

Abstract
Bovine herpesvirus 1 (BHV-1) infection induces clinical symptoms in the upper respiratory tract, inhibits immune responses, and can lead to life-threatening secondary bacterial infections. Following acute infection, BHV-1 establishes latency in sensory neurons within trigeminal ganglia, but stress can induce reactivation from latency. The latency-related (LR) RNA is the only viral transcript abundantly expressed in latently infected sensory neurons. An LR mutant virus with stop codons at the amino terminus of the first open reading frame (ORF) in the LR gene (ORF2) is not reactivated from latency, in part because it induces higher levels of apoptosis in infected neurons. ORF2 inhibits apoptosis in transiently transfected cells, suggesting that it plays a crucial role in the latency-reactivation cycle. ORF2 also interacts with Notch1 or Notch3 and inhibits its ability to trans activate certain viral promoters. Notch3 RNA and protein levels are increased during reactivation from latency, suggesting that Notch may promote reactivation. Activated Notch signaling interferes with neuronal differentiation, in part because neurite and axon generation is blocked. In this study, we demonstrated that ORF2 promotes neurite formation in mouse neuroblastoma cells overexpressing Notch1 or Notch3. ORF2 also interfered with Notch-mediated trans activation of the promoter that regulates the expression of Hairy Enhancer of Split 5, an inhibitor of neurite formation. Additional studies provided evidence that ORF2 promotes the degradation of Notch3, but not that of Notch1, in a proteasome-dependent manner. In summary, these studies suggest that ORF2 promotes a mature neuronal phenotype that enhances the survival of infected neurons and consequently increases the pool of latently infected neurons.
INTRODUCTION
Bovine herpesvirus 1 (BHV-1) is a significant viral pathogen of cattle (1) that suppresses immune responses. BHV-1, because of its immunosuppressive properties, can initiate a polymicrobial respiratory tract disease commonly referred to as bovine respiratory disease complex (reviewed in references 2–4). As for other Alphaherpesvirinae subfamily members, the primary site for BHV-1 latency is sensory neurons within trigeminal ganglia (TG). Lytic-cycle viral gene expression (5) and infectious virus (6) are detected in TG during acute infection, but latency is subsequently established. Increased corticosteroid levels, as a result of stress, can initiate BHV-1 reactivation from latency (3). Administration of the synthetic corticosteroid dexamethasone (DEX) to calves or rabbits latently infected with BHV-1 reproducibly leads to reactivation from latency and virus shedding (6–11). Induction of lytic-cycle viral gene expression is consistently detected in TG neurons of calves latently infected with BHV-1 following DEX treatment (11–13).
The BHV-1 latency-related (LR) RNA is the only abundant viral transcript expressed in latently infected sensory neurons (14, 15). The LR gene contains two major open reading frames (ORFs), ORF2 and ORF1, and two reading frames that lack an initiating methionine (RF-C and RF-B) (16). Two microRNAs encoded within the LR gene inhibit BHV-1 infected cell protein 0 (bICP0) expression (17) and promote cell survival (18). A mutant BHV-1 strain with three stop codons at the N terminus of ORF2 (LR mutant virus) does not express detectable levels of ORF2 or RF-C (19) but expresses reduced levels of ORF1 during the productive infection of cultured cells (20). The LR mutant virus grows less efficiently in the ocular cavity or TG but is not reactivated from latency following DEX treatment (6, 21), suggesting that expression of LR proteins is required for the latency-reactivation cycle in cattle. During the establishment of latency, the LR mutant virus induces higher levels of apoptosis in TG neurons of infected calves (22) and plasmids with the same stop codon mutations exhibit little or no antiapoptosis activity (23, 24).
ORF2 protein expression, in the absence of other viral genes, inhibits apoptosis in transiently transfected cells (25), suggesting that ORF2 is a dominant function encoded by the LR gene. ORF2 also interacts with the intracellular domain (ICD) of Notch1 and Notch3, components of the Notch signaling pathway (26). Notch1, but not Notch3, enhances productive BHV-1 infection plus trans activates the BHV-1 immediate-early transcription unit 1 (IEtu1) and bICP0 early promoters, whereas both Notch1 and Notch3 trans activate the late BHV-1 glycoprotein C (gC) promoter. ORF2 interferes with the ability of Notch1 to trans activate the bICP0 early promoter and Notch1- or Notch3-mediated activation of the gC promoter (26), suggesting that this function promotes the establishment and/or maintenance of latency.
