

Abstract
West Nile virus (WNV) is a mosquito-transmitted pathogen that can cause serious disease in humans. Our laboratories are focused on understanding how interactions between WNV proteins and host cells contribute to virus replication and pathogenesis. WNV replication is relatively slow, and on the basis of earlier studies, the virus appears to activate survival pathways that delay host cell death during virus replication. The WNV capsid is the first viral protein produced in infected cells; however, its role in virus assembly is not required until after replication of the genomic RNA. Accordingly, from a temporal perspective, it is perfectly suited to block host cell apoptosis during virus replication. In the present study, we provide evidence that the WNV capsid protein blocks apoptosis through a phosphatidylinositol (PI) 3-kinase-dependent pathway. Specifically, expression of this protein in the absence of other viral proteins increases the levels of phosphorylated Akt, a prosurvival kinase that blocks apoptosis through multiple mechanisms. Treatment of cells with the PI 3-kinase inhibitor LY294002 abrogates the protective effects of the WNV capsid protein.
INTRODUCTION
West Nile virus (WNV) is an important human pathogen that can cause severe neurological disease (reviewed in reference 1). As a member of the genus Flavivirus, WNV is related to several other medically important arboviruses, including Japanese encephalitis virus (JEV), yellow fever virus, and dengue virus (DENV). WNV was first identified in Uganda 75 years ago (2) but has a wide distribution throughout the globe. However, until 1999, this virus was not known to circulate in North America (3). The 1999 outbreak which originated in New York City was of particular significance because the WNV strain isolated from infected patients was much more neuroinvasive than most Old World strains. WNV is now the most important vector-transmitted virus in North America. Currently, there are no vaccines or therapies approved for use in humans.
With respect to understanding the molecular basis of WNV disease and developing antiviral therapies, it is critical to know how interactions between viral proteins and host proteins affect virus replication and cell physiology. The WNV virion is composed of a nucleocapsid core surrounded by a host-derived membrane that contains two virus-encoded membrane proteins, E and M (reviewed in reference 4). The nucleocapsid consists of a single-stranded RNA genome of approximately 11 kb complexed with capsid protein. The viral genome encodes a single open reading frame which, when translated, produces a polyprotein that is processed into 3 structural and 7 nonstructural proteins. Recent research suggests that additional viral proteins may be produced by ribosomal frameshifting (5, 6). Manipulation of multiple host cell factors by a relatively small number of viral proteins is critical for virus replication and spread. Due to the limited coding capacity of the WNV genome, its protein products must be multifunctional in order to counter host cell antiviral defenses. Although originally thought to serve purely structural roles, capsid proteins of RNA viruses are emerging as important players at the virus-host interface. Our research group and others have shown that capsid proteins of WNV and other RNA viruses are involved in a variety of nonstructural roles during the replication cycle, including RNA replication, translational regulation, modulation of virus infectivity, and cell survival (7–13). With respect to cell survival, the ability of viral proteins to affect the onset of apoptotic processes in infected cells may be a critical aspect of viral pathogenesis. If this is so, gaining a mechanistic understanding of how WNV alters host cell survival may facilitate the development of vaccines and therapeutics for use in humans.
As is the case with many RNA viruses, infection of mammalian cells with WNV has been reported to cause apoptosis (14–18). In most cell types, WNV-induced cell death occurs following several rounds of replication, often days after infection. This is in contrast to other RNA viruses, such as Sindbis virus and vesicular stomatitis virus (VSV), which replicate rapidly and trigger cell death pathways much earlier than flaviviruses (19–21). Regarding the mechanism of WNV-induced cell death, numerous studies have implicated the capsid protein in this process (22–25). However, unlike Sindbis virus and VSV, replication of WNV and other flaviviruses is relatively slow, and therefore, it is not seemingly beneficial to the virus if the first viral protein produced in an infected cell induces programmed cell death. In contrast, during the early stages of flavivirus infection, apoptosis appears to be inhibited by a process that involves activation of the prosurvival kinase Akt (26–28). In cells infected with WNV, DENV, or JEV, elevated Akt activity can be detected within an hour. Prevention of Akt phosphorylation by pharmacologically blocking the upstream kinase phosphatidylinositol 3-kinase (PI3K) results in increased activation of the critical apoptotic effector caspase-3 and decreased viral titers. Moreover, significant caspase-3 activation does not generally occur in infected cells until 48 to 72 h postinfection. The ability of WNV to inhibit cell death likely serves to counter proapoptotic mechanisms of the innate immune system, such as Fas ligation by CD8+ T cells, to allow sufficient time for replication. Indeed, Fas ligation appears to play an important role in controlling WNV infection (29, 30), particularly in the killing of infected neurons of the central nervous system. A common cellular response to virus infection is increased Fas/CD95 expression and/or sensitivity to Fas-mediated apoptosis (31). This response is thought to function in clearance of virus-infected cells and can be triggered by the presence of double-stranded RNA and possibly specific viral proteins. Thus, capsid-dependent inhibition of Fas-dependent apoptosis would allow the virus to better elude the innate immune response, thereby allowing replication and spread.
In apparent contrast to the idea that WNV capsid is proapoptotic (22–25), it has been reported that this protein can be stably expressed in mammalian cell lines (32). With respect to the latter publication, a 105-amino-acid-residue isoform of the WNV capsid was utilized. Conversely, in studies where capsid was found to be proapoptotic, a 123-amino-acid-residue isoform of capsid which includes the 18-amino-acid-residue signal peptide of prM was used for the experiments. Several reports have established that the mature flavivirus capsid is in fact 105 amino acid residues in size, the result of coordinated cleavage of the polyprotein between capsid and prM by the NS2B/3 protease (33–35). Here we describe how expression of the mature 105-amino-acid-residue isoform of the WNV capsid affects apoptosis in mammalian cells. Our findings indicate that WNV capsid blocks apoptosis through a pathway that requires PI3K activity.
MATERIALS AND METHODS
Cell lines and viruses.
All mammalian cell lines employed were maintained at 37°C in a 5% CO2 atmosphere. Human alveolar epithelial carcinoma (A549) and human embryonic kidney (HEK293T) cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and cultured in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS; Gibco Invitrogen, Carlsbad, CA) and 20 mM HEPES sodium salt. African green monkey kidney (Vero76) and baby hamster kidney (BHK-21) cells (ATCC) were cultured as described above, with the exception that the growth medium contained 5% FBS. Human embryonic lung (HEL/18) fibroblasts obtained from Eva Gönczöl (Wistar Institute, Philadelphia, PA) were cultured in RPMI 1640 medium with 10% FBS and nonessential amino acids (Life Technologies Inc., Burlington, ON, Canada) as described previously (36).
