

Abstract
Vaccinia virus membrane biogenesis requires the A14 and A17 proteins. We show here that both proteins can associate with membranes co- but not posttranslationally, and we perform a structure function analysis of A14 and A17 using inducible recombinants. In the absence of A14, electron-dense virosomes and distinct clusters of small vesicles accumulate; in the absence of A17, small vesicles form a corona around the virosomes. When the proteins are induced at 12 h postinfection (hpi), crescents appear at the periphery of the electron-dense virosomes, with the accumulated vesicles likely contributing to their formation. A variety of mutant alleles of A14 and A17 were tested for their ability to support virion assembly. For A14, biologically important motifs within the N-terminal or central loop region affected crescent maturation and the immature virion (IV)→mature virion (MV) transition. For A17, truncation or mutation of the N terminus of A17 engendered a phenotype consistent with the N terminus of A17 recruiting the D13 scaffold protein to nascent membranes. When N-terminal processing was abrogated, virions attempted to undergo the IV-to-MV transition without removing the D13 scaffold and were therefore noninfectious and structurally aberrant. Finally, we show that A17 is phosphorylated exclusively within the C-terminal tail and that this region is a direct substrate of the viral F10 kinase. In vivo, the biological competency of A17 was reduced by mutations that prevented its serine-threonine phosphorylation and restored by phosphomimetic substitutions. Precleavage of the C terminus or abrogation of its phosphorylation diminished the IV→MV maturation; a block to cleavage spared virion maturation but compromised the yield of infectious virus.
INTRODUCTION
Poxviruses are complex DNA viruses that are ubiquitous in nature; the orthopoxvirus family includes variola virus, the etiologic agent of smallpox, and the closely related monkeypox virus, which is endemic in Africa and also causes a severe, acute febrile illness (1, 2). The prototypic poxvirus for experimental study is the closely related vaccinia virus, which has played an eminent role in public health as the vaccine strain used in the successful eradication of smallpox. Poxviruses are unique among DNA viruses in that they replicate exclusively in the cytoplasm of infected cells (2). This physical autonomy is enabled by the presence of ∼200 genes in the viral genome that encode the machinery for virion entry, gene expression, genome replication and maturation, virion assembly and maturation, and virion egress. The assembly of nascent virions is one of the most unique facets of poxviral infection (3). Unlike most other enveloped viruses, vaccinia virus does not acquire its delimiting membrane by budding into an organelle or through the plasma membrane. Instead, the membrane develops within the cytoplasm, seeded by small patches of membrane that grow into characteristic crescents. As these nascent membrane sheets develop, they engulf virosomal proteins destined for encapsidation in the virion core. The exterior face of the developing membrane is supported and shaped by a lattice composed of the D13 protein; the intrinsic curvature of this lattice determines the size and shape of the oval, immature virions (IVs). These immature virions contain the viral genome and a plethora of structural and enzymatic proteins.
The earliest stages of membrane formation are regulated by a number of viral proteins, including the F10 protein kinase and the H7, A11, L2, and A6 nonstructural proteins (4–12). It has been previously shown by our laboratory, as well as others, that the vaccinia virus A14 and A17 transmembrane proteins are also required for the biogenesis of the virion membrane (13–18). These two proteins are thought to adopt hairpin confirmations and span the membrane twice, albeit with opposite polarities (3). Portions of the A17 protein are exposed on the virion surface, as shown by immunogold staining of negatively stained virions and by the ability of anti-A17 sera to neutralize virion infectivity (19, 20). Neither the origin of the seed membrane nor the mechanism by which the membrane is diverted for maturation into the virion membrane is understood. However, both of the A14 and A17 proteins are thought to be synthesized in the endoplasmic reticulum (ER), despite lacking conventional signal sequences (21, 22). Inducible recombinants that express A14 in a tetracycline (TET)-dependent and A17 in an isopropyl-β-d-thiogalactopyranoside (IPTG)-dependent manner have been invaluable in studying the role of these proteins in membrane biogenesis. Repression of A14 expression leads to the accumulation of large electron-dense virosomes containing proteins destined for encapsidation in the core and, at a distance, numerous clusters of 25-nm-diameter vesicles. Occasionally, a few detached crescents are seen at the periphery of the virosomes (16, 17). Repression of A17 leads to the accumulation of large electron-dense virosomes that are surrounded by a corona of 25-nm-diameter vesicles (14, 15, 18). Crescents and immature or mature virions (MVs) are not observed in either case.
The A14 and A17 proteins are known to undergo several posttranslational modifications. In vivo, A14 is phosphorylated on ser85; mutation of this residue does not compromise the ability of A14 to support virion morphogenesis (13). Moreover, this residue is thought to reside within the ER lumen during A14 synthesis. A14 can also undergo glycosylation on an N83HS motif that overlaps the phosphorylation site (13). The glycosylated species is not found in mature virions, and the number of A14 molecules that undergo glycosylation increases dramatically when virion assembly is blocked at a very early stage. Thus, the A14 destined for inclusion in the virion membrane probably exits the ER prior to glycosylation. A17 also undergoes phosphorylation (23, 24), and this modification is addressed here. In addition, A17 is known to undergo posttranslational cleavage at both its N and C termini at the AG↓X motifs targeted by the viral I7 protease (23, 25). These cleavage events are also addressed here. A17 has been shown to interact with the D13 protein (26), and A14 and A17 can be coimmunoprecipitated, although whether this interaction is direct or indirect remains unknown (23, 27). A17 also interacts with A27 (molecular weight [MW], 14K) (28), a peripheral protein that is not involved in the assembly of MV but is important for the wrapping of a subset of these virions in an additional membrane and their egress from the cell as enveloped virions (EV) that mediate proximal and distal spread (29).
In this report, we have analyzed the ability of a variety of mutant alleles of A14 and A17 to support virion assembly. The mutant alleles contain substitutions that target the cleavage sites of A17, the putative phosphorylation sites of A17, and the hydrophilic tails of A17 exposed on the virion surface. They also target motifs within A14 that we have previously shown to be important for the biological competency of A14 (13). Premature truncation or amino acid substitutions in the N terminus of A17 render a phenotype that is similar to that seen with the drug rifampin (RIF), supporting the conclusion (26) that recruitment of the D13 scaffolding protein to the nascent membrane is mediated by the N terminus of A17. Conversely, mutations that prevent the subsequent cleavage of the N terminus of A17 also impair virion assembly by preventing the removal of the D13 exoskeleton at late times of virion maturation. We have also shown that the C terminus of A17 is a direct substrate of the viral F10 kinase and that mutation of these sites of phosphorylation compromises the biological activity of A17 and impairs virion maturation. Finally, we have shown that previously identified motifs of A14 are required for the immature virion (IV)→mature virion (MV) transition but do not block the A14-A17 interaction. Together, these data shed light on the role(s) of A14 and A17 in membrane biogenesis and virion maturation.
MATERIALS AND METHODS
Materials.
Restriction enzymes, T4 DNA ligase, calf intestinal alkaline phosphatase (CIP), pancreatic RNase, Taq polymerase, and isopropyl-b-d-thiogalactoside (IPTG) were purchased from Roche Diagnostics (Indianapolis, IN). Protein A Sepharose, V5 agarose affinity gel, and araC (cytosine arabinoside) were acquired from Sigma-Aldrich (St. Louis, MO). γ-32P-ATP, Easy Tag methionine (met) L-[35S], and EXPRE35S35S protein labeling mix were purchased from PerkinElmer Life Sciences (Boston, MA). Lipofectamine 2000, Lipofectamine LTX, glutamine, and molecular weight standards were purchased from Invitrogen (Carlsbad, CA). Supersignal chemiluminescent substrate was acquired from Pierce (Rockford, IL). Protran nitrocellulose was obtained from Whatman, Inc. (Dassel, Germany). The anti-A17 polyclonal serum was generated in our laboratory (described here); the A17L-N antibody was kindly provided by B. Moss (NIAID, NIH) (26). Opti-MEM, Dulbecco's modified Eagle's medium (DMEM), and DMEM lacking phosphate or methionine were purchased from MP Biomedicals (Solon, OH). Anti-phosphotyrosine antibody was purchased from Zymed Laboratories (a division of Invitrogen). Maltose binding protein was acquired from New England BioLabs (Ipswich, MA). The kit for IVTT (in vitro transcription and translation) was purchased from Promega Corporation (Madison, WI). Glutaraldehyde was obtained from Electron Microscopy Sciences (Hatfield, PA). All oligonucleotides were purchased from Integrated DNA Technologies (IDT; Coralville, IA).
Cells and viruses.
Confluent monolayers of BSC-40 green monkey kidney cells were maintained in DMEM supplemented with 5% fetal bovine serum (Invitrogen). Wild-type (WT) vaccinia virus (WR strain), vTF7.3 (30), vindA14 (17), vindA17 (18) (kindly provided by B. Moss), the temperature-sensitive (ts) virus ts28 (11, 12), and tsH5-4 (31) were propagated on monolayers of BSC-40 cells; viral stocks were prepared by ultracentrifugation of clarified cytoplasmic lysates through 36% sucrose cushions. Titers were determined by plaque assays that were performed on BSC-40 cells stained with crystal violet at 48 h postinfection (hpi). For vindA17, IPTG was used at 5 mM; for vindA14, tetracycline was added for a final concentration of 1 μg/ml. Infections were performed at 37°C unless indicated otherwise.
Preparation of anti-A17 serum.
Full-length A17 (encoding amino acids [aa] 1 to 203) was amplified by PCR using WT genomic DNA (WR strain) as the template. The upstream primer (TrpU) (5′-CCCGCGTCTAGAATGAGTTATTTAAGATA-3′) introduced an XbaI site (underlined), and the downstream primer (TrpD) (5′-GGGGGATCCTTAATAATCGTCAGTAT-3′) introduced a BamHI (bold) site. The 612-bp PCR product was digested with XbaI and BamHI and ligated into pATH23 (32), a plasmid DNA that had previously been similarly digested and treated with calf intestinal alkaline phosphatase. The resultant plasmid placed the A17 open reading frame (ORF) downstream of, and in frame with, the Escherichia coli trpE gene (pATH23:A17). To induce expression of the fusion protein, E. coli transformants carrying pATH23:A17 were starved of tryptophan and treated with indoleacrylic acid. Insoluble inclusion bodies were collected and resolved by SDS-PAGE. The 59-kDa fusion protein was visualized with 0.3 M CuCl2; the gel slice was excised and used to immunize rabbits.