Notch receptors (Notch1 to Notch4) are membrane-tethered transcription factors that regulate the differentiation and development of nearly all cell types (reviewed in references 27–30). When the Notch receptor is engaged by one of its five transmembrane ligands (Jagged1, Jagged2, Delta-like1, Delta-like3, and Delta-like4), the Notch ICD is cleaved by specific proteases and the Notch ICD translocates to the nucleus and is considered to be “active.” The Notch ICD interacts with members of the CSL family of transcriptional repressors, CBF1, Su(H), or Lag1 (also referred to as RBPjκ for mammalians), and mastermind (MAML). The Notch-CSL-MAML complex binds to specific DNA sequences, RNA polymerase II coactivators are recruited to the Notch-CSL-MAML complex, and consequently transcription occurs (27, 29). In the absence of an activated Notch family member, a CSL complex, which includes repressor proteins, inhibits transcription. Activation of Notch signaling in postmitotic neurons or neuroblastoma cells inhibits neurite sprouting (31–34) and axon repair (35), which can lead to neuronal degeneration and apoptosis (36–38). Conversely, neurite sprouting correlates with the regeneration of damaged axons and dendrites (35). During neural development, activation of Notch is crucial for the maintenance of neural progenitors and suppression of neuronal differentiation (30, 39). Consequently, disruption of Notch1 in mice leads to the depletion of neural progenitor cells (40, 41).
In this study, we found that Notch1 or Notch3 inhibits neurite formation in serum-arrested mouse neuroblastoma (Neuro-2A) cells. When ORF2 is cotransfected with Notch1 or Notch3, neurite formation is restored, suggesting that ORF2 impairs Notch-dependent signaling. ORF2 also interfered with Notch-mediated trans activation of the promoter that encodes Hairy Enhancer of Split 5 (HES5), which interferes with neurite formation when overexpressed. Certain transposon mutant constructs and the nuclear localization signal in ORF2 were important for inhibition of neurite formation in the presence of Notch. Mutation of five consensus protein kinase A and/or protein kinase C phosphorylation sites within ORF2 did not affect the ability of ORF2 to stimulate neurite formation. We suggest that the ability of ORF2 to promote neurite formation in the presence of activated Notch promotes normal neural functions following infection.
MATERIALS AND METHODS
Cells.
Murine neuroblastoma (Neuro-2A) cells were grown in Earle's modified Eagle's medium (EMEM) supplemented with 5% fetal calf serum (FCS), penicillin (10 U/ml), and streptomycin (100 μg/ml).
Plasmids and transient transfections.
The ORF2 wild-type (WT) and mutant constructs were cloned into the pCMV-Tag-2A or pCMV-Tag-2B vector (Stratagene, La Jolla, CA) downstream of a Flag epitope by using BamHI-HindIII restriction enzymes as previously described (25). In pCMV-TAG-2A, ORF2 is cloned as a single nucleotide frameshift, which expresses the mRNA but not the protein (25). Notch1 and Notch3 ICD constructs were kindly provided by U. Lendahl, Karolinska Institute, Stockholm, Sweden (42). A plasmid containing the firefly luciferase gene downstream of the HES5 promoter was a kind gift from (R. Kopan, Washington University, St. Louis, MO). All plasmids were transfected into Neuro-2A cells in 60-mm dishes as indicated, by using TransIT Neural (MIR2145; Mirus) according to the manufacturer's instructions.
Immunohistochemistry analysis.