Virus infection protocols.
Stocks of WNV strain NY99-385 were produced via transfection of plasmid-launched infectious clone pCMVNY99, as described below (37). All procedures involving infectious WNV were carried out in the Glaxo biosafety level 3 lab at the University of Alberta. To prepare viral stocks, HEK293T cells were plated at a density of 2 × 105 cells per well of a 6-well plate 24 h before transfection. Subsequently, 1 μg of pCMVNY99 plasmid DNA was transfected into cells in 6-well plates using TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI). Following incubation for 72 h, culture supernatants were recovered and cellular debris was removed by centrifugation at 1,500 × g for 15 min. The number of infectious particles was determined by plaque assay as described previously (8). Following determination of virus titer, Vero 76 cells in 150-mm plates were infected at a multiplicity of infection (MOI) of 0.1. The virus inoculum was adjusted to 10 ml with serum-free medium and then added to cells, which were incubated for 60 min at 37°C with agitation every 15 min. Following aspiration of the virus inoculum, cells were washed with phosphate-buffered saline (PBS), after which 15 ml of complete growth medium containing 2% FBS was added. Culture supernatants were collected at 72 h postinfection, and cellular debris was pelleted by centrifugation. After determination of virus titers by plaque assay, the WNV stocks were aliquoted and frozen at −80°C until needed.
Infection of cells with VSV (Indiana strain), produced by infection of Vero76 cell monolayers at an MOI of 0.1, and subsequent harvesting of cell culture supernatants have been described previously (10).
Expression plasmids.
With the exception of pCMVNY99 (37), all plasmids were propagated in Escherichia coli DH5α under standard growth conditions in Luria-Bertani (LB) medium with the appropriate antibiotic. The WNV infectious clone plasmid pCMVNY99 was amplified in E. coli strain HB101 as described previously (37). Plasmids for production of recombinant lentiviruses (pTRIP-CMV-IVSb-IRES-RFP, pHCMV-VSV.G, and pGag-Pol) were a generous gift from Charles Rice (Rockefeller University, New York, NY). To produce pTRIP-CMV-MCS-IRES-tagRFP, the vector pTRIP-CMV-IVSb-IRES-tagRFP (38) was digested with SpeI and XhoI restriction enzymes to remove the Gateway destination cassette. Subsequently, two annealed oligonucleotides [MCS (+) and MCS (−)], which contained restriction enzyme sites for SpeI, BamHI, MluI, SalI, ClaI, and XhoI, were ligated into the cut vector to produce pTRIP-CMV-MCS-IRES-tagRFP. In order to replace the tagRFP (red fluorescent protein) cDNA sequence in pTRIP-CMV-MCS-IRES-tagRFP with green fluorescent protein (AcGFP), the AcGFP-coding sequence was amplified from pIRES2-AcGFP1 using primers AcGFP-NheI and AcGFP-SacII (Table 1), digested with NheI and SacII, and ligated into the cut vector. This plasmid, pTRIP-CMV-MCS-IRES-AcGFP, was used for expression cloning and all subsequent lentiviral experiments in this study. It is referred to herein as pTRIP-AcGFP for ease of reference. Two cDNAs encoding the 105-amino-acid isoform of the WNV capsid were produced by PCR using the primers WNV-Cap-EcoRI and WNV-Cap-BamHI or WNV-Cap-SpeI and WNV-Cap-XhoI (Table 1) and pCMVNY99 as the template. The resulting capsid cDNA was digested with either EcoRI and BamHI or SpeI and XhoI before ligation into pIRES2-AcGFP1 or pTRIP-AcGFP to produce pIRES2-AcGFP1-WNV-Cap and pTRIP-AcGFP-WNV-Cap, respectively. The plasmids pCMV5-Cap and pCMV5-aCap, encoding the 105- and 123-amino-acid-residue isoforms of WNV capsid, respectively, have been described previously (8).
Table 1.
Oligonucleotide primers
| Name | Sequence |
|---|---|
| WNV-Cap-SpeI | 5′-GTA CAC TAG TGC CAC CAT GTC TAA GAA ACC AGG AGG-3′ |
| WNV-Cap-XhoI | 5′-GAT CCT CGA GTT ATC TTT TCT TTT GTT TTG AGC-3′ |
| WNV-Cap-EcoRI | 5′-GAT CGA ATT CGC CAC CAT GTC TAA GAA ACC AGG AGG-3′ |
| WNV-Cap-BamHI | 5′-GTA CGG ATC CTT ATG CTC CTA CGC TGG CGA TCA GGC C-3′ |
| MCS (+) | 5′-GTA CAC TAG TAC TGG ATC CAC TAC GCG TAT AGT CGA CAA GAT CGA TAT ACT CGA GCA TG-3′ |
| MCS (−) | 5′-CAT GCT CGA GTA TAT CGA TCT TGT CGA CTA TAC GCG TAG TGG ATC CAG TAC TAG TGT AC-3′ |
| AcGFP-NheI | 5′-GAT CGC TAG CAT GGT GAG CAA GGG CGC CGA-3′ |
| AcGFP-SacII | 5′-GTA CCC GCG GTC ACT TGT ACA GCT CAT CCA-3′ |
Antibodies.
Antibodies were from the following sources: rabbit anti-human activated caspase-3 antibody for flow cytometric analyses was from BD Biosciences (Franklin Lakes, NJ); rabbit anti-human activated caspase-8 (D391), Akt, and phospho-Akt (S473) antibodies were from Cell Signaling Technology (Beverly, MA); rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) and mouse anti-p53 antibodies were from Abcam (Cambridge, MA); mouse IgM anti-human Fas was from Millipore (Billerica, MA); donkey anti-rabbit horseradish peroxidase (HRP) and goat anti-mouse HRP antibodies were from Jackson ImmunoResearch (West Grove, PA); goat anti-mouse Alexa Fluor 680 was from Invitrogen (Burlington, ON, Canada); donkey anti-rabbit IRDye 800 antibody was from LiCor Biosciences (Lincoln, NE); and rabbit anti-GFP was from Eusera (Edmonton, AB, Canada). Mouse monoclonal antibodies to the vesicular stomatitis virus G protein were purified from the BW8G65 hybridoma (ATCC). Rabbit anti-WNV capsid antibody was produced in-house as described previously (8). Guinea pig antibodies to WNV capsid protein were produced by Pocono Rabbit Farm & Laboratory (Canadensis, PA). To produce guinea pig antibodies against the WNV capsid, animals were immunized with recombinant WNV capsid protein purified from E. coli, a gift from M. G. Rossmann (Purdue University, West Lafayette, IN).
SDS-PAGE and immunoblotting.