Construction of A17 and D13 mutants.
Construction of pUC1246 (WT A17, Y3,6,7F, and Y203F) has been previously described (24). The remaining pUC1246 or pTM1 A17 mutants were generated by overlap PCR using viral genomic DNA or previously generated alleles as templates. A complete listing of the primers used to introduce the internal mutations and a description of the PCR scheme are available upon request.
To construct pTM1-V5D13, an ∼1,200-bp DNA fragment encoding D13 was amplified using viral genomic DNA as a template. The upstream primer, 5′-GGGGATCCACATGTTGGGTAAACCTATCCCTAACCCTTTGTTGGGACTCGACTCTACAAATAATACTATCATTAATTC-3′, introduced BamHI (italics) and AflIII (underlined) sites upstream of sequences encoding the V5 epitope tag (bold) and the N terminus of the D13 protein. The downstream primer, 5′-CCGGAATTCGGATCCTTAGTTATTATC-3′, introduced EcoRI (underlined) and BamHI (italics) sites downstream of the stop codon of the D13 ORF. Here and throughout our studies, plasmids were subjected to DNA sequencing to confirm the presence of the desired sequence and rule out the presence of spurious mutations. The insert was sequentially digested with AflIII and BamHI and ligated to pTM1 (33) plasmid DNA that had been digested with NcoI and BamHI and treated with calf intestinal alkaline phosphatase.
Generation of an MBP (maltose binding protein):A17(161–203) fusion protein.
(i) For plasmid construction, a 126-bp fragment representing residues 161 to 203 of the A17 ORF was amplified using primers U [5′-CGGGATCCAGACAGGACTATAGAAG-3′] and D [5′-CGTCTAGATTAATAATCGTCAGTAT-3′]. The PCR product was digested with BamHI (bold and italicized) and XbaI (bold) prior to being ligated into pMAL-P-1 plasmid DNA that had been similarly digested and treated with calf intestinal alkaline phosphatase. This construct encoded a protein containing the C terminus of A17 (aa 161 to 203) fused to the maltose binding protein. (ii) For protein expression and purification, E. coli transformants [BL21(DE3) strain] carrying the pMAL:MBP:A17(161–203) plasmid were grown in the presence of 0.2% glucose and induced by the addition of 0.5 mM IPTG for 2 h at 37°C. Bacteria were solubilized in 0.2 mM NaCl–20 mM Tris-HCl (pH 7.4)–0.5 mM EDTA; lysates were applied to amylose resin (prepared according to the manufacturer's instructions) and eluted by the addition of 10 mM maltose. Fractions were analyzed by SDS-PAGE, visualized by Coomassie blue staining, quantitated by silver staining, and used as the substrate in an in vitro kinase assay.
Transient-complementation assay.
Dishes (60-mm diameter) of confluent BSC-40 cells were infected with vindA17 (18) (multiplicity of infection [MOI] = 10) or vindA14 (17) (MOI = 5) in the presence or absence of IPTG or tetracycline, respectively. A total of 3 μg (Lipofectamine 2000) or 4 μg (Lipofectamine LTX) of plasmid DNA was introduced at 3 hpi (Lipofectamine 2000) or after removal of the initial inoculum (30 min postinfection) (Lipofectamine LTX). Cells were harvested at 18 to 24 hpi and subjected to immunoprecipitation or immunoblot analysis, to plaque assay titration for the determination of viral yield by titration, or to visualization by electron microscopy (EM). The relevant sections of a single immunoblot are shown (see Fig. 5B and 6B; the positive and negative controls are included in Fig. 5B).
Fig 5.
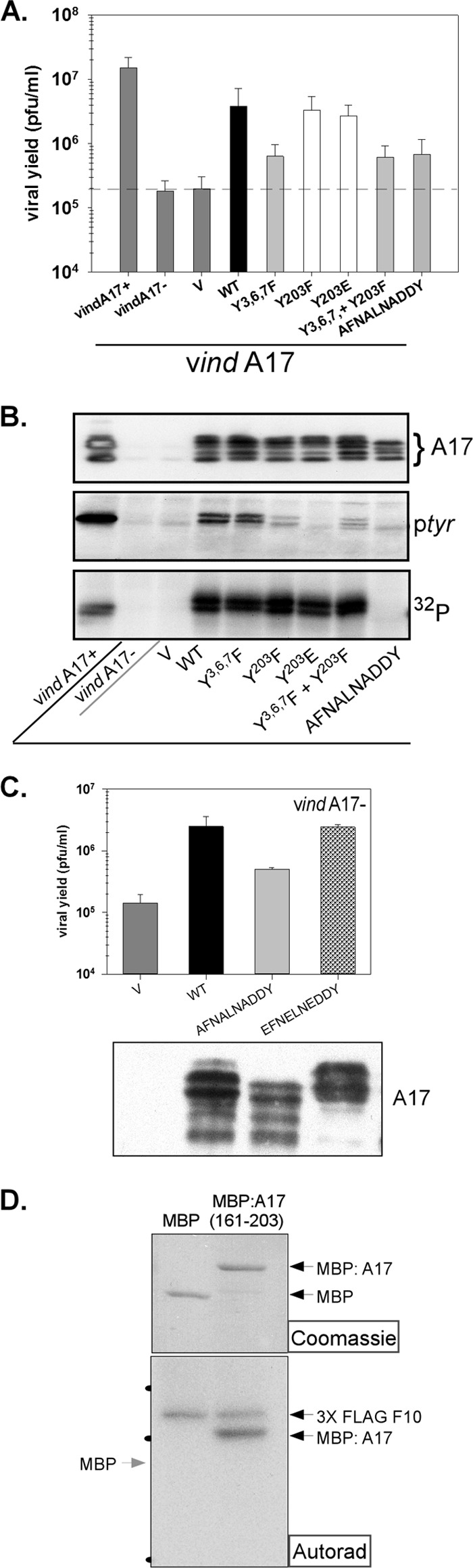
Phosphorylation of A17: identification of target sites and assessment of their importance. (A) BSC-40 cells were infected with vindA17 with or without IPTG (two leftmost samples) and transfected with empty vector (V) or plasmids encoding WT or mutant forms of A17. Viral yield was determined by a plaque assay; data representing averages of the results determined for 4 biological replicates (with standard errors) are shown graphically. (B) Lysates from an experiment comparable to that described for panel A were resolved electrophoretically and subjected to immunoblot analysis to visualize total A17 (top; multiple species reflecting differential proteolytic processing and phosphorylation are indicated by the bracket). A17 was also retrieved from the lysates by immunoprecipitation, and tyrosine phosphorylation of A17 was visualized by immunoblot analysis (ptyr) (middle). Alternatively, cells were radiolabeled with 32PPi and the phosphorylation of immunoprecipitated A17 was visualized by autoradiography (32P) (bottom). (C) Transient-complementation assays were performed as described for panel A to compare the biological competence of WT A17, a mutant lacking the C-terminal sites of ser-thr phosphorylation (AFNALNADDY), to that of a mutant containing phosphomimetic residues instead (EFNELNEDDY). Viral yield was assessed by a plaque assay, and data representing averages of the results of two experiments are shown (with standard errors); the accumulation of the A17 proteins was assessed by immunoblot analysis (bottom). (D) To assess whether the C-terminal tail of A17 is a direct substrate of F10, 10 to 20 ng of purified 3XFLAG-F10 was incubated with 2.5 μg of purified MBP or MBP:A17(161-203) for 30 min in the presence of [γ-32P]ATP. The reactions were resolved by SDS-PAGE; the reaction components were visualized by Coomassie blue staining (top) and autoradiography (bottom). The migrations of the 66-, 46-, and 30K protein standards are indicated by the black dots at the left of the autoradiograph (Autorad).
Fig 6.
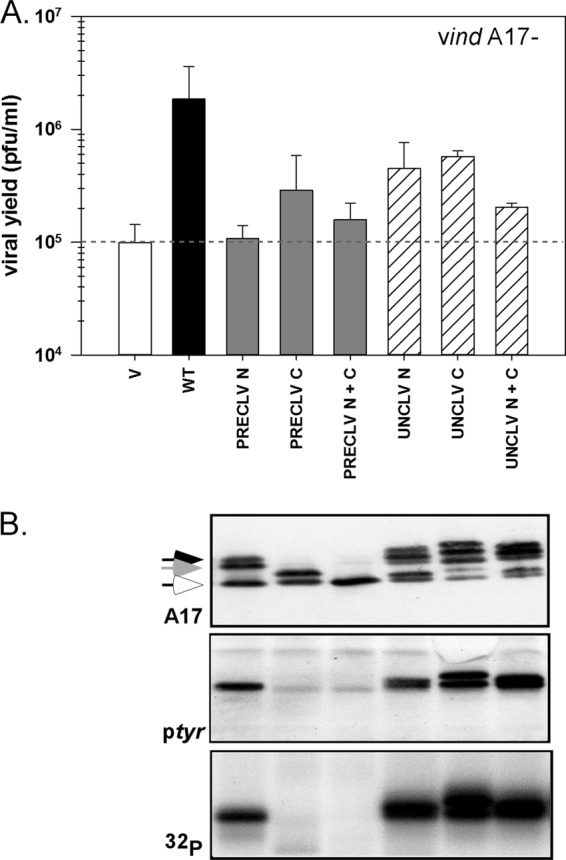
Structure function analysis of the N and C termini of A17. (A) Transient complementation was performed as described previously; cells were infected with vindA17-IPTG and transfected with empty vector (V) or plasmids encoding WT or mutant forms of A17. Viral yield was assessed by a plaque assay; data representing averages of the results of two experiments (with standard errors) are shown. (B) Lysates were prepared from the experiment described for panel A. Samples were analyzed as described for Fig. 5B; the accumulations of total A17 (arrows indicate differentially phosphorylated and/or cleaved species), tyrosine-phosphorylated A17, and phosphorylated A17 were assessed. Immunoblot results for cells transfected with empty vector (V) or WT A17 are shown in Fig. 5B.