Immunohistochemistry analysis was performed by using an ABC kit (Vector Laboratories) according to the manufacturer's specifications. TG were collected from calves that were mock infected, latently infected (at least 60 days after initial infection), or latently infected and treated with DEX to induce reactivation from latency as previously described (43). TG were fixed with formalin and then embedded in paraffin. Thin sections (4 to 5 μm) of TG were cut and mounted on glass slides. Slides were first incubated at 65°C for 20 min and then washed twice in xylene for 10 min each time, twice in 100% ethanol (EtOH) for 5 min, once in 90% EtOH for 5 min, once in 70% EtOH for 5 min, twice in distilled H2O for 5 min, and three times in 1× Tris-buffered saline (TBS) for 5 min each time. To block any endogenous peroxidase activity, sections were incubated in hydrogen peroxide (0.03% in phosphate-buffered saline [PBS], pH 7.4) for 20 min at room temperature. Tissue sections were then washed three times in 1× TBS for 5 min at room temperature, followed by digestion with proteinase K (S3020; Dako) for 20 min at 37°C. Tissue sections were then blocked with 5% normal serum diluted in 1× TBS containing 0.25% bovine serum albumin for 45 min at room temperature in a humidified chamber. Slides were incubated with Notch1 (3268S; Cell Signaling), Notch3 (SC-5593; Santa Cruz Biotechnology), or ORF2 rabbit polyclonal antibody at a 1:500 dilution overnight in a humidified chamber at 4°C. The next day, slides were washed in 1× TBS and incubated in biotinylated goat anti-rabbit IgG (PK-6101; Vector Laboratories) for 30 min at room temperature in a humidified chamber. Avidin-biotinylated enzyme complex was added to the slides for 30 min of incubation at room temperature. After three washes in 1× TBS, slides were incubated with freshly prepared substrate (SK-4800; Vector Laboratories), rinsed with distilled water, and counterstained with hematoxylin for 1 min. Thin sections from mock-infected calves were used as a negative control.
Neurite formation assay.
Neuro-2A cells grown in 60-mm plates were cotransfected with a human cytomegalovirus promoter plasmid expressing the Notch1 or Notch3 ICD (1 μg DNA), a pCMV-Tag plasmid expressing Flag-tagged ORF2 or mutant ORF2 constructs (1 μg DNA), and a pCMV-β-Gal plasmid (1 μg DNA). To induce neurite sprouting, 24 h after transfection, cells were seeded into new plates at a low density of 2,000/cm2 and starved in medium with 0.5% serum for 3 days. Cells were then fixed and stained, and a β-galactosidase (β-Gal) assay was performed as previously described (44). The percentage of cells with β-Gal+ neurites was calculated by dividing the number of β-Gal+ cells with a neurite length at least twice the diameter of the cell by the total number of β-Gal+ cells. The results are an average of three independent experiments.
Dual-luciferase reporter assay.
Neuro-2A cells (8 × 105) were seeded into 60-mm dishes containing EMEM with 10% FCS at 24 h prior to transfection. Two hours before transfection, the medium was replaced with fresh EMEM containing 0.5% FCS to lower the basal levels of HES5 promoter activity. Cells were cotransfected with a plasmid containing the firefly luciferase gene downstream of the HES5 promoter (1 μg DNA), a plasmid encoding Renilla luciferase under the control of the herpesvirus thymidine kinase (TK) promoter (50 ng), the indicated Notch1 or Notch3 plasmid (1 μg DNA), and a WT or mutant ORF2 plasmid (1 μg DNA). To maintain equal plasmid amounts in the transfection mixtures, the empty expression vector was added as needed. Forty hours after transfection, cells were harvested and protein extracts were subjected to a dual-luciferase assay by using a commercially available kit (E1910; Promega) according to the manufacturer's instructions. Luminescence was measured by using a GloMax 20/20 luminometer (E5331; Promega).
Western blot assay.
Neuro-2A cells in 60-mm plates were cotransfected with a plasmid expressing the Notch3 ICD (2 μg DNA) and the designated ORF2 plasmids (2 μg DNA), and cultures were washed once with 1× PBS and then suspended in radioimmunoprecipitation assay buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1% Triton X-100, protease inhibitor) and incubated at 4°C for 30 min with rotation. Lysate was cleared by centrifugation at 14,000 rpm for 10 min. Protein concentration was measured by using the standard Bradford assay. Equal amounts of protein were boiled in Laemmli sample buffer for 5 min and separated on a 10% SDS-polyacrylamide gel. A mouse anti-Flag antibody (F1804; Sigma) at 1:500 was used to detect WT and mutant ORF2, while Notch3 was detected by using a rabbit anti-Notch3 antibody (SC-5593; Santa Cruz Biotechnology). A goat anti-β-actin antibody (SC-1616; Santa Cruz Biotechnology) was used as a loading control. To block protein synthesis, 40 h after transfection, cells were treated with cycloheximide (CHX; 100 μM) at the indicated time points. To block proteasome activity, cells were treated with 15 μM lactacystin (70980; Cayman Chemical) 10 h prior to CHX treatment. Cells were subsequently processed for Western blot analysis.