Cells were lysed in SDS-PAGE loading buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.02% bromophenol blue), which was then heated to 80°C for 5 min. Subsequently, samples were incubated with 500 U of Benzonase nuclease (EMD Biosciences, Darmstadt, Germany) for 10 min at room temperature, before freezing at −80°C or immediate loading onto polyacrylamide gels. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes at 100 V for 2 h using Towbin transfer buffer (25 mM Tris-HCl, pH 8.3, 192 mM glycine, 20% methanol). Membranes were then blocked for at least 1 h in TBST (20 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 0.1% Tween 20) and 5% skim milk or 5% bovine serum albumin (BSA), prior to incubation with primary antibodies for at least 1 h. After washing with TBST, membranes were incubated with secondary antibodies for 30 min and then washed again with TBST. Proteins were visualized using chemiluminescent Supersignal ECL (Thermo Scientific, Rockford, IL) or fluorescent (LiCor Odyssey system; LiCor Biosciences, Lincoln, NE) detection systems.
Production of lentiviral particles and transduction of cells.
Pseudotyped lentiviruses for expression of WNV capsid were recovered from transfected HEK293T cells. Briefly, 1.6 μg of pHCMV-VSV.G, 5.6 μg of pGag-Pol, and 5.6 μg of pTRIP-AcGFP or pTRIP-AcGFP-WNV-Cap were mixed and allowed to incubate at room temperature for 5 min. Concurrently, 48 μl of TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI) was diluted in 800 μl of Opti-MEM I medium (Gibco Invitrogen, Carlsbad, CA) and allowed to incubate at room temperature for 5 min. The DNA and transfection reagent mixtures were combined and incubated for 20 min at room temperature, after which the resulting complexes were added to HEK293T cells (2.5 × 106 cells) in 10-cm dishes. At 6 h prior to transfection, the HEK293T culture medium was changed to 7 ml of DMEM with 3% FBS. Following 6 h of incubation with the transfection mixture, the medium was replaced with 10 ml of fresh DMEM containing 3% FBS and cells were incubated overnight. Lentivirus-containing culture medium was harvested twice, first at 48 h posttransfection and then again at 72 h. The combined lentivirus-containing media were adjusted with Polybrene (4 μg/ml), passed through surfactant-free cellulose acetate filters (pore size, 0.45 μm), aliquoted, and then stored at −80° until needed.
Prior to use in experiments, the titers of lentiviral stocks were determined on A549 cells. Briefly, dilutions of lentiviral stocks prepared in DMEM containing 3% FBS and 4 μg/ml Polybrene were added to cells (3 × 105 cells) in 6-well dishes. The plates were then centrifuged at 1,000 × g in a swinging-bucket rotor for 60 min, before incubation at 37°C for 6 h. The lentivirus-containing media were removed and replaced with DMEM containing 10% FBS. Titers were determined from samples in which the percentage of AcGFP-positive cells was 15% or less. Unless otherwise indicated, for experimental procedures, cells were transduced at a multiplicity of transduction of 3 and then analyzed 48 h later.
TMRM assays.
A549 cells were seeded at a density of 2 × 105 cells/well in 6-well plates in antibiotic-free medium. Cells were then transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) mixed with 1 μg of pIRES2-AcGFP1-WNV-Cap or pIRES2-AcGFP1. Plasmid DNA was diluted in Opti-MEM I medium (1 μg/well), as was Lipofectamine 2000 (1 μl/well). After mixing and incubation for 20 min, complexes were added to cells in a total volume of 1 ml of Opti-MEM I medium. Following 6 h of incubation, the complexes were removed by aspiration and 2 ml of complete growth medium was added to each well. At a total of 6 h before sample harvesting, cells were treated or mock treated with 250 ng/ml anti-Fas-activating antibody and 1 μg/ml cycloheximide for 6 h. To analyze the apoptotic status of treated cells, the membrane-permeant fluorescent dye tetramethyl rhodamine methyl ester (TMRM), which was prepared as a dimethyl sulfoxide (DMSO) stock, was added to a concentration of 1 μM. After 30 min, samples were prepared for live-cell flow cytometric analysis. Culture supernatants were collected and combined with trypsinized cells from each well prior to analyses. Cells were collected by centrifugation at 600 × g for 5 min and washed twice in PBS containing 1% FBS. After resuspension in 500 μl PBS containing 1% FBS, samples were analyzed on a FACScan flow cytometer (BD Biosciences).
Detection of caspase-3 and -8 activation in infected or transduced cells.
At 24 h before infection with WNV (MOI = 0.1), A549 and HEL/18 cells were plated at a density of 2 × 105 and 1.5 × 105 cells/well, respectively, in 6-well plates. For experiments in which WNV capsid was expressed via lentiviral transduction, cells were transduced for 48 h as described above. Cells were then challenged with 250 ng/ml of anti-Fas antibody and 1 μg/ml cycloheximide for 6 to 8 h following a medium change. To detect caspase-3 activation during viral infection, A549 cells were mock treated or infected with VSV or WNV at an MOI of 0.1. At the indicated time points, the culture media were collected and cell monolayers were washed in PBS and then detached from the plates by incubations with PBS containing 0.05% trypsin and 0.02% EDTA. Cells were then resuspended in DMEM containing 10% FBS and combined with the collected culture supernatant, after which cells were pelleted at 500 × g for 5 min. Following aspiration of the medium, cells were resuspended in PBS containing 2% paraformaldehyde and placed on ice for a minimum of 1 h. Cells were pelleted again, washed once with PBS, and permeabilized with PBS containing 0.2% Triton X-100 for 12 min. After washing in PBS, cells were incubated with 0.25 μg of either rabbit anti-active caspase-3 or rabbit anti-active caspase-8 antibody in 50 μl PBS containing 1% BSA for 60 min. Cells were then washed in PBS and then incubated for 30 min with 0.1 μg of the appropriate fluorescent secondary antibody in PBS containing 1% BSA. Following a single wash in PBS containing 0.1% Triton X-100, cells were resuspended in PBS containing 1% BSA and analyzed via flow cytometry utilizing a FACSCanto II flow cytometer (BD Biosciences).
Detection of caspase-3 activation in transduced cells treated with LY294002.
Transduction of A549 cells was carried out as described above with pTRIP-AcGFP or pTRIP-AcGFP-WNV-Cap lentiviruses. In addition to treatment with anti-Fas antibody and cycloheximide or mock treatment, cells were treated with either 50 μM LY294002 or DMSO alone and incubated for 6 to 8 h before fixation and staining with anti-caspase-3 antibody, followed by fluorescence-activated cell sorter (FACS) analysis. Duplicate samples were subjected to lysis in SDS-PAGE lysis buffer and treatment with Benzonase, before storage at −80°C until needed for analysis.