Iodixanol gradient fractionation of cell lysates.
Three 15-cm-diameter dishes of BSC-40 cells were infected (MOI = 2) with either vindA14-TET or vindA17-IPTG. Cells were harvested at 17 hpi, rinsed 3 times with phosphate-buffered saline (PBS) and 2 times with wash buffer (WB) (0.25 M sucrose, 10 mM triethanolamine [pH 7.8 with acetic acid]). Homogenization buffer (HB) (0.25 M sucrose, 10 mM triethanolamine [pH 7.8], 1 mM EDTA [pH 7.8 with acetic acid]) (4°C) was then added; cells were removed by scraping and subjected to centrifugation at 800 × g and 4°C for 10 min. The cell pellet was resuspended in fresh HB and homogenized by 10 to 25 strokes in a Dounce homogenizer. Sequential rounds of sedimentation at 1,000 × g and 4°C for 10 min followed by 3,000 × g and 4°C for 10 min yielded a postmitochondrial supernatant (PMS) (per Opti-Prep applications manual). This material was loaded onto a 10% to 40% iodixanol gradient and sedimented at 100,000 × g for 18 h at 4°C. Fractions were collected, and aliquots were resolved by SDS-PAGE and subjected to immunoblot analysis.
Metabolic labeling of infected cultures.
(i) For 32PPi labeling, confluent monolayers of BSC-40 cells (in 60-mm-diameter dishes) were infected with vindA17 (18) (MOI = 10) in the presence or absence of IPTG (5 mM). At 3 hpi, cells were transfected with 3 μg of plasmid DNA encoding WT or mutant forms of A17 using Lipofectamine 2000 and metabolically labeled with 100 μCi/ml of 32PPi for 18.5 h before being harvested. Metabolic labeling was performed in the presence of DMEM lacking phosphate supplemented with 5% fetal calf serum that had been dialyzed against Tris-buffered saline (50 mM Tris-HCl [pH 7.4], 150 mM NaCl). (ii) For [35S]methionine labeling, confluent monolayers of BSC-40 cells were infected with either vindA17 with or without IPTG or vindA14 with or without TET (MOI = 10). At 1 hpi, cells were transfected with 8 μg of the indicated plasmid and metabolically labeled with 100 μCi/ml of [35S]methionine for 18 h. Metabolic labeling was performed in DMEM lacking methionine that had been supplemented with 5% dialyzed fetal calf serum.
Immunoprecipitation analysis.
For most experiments, cells were lysed on ice in 1× phospholysis buffer (10 mM NaPO4 [pH 7.4], 100 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) in the presence of phosphatase inhibitors (40 mM β-glycerolphosphate, 1 mM sodium orthovanadate, 1 mM sodium fluoride). For the analysis of the interaction of A14 and A17 (see Fig. 3B), the lysis buffer contained 50 mM Tris [pH 7.4], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 1% SDS, and 1 mM EDTA. Clarified lysates were incubated with anti-A17 (described here) or anti-A14 (13, 17) sera for 4 h, at which time protein A Sepharose beads were added and incubation was allowed to proceed for 1.5 h at 4°C with end-over-end mixing. Complexes were washed repeatedly with vigorous vortexing; immunoprecipitates were analyzed by SDS-PAGE and visualized by autoradiography or by immunoblot analysis, as indicated.
Fig 3.

Structure function analysis of A14: key motifs and the A14-A17 interaction. (A) The topology of A14 within the viral membrane is shown schematically; cys71 enables A14 to form disulfide-linked dimers (-S-S-) which enhance virion integrity and stability. A14 can undergo phosphorylation and glycosylation within the N83HS85 motif; however, neither modification is important in viral infection. In contrast, mutation of N7YF→AAA, S10→A, or P39TRTWK→AAAAAA or →PARTAA has been shown to ablate its ability to support viral infection. (B) BSC-40 cells were infected with vindA14 with (+) TET (lane 1) or without (−) TET (lanes 2 to 6) (MOI = 10) and transfected with plasmids encoding WT or mutant forms of A14. Cells were metabolically labeled with [35S]methionine, and lysates were subjected to immunoprecipitation with anti-A17 serum. A17 and any coprecipitating A14 are indicated by the diamond and arrow, respectively. (C and D) Cells were infected with vindA14-TET and transfected with empty vector (V) or with plasmids encoding WT or mutant forms of A14. At 18 hpi, samples were processed for electron microscopy. For each sample, 20 cells were chosen randomly and scored for their profile of morphogenesis (C). The most advanced and predominant stage of morphogenesis seen in each sample is shown graphically, using four categories: vesicles and virosomes, crescents and virosomes, IV and IVN, and MV. Representative images of the cells described are shown in panel D; scale bars represent 0.5 μm.
Affinity purification of V5-D13 and associated A17.
Dishes (60-mm diameter) of BSC-40 cells were infected with vTF7.3 (30) (MOI = 5) in the presence of 20 μM araC. At 1 hpi, the inoculum was removed and cells were transfected with 4 μg of the indicated pTM1 constructs using Lipofectamine LTX. At 24 hpi, cells were harvested; lysates were prepared in 50 mm Tris-HCl (pH 8)–150 mm NaCl–1% NP-40–0.1% SDS, clarified, and incubated with 40 μl of V5 affinity agarose beads (prepared according to the instructions of the manufacturer) overnight at 4°C with end-over-end mixing. The beads were washed 3 times with lysis buffer and once with 1× PBS; proteins were liberated from the beads with Laemmli buffer (50 mM Tris [pH 6.8], 1% SDS, 1% β-mercaptoethanol, 10% glycerol), resolved electrophoretically, and subjected to immunoblot analysis.
Kinase assay.
Purified 3XFLAG-F10 (10 to 20 ng) (7, 34) was added to a reaction mixture containing 50 mM Tris (pH 7.4), 10 mM MgCl2, 5 μM [γ-32P]ATP, 1 mM dithiothreitol, and 2.5 μg of purified maltose binding protein (MBP) or A17161–203-MBP. Reaction mixtures were incubated at room temperature for 30 min and stopped by the addition of Laemmli buffer, resolved by SDS-PAGE, and analyzed by autoradiography.
In vitro transcription-translation reactions: assessment of membrane association.
Coupled in vitro transcription and translation (IVTT) reactions programmed with pTM1-A17 or -A14 constructs were carried out using a TNT-T7-coupled reticulocyte lysate system from Promega (Madison, WI). Reactions using a 50-μl reaction mixture were performed in the presence of 20 μCi [35S]methionine, with or without canine pancreatic microsomal membranes (MM), according to the manufacturer's instructions. For those reactions in which the MM were added “posttranslationally,” cycloheximide was added at 200 μg/ml, after the IVTT was complete. Microsomal membranes were added and the reactions incubated for an additional 30 min. A 5-μl volume of each reaction mixture was set aside for analysis as the input. The remainder was subjected to centrifugation at 16,100 × g for 20 min at 4°C; the pellet was washed and resuspended in 1× PBS and layered on a 0.5 M sucrose cushion for centrifugation at 16,100 × g for 20 min at 4°C. The pellet obtained from the final sedimentation was retained as the membrane-associated fraction. The input and membrane-associated fractions were resolved by SDS-PAGE and visualized by autoradiography.
Immunoblot analysis.
Immunoprecipitates or lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Whatman) in 10 mM CAPS buffer {[3-(cyclohexylamino)-1-propane-sulfonic acid] [pH 11.3] with 10% methanol}. Membranes were incubated with polyclonal sera directed against A17, A17L-N, A14, or ptyr or with a monoclonal antibody specific for the V5 epitope tag. A14 and A17 migrate with apparent MWs of 16K and 21- to 25K, respectively. Secondary antibodies were horseradish peroxidase (HRP) conjugated; blots were developed with chemiluminescent reagent and visualized on Kodak MR film and quantitated using a FluorChem HD2 imager.
Electron microscopy.
Confluent 60-mm-diameter dishes of BSC-40 cells were infected with either vindA14-TET or vindA17-IPTG. At 1 hpi, the inoculum was removed and cells were fed with fresh medium and transfected with 8 μg of the indicated plasmid using Lipofectamine LTX. The medium was changed at 4 to 5 h posttransfection. At 18 hpi, cells were fixed in situ with 1% glutaraldehyde in 0.1 M Sorensen's phosphate buffer (pH 7.4) and processed for epoxy embedding and conventional transmission electron microscopy or washed with phosphate buffer and then fixed with 0.1% glutaraldehyde–4% paraformaldehyde and processed for Lowicryl K4M embedding (35) for immunoelectron microscopy. Sections were examined on either a Hitachi H-600 or a JEOL JEM 2100 electron microscope, and digital images were acquired. For each sample, 20 cells were scored for their morphogenesis phenotype, based on the most advanced assembly intermediates observed.
Preparation of digital figures.
Original data were scanned on an Epson Perfection scanner (Long Beach, CA) and were adjusted with Photoshop software (Adobe Systems Inc., San Jose, CA). Images from immunoblot analysis were acquired using the Kodak film or FluorChem HD2 documentation system (ProteinSimple, Santa Clara, CA). Final figures were assembled and labeled with Canvas software (Deneba Systems, Miami, FL). A solid vertical line (see Fig. 2B and 4B) represents the juxtaposition of samples that were analyzed on the same gel but were not originally adjacent to each other. Dashed horizontal lines demark where nitrocellulose membranes were sectioned for probing with various antibodies for immunoblot analysis (Fig. 1B). Dashed vertical lines separate lanes based on infections with various viruses (Fig. 2).
Fig 2.