RESULTS
Expression of Notch and ORF2 in TG during the latency-reactivation transition.
A previous study demonstrated that Notch3 RNA levels were increased in TG during DEX-induced reactivation from latency (26). Immunohistochemistry analysis was used to determine if Notch protein expression occurs in sensory neurons within TG during DEX-induced reactivation from latency. Notch3-positive neurons were detected in a subset of TG neurons at 3 or 6 h after DEX treatment (Fig. 1A). Even at 90 min after DEX treatment, Notch3-positive neurons could be detected (data not shown). Conversely, Notch3-positive neurons were not readily detected in mock-infected TG sections, in latently infected TG, or at 24 h after DEX treatment (data not shown). Although Notch1 was detected in TG after DEX treatment (Fig. 1B), in general, fewer neurons were positive and the signal was weak.
Fig 1.

Expression of Notch and ORF2 in TG. TG were collected from calves that were mock infected, latently infected (at least 60 days postinfection), or latently infected and treated with DEX at the indicated times (hours [H]) after DEX treatment to induce reactivation from latency. Thin sections were cut and subjected to immunohistochemistry analysis with antibodies that specifically recognize Notch3 (A), Notch1 (B), or ORF2 (C) as described in Materials and Methods. Biotinylated goat anti-rabbit IgG was used as a secondary antibody. Thin sections from mock-infected calves were used as a negative control. Arrows indicate positive neurons.
As a comparison, we examined ORF2 protein expression in TG samples prior to and during the early stages of DEX-induced reactivation from latency. In contrast to TG from mock-infected calves, ORF2 was readily detected in TG sections of latently infected calves (Fig. 1C). Since LR RNA expression is reduced during DEX-induced reactivation from latency (11), it was not surprising to find that the number of ORF2-positive neurons appeared to go down at 6 and 24 h after DEX treatment (Fig. 1C). In summary, these results provide evidence that Notch3, but not ORF2, protein expression was induced by DEX in TG neurons.
ORF2 antagonizes Notch-mediated inhibition of neurite formation.
ORF2 stably interacts with Notch3 or Notch1, and these interactions impair the ability of Notch to stimulate viral gene expression and productive infection (26, 44). Although these studies indicated that ORF2 interferes with the ability of Notch family members to stimulate viral promoters and productive infection, they did not address whether ORF2 influences activated Notch in the context of known cellular functions. Activated Notch signaling in neurons inhibits neurite sprouting (31–34) and axon repair (35), which can result in neuronal cell death (36, 37). Conversely, neurite sprouting is synonymous with the regeneration of damaged axons and dendrites (35). Although many of these studies were performed with neurons derived from the central nervous system (CNS) or in the CNS itself, activated Notch also interferes with neuronal differentiation in the ophthalmic branch of TG (45).
To test whether ORF2 affects Notch functions in mammalian cells, mouse neuroblastoma cells (Neuro-2A) were transfected with a plasmid that expresses Notch1 or Notch3 and the effect ORF2 has on neurite formation was examined. For all of these studies, the plasmids used to express Notch1 or Notch3 express only the Notch ICD, these proteins are constitutively active, and they function regardless of the Notch ligand or receptor status of surrounding cells. When Neuro-2A cells were transfected with a LacZ expression vector and growth factors were removed, β-Gal+ neurites were readily detected when cells were seeded at low density and then serum starved for 3 days (Fig. 2A, control panel). As expected (32, 33), Notch1 or Notch3 overexpression inhibited neurite formation after growth factor withdrawal (Notch1 or Notch3 panels). When ORF2 was cotransfected with the Notch1 or Notch3 expression plasmid, β-Gal+ Neuro-2A cells with extended neurites were frequently detected. We detected β-Gal+ Neuro-2A cells with extended neurites that were touching each other after cotransfection with Notch1 or Notch3 and ORF2, as well as single β-Gal+ Neuro-2A cells. Approximately half of the β-Gal+ cells consistently sprouted neurites when Neuro-2A cells were cotransfected with Notch1 or Notch3 and ORF2, which was a significantly larger proportion than that of Neuro-2A cells transfected with Notch1 or Notch3 and the empty expression vector (Fig. 2B).