Determination of phospho-Akt serine 473 levels in capsid-expressing A549 cells.
The day after plating at a density of 2 × 105 cells/well, A549 cells were transduced with lentiviral stocks prepared using pTRIP-AcGFP or pTRIP-AcGFP-WNV-Cap lentiviruses. At 24 h postransduction, cells were shifted to DMEM containing 2% FBS overnight. On the following day, cells were treated with either DMSO or graded concentrations of LY294002 (6.25 μM, 12.5 μM, or 25 μM) for 1 h. Following incubation, cells were lysed with 1× SDS-PAGE loading buffer, treated with Benzonase, and frozen at −80°C prior to analyses.
RESULTS
A single isoform of WNV capsid protein is detectable in infected cells.
Although it has been reported that expression of WNV capsid induces apoptosis (22–25), it is important to point out that these studies employed a 123-amino-acid-residue form of capsid. Based on the spatial and temporal aspects of viral polyprotein processing in yellow fever virus- and Murray Valley encephalitis virus-infected cells (33–35), we expected that the larger (123-amino-acid-residue) isoform of WNV capsid would be very transient or undetectable in infected cells. To test this directly, we expressed both the 105-amino-acid and 123-amino-acid isoforms of WNV capsid in transfected cells and compared the electrophoretic mobilities of these proteins to the mobility of the capsid protein in infected A549 and HEK293T cells (Fig. 1). At all time points examined, the only species of capsid in the WNV-infected cells that was detectable by immunoblotting comigrated with the 105-amino-acid-residue isoform. Based on these observations, we conclude that the reported proapoptotic effects of the 123-amino-acid isoform of WNV capsid are of questionable relevance to WNV biology and that further investigation of how the 105-amino-acid isoform functions in cellular death signaling is warranted.
Fig 1.
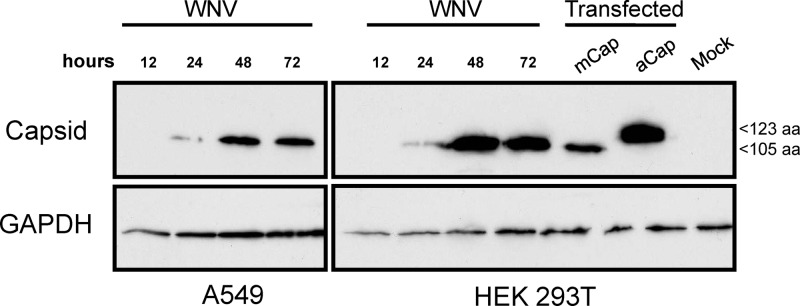
Only the 105-amino-acid form of capsid protein is detected in WNV-infected cells. A549 and HEK293T cells were infected with WNV at an MOI of 5, and at the indicated time points, cell lysates were prepared and analyzed by immunoblotting with antibodies to capsid and GAPDH (loading control). As indicated, cells were transfected with plasmid pCMV5-mCap, which encodes the 105-amino-acid (105 aa) form of capsid, or pCMV5-aCap, which encodes the 123-amino-acid (123 aa) form of capsid.
Cells infected with WNV do not undergo caspase-dependent cell death until late in infection.
Apoptosis in WNV-infected cell lines has been previously reported, and the kinetics of apoptosis depend on the cell line and MOI used (16, 39, 40). To determine the kinetics of apoptosis induction in WNV-infected A549 cells, we performed a time course experiment where caspase activation was monitored at various times postinfection. Cells were infected with WNV and VSV at various MOIs. Infection of A549 cells with VSV is known to cause extensive cell death in less than 24 h (41), and therefore, this virus served as a positive control for rapid induction of apoptosis. Mock-infected and infected cells were harvested at 24, 48, and 72 h postinfection, fixed, permeabilized, and stained with an antibody that recognizes activated caspase-3 followed by an Alexa Fluor 647-labeled secondary antibody. Samples were then subjected to analyses by flow cytometry. Figure 2A shows that regardless of the MOI, WNV antigen was detectable at all time points, with the exception of 24 h when cells were infected with an MOI of 0.1. For WNV samples, even though the majority of the cells were infected at 48 h, the proportion of cells which were positive for active caspase-3 was below ∼20%, even at an MOI of 3. This is in contrast to the data in Fig. 2C, which show that by 24 h postinfection, ∼30% of the VSV-infected cell population was positive for active caspase-3. None of these cells survived past the 48-h time point, and thus, data for only one time point are shown. In contrast, less than 5% of WNV-infected cells were caspase-3 positive at 24 h postinfection. Only after 72 h did the level of apoptosis in WNV-infected samples approach 30% (Fig. 2D). These data suggest that although WNV-infected A549 cells eventually undergo apoptosis, this does not occur until late in the infection cycle.
Fig 2.

WNV does not cause significant activation of caspase-3 until late in infection. A549 cells were infected with WNV at an MOI of 0.1, 1, or 3 or VSV at an MOI of 0.1. At 24, 48, and 72 h postinfection, WNV-infected cells were harvested. VSV-infected cells were harvested only at 24 h postinfection because shortly after this time point, the majority of cells were dead. (A, B) Following fixation, WNV-infected cells were incubated with mouse NS2B/3 (to detect virus antigen) and rabbit active caspase-3 and then anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 647. Samples were then subjected to flow cytometric analysis. Histogram overlays were constructed from representative experiments in which the fluorescent intensity distribution of mock-infected samples (unfilled histograms) is shown together with that of infected samples (gray histograms). An interval gate (vertical line) is indicated in each plot. Events to the right of the interval gate are considered positive for the given antigen. (C) VSV-infected cells were stained with a mouse monoclonal antibody (BW8G65) to VSV glycoprotein (virus antigen) or rabbit anti-active caspase-3, and analyses were performed as described for panels A and B. The results of three independent experiments are summarized in panel D.
Expression of WNV capsid protects cells from Fas- but not staurosporine-dependent apoptosis.