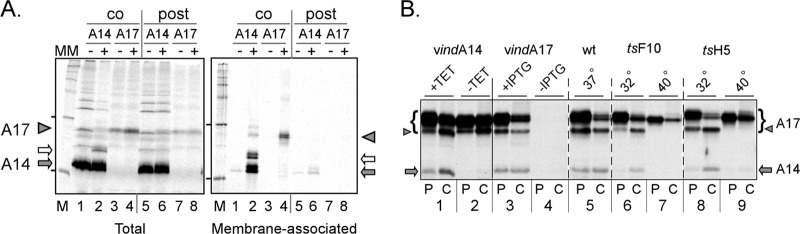
A14 and A17 associate with membranes cotranslationally in vitro and with each other in vivo. (A) A14 and A17 are cotranslationally inserted into membranes in vitro. IVTT reactions were programmed with plasmids expressing A14 (lanes 1, 2, 5, and 6) or A17 (lanes 3, 4, 7, and 8) and performed in the absence (lanes 1, 3, and 5 to 8) or presence (lanes 2 and 4; cotranslational [co] MM) of microsomal membranes (MM). MM were added posttranslationally (post) to samples 6 and 8. After removal of an aliquot of the reaction for subsequent analysis, membranes and associated proteins were purified by sedimentation through a sucrose cushion. The total and membrane-associated samples were resolved by SDS-PAGE (along with molecular weight standards; lanes M) and visualized by autoradiography. A17 is indicated by the filled triangles; the filled and open arrows indicate unmodified and glycosylated A14, respectively. The leftmost lane of each panel represents molecular weight standards, with the 30K and 14.3K proteins indicated by dashes. (B) A14 and A17 associate in vivo in a manner that is dependent upon virion morphogenesis. Cells were infected (MOI = 10) with vindA14 with or without TET (lanes 1 and 2), vindA17 with or without IPTG (lanes 3 and 4), WT virus (lanes 5), ts28 at 31.5 or 40°C (lanes 6 and 7), or tsH5 at 31.5 or 40°C (lanes 8 and 9). At 5.5 hpi, duplicate cultures were labeled with [35S]methionine for 1 h; samples were either harvested immediately (P [pulse]) or fed with methionine-containing complete medium and incubated for an additional 2.5 h before being harvested (C [chase]). Lysates were immunoprecipitated with antisera specific to A17, resolved by SDS-PAGE, and visualized by autoradiography. A17 is shown by a bracket, with the proteolytically processed form indicated by a triangle. The A14 protein that was coprecipitated in some samples is shown by an arrow.
Fig 4.
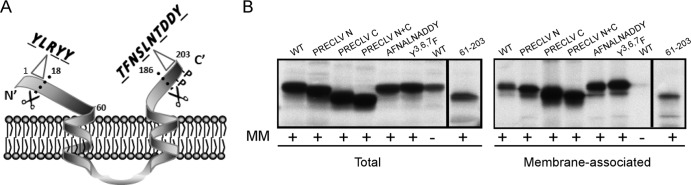
Structure function analysis of A17: key motifs and the association of A17 with membranes in vitro. (A) The presumed topology of A17 within the viral membrane is shown. A17 undergoes phosphorylation (P) and proteolytic processing (scissors). Subsequent figures focus on assessing the localization and importance of phosphorylation, the role of three conserved tyr residues in the N terminus (Y3LRYY), the role of ser, thr, and tyr residues in the C terminus (T194FNSLNTDDY), and the impact of precleaving or abrogating the cleavage of the N and C termini. (B) IVTT reactions were programmed with plasmids encoding WT A17 or the indicated A17 mutants in the presence (+) of microsomal membranes (MM); one reaction programmed to express WT A17 was performed in the absence (−) of membranes as a control. Total and membrane-associated fractions were prepared and analyzed as described for Fig. 2A. All of the A17 variants analyzed retained the ability to associate cotranslationally with membranes in vitro.
Fig 1.

Visualization and characterization of the vesicles that accumulate in the absence of either the A14 or A17 protein. (A) Cells infected with WT virus, vindA14-TET, or vindA17-IPTG were harvested at 18 hpi and processed for electron microscopy. The IV (immature virions; open triangles), IVN (immature virions with nucleoids; filled triangles) and MV (mature virions; arrows) that accumulate during WT infections are shown in the top panels. The middle panel shows the typical phenotype seen when A17 is not expressed: numerous vesicles (asterisks) form a corona (also see inset) around large, electron-dense virosomes (V). The bottom panel shows the typical phenotype seen when A14 is not expressed: numerous vesicles accumulate in distinct clusters (asterisk) (also see inset) at a distance from the periphery of large, electron-dense virosomes (V). (B) Postmitochondrial supernatants were prepared from cells infected with vindA17-IPTG (top and middle panels) or vindA14-TET (bottom panel) (MOI = 2; 17 hpi), and loaded onto 10% to 40% iodixanol gradients, which were sedimented at 100,000 × g for 18 h. Fractions were subjected to immunoblot analysis to monitor the cellular organelles and viral proteins indicated. The four fractions containing the peak of the A14-positive (A14+), A13+ vesicles that accumulate during A17-deficient infections are outlined with a dashed box, as are those that contain the peak of the A17+, A13+ vesicles that accumulate during A14-deficient infections. The unmodified and glycosylated forms of A14 are shown with filled and open arrows, respectively; the precursor and cleaved forms of A17 are shown with open and filled triangles, respectively. (C) Cells were infected with vindA14-TET or vindA17-IPTG for 12 h, and then the appropriate inducer was added; cells were processed for EM at 2.5 and 5 h thereafter. Representative images are shown; crescents are indicated by arrows. The white triangles (bottom right panel) indicate the tubular structures that appear to be capped with a vesicle. All scale bars in panels A and C represent 0.5 μm.
RESULTS
Repression of A14 or A17 leads to the accumulation of distinct populations of vesicles that contain other viral membrane proteins; morphogenesis recovers after addition of inducer.
At 17 hpi with WT virus (Fig. 1A), both immature virions (IV; triangles) and mature virions (arrows) are readily visualized by electron microscopy. However, as has been seen before, morphogenesis is arrested at an early stage when either A14 or A17 is absent (14–17). During vindA17-IPTG infections, large electron-dense virosomes (V) accumulate; these virosomes are surrounded by a corona of small vesicular membranes (asterisks). During vindA14-TET infections, the same large electron-dense virosomes (V) are seen; in some cases, aberrant crescents that seem detached from the virosomal contents are seen at their periphery (not shown). Large numbers of membrane vesicles accumulate in clusters at a distance from the virosomes (asterisk). These A14-deficient vesicles appear somewhat denser and smaller than those seen in the vindA17-deficient infections (compare insets).
Thus, although the phenotypes seen upon repression of A14 or A17 are distinct, the accumulation of large numbers of ∼25-nm-diameter vesicles is a hallmark in both cases. We do not yet know whether these vesicles are a normal, transient intermediate in morphogenesis that accumulate when A14 or A17 is repressed or whether they represent an aberrant intermediate seen only under these nonpermissive conditions. To better understand the nature of these vesicles, postmitochondrial supernatants (PMS) from vindA17-IPTG- or vindA14-TET-deficient infections were subjected to sedimentation on 10% to 40% iodixanol gradients (Fig. 1B). The sedimentation profiles of the ER, ER-Golgi intermediate compartment (ERGIC), and Golgi membranes were visualized using antisera to calnexin (apparent MW, 90K), ε-cop (apparent MW, 36K), and 14-3-3(3C8) (apparent MW, 30K), respectively; comparable profiles were seen for uninfected cells (not shown), cells infected with vindA17-IPTG (Fig. 1B, top panels), or vindA14-TET (not shown). When the PMS from A17-deficient infections was analyzed (middle panels), the A14 protein was seen in two distinct groups of fractions. The glycosylated form of A14 (gray arrow), which is excluded from nascent virions (13), cosedimented with the ER. The unglycosylated form of A14 (black arrow) sedimented at a denser region of the gradient; these fractions (gray dashed box) also contained the viral A13 protein, another essential component of the virion membrane (36). These fractions almost certainly represent the vesicles that are the hallmark of this nonpermissive infection. When the PMS from A14-deficient infections was analyzed, a distinct profile was observed. The fractions that cosedimented with the ER were enriched in the uncleaved form of A17 (white triangle). The cleaved form of A17 (black triangle) was predominant in a group of fractions (gray-dashed box) that also contained the A13 protein. These fractions almost certainly represent the vesicles that accumulate upon A14 repression. The different densities of the A17-deficient and A14-deficient vesicles were reproducible; when the two PMS samples were mixed and resolved on the same gradient, the distinct sedimentation profiles of the two pools of vesicles were confirmed (not shown). The different densities of the two vesicle populations may reflect the different sizes of the A14 and A17 proteins (90 and 203 aa, respectively) and/or the relative protein/lipid ratios of these vesicles.
To better understand the nature of the vesicles that accumulate during A14-deficient and A17-deficient infections, we infected cells with the inducible recombinants in the absence of inducer for 12 h and then added the appropriate inducer (TET or IPTG) and processed cells at 2.5 or 5 h postinduction for analysis by EM (Fig. 1C). This experimental design enabled us to visualize the synchronous recovery of morphogenesis and to assess whether the accumulated vesicles participated in crescent and IV formation. In the vindA14 infections, crescent formation was evident in approximately 50% of the cells at 2.5 h postinduction and in essentially all of the cells at 5 hpi (Fig. 1C). In some cases, clusters of vesicles remained at a distance from the virosomes. However, in many cases, vesicles were seen immediately adjacent to the edges of growing crescents at the periphery of the dense virosomes. These data suggest that the vesicles, which contain A17 (but not A14), may be able to “fuse” with growing crescents and become incorporated into nascent IV.
In the vindA17 infections, recovery was more rapid after the addition of inducer (Fig. 1C), with most cells showing robust crescent formation at 2.5 h postinduction and many cells showing fully formed IV by 5 h postinduction. The vesicles (which accumulate as a corona surrounding the virosomes) were frequently seen at the edges of forming crescents. As crescent formation became abundant and advanced around a given virosome, the corona was no longer seen. These data strongly suggest that the vesicles (which contain A14 but not A17), may be able to “fuse” with growing crescents and become incorporated into nascent IV. Finally, the “recovering” vindA17 infections showed a previously undescribed feature near virosomes in which crescent formation was actively ongoing. Numerous linear structures with an apparent lumen were seen and frequently appeared to be capped by a terminal vesicle (white triangles). The diameter of the structure was significantly smaller than that of the vesicles that accumulated around the virosome, and as yet we have no direct evidence that these elements are delimited by the membrane, although they do appear to have a “hollow” center.
