WS, Simon I, Yao Y, Rose JK, Ghosh P. 2009. Viral strategies for studying the brain, including a replication-restricted self-amplifying delta-G vesicular stomatitis virus that rapidly expresses transgenes in brain and can generate a multicolor Golgi-like expression. J. Comp. Neurol. 516:456–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flanagan EB, Zamparo JM, Ball LA, Rodriguez LL, Wertz GW. 2001. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J. Virol. 75:6107–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flanagan EB, Schoeb TR, Wertz GW. 2003. Vesicular stomatitis viruses with rearranged genomes have altered invasiveness and neuropathogenesis in mice. J. Virol. 77:5740–5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barr JN, Wertz GW. 2001. Polymerase slippage at vesicular stomatitis virus gene junctions to generate poly(A) is regulated by the upstream 3′-AUAC-5′ tetranucleotide: implications for the mechanism of transcription termination. J. Virol. 75:6901–6913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wertz GW, Moudy R, Ball LA. 2002. Adding genes to the RNA genome of vesicular stomatitis virus: positional effects on stability of expression. J. Virol. 76:7642–7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. 2005. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J. Virol. 79:15043–15053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davis JN, van den Pol AN. 2010. Viral mutagenesis as a means for generating novel proteins. J. Virol. 84:1625–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schnell MJ, Buonocore L, Whitt MA, Rose JK. 1996. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol. 70:2318–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gallione CJ, Greene JR, Iverson LE, Rose JK. 1981. Nucleotide sequences of the mRNAs encoding the vesicular stomatitis virus N and NS proteins. J. Virol. 39:529–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rose JK, Gallione CJ. 1981. Nucleotide sequences of the mRNAs encoding the vesicular stomatitis virus G and M proteins determined from cDNA clones containing the complete coding regions. J. Virol. 39:519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schubert M, Harmison GG, Meier E. 1984. Primary structure of the vesicular stomatitis virus polymerase (L) gene: evidence for a high frequency of mutations. J. Virol. 51:505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schubert M, Harmison GG, Richardson CD, Meier E. 1985. Expression of a cDNA encoding a functional 241-kilodalton vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 82:7984–7988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dalton KP, Rose JK. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279:414–421 [DOI] [PubMed] [Google Scholar]
- 41. Publicover J, Ramsburg E, Rose JK. 2004. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J. Virol. 78:9317–9324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 28:1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ellis E, Delbruck M. 1939. The growth of bacteriophage. J. Gen. Physiol. 22:365–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kretzschmar E, Peluso R, Schnell MJ, Whitt MA, Rose JK. 1996. Normal replication of vesicular stomatitis virus without C proteins. Virology 216:309–316 [DOI] [PubMed] [Google Scholar]
- 45. Paglino JC, van den Pol AN. 2011. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J. Virol. 85:9346–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, Broberger C. 2009. Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. J. Neurosci. 29:4622–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van den Pol AN, Acuna-Goycolea C, Clark KR, Ghosh PK. 2004. Physiological properties of hypothalamic MCH neurons identified with selective expression of reporter gene after recombinant virus infection. Neuron 42:635–652 [DOI] [PubMed] [Google Scholar]
- 48. Felts K, Rogers B, Chen K, Ji H, Sorge J, Vaillancourt P. 2000. Recombinant Renilla reniformis GFP displays low toxicity. Stratagene Strategies 13:85–87 [Google Scholar]
- 49. Ball LA, Pringle CR, Flanagan B, Perepelitsa VP, Wertz GW. 1999. Phenotypic consequences of rearranging the P, M, and G genes of vesicular stomatitis virus. J. Virol. 73:4705–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wertz GW, Perepelitsa VP, Ball LA. 1998. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc. Natl. Acad. Sci. U. S. A. 95:3501–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lundh B, Löve A, Kristensson K, Norrby EJ. 1988. Non-lethal infection of aminergic reticular core neurons: age-dependent spread of ts mutant vesicular stomatitis virus from the nose. Neuropathol. Exp. Neurol. 47:497–506 [DOI] [PubMed] [Google Scholar]