Having established that induction of apoptosis is delayed in WNV-infected A549 cells, we next determined whether expression of capsid, which is the first viral protein produced, affects the onset of apoptosis. We chose to assess whether capsid expression influenced cell death triggered by Fas (CD95) ligation. This pathway is well characterized and plays a physiologically relevant role in elimination of virus-infected cells (reviewed in reference42). At 24 and 48 h after transfection of A549 cells with pIRES2-AcGFP1 or pIRES2-AcGFP1-capsid plasmids, cells were challenged with anti-Fas and cycloheximide for 6 h. The membrane-permeant dye TMRM was used to detect the loss of mitochondrial membrane potential, which is indicative of commitment to apoptosis (43). After staining with TMRM, transfected cells were analyzed by flow cytometry. Gating on AcGFP-expressing cells was used to differentiate transfected cells from nontransfected cells (Fig. 3C). TMRM staining is the brightest in cells that have an intact mitochondrial membrane potential, and the vertical line in the histogram represents the threshold between viable and apoptotic cells, with apoptotic cells grouping to the left. The primary and summarized data in Fig. 3A and D show that at the 24- and 48-h time points, expression of WNV capsid reduced the onset of Fas-induced apoptosis by 43% and 50%, respectively, compared to that in cells expressing AcGFP only. In these samples, treatment with anti-Fas antibody caused a negligible increase in apoptotic cells during the time course examined. These data suggest that capsid blocks execution of the Fas-dependent apoptotic program by delaying the loss of mitochondrial membrane potential and the subsequent release of proapoptotic factors. Interestingly, WNV capsid failed to protect cells from mitochondrial membrane potential loss during treatment with staurosporine (2 μM), a potent and pan-specific kinase inhibitor (Fig. 3B). This suggests that the antiapoptotic activity of WNV capsid is limited to pathways that rely upon the activity of specific kinases.
Fig 3.

WNV capsid protects against anti-Fas challenge but not staurosporine treatment. A549 cells were transfected with plasmids encoding AcGFP alone (Control) or AcGFP and WNV capsid protein (Capsid). At 24 and 48 h posttransfection, cells were treated with DMSO, staurosproine (ST), mock treated, or treated with anti-Fas and cycloheximide (Fas) for 6 h. Cells were then incubated with the fluorescent mitochondrial dye TMRM and processed for live-cell flow cytometry. Following FACS analyses, histogram overlays were constructed from a representative experiment in which capsid-expressing cells (unfilled histograms) were compared to AcGFP-only-expressing cells (gray-filled histograms). Results from analyses with anti-Fas (A) and staurosporine (B) are shown. Individual events to the left of the interval gates in panels A and B were considered positive for cell death. (C) Distribution of AcGFP fluorescence in transfected cells. All points to the right of the vertical line were considered positive for AcGFP. (D, E) The percentage of dead mock-treated cells was subtracted from the percentage of dead cells in anti-Fas or staurosporine-treated samples. The resultant percentage was referred to as relative specific cell death. Results are averages from 3 experiments. Standard error bars are indicated. Note that only AcGFP-positive events were analyzed for TMRM fluorescence. Statistical analyses (paired t test) were performed, and statistically significant differences are indicated (*, P ≤ 0.05; ***, P ≤ 0.001).
Next, we compared the relative activation of initiator (caspase-8) and executioner (caspase-3) caspases in control and WNV capsid-expressing cells that had been challenged with anti-Fas. For these experiments, we used recombinant lentiviruses instead of plasmid-based transfection to mediate capsid expression. Transduced A549 cells were fixed and stained sequentially with anti-active caspase-8 or -3 antibodies and then Alexa Fluor 647 secondary antibody prior to flow cytometric analyses. Analyses revealed that expression of WNV capsid reduced activation of caspase-8 and caspase-3 in response to anti-Fas by 43% and 46%, respectively (Fig. 4). These results are consistent with the data in Fig. 3 showing that capsid protects cells against loss of mitochondrial membrane potential. To ensure that the observed effect of capsid on cell death was not a cell line-specific effect, we also assayed the activation of caspase-3 in primary human fibroblasts. Similar to what was observed in A549 cells, capsid expression in HEL/18 cells reduced activation of caspase-3 by 43% in response to anti-Fas challenge (Fig. 5).
Fig 4.

WNV capsid inhibits Fas-dependent activation of caspase-3 and -8. A549 cells were transduced with lentiviruses encoding AcGFP alone (AcGFP) or AcGFP and WNV capsid protein (Capsid) at a multiplicity of transduction of 3. At approximately 48 h postransduction, cells were treated with anti-Fas (Fas) or mock treated (Mock). Samples were then processed for intracellular staining with antibodies to active caspase-3 or caspase-8, followed by staining with anti-rabbit Alexa Fluor 647. Samples were then analyzed via FACS, and histogram overlays (A) were constructed from a representative experiment in which capsid-expressing cells (unfilled histograms) were compared to AcGFP-only-expressing cells (gray-filled histograms). Individual events to the right of the interval gate were considered positive for the given active caspase. (B) Mock-treated samples were used to establish a baseline for activated caspase staining. Note that only AcGFP-positive cells were analyzed for the presence of active caspase. Results averaged from 3 independent experiments are shown. Standard error bars are indicated. Statistical analyses (paired t test) were performed, and statistically significant differences are indicated (**, P ≤ 0.01). (C) Histograms display the levels of AcGFP expression and thus transduction in experimental samples. Events to the right of the interval gate are considered positive for AcGFP. (D) Immunoblot data show that transduction of cells with lentiviruses encoding WNV capsid results in robust expression.
Fig 5.

WNV capsid inhibits Fas-mediated apoptosis in HEL/18 cells. HEL/18 cells were transduced with lentiviruses encoding AcGFP alone (AcGFP) or AcGFP and WNV capsid protein (Capsid) at a multiplicity of transduction of 3. At approximately 48 h postransduction, cells were treated with or without anti-Fas (Fas). Samples were then processed for intracellular staining with antibodies to active caspase-3, followed by staining with anti-rabbit Alexa Fluor 647. (A) Samples were then analyzed via FACS, and histogram overlays were constructed from a representative experiment in which capsid-expressing cells (unfilled histograms) were compared to AcGFP-only-expressing cells (gray-filled histograms). (B) Results averaged from 3 independent experiments are shown. Standard error bars are indicated. Statistical analyses (paired t test) were performed, and statistically significant differences are indicated (*, P ≤ 0.05). (C) Histograms display the levels of AcGFP expression and thus transduction in experimental samples. (D) Immunoblot data show that transduction of cells with lentiviruses encoding WNV capsid results in robust expression.
Expression of WNV capsid increases Akt phosphorylation at serine 473, and its protective effects are dependent on PI3K activity.
Recent studies revealed that antiapoptotic/survival signaling is activated shortly after flavivirus infection (27). This signaling process involves the kinase Akt, which is activated by PI3K-dependent phosphorylation on serine 473. To determine if WNV capsid is involved in induction of survival signaling, we monitored the phosphorylation of Akt in capsid-expressing cells. Immunoblot analyses of these samples showed that WNV capsid increased the phosphorylation of serine 473 by 2-fold (Fig. 6A). Capsid-induced phosphorylation of Akt was abrogated in the presence of the PI3K inhibitor LY294002 (Fig. 6B). We next asked whether PI3K activity is required for the protective effect of WNV capsid. Transduced A549 cells expressing AcGFP only or WNV capsid were challenged with anti-Fas and cycloheximide and/or LY294002. Immunoblot analyses confirmed that complete abrogation of Akt phosphorylation at serine 473 was achieved by treatment with LY294002 (Fig. 7A). Data in Fig. 7B show that in cells treated with LY294002, WNV capsid did not protect against anti-Fas (Fig. 7C).