A14 and A17 undergo cotranslational association with membranes in vitro; in vivo, the A14-A17 interaction requires the F10 kinase and/or ongoing morphogenesis.
Although A14 and A17 do not possess any recognizable signal sequences, they are thought to be synthesized in the ER (21, 22, 37, 38), after which they can be detected in crescents and in the membranes of immature and mature virions. As an additional means of addressing membrane association, we utilized in vitro transcription-translation reactions (IVTT) to test whether A14 and A17 could undergo co- and/or posttranslational insertion into microsomal membranes (MM). As shown in Fig. 2A, both proteins associated with membranes in a cotranslational fashion (lanes 2 and 4), but neither showed any significant association with membranes when they were added posttranslationally (lanes 6 and 8). As expected, little if any protein sedimented through the sucrose cushion (evidence of membrane association) in the absence of MM (lanes 1 and 3).
Attempts to coprecipitate A14 and A17 from microsomal membranes when both proteins were synthesized in the same reactions (or when reactions were mixed) were not successful (data not shown), suggesting that the previously observed interaction between these proteins might be indirect or might occur only within the context of infected cells. To address the latter issue, we assessed the ability of A14 to be coprecipitated with A17 from cells infected under various conditions (Fig. 2B). In each case, infected cells were pulse-labeled with [35S]met for 60 min at 5.5 hpi and either harvested immediately (P) or chased for 2.5 h in the presence of complete medium (C). All sample sets were subjected to immunoprecipitation with an α-A17 antiserum. As shown in samples 1 and 3, A14 (arrow) is coprecipitated with A17 (bracket) when both proteins are expressed. The specificity of the assay is validated by the fact that no A14 is retrieved when either A14 (sample 2) or A17 (sample 4) is not expressed. The interaction appears to be rapid, since retrieval is seen in both the P and C lanes. Analogously, coretrieval of A14 with A17 is seen during a WT infection (sample 5). The same experiment was performed using two temperature-sensitive (ts) viruses in which the biochemical stages of infection proceed normally but morphogenesis arrests prior to membrane formation, at the nonpermissive temperature. The tsF10 mutant (ts28) (7, 11, 12) encodes an impaired F10 protein kinase; the tsH5 mutant (tsH5-4) (31) encodes a dominant-negative form of the abundant H5 phosphoprotein. At the permissive temperature, A14 is coretrieved with A17 in both infections (samples 6 and 8), although the interaction appears to be slower at 32°C, since coretrieval is seen only in the C samples. However, at the nonpermissive temperature, no coretrieval of A14 is seen in either case (samples 7 and 9); the lack of A17 cleavage (arrowhead) under these conditions verifies the early block to morphogenesis. These results suggest either that A14 can interact only with the cleaved form of A17 or that A14-A17 interaction can occur only in the context of ongoing membrane formation.
Structure function analysis of the A14 protein: analysis of mutant proteins for their interaction with A17 and visualization of their ability to support morphogenesis.
We have previously published a structure function analysis of the A14 protein (13); some of the key motifs analyzed in that prior study and here are shown in Fig. 3A. We reported that A14 undergoes intermolecular disulfide bond formation via cys71; this dimerization reaction imparts detergent resistance to virions. We also demonstrated that A14 can also undergo glycosylation at an N83HS motif. However, glycosylation does not appear to be relevant to the production of MV, since the levels of glycosylated A14 increase when proper trafficking of A14 to nascent membranes is blocked, and glycosylated A14 is not seen in virions (13). Ser85 is also the site on which A14 undergoes phosphorylation, but this modification has no impact on A14's biological competency. Finally, we identified two motifs that were crucial for the biological competency of A14: N9YF, which lies within a short hydrophilic region at the N terminus of the protein, and P39TRTWK, which is within a hydrophilic loop of the protein presumed to be exposed on the external surface of the virion membrane. Here, we extended our earlier study by examining whether these regions were important for the interaction of A14 and A17 in vivo and also by assessing the specific morphogenetic defect(s) seen when these motifs were mutated. To address the former issue, cells were infected with vindA14 with or without TET in the presence of [35S]met; duplicate dishes infected in the absence of TET were also transfected with plasmids expressing WT A14 or mutant forms of A14 containing NYF→AAA, PTRTWK→AAAAAA, or PTRTWK→PARTAA substitutions. A17 was retrieved by immunoprecipitation, and the coprecipitation of A14 was assessed (Fig. 3B). As expected, A17 was retrieved in all samples (diamond); as seen in the first 2 lanes, A14 (arrow) was coretrieved when the infection was performed in the presence, but not the absence, of TET. Each of the A14 variants expressed from the transfected plasmids was coretrieved in the immunoprecipitation, indicating that neither the NYF nor PTRTWK motif plays a role in this interaction.
Cells infected with vindA14-TET and transfected with empty plasmid or plasmids encoding the WT or mutant forms of A14 were also examined by electron microscopy. In each case, 20 cells were scored for the morphogenesis phenotype(s) observed; a graphical representation of the scoring is shown in Fig. 3C. Representative images are shown in Fig. 3D. For the sample that received empty vector (V), 80% of the cells contained only large virosomes and clusters of numerous vesicles. For cells transfected with a plasmid encoding WT A14, 65% of the cells completed morphogenesis and contained numerous mature virions (MV), and 0% retained the virosome/vesicle phenotype. A total of 60% of the cells receiving the NYF→AAA mutant contained abundant IV and immature virions with nucleoids (IVN) but not MV. In contrast, only 10% of the cells receiving the PTRTWK→AAAAAA mutant contained either IV or MV, with 35% retaining the virosome/vesicle phenotype and 55% containing virosomes and crescents. A total of 75% of the cells expressing the PTRTWK→PARTAA mutant contained either IV or MV. Taken together, these data suggest that the PTRTWK motif is important both for crescent enlargement and for MV production, whereas the NYF motif is more important for the maturation of IV to MV.
Structure function analysis of A17: impact on association with membranes in vitro.
A17 is the other major membrane protein that is essential for membrane biogenesis. The current view of the structure of the 203-aa A17 protein is depicted in Fig. 4A. It is generally accepted that the N and C termini are exposed on the outer face of the virion membrane (3, 20). The N terminus undergoes cleavage at an AG↓X motif early during morphogenesis. The impact of expressing only an N-terminally “precleaved” (PRECLV) or “uncleavable” (UNCLV) version of A17 is assessed in the following figures. Three highly conserved tyr residues (Y3,6,7) are present within this N-terminal hydrophilic tail, and their importance (Y3,6,7 →F) is also discussed below. The central portion of A17 is highly hydrophobic, and there have been conflicting reports as to whether this region comprises two long or four standard transmembrane domains (37, 38). The two-membrane spanning model is shown here. The C terminus of the protein also undergoes morphogenesis-associated cleavage at an AG↓X motif. The impact of expressing only a C-terminally precleaved (PRECLV) or uncleavable (UNCLV) version of A17 is assessed in the following figures. Tyr203, the last amino acid in the protein, has been previously shown to undergo phosphorylation (24), and the impact of altering this residue (Y203→F, Y203→E) is tested in the following figures, as is the impact of altering ser and thr residues within the C-terminal tail (TFNSLNTDDY→AFNALNADDY, TFNSLNTDDY→EFNELNEDDY).
As mentioned above, the A17 protein undergoes cotranslational insertion into microsomal membranes in vitro (Fig. 2A) and is thought to be synthesized in the ER in vivo, although no conventional signal sequence is present. To test whether the N- or C-terminal hydrophilic tails (or conserved motifs within them) play a role in membrane association, we generated plasmids encoding variant alleles of A17 and tested them for cotranslational association with membranes in vitro (Fig. 4B). Variants of A17 with precleaved N and/or C termini, and even a variant in which the entire N-terminal hydrophilic extension was removed (leaving aa 61 to 203), retained the ability to associate with membranes. Similarly, mutating the Y3,6,7 or TFNSLNTDDY motif had no inhibitory effect on membrane association either.
A17 undergoes F10-mediated, biologically important phosphorylation on Ser, Thr, and Tyr residues within the C-terminal tail.
A17 has been shown to be phosphorylated on ser, thr, and tyr residues (23, 24), and this phosphorylation has been shown to be genetically dependent upon the F10 kinase. However, the detailed mapping of these sites, the biological importance of these modifications, and the determination of whether F10 is indeed the kinase involved have not been assessed. We therefore took advantage of an inducible recombinant in which A17 expression is IPTG dependent (18) (see Fig. 1A for EM). Cells were infected with this inducible recombinant (denoted vindA17 here) in the presence (as a control) or absence of IPTG and then transfected with empty vector (V) or with plasmids expressing WT or mutant forms of A17. At 24 hpi, cells were harvested, the viral yield was quantified by a plaque assay, and accumulation of A17 was monitored by immunoblot analysis. Parallel dishes were metabolically labeled with 32PPi, and the phosphorylation of the immunoprecipitated A17 protein was studied both by autoradiography and by immunoblot analysis with an anti-ptyr antibody. These results are shown in Fig. 5A and B. The 32P labeling and ptyr modification of the WT A17 protein were readily seen (Fig. 5B). Transient expression of WT A17 led to an ∼20-fold increase in viral yield relative to that obtained after transfection of empty vector. The Y3,6,7→F variant was diminished in biological activity, since the rescue of viral yield was only ∼3-fold, but neither the 32P labeling nor anti-ptyr reactivity was affected. In contrast, the Y203→F mutation, which would prevent phosphorylation of tyr203, had no effect on biological competency and no effect on bulk 32P labeling but reduced the anti-ptyr reactivity to near-background levels. The same was true of the phosphomimetic mutant Y203→E. A mutant in which Y3,6,7 and Y203 were simultaneously mutated retained the reduction in biological competency seen with Y3,6,7 and the loss of tyr phosphorylation seen with Y203. Finally, mutation of the ser and thr (but not the tyr) residues within the C-terminal tail (TFNSLNTDDY→AFNALNADDY) eliminated 32P labeling of the protein as well as eliminating the reactivity with the anti-ptyr antibody. Thus, all of the phosphorylation of A17 involves residues within this C-terminal tail, and it appears that modification of the ser and thr residues is a prerequisite for modification of the tyr residue. Moreover, mutation of the ser and thr residues causes a significant decrease in the biological competence of A17; the rescue of viral yield is only 3-fold, compared to 20-fold for the WT protein.