- 52. Ayala-Breton C, Barber GN, Russell SJ, Peng KW. 2012. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum. Gene Ther. 23:484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Muik A, Dold C, Geiss Y, Volk A, Werbizki M, Dietrich U, von Laer D. 2012. Semireplication-competent vesicular stomatitis virus as a novel platform for oncolytic virotherapy. J. Mol. Med. (Berl.). 90:959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, Grdzelishvii VZ. 2012. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J. Virol. 86:3073–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Naik S, Nace R, Barber GN, Russell SJ. 2012. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-β. Cancer Gene Ther. 19:443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stephenson KB, Barra NG, Davies E, Ashkar AA, Lichty BD. 2012. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 19:238–246 [DOI] [PubMed] [Google Scholar]
- 57. Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shut down innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275 [DOI] [PubMed] [Google Scholar]
- 58. Drake JW, Holland JJ. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 96:13910–13913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barr JN, Whelan SPJ, Wertz GW. 2002. Transcriptional control of the RNA-dependent RNA polymerase of vesicular stomatitis virus. Biochim. Biophys. Acta 1577:337–353 [DOI] [PubMed] [Google Scholar]
- 60. Iverson LE, Rose JK. 1981. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell 23:477–484 [DOI] [PubMed] [Google Scholar]
- 61. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6:821–825 [DOI] [PubMed] [Google Scholar]
- 62. Wollmann G, Tattersall P, van den Pol AN. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wollmann G, Robek MD, van den Pol AN. 2007. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J. Virol. 81:1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, Kottke T, Thompson J, Melcher A, Vile R. 2011. Activating systemic T-cell immunity against self-tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum. Gene Ther. 22:1343–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tesh RB, Peralta PH, Johnson KM. 1969. Ecological studies of vesicular stomatitis virus. I. Prevalence of infection among animals and humans living in an area of endemic VSV activity. Am. J. Epidemiol. 90:255–261 [DOI] [PubMed] [Google Scholar]
- 66. Ali S, King GD, Curtain JF, Candolfi M, Xiong W, Liu C, Puntel M, Cheng Q, Prieto J, Ribas A, Kupiec-Weglinski J, van Rooijen N, Lassmann H, Lowenstein PR, Castro MG. 2005. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 65:7194–7204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Barzon L, Pacenti M, Franchin E, Colombo F, Paula G. 2009. HSV-TK/IL-2 gene therapy for glioblastoma multiforme. Methods Mol. Biol. 542:529–549 [DOI] [PubMed] [Google Scholar]
- 68. Breitbach CJ, Thorne SH, Bell JC, Kirn DH. 2012. Targeted and armed oncolytic poxviruses for cancer: the lead example of JX-594. Curr. Pharm. Biotechnol. 13:1768–1772 [DOI] [PubMed] [Google Scholar]
- 69. Hiramatsu N, Kasai A, Meng Y, Hayakawa K, Yao J, Kitamura M. 2005. Alkaline phosphatase vs luciferase as secreted reporter molecules in vivo. Anal. Biochem. 339:249–256 [DOI] [PubMed] [Google Scholar]
- 70. Peng KW, Facteau S, Wegman T, O'Kane D, Russell SJ. 2002. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat. Med. 8:527–531 [DOI] [PubMed] [Google Scholar]
- 71. Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, Mishra PK, Macura SI, Russell SJ, Galanis EC. 2003. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 63:2462–2469 [PubMed] [Google Scholar]
- 72. Shashkova EV, Kuppuswamy MN, Wold WS, Doronin K. 2008. Anticancer activity of oncolytic adenovirus vector armed with IFN-alpha and ADP is enhanced by pharmacologically controlled expression of TRAIL. Cancer Gene Ther. 15:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ahmed M, Lyles DS. 1997. Identification of a consensus mutation in M protein of vesicular stomatitis virus from persistently infected cells that affects inhibition of host-directed gene expression. Virology 237:378–388 [DOI] [PubMed] [Google Scholar]