Fig 6.

WNV capsid protein enhances phosphorylation of Akt at serine 473. A549 cells were transduced with lentiviruses expressing WNV capsid or AcGFP alone. After incubation under reduced-serum conditions for 24 h, cells were treated with increasing concentrations of LY294002 (6.25, 12.5, 25 μM) or DMSO as a control. (A) Cell lysates were collected and subjected to SDS-PAGE and immunoblotting for phospho-Akt-S473, WNV capsid, and actin as a loading control. (B) Levels of phospho-Akt relative to total Akt were quantified, and the results are expressed as the fold change with respect to the result for the control. The results of three independent experiments are summarized. Statistical analyses (paired t test) were performed, and significant differences are indicated (**, P ≤ 0.01).
Fig 7.

Inhibition of PI3 kinase abrogates protection by WNV capsid protein. A549 cells were transduced with lentiviruses encoding WNV capsid protein or AcGFP alone, before challenge with anti-Fas with or without addition of LY294002. (A) Resulting cell lysates were subjected to SDS-PAGE and immunoblotting with antibodies to phospho-Akt, WNV capsid, and GAPDH (loading control). Note that treatment with the PI3 kinase inhibitor (LY294002) reduces phospho-Akt regardless of whether capsid protein is expressed or not. At 48 h postransduction, cells expressing AcGFP or WNV capsid were treated with anti-Fas and LY294002 or DMSO. Samples were then processed for intracellular staining with antibodies to active caspase-3, followed by staining with anti-rabbit Alexa Fluor 647. (B) Cells were then analyzed via FACS, and histogram overlays were constructed from a representative experiment in which capsid-expressing cells (unfilled histograms) were compared to AcGFP-only-expressing cells (gray-filled histograms). Events to the right of the vertical line are considered positive. (C) Results from 3 independent experiments were averaged and plotted. Standard error bars are indicated. Statistical analyses (paired t test) were performed, and statistically significant differences are indicated (*, P ≤ 0.05). (D) Histograms display the levels of AcGFP expression and thus transduction in experimental samples.
DISCUSSION
RNA viruses cause the majority of cases of viral disease in humans and are therefore responsible for large economic and medical burdens throughout the world. The flavivirus family of RNA viruses contains a large proportion of medically important viruses, such as hepatitis C virus (HCV), WNV, and DENV. There are few vaccine options, and with the exception of HCV, no highly effective treatments exist for these important pathogens. Although there has been much progress in understanding how flaviviruses cause disease, there remains a paucity of information on the ways in which these pathogens interact with host cells during virus replication and spread. Specifically, we know comparatively little about how individual flavivirus proteins interact with host cell factors and how these interactions may lead to human disease. Of particular note are the interactions that affect the survival of host cells, as these play major roles in the replicative success of viruses in vivo. Infected hosts attempt to kill and clear virus-infected cells, but some viruses are known to encode proteins that allow evasion of this ubiquitous antiviral response. With few exceptions, most studies regarding virus-encoded antiapoptotic proteins have focused on DNA viruses. The known exceptions are the picornavirus-encoded proteins leader and 2BC (44, 45), as well as the rubella virus capsid protein (9). Although infection with these viruses induces apoptosis in many cell lines, this is generally observed late in the infection process.
In this report, we demonstrate that the major isoform of WNV capsid protein in infected cells functions to block apoptosis. The observation that but a single capsid species predominates in WNV-infected cells is supported by previous studies of flavivirus polyprotein processing kinetics (33–35). Importantly, the fact that the mature cleaved form of capsid protein predominates in infected cells can explain why our observations differ from those of other studies reporting that the larger isoform of WNV capsid protein induces apoptosis (22–25). The fact that PIK3 activity is important for the antiapoptotic function of a flavivirus capsid protein is consistent with our observation that capsid-expressing cells are not resistant to cell death induced by the pan-specific kinase inhibitor staurosporine. The maximum antiapoptotic activity of Akt requires sequential phosphorylation on S308 and S473 by PI3K. Phospho-Akt is then able to phosphorylate and inhibit proapoptotic substrates, including Bad, caspase-9, and glycogen synthase kinase 3β (46–48).
The idea that WNV capsid is a proapoptotic protein (22–25) does not fit well with what is known about the biology of this virus. Specifically, several publications have shown that depending upon the specific flavivirus and cell type involved, PI3K/Akt signaling is activated within minutes (or hours) of infection with WNV, DENV, and JEV (26, 27, 49). Moreover, caspase-3 activation does not occur until later in infection, after the activation of PI3K/Akt has subsided. Enhanced signaling through this critical survival pathway is likely important for virus replication and pathogenesis. Inhibition of PI3K by LY294002 leads to earlier activation of caspase-3 and enhanced cell death during flavivirus infection in vitro (27). As mentioned above, the kinetics of virus-induced survival signaling varies among cell lines and flaviviruses, and as a consequence, the extent of apoptosis is affected. With regard to WNV, Scherbik and Brinton (28) reported that infection of MEFs significantly increases phospho-Akt within 2 h of infection and this effect is sustained for more than 24 h. Similarly, JEV infection of RBA-1 cells activates the PI3K/Akt pathway, resulting in stimulatory phosphorylation of Akt within minutes (49). In contrast, WNV infection of neuro2A cells, a mouse neuroblastoma cell line, results in extensive cell death in less than 72 h, even at very low MOIs (50). Together, these observations may explain the differences in the extent of apoptosis observed between WNV-infected neuronal and nonneuronal cells.
The importance of the PI3K/Akt pathway in virus biology has been studied extensively in the context of both RNA and DNA virus infections. Through this pathway, viruses are able to affect several key cellular processes, such as mRNA translation, glucose metabolism, and cell survival. With respect to cell survival, Akt activity is particularly relevant in modulating Fas-mediated apoptosis. Among the multiple ways in which this kinase antagonizes death receptor-dependent apoptosis is through phosphorylation of FKHRL1, a transcription factor whose downstream targets include Fas ligand (51). Indeed, elevated PI3K-dependent activation of Akt has been shown to protect against Fas-mediated apoptosis in multiple cell types, including those derived from tumors (52). Thus, WNV capsid exploits a pathway that is commonly altered in transformed cells to promote cell survival in the presence of apoptotic stimuli.