The fact that mutation of the ser and thr residues to ala within the C-terminal tail of A17 eliminated phosphorylation and reduced biological competency suggested that phosphorylation of A17 is vital for its function. To strengthen this conclusion, we generated another mutant allele in which the same residues were changed to glu, rather than ala (TFNSLNTDDY→EFNELNEDDY), to introduce negative charges that could serve as phosphomimetics. The WT, AFNALNADDY, and EFNELNEDDY alleles were compared in a transient-complementation assay (Fig. 5C); indeed, the EFNELNEDDY variant was as biologically competent as the WT A17. These data strongly suggest that the phosphorylation of the C-terminal tail of A17 plays a key role.
Having established that the C-terminal tail of A17 includes all of the sites on which the protein is phosphorylated in vivo and that this phosphorylation is biologically important, we wanted to move beyond the previous demonstration that phosphorylation is genetically dependent upon F10 (23, 24) to testing whether F10 can directly phosphorylate A17. We therefore generated a fusion protein in which residues 161 to 203 of A17 were fused to the maltose binding protein (MBP). MBP and MBP:A17(161–203) were expressed in E. coli, purified, and used as substrates in an in vitro kinase assay (Fig. 5D). The Coomassie-stained gel shown in the upper panel confirms the presence of equivalent levels of the two substrates in the kinase assay; the autoradiograph shown in the lower panel confirms that F10 undergoes autophosphorylation, as expected (7, 12, 34, 39). Moreover, the autoradiograph shows clearly that the MBP:A17(161–203) fusion protein, but not MBP alone, is a substrate for F10-mediated phosphorylation.
The dynamic cleavage of the N and C termini is important during infection; precleaved or uncleavable alleles show reduced biological competency.
As described above, A17 is known to undergo cleavage at both the N and C termini, leading to the removal of 16 and 18 aa, respectively. These cleavages occur early during morphogenesis, unlike the proteolytic processing of the major core proteins of the virion, which occurs only during the IV→MV transition. However, when this work was initiated, little was known about the roles played by the terminal regions of the protein and why, or if, removal of these termini is important for the progression of morphogenesis. We therefore generated precleaved alleles in which the N and/or C termini resembled those seen after the AG↓X processing events. Conversely, we also generated uncleavable alleles by mutating the AG↓X motifs to AA↓X (34, 40, 41), thereby preventing processing in vivo. Plasmids encoding these alleles were used in transient-complementation assays, and the 24-h viral yield, as well as accumulation and phosphorylation of A17, were assessed (Fig. 6). Whereas transient expression of WT A17 in the context of a vindA17-IPTG infection led to a 20-fold increase in virus production, the precleaved N protein was incapable of effecting any rescue. The precleaved C mutant was severely impaired; its expression increased viral yield only 3-fold. Once again, when the C terminus was prematurely cleaved, phosphorylation was interrupted; 32P labeling was absent, and no tyrosine phosphorylation is observed. Thus, the N and C termini play essential and important roles during infection, respectively. Moreover, these roles must be highly regulated, because preventing the cleavage of the termini also had an adverse effect. The uncleavable N, C, and N+C mutants were able to increase viral yield 4.5-, 6-, and 2-fold, respectively. These data suggest that the presence and subsequent removal of the exposed termini of the A17 protein may regulate temporal and/or spatial steps in virion assembly.
The N terminus of A17 regulates the association and subsequent disassociation of the D13 protein with the virion membrane as assembly progresses.
To better understand the role(s) played by the N terminus of the A17 protein, we turned to electron microscopy to visualize how the Y3,6,7→F substitutions, the absence of the terminal region, or the inability to remove this region by proteolytic processing affected virion assembly. Transient-complementation experiments were performed as described above (Fig. 5 and 6); one sample was analyzed by a plaque assay to confirm that the viral yield was consistent with prior experiments (data not shown), and the other was analyzed by conventional electron microscopy. For each EM sample, 20 cells were scored for the latest stage of assembly observed; representative images, and the scoring, are shown in Fig. 7A and B. In the absence of A17 expression, the defining phenotype of large virosomes surrounded by a corona of vesicles was seen in every cell. WT A17 restored the full morphogenetic program, with most cells progressing to the formation of numerous MV. In contrast, expression of either the Y3,6,7→F mutant or the precleaved N mutant led to a striking phenotype that phenocopies what is seen in the presence of the drug rifampin (RIF) (36, 42–46). This phenotype is characterized by the presence of small foci of viroplasm that are surrounded by sheets of flaccid membrane; these membranes are not supported by an external lattice of the D13 protein and hence do not have the rigid, spiked, and curved appearance of crescent membranes. These data were supported by immunoelectron microscopy (Fig. 7C). When only the precleaved N A17 was present, it localized to membranes surrounding viroplasmic foci, whereas the D13 protein was absent from these membranes and instead present in inclusion bodies (star) (9, 26). Again, this is the phenotype that mirrors what is seen in the presence of RIF.
Fig 7.

Structure function analysis of the role of the A17 N terminus in virion morphogenesis. (A and B) BSC-40 cells were infected with vindA17-IPTG and transfected with empty vector (V) or plasmids encoding WT or mutant alleles of A17. Cells were harvested at 18 to 24 hpi and processed for conventional electron microscopy. Representative images of the most advanced and most predominant phenotypes are shown in panel A. A total of 20 cells were examined and scored as described for Fig. 3C. The scoring categories were vesicles and virosomes, crescents and virosomes, RIF-like membranes, IV and IVN, MV, and aberrant MV; data representing averages of the results of two independent experiments are shown graphically in panel B. In panel A, the arrows indicate the RIF-like membranes (flaccid sheets of membranes surrounding virosomes) seen in the presence of the PRECLV N or Y3,6,7F mutants, and the triangles indicate aberrant MV (spherical MV with internal core) seen in the presence of the UNCLV N mutant. (C) To complement the conventional EM analysis, cells infected with vindA17-IPTG and transfected with plasmids encoding the PRECLV N or UNCLV N mutants were also examined by immunoelectron microscopy using antisera to the D13 or A17 proteins. The star marks D13-containing inclusion bodies. (D) Cells were infected in the presence of araC and transfected with plasmids encoding WT A17 or the PRECLV N or Y3,6,7F mutants, alone or with V5-D13; cells transfected with V5-D13 alone or with empty vector (V) were also included as controls. At 24 hpi, cell lysates were prepared. An aliquot was removed for analysis as the input, and the remainder of the D13 protein was purified on anti-V5 beads along with any interacting proteins. The V5-D13 (arrowhead) and A17 (triangle) in the input and pulldown samples were assessed by immunoblot analysis. (The IgG heavy chain [diamond] was also seen in the pulldown samples.) The dashed lines indicate where the immunoblots were cut prior to being probed with different antibodies; the numbers at the left of each panel indicate the MW (× 10−3) of the protein standards.
The phenotype seen upon expression of the A17 variant in which the N terminus was uncleavable (UNCLV N) was radically different from what had been observed for the Y3,6,7→F or precleaved N mutants (Fig. 7A and B). In this case, morphogenesis appeared to proceed normally, with the presence of numerous IVs. However, the maturation of these IVs yielded aberrant spherical virions in which a rigid, spherical membrane surrounded the core. This core appeared oblong, rather than being compressed into the dumbbell shape seen in WT virions; moreover, there was a considerable gap between the spherical membrane and the core. The rigid membrane appeared to retain its D13 coat, which is usually lost at the IV→MV transition; this conclusion was supported by immunoelectron microscopy (Fig. 7C). These data suggest that proteolytic removal of the N terminus of A17 is required for the removal of the D13 scaffold at the IV→MV transition but also suggest that the removal of this scaffold is not a prerequisite for the initiation of core maturation.
These data support the conclusion that the N-terminal 16 aa of A17, including tyr3,6,7, are needed for the interaction of A17 with D13. To address this hypothesis further, we infected cells in the presence of araC to prevent endogenous late gene expression and then transiently expressed WT A17 or the Y3,6,7→F or precleaved N mutants, either alone or along with V5-D13. V5-D13 was retrieved on anti-V5 beads, and the presence of A17 was assessed by immunoblot analysis (Fig. 7D). The specificity of the assay was confirmed by the absence of A17 in any of the pulldowns when V5-D13 was not coexpressed. The precleaved N-terminal mutant was retrieved at modestly decreased levels relative to WT A17 (75% ± 15%), but retrieval of the Y3,6,7→F protein was markedly less efficient (44% ± 2%). These data suggest that the N terminus contributes to, but is not necessary for, the binary A17-D13 interaction and that, when the N terminus is present, the mutation of tyr3,6,7 may cause a conformational change that destabilizes the interaction. As designed, this experiment assesses the D13:A17 interaction in the absence of membrane biogenesis. In vivo, the N-terminal tail may play a significantly greater role in mediating or stabilizing the interaction of D13 with A17 when A17 is embedded in the growing virion membrane.
The C terminus of A17 plays a role in the maturation of IV to MV and in virion infectivity.