Fig 2.

ORF2 antagonizes Notch inhibition of neurite formation. (A) Neuro-2A cells were cotransfected with a plasmid expressing the Notch1 or Notch3 ICD, a plasmid expressing ORF2, and a plasmid expressing the LacZ gene (transfection control). To induce neurite sprouting, 24 h after transfection, cells were seeded into new plates at a low density (2,000/cm2) and then starved in medium with 0.5% serum for 3 days. Cells were then fixed, and β-Gal+ cells were identified as described in Materials and Methods. (B) The percentage of β-Gal+ cells containing neurites was calculated by dividing the number of β-Gal+ cells with a neurite length at least twice the diameter of the cell by the total number of β-Gal+ cells. The average of at least three independent experiments is shown with the respective standard deviation. An asterisk denotes significant differences (P < 0.05) in β-Gal+ Neuro-2A cells containing neurites following cotransfection with the ORF2 reporter and a Notch1 or Notch3 expression plasmid relative to β-Gal+ Neuro-2A cells with neurites following transfection with a plasmid expressing Notch1 or Notch3 plus the empty vector, as determined by the Student t test.
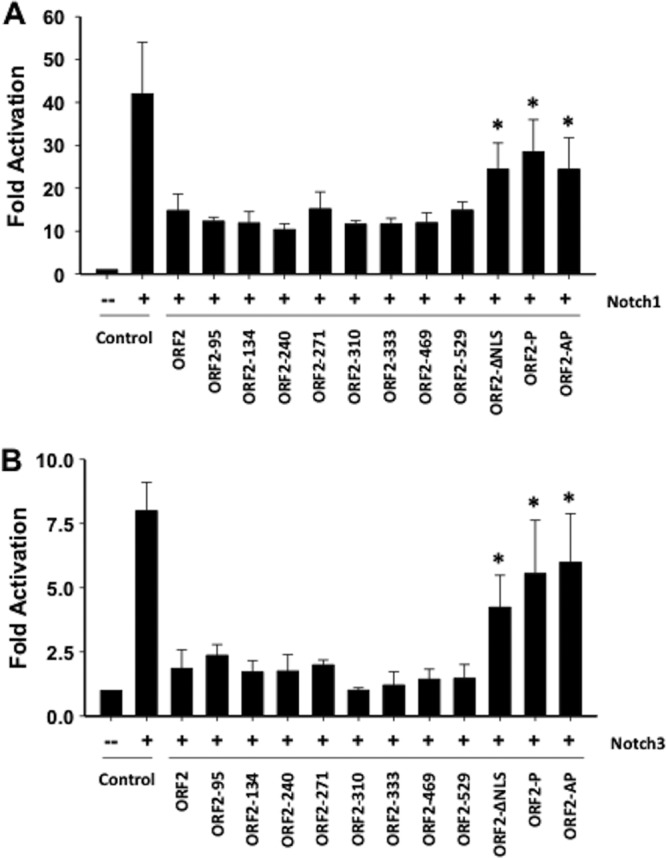
To localize domains in ORF2 that interfere with Notch inhibition of neurite formation, the neurite formation assay was performed with Neuro-2A cells and a panel of mutant constructs previously described (44) (schematics of the respective mutant ORF2 constructs are shown in Fig. 3A and B). Compared to WT ORF2, four transposon mutant constructs, ORF2-95, ORF2-134, ORF2-240, and ORF2-271, did not restore neurite formation as efficiently (Fig. 4A and B). A significant difference between the four transposon mutant constructs and WT ORF2 was observed (P < 0.05). Since ORF2-95, unlike the other transposon mutant constructs, expresses lower protein levels in Neuro-2A cells than WT ORF2 does (44), it is difficult to distinguish whether its impaired function was due to protein expression levels or if an essential inhibitory domain was disrupted. The remainder of the transposon mutant constructs restored neurite formation similar to that seen with WT ORF2, regardless of whether they were cotransfected with Notch1 or Notch3 (Fig. 4A and B). The phosphorylation mutant constructs ORF2-P and ORF2-AP, but not ORF2-ΔNLS, promoted neurite formation in the presence of Notch1 or Notch3 with an efficiency similar to that of WT ORF2, suggesting that nuclear localization, but not the phosphorylation status of ORF2, was important. In conclusion, these studies provided evidence that ORF2 interfered with activated Notch1 or Notch3 signaling and promoted neurite formation in Neuro-2A cells. Certain transposon mutant constructs and nuclear localization signals within ORF2 were important for this function.