Activation of Akt can also lead to phosphorylation and subsequent upregulation of the translational activity of the 4B subunit of eukaryotic initiation factor (53). In turn, cap-dependent translation is increased. The availability of UDP glucose, which is required for protein N-glycosylation and proper protein folding in the endoplasmic reticulum, is also enhanced following activation of PI3K/Akt. This is mediated by phosphorylation and activation of ENTPD5, a uridine diphosphatase that promotes UDP hydrolysis and subsequent N-glycosylation and protein folding (54, 55). Several WNV proteins are heavily glycosylated, and increasing the capacity of this organelle to N-glycosylate and fold viral proteins is a likely way to increase viral fitness and minimize the effects of the unfolded protein response. Thus, in addition to blocking apoptosis, WNV capsid protein may serve to modulate the cellular environment through other mechanisms to make it more favorable for virus replication.
In summary, we have reported for the first time that WNV capsid protein possesses antiapoptotic activity. Importantly, the antiapoptotic effects of WNV capsid are not limited to a single cell line, as they were observed in both continuous and primary cell lines. The protective capacity of capsids is dependent upon PI3K activity, a situation that is consistent with earlier studies showing that activating phosphorylation of Akt on serine 473 occurs shortly after cells are infected with flaviviruses (26, 27, 49). These data strongly suggest that capsid is the viral protein that mediates the increase in prosurvival signaling seen during virus infection.
ACKNOWLEDGMENTS
We thank Eileen Reklow and Valeria Mancinelli for technical support.
M.D.U. is the recipient of a Frederick Banting & Charles Best Canada Graduate Doctoral Scholarship from the Canadian Institutes of Health Research (CIHR). T.C.H. holds a scientist award from Alberta Innovates Health Solutions and is a Canada Research Chair. This work was supported by operating funds from the CIHR.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Diamond MS. 2009. Progress on the development of therapeutics against West Nile virus. Antiviral Res. 83:214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smithburn KC, Hughes TP, Burke AW, Paul JH. 1940. A neurotropic virus isolated from the blood of a native of Uganda. Am. J. Trop. Med. Hyg. 20:471–492 [Google Scholar]
- 3. Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Crise B, Volpe KE, Crabtree MB, Scherret JH, Hall RA, MacKenzie JS, Cropp CB, Panigrahy B, Ostlund E, Schmitt B, Malkinson M, Banet C, Weissman J, Komar N, Savage HM, Stone W, McNamara T, Gubler DJ. 1999. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286:2333–2337 [DOI] [PubMed] [Google Scholar]
- 4. Brinton MA. 2002. The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu. Rev. Microbiol. 56:371–402 [DOI] [PubMed] [Google Scholar]
- 5. Firth AE, Atkins JF. 2009. A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1′ may derive from ribosomal frameshifting. Virol. J. 6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Melian EB, Hinzman E, Nagasaki T, Firth AE, Wills NM, Nouwens AS, Blitvich BJ, Leung J, Funk A, Atkins JF, Hall R, Khromykh AA. 2010. NS1′ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 84:1641–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beatch MD, Everitt JC, Law LJ, Hobman TC. 2005. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J. Virol. 79:10807–10820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunt TA, Urbanowski MD, Kakani K, Law LM, Brinton MA, Hobman TC. 2007. Interactions between the West Nile virus capsid protein and the host cell-encoded phosphatase inhibitor, I2PP2A. Cell. Microbiol. 9:2756–2766 [DOI] [PubMed] [Google Scholar]
- 9. Ilkow CS, Goping IS, Hobman TC. 2011. The rubella virus capsid is an anti-apoptotic protein that attenuates the pore-forming ability of Bax. PLoS Pathog. 7:e1001291 doi:10.1371/journal.ppat.1001291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ilkow CS, Mancinelli V, Beatch MD, Hobman TC. 2008. Rubella virus capsid protein interacts with poly(A)-binding protein and inhibits translation. J. Virol. 82:4284–4294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ilkow CS, Weckbecker D, Cho WJ, Meier S, Beatch MD, Goping IS, Herrmann JM, Hobman TC. 2010. The rubella virus capsid protein inhibits mitochondrial import. J. Virol. 84:119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tzeng WP, Frey TK. 2005. Rubella virus capsid protein modulation of viral genomic and subgenomic RNA synthesis. Virology 337:327–334 [DOI] [PubMed] [Google Scholar]
- 13. Xu Z, Anderson R, Hobman TC. 2011. The capsid-binding nucleolar helicase DDX56 is important for infectivity of West Nile virus. J. Virol. 85:5571–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kleinschmidt MC, Michaelis M, Ogbomo H, Doerr HW, Cinatl J., Jr 2007. Inhibition of apoptosis prevents West Nile virus induced cell death. BMC Microbiol. 7:49 doi:10.1186/1471-2180-7-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kobayashi S, Orba Y, Yamaguchi H, Kimura T, Sawa H. 2011. Accumulation of ubiquitinated proteins is related to West Nile virus-induced neuronal apoptosis. Neuropathology 32:398–405 [DOI] [PubMed] [Google Scholar]
- 16. Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, Defilippis V, Fruh K, Mason PW, Nikolich-Zugich J, Nelson JA. 2007. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81:10849–10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parquet MC, Kumatori A, Hasebe F, Morita K, Igarashi A. 2001. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 500:17–24 [DOI] [PubMed] [Google Scholar]
- 18. Smith JL, Grey FE, Uhrlaub JL, Nikolich-Zugich J, Hirsch AJ. 2012. Induction of the cellular microRNA, Hs_154, by West Nile virus contributes to virus-mediated apoptosis through repression of antiapoptotic factors. J. Virol. 86:5278–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gadaleta P, Perfetti X, Mersich S, Coulombie F. 2005. Early activation of the mitochondrial apoptotic pathway in vesicular stomatitis virus-infected cells. Virus Res. 109:65–69 [DOI] [PubMed] [Google Scholar]
- 20. Lopez-Herrera A, Ruiz-Saenz J, Goez YP, Zapata W, Velilla PA, Arango AE, Urcuqui-Inchima S. 2009. Apoptosis as pathogenic mechanism of infection with vesicular stomatitis virus. Evidence in primary bovine fibroblast cultures. Biocell 33:121–132 [PubMed] [Google Scholar]