The data shown in Fig. 5 indicated that mutation of the targets of phosphorylation within the C-terminal tail of A17, precleavage of A17, and prevention of cleavage of A17 all diminished the ability of A17 to support the production of infectious virus. To gain further insight into the role(s) played by this region of the protein, we again utilized EM to visualize the impact of mutating the C-terminal tail of A17 on morphogenesis (Fig. 8). As described for Fig. 7, transient complementation analyses were performed in duplicate, so that viral yield could be quantified in parallel with the EM analyses to confirm that the outcome of the experiment was consistent with previous studies (not shown). As described for Fig. 3 and 7, 20 cells were scored for the latest intermediate in morphogenesis that was seen; representative images are shown in Fig. 8A and the scoring in Fig. 8B. As seen before, expression of WT A17 enabled 85% of the cells to generate the full range of assembly intermediates and progress to the formation of numerous MV. In contrast, when either the TFNSLNTDDY→AFNALNADDY mutant or the precleaved C mutant was expressed, although the majority (50 to 60%) of cells formed normal IV, the maturation to MV occurred in only 25% of the cells. These data suggest that the C-terminal tail of A17, and its phosphorylation, is important in enabling the IV→MV transition. In contrast, when the C-terminal tail of A17 was rendered uncleavable, a WT phenotype was seen by EM: the full range of assembly intermediates was seen, and abundant MV were observed. Nevertheless, there was a (6-fold) reduction in infectious viral yield, implying that retention of the C-terminal tail causes a diminution either in the number of mature virions produced or in the specific infectivity of these virions.
Fig 8.

Structure function analysis of the role of the A17 C terminus in virion morphogenesis. (A and B) BSC-40 cells were infected with vindA17-IPTG and transfected with empty vector or plasmids encoding WT or mutant forms of A17. Cells were harvested at 18 to 24 hpi and processed for conventional electron microscopy. Representative images of the most advanced and most predominant phenotypes are shown in panel A; scale bars represent 0.5 μm. A total of 20 cells were examined and scored as described for Fig. 3C. The scoring categories were vesicles and virosomes, crescents and virosomes, IV and IVN, and MV; data representing averages of the results of two independent experiments (with standard errors) are shown graphically in panel B.
DISCUSSION
The biogenesis of the vaccinia virus membrane remains one of the most intriguing and complex aspects of the infectious cycle of this prototypic poxvirus. The A14 and A17 proteins, which are major components of this membrane and are essential for its formation (13–18), are thought to be synthesized in the ER despite the absence of signal sequences. They, along with components of the ER membrane, are diverted to sites of membrane biogenesis; indeed, it is the absence of specific trafficking signals that enables their inclusion in the virion membrane as opposed to their progression through the secretory pathway (21, 22). Membrane biogenesis is also dependent upon the viral F10 kinase (7, 10–12, 23), although how F10 mediates the early stages of assembly remains unknown. L2, A11, H7, and A6 are nonstructural viral proteins that also regulate membrane biogenesis (4–6, 8, 9, 47, 48). Again, their mechanism of action remains unknown.
A14 and A17 are small proteins of 90 and 203 aa, respectively. The work described here has added to our understanding of the structure and function of these proteins by combining genetic manipulation, transient complementation, and electron microscopy. Our data show that in vitro, A14 and A17 can associate with membranes co- but not posttranslationally. This finding has implications for how crescent membranes enlarge, since the lipids incorporated into growing crescents must contain preinserted A14 and A17 so as to maintain equivalent concentrations of these proteins. However, the understanding of how membrane insertion occurs remains elusive, since none of the mutants tested are impaired in their ability to associate with membranes.
For A14, our data also indicate that the vesicles that accumulate when A14 is absent (16, 17) contain the viral A13 and A17 proteins and can be purified on iodixanol gradients. When A14 is induced after 12 h of a nonpermissive infection (vindA14-TET), membrane assembly initiates at the periphery of the existing dense virosomes. The growing crescents appear to interact with, and incorporate, the existing vesicles. Two motifs were previously found to be essential for the biological competency of A14: the NYFS motif at the junction of the N-terminal tail (thought to be exposed on the inner surface of the virion membrane) and the first transmembrane domain, and the PTRTWK motif in the hydrophilic loop exposed on the outer surface of the virion (see schematic in Fig. 3A). Inactivating mutations in these domains, however, do not compromise the ability of A14 to interact with A17. How these two proteins interact, and indeed whether the interaction is direct, is not yet known. We have expressed each of the hydrophilic portions of the proteins in E. coli as GST (glutathione S-transferase), MBP (maltose binding protein), or CBP (chitin binding protein) fusion proteins [for A14, GST:A14(1–12), GST:A14(32–44), and GST:A14(65–90); for A17, CBP:A17(1–60), MBP:A17(77–140), and MBP:A17(161–203)] and used them in pairwise interaction studies. We have not been able to detect any interaction between any of the subregions of A14 and A17 (not shown). These data suggest that either A14 and A17 interact via their transmembrane domains or that the interaction observed in immunoprecipitation analyses (as described here and in references 23 and 27) is indirect. Either of these interpretations is consistent with the fact that the coimmunoprecipitation of A14 and A17 is prevented when early morphogenesis is prevented during nonpermissive infections with tsF10 or tsH5 (Fig. 2B), both of which arrest prior to the formation of any observable membrane biogenesis.
Throughout these studies, we performed electron microscopic analysis on cells infected under conditions in which A14 or A17 was repressed and WT or mutant proteins were transiently expressed from transfected plasmids. Categories of morphogenesis were established and cells were scored to obtain a quantitative assessment of the morphogenesis phenotype. A14 mutants containing altered NYF or PTRTWK motifs retained the ability to support crescent biogenesis in our transient-complementation assays. Mutation of the PTRTWK motif to AAAAAA appeared to impair the maturation of crescents to IV; it will be of interest to determine how this region of the protein participates in crescent growth or curvature. Mutation of the NYF motif, in contrast, appeared to significantly impair the subsequent IV→MV transition. Understanding the mechanism underlying this defect will also be of interest for future study.
One of the goals of our structure function analysis of A17 was to determine the site(s) and importance of phosphorylation. We had previously shown that the terminal amino acid of A17, tyr203, was modified in vivo, although this modification was rare compared to phosphorylation on ser and thr residues (24). Here we show that preventing this modification by introducing a Y203→F mutation had no impact on the biological competence of A17 (Fig. 5A). More importantly, we demonstrated that changing all of the ser and thr residues in the C-terminal tail of A17, which undergoes proteolytic cleavage during morphogenesis, eliminated all phosphorylation of A17 in vivo. Interestingly, eliminating the target ser and thr residues also eliminated tyr phosphorylation of A17. We also showed definitively that the C-terminal region of A17 is a direct substrate of the F10 kinase in vitro.
Eliminating the target ser and thr residues within A17 had a significant biological effect. The TFNSLNTDDY→AFNALNADDY allele increased viral yield only 3-fold over that seen with empty vector, compared to the 20-fold increase seen upon transient expression of WT A17. The fact that a phosphomimetic variant (TFNSLNTDDY→EFNELNEDDY) was equivalent to WT A17 in its biological activity suggests that the negative charge of this C-terminal tail plays a key role during infection (Fig. 5C). This role must be transient, however, since this region is removed proteolytically. Our EM analysis suggested that elimination of the target ser and thr residues, or elimination of the C-terminal 18 aa (PRECLV C), impaired the IV→MV transition. It will be of interest to determine how this charged tail facilitates virion maturation, which is accompanied by loss of the D13 coat, activation of core protein cleavage, appearance of a visible core, and a spherical→oblong shape transition. In contrast, preventing the cleavage of the C-terminal tail had no impact on the progression of morphogenesis but decreased the yield of infectious virus (Fig. 6 and 8). Although we have not yet determined whether there is a quantitative change in virion production, we think it likely that the retention of the C-terminal tail reduces virion infectivity. One reasonable hypothesis is that retention of the tail may interfere with virion binding or entry.
These observations regarding the phosphorylation of A17 dovetailed well with the other major goal of our A17 studies, which was to analyze the role of the proteolytic cleavages of the N and C termini. As for the study of the C-terminal tail described above, analysis of the N terminus was fruitful: precleavage eliminated the biological competency of A17 (Fig. 6 and7). Our studies confirmed and extended the work previously reported by the Moss group regarding the interaction of the N-terminal region of A17 with the D13 scaffold protein (26). Precleavage of the N terminus of A17 (with removal of 16 aa) gives a phenotype similar to that seen when infections are performed in the presence of RIF (rifampin), which blocks the A17-D13 interaction (Fig. 7D) (26, 36, 42, 45, 46). Membrane sheets accumulate around virosomes, and the D13 protein is found in inclusion bodies. These data imply that the N-terminal tail of A17 is important for the D13-A17 interaction, but, like Bisht et al. (26), we find no dramatic differences between the abilities of the WT and PRECLV N forms of A17 to interact with V5-D13 when transiently expressed in the absence of other late proteins or virion morphogenesis. We propose that the direct site of interaction lies elsewhere. In fact, Bisht et al. reported that removal of aa 16 to 38 from A17 does eliminate the interaction and that the sequence is likely to contain the D13 binding site (26). Interestingly, when we mutated 3 highly conserved tyr residues within the extreme N terminus (Y3,6,7→F), the rescue of viral yield was only 3-fold above that seen with vector alone and we observed the same RIF-like phenotype seen with precleavage of the 16-aa tail. Significantly, the Y3,6,7→F protein showed a significantly decreased ability to interact with V5-D13 when transiently expressed in vivo, suggesting that even these conserved amino acid changes had a dramatic impact on the structure of the N terminus in vivo.
Since the IV→MV transition is accompanied by the loss of the D13 scaffold, retention of the N-terminal tail might be expected to affect this transition. Mutation of the cleavage site to generate an uncleavable N terminus (UNCLV N) reduced the biological activity of the A17 protein; the rescue of viral yield above that seen with vector alone was only 5-fold. The EM images were striking, with the appearance of aberrant MV in which a normal-looking core was contained within a balloon-like circular membrane that retained its D13 coat (Fig. 7). These data again confirm and extend the work of Bisht et al. (26). Importantly, they also provide a new insight into the IV→MV transition: removal of D13 accompanies the transition but does not drive the transition. Even when virions retain an uncleaved N terminus of A17 that (presumably) prevents the release of D13, the dramatic reorganization of the interior of the virion occurs. The homogeneous appearance of IV is replaced by the appearance of an oblong core with a defined wall. Thus, there may be some other signal that senses that IV formation is complete and the genome has been encapsidated and then triggers both core formation and D13 release. Again, this is a topic for further study.
Examination of the phenotype(s) seen upon repression of A14 and A17, and then of the synchronous resumption of morphogenesis seen upon induction, has provided some new tools and insights. In the absence of either protein, dense virosomes (containing proteins destined for inclusion in the virion interior) accumulate, as do large numbers of membrane vesicles (Fig. 1) (14–17). Here, we report that these vesicles can be purified on iodixanol gradients and have distinct densities. Our side-by-side EM analyses also indicate that these vesicles exhibit several morphogenetic differences. The A14-deficient vesicles accumulate in isolated clusters and are very dense; it is difficult to discern an interior lumen. These vesicles often appear to be “lining up” as if to form a crescent; perhaps the A17 which is on the surface of these vesicles recruits a D13 scaffold that bridges the vesicles. The fact that the vesicles do not contact the virosomes implies that A14 may “bridge” crescents and virosomes by interacting with one or more virosomal proteins. When A14 is induced after 12 h of a nonpermissive infection, crescents develop at the periphery of the accumulated virosomes, and the vesicles often appear to be joining developing crescents.
The A17-deficient vesicles surround the virosomes, again suggesting that A14 may provide a bridge to the virosomal proteins. These vesicles are less dense than the A14-deficient vesicles and are larger in diameter and have a distinct lumen. Upon induction of A17 after 12 h of a nonpermissive infection, crescent formation is induced rapidly, and by 5 hpi IV are numerous and the corona of vesicles is no longer seen. Again, the vesicles appear to be positioned so as to join in during crescent biogenesis.
Many interesting questions remain about A14 and A17 and their role in membrane biogenesis. Understanding the real topology of these proteins within the membrane, and their ability to induce membrane curvature and/or fusion, is of great interest. Do the vesicles seen when one or the other protein is repressed reflect the stabilization of transient intermediates, which normally fuse rapidly when brought together? Or do these vesicles represent collapsed membranes that have unfavorable conformations or unstable termini in the absence of the two proteins? Do A14 and A17 interact directly, and if so, is it via their hydrophobic regions? There are published data regarding the fusogenic properties of A17 (49), and this possibility needs to be revisited in the context of membrane biogenesis. Our demonstration that these vesicles can be purified on iodixanol gradients (Fig. 1B) sets the stage for more in-depth study of these questions. The fact that the A14-deficient vesicles fail to contact virosomes (16, 17) and our observation that A14 mutants with substitutions in the NYF and PTRTWK motifs have defects post-crescent formation suggest that the A14 protein may play roles in morphogenesis beyond the initial stages of membrane biogenesis. In particular, it is important to determine whether A14 interacts with other membrane and/or core components. There is no doubt that the biogenesis of the poxvirus membrane remains an unsolved mystery of great cell biological interest and significant biomedical importance.
ACKNOWLEDGMENTS
We thank Clive Wells for expert assistance with electron microscopy and R. Topaz for help in preparing the anti-A17 antibody.
This work was supported, in part, by NIH grant AI 9R01 A1063620 awarded to P.T.
Footnotes
Published ahead of print 7 November 2012
REFERENCES
- 1. Damon I. 2007. Poxviruses, p 2947–2976 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2946 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 4. Maruri-Avidal L, Domi A, Weisberg AS, Moss B. 2011. Participation of vaccinia virus l2 protein in the formation of crescent membranes and immature virions. J. Virol. 85:2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maruri-Avidal L, Weisberg AS, Moss B. 2011. Vaccinia virus L2 protein associates with the endoplasmic reticulum near the growing edge of crescent precursors of immature virions and stabilizes a subset of viral membrane proteins. J. Virol. 85:12431–12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meng X, Embry A, Rose L, Yan B, Xu C, Xiang Y. 2012. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J. Virol. 86:5603–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Punjabi A, Traktman P. 2005. Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J. Virol. 79:2171–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Satheshkumar PS, Weisberg A, Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szajner P, Weisberg AS, Moss B. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Traktman P, Caligiuri A, Jesty SA, Liu K, Sankar U. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J. Virol. 69:6581–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S, Shuman S. 1995. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase 2. J. Virol. 69:6376–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mercer J, Traktman P. 2003. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J. Virol. 77:8857–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodríguez D, Esteban M, Rodriguez JR. 1995. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J. Virol. 69:4640–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodríguez D, Risco C, Rodriguez JR, Carrascosa JL, Esteban M. 1996. Inducible expression of the vaccinia virus A17L gene provides a synchronized system to monitor sorting of viral proteins during morphogenesis. J. Virol. 70:7641–7653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodríguez JR, Risco C, Carrascosa JL, Esteban M, Rodriguez D. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Traktman P, Liu K, DeMasi J, Rollins R, Jesty S, Unger B. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wolffe EJ, Moore DM, Peters PJ, Moss B. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Unger B, Nichols RJ, Stanitsa ES, Traktman P. 2008. Functional characterization of the vaccinia virus I5 protein. Virol. J. 5:148 doi:10.1186/1743-422X-5-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wallengren K, Risco C, Krijnse-Locker J, Esteban M, Rodriguez D. 2001. The A17L gene product of vaccinia virus is exposed on the surface of IMV. Virology 290:143–152 [DOI] [PubMed] [Google Scholar]
- 21. Husain M, Weisberg AS, Moss B. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. U. S. A. 103:19506–19511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Husain M, Weisberg AS, Moss B. 2007. Sequence-independent targeting of transmembrane proteins synthesized within vaccinia virus factories to nascent viral membranes. J. Virol. 81:2646–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Betakova T, Wolffe EJ, Moss B. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L kinase. J. Virol. 73:3534–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Derrien M, Punjabi A, Khanna M, Grubisha O, Traktman P. 1999. Tyrosine phosphorylation of A17 during vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J. Virol. 73:7287–7296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Betakova T, Moss B. 2000. Disulfide bonds and membrane topology of the vaccinia virus A17L envelope protein. J. Virol. 74:2438–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bisht H, Weisberg AS, Szajner P, Moss B. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodriguez D, Rodriguez JR, Esteban M. 1993. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J. Virol. 67:3435–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vázquez MI, Rivas G, Cregut D, Serrano L, Esteban M. 1998. The vaccinia virus 14-kilodalton (A27L) fusion protein forms a triple coiled-coil structure and interacts with the 21-kilodalton (A17L) virus membrane protein through a C-terminal alpha-helix. J. Virol. 72:10126–10137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez JF, Smith GL. 1990. IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucleic Acids Res. 18:5347–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DeMasi J, Traktman P. 2000. Clustered charge-to-alanine mutagenesis of the vaccinia virus H5 gene: isolation of a dominant, temperature-sensitive mutant with a profound defect in morphogenesis. J. Virol. 74:2393–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koerner TJ, Hill JE, Myers AM, Tzagoloff A. 1991. High-expression vectors with multiple cloning sites for construction of trpE fusion genes: pATH vectors. Methods Enzymol. 194:477–490 [DOI] [PubMed] [Google Scholar]
- 33. Elroy-Stein O, Fuerst TR, Moss B. 1989. Cap-independent translation of mRNA conferred by encephalomyocarditis virus 5′ sequence improves the performance of the vaccinia virus/bacteriophage T7 hybrid expression system. Proc. Natl. Acad. Sci. U. S. A. 86:6126–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mercer J, Traktman P. 2005. Genetic and cell biological characterization of the vaccinia virus A30 and G7 phosphoproteins. J. Virol. 79:7146–7161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burleigh BA, Wells CW, Clarke MW, Gardiner PR. 1993. An integral membrane glycoprotein associated with an endocytic compartment of Trypanosoma vivax: identification and partial characterization. J. Cell Biol. 120:339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Unger B, Traktman P. 2004. Vaccinia virus morphogenesis: A13 phosphoprotein is required for assembly of mature virions. J. Virol. 78:8885–8901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Betakova T, Wolffe EJ, Moss B. 1999. Membrane topology of the vaccinia virus A17L envelope protein. Virology 261:347–356 [DOI] [PubMed] [Google Scholar]
- 38. Salmons T, Kuhn A, Wylie F, Schleich S, Rodriguez JR, Rodriguez D, Esteban M, Griffiths G, Locker JK. 1997. Vaccinia virus membrane proteins p8 and p16 are cotranslationally inserted into the rough endoplasmic reticulum and retained in the intermediate compartment. J. Virol. 71:7404–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin S, Broyles SS. 1994. Vaccinia protein kinase 2: a second essential serine/threonine protein kinase encoded by vaccinia virus. Proc. Natl. Acad. Sci. U. S. A. 91:7653–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Byrd CM, Hruby DE. 2006. Vaccinia virus proteolysis—a review. Rev. Med. Virol. 16:187–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whitehead SS, Hruby DE. 1994. Differential utilization of a conserved motif for the proteolytic maturation of vaccinia virus proteins. Virology 200:154–161 [DOI] [PubMed] [Google Scholar]
- 42. Miner JN, Hruby DE. 1989. Rifampicin prevents virosome localization of L65, an essential vaccinia virus polypeptide. Virology 170:227–237 [DOI] [PubMed] [Google Scholar]
- 43. Mohandas AR, Dales S. 1995. Involvement of spicules in the formation of vaccinia virus envelopes elucidated by a conditional lethal mutant. Virology 214:494–502 [DOI] [PubMed] [Google Scholar]
- 44. Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y, Moss B. 1992. Immature viral envelope formation is interrupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology 187:643–653 [DOI] [PubMed] [Google Scholar]
- 47. Meng X, Embry A, Sochia D, Xiang Y. 2007. Vaccinia virus A6L encodes a virion core protein required for formation of mature virion. J. Virol. 81:1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu X, Meng X, Yan B, Rose L, Deng J, Xiang Y. 2012. Vaccinia virus virion membrane biogenesis protein A11 associates with viral membranes in a manner that requires the expression of another membrane biogenesis protein A6. J. Virol. 86:11276–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kochan G, Escors D, Gonzalez JM, Casasnovas JM, Esteban M. 2008. Membrane cell fusion activity of the vaccinia virus A17-A27 protein complex. Cell Microbiol. 10:149–164 [DOI] [PubMed] [Google Scholar]