- 21. Pearce AF, Lyles DS. 2009. Vesicular stomatitis virus induces apoptosis primarily through Bak rather than Bax by inactivating Mcl-1 and Bcl-XL. J. Virol. 83:9102–9112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhuvanakantham R, Cheong YK, Ng ML. 2010. West Nile virus capsid protein interaction with importin and HDM2 protein is regulated by protein kinase C-mediated phosphorylation. Microbes Infect. 12:615–625 [DOI] [PubMed] [Google Scholar]
- 23. Oh W, Yang MR, Lee EW, Park KM, Pyo S, Yang JS, Lee HW, Song J. 2006. Jab1 mediates cytoplasmic localization and degradation of West Nile virus capsid protein. J. Biol. Chem. 281:30166–30174 [DOI] [PubMed] [Google Scholar]
- 24. Yang JS, Ramanathan MP, Muthumani K, Choo AY, Jin SH, Yu QC, Hwang DS, Choo DK, Lee MD, Dang K, Tang W, Kim JJ, Weiner DB. 2002. Induction of inflammation by West Nile virus capsid through the caspase-9 apoptotic pathway. Emerg. Infect. Dis. 8:1379–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang MR, Lee SR, Oh W, Lee EW, Yeh JY, Nah JJ, Joo YS, Shin J, Lee HW, Pyo S, Song J. 2008. West Nile virus capsid protein induces p53-mediated apoptosis via the sequestration of HDM2 to the nucleolus. Cell. Microbiol. 10:165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Das S, Chakraborty S, Basu A. 2010. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J. Neurochem. 115:537–549 [DOI] [PubMed] [Google Scholar]
- 27. Lee CJ, Liao CL, Lin YL. 2005. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J. Virol. 79:8388–8399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scherbik SV, Brinton MA. 2010. Virus-induced Ca2+ influx extends survival of West Nile virus-infected cells. J. Virol. 84:8721–8731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shrestha B, Diamond MS. 2007. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J. Virol. 81:11749–11757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10:1366–1373 [DOI] [PubMed] [Google Scholar]
- 31. O'Donnell D, Milligan RL, Stark JM. 1999. Induction of CD95 (Fas) and apoptosis in respiratory epithelial cell cultures following respiratory syncytial virus infection. Virology 257:198–207 [DOI] [PubMed] [Google Scholar]
- 32. Medigeshi GR, Hirsch AJ, Brien JD, Uhrlaub JL, Mason PW, Wiley C, Nikolich-Zugich J, Nelson JA. 2009. West Nile virus capsid degradation of claudin proteins disrupts epithelial barrier function. J. Virol. 83:6125–6134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amberg SM, Rice CM. 1999. Mutagenesis of the NS2B-NS3-mediated cleavage site in the flavivirus capsid protein demonstrates a requirement for coordinated processing. J. Virol. 73:8083–8094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lobigs M, Lee E, Ng ML, Pavy M, Lobigs P. 2010. A flavivirus signal peptide balances the catalytic activity of two proteases and thereby facilitates virus morphogenesis. Virology 401:80–89 [DOI] [PubMed] [Google Scholar]
- 35. Stocks CE, Lobigs M. 1998. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 72:2141–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Megyeri K, Berencsi K, Halazonetis TD, Prendergast GC, Gri G, Plotkin SA, Rovera G, Gonczol E. 1999. Involvement of a p53-dependent pathway in rubella virus-induced apoptosis. Virology 259:74–84 [DOI] [PubMed] [Google Scholar]
- 37. Borisevich V, Seregin A, Nistler R, Mutabazi D, Yamshchikov V. 2006. Biological properties of chimeric West Nile viruses. Virology 349:371–381 [DOI] [PubMed] [Google Scholar]
- 38. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chu JJ, Ng ML. 2003. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 84:3305–3314 [DOI] [PubMed] [Google Scholar]
- 40. Samuel MA, Morrey JD, Diamond MS. 2007. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 81:2614–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sharif-Askari E, Nakhaei P, Oliere S, Tumilasci V, Hernandez E, Wilkinson P, Lin R, Bell J, Hiscott J. 2007. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology 365:20–33 [DOI] [PubMed] [Google Scholar]
- 42. Keckler MS. 2007. Dodging the CTL response: viral evasion of Fas and granzyme induced apoptosis. Front. Biosci. 12:725–732 [DOI] [PubMed] [Google Scholar]
- 43. Ly JD, Grubb DR, Lawen A. 2003. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 8:115–128 [DOI] [PubMed] [Google Scholar]
- 44. Romanova LI, Lidsky PV, Kolesnikova MS, Fominykh KV, Gmyl AP, Sheval EV, Hato SV, van Kuppeveld FJ, Agol VI. 2009. Antiapoptotic activity of the cardiovirus leader protein, a viral “security” protein. J. Virol. 83:7273–7284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salako MA, Carter MJ, Kass GE. 2006. Coxsackievirus protein 2BC blocks host cell apoptosis by inhibiting caspase-3. J. Biol. Chem. 281:16296–16304 [DOI] [PubMed] [Google Scholar]
- 46. Diez H, Garrido JJ, Wandosell F. 2012. Specific roles of Akt iso forms in apoptosis and axon growth regulation in neurons. PLoS One 7:e32715 doi:10.1371/journal.pone.0032715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. 2003. PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998 [DOI] [PubMed] [Google Scholar]
- 48. Kapoor V, Zaharieva MM, Das SN, Berger MR. 2012. Erufosine simultaneously induces apoptosis and autophagy by modulating the Akt-mTOR signaling pathway in oral squamous cell carcinoma. Cancer Lett. 319:39–48 [DOI] [PubMed] [Google Scholar]
- 49. Yang CM, Lin CC, Lee IT, Lin YH, Chen WJ, Jou MJ, Hsiao LD. 2012. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J. Neuroinflammation 9:12 doi:10.1186/1742-2094-9-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lucas M, Mashimo T, Frenkiel MP, Simon-Chazottes D, Montagutelli X, Ceccaldi PE, Guenet JL, Despres P. 2003. Infection of mouse neurones by West Nile virus is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immunol. Cell Biol. 81:230–236 [DOI] [PubMed] [Google Scholar]
- 51. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868 [DOI] [PubMed] [Google Scholar]
- 52. Hausler P, Papoff G, Eramo A, Reif K, Cantrell DA, Ruberti G. 1998. Protection of CD95-mediated apoptosis by activation of phosphatidylinositide 3-kinase and protein kinase B. Eur. J. Immunol. 28:57–69 [DOI] [PubMed] [Google Scholar]
- 53. Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. 2006. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25:2781–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, Wang X. 2010. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 143:711–724 [DOI] [PubMed] [Google Scholar]
- 55. Shen Z, Huang S, Fang M, Wang X. 2011. ENTPD5, an endoplasmic reticulum UDPase, alleviates ER stress induced by protein overloading in AKT-activated cancer cells. Cold Spring Harbor Symp. Quant. Biol. 76:217–223 [DOI] [PubMed] [Google Scholar]