Fig 3.
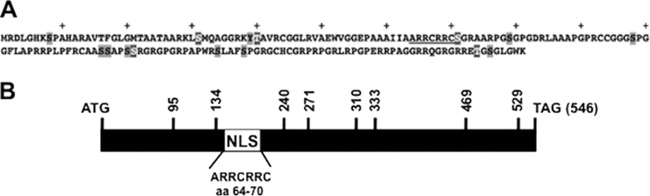
Schematics of the ORF2 and mutant ORF2 constructs used for this study. (A) Amino acid sequence of ORF2. The nuclear localization signal (underlined), 15 putative phosphorylation sites (gray-shaded amino acids) and five consensus protein kinase A and/or protein kinase C phosphorylation sites (gray-shaded amino acids with white lettering) are shown. The plus signs denote every 10th amino acid in ORF2. (B) ORF2 coding sequences (BamHI-SalI) were cloned into the pUC57 vector, and transposon linker insertion reactions were performed as previously described (44). Vertical lines with the respective numbers indicate the nucleotide position of the respective transposon insertions. The relative position of the consensus nuclear localization signal (NLS) is denoted by the white rectangle.
Fig 4.

Localization of ORF2 sequences necessary to inhibit neurite formation in the presence of Notch1 or Notch3. Neuro-2A cells were cotransfected with a plasmid expressing the Notch1 (A), or Notch3 (B) ICD, a plasmid expressing WT ORF2 or the designated mutant ORF2 construct, and a plasmid expressing LacZ. Neurite formation was measured as described in the legend to Fig. 2 after serum starvation and the identification of β-Gal+ Neuro-2A cells. The results represent the average of at least three independent experiments with the respective standard deviation. An asterisk denotes significant differences (P < 0.05) in β-Gal+ Neuro-2A cells containing neurites that were cotransfected with the ORF2 reporter and a Notch1 or Notch3 expression plasmid relative to β-Gal+ Neuro-2A cells containing neurites after transfection with a plasmid expressing Notch1 or Notch3 plus the empty vector, as determined by the Student t test.
ORF2 inhibits activation of the HES5 promoter, a downstream target of Notch.
To further examine the ability of ORF2 to interfere with Notch signaling, we tested whether ORF2 influences the Notch-mediated trans activation of the HES5 promoter. The HES family of transcription factors contains basic helix-loop-helix domains that generally repress transcription, and HES promoters are trans activated by Notch (reviewed in references 27–29 and 46). Since Hes1 and Hes5 are transcription factors that inhibit neuronal differentiation (47), examination of the effect of ORF2 on HES5 promoter activity is relevant to the finding that ORF2 promoted neurite formation in the presence of Notch1 or Notch3. A dual-luciferase assay was performed with a reporter plasmid that contains the HES5 promoter upstream of the firefly luciferase gene. Increasing amounts of Notch1 (Fig. 5A) and Notch3 (Fig. 5B) increased HES5 promoter activity in a dose-dependent fashion. In general, Notch1 is a more efficient trans activator than Notch3 (42), which was consistent with the results obtained with Neuro-2A cells.
Fig 5.

ORF2 interferes with Notch-mediated trans activation of the HES5 promoter. Neuro-2A cells were cotransfected with a plasmid containing the firefly luciferase gene downstream of the HES5 promoter, a plasmid expressing the Notch1 (A, C) or Notch3 (B, D) ICD, and increasing amounts of a plasmid expressing ORF2 (C, D). Promoter activity was measured by using a dual-luciferase assay. A plasmid expressing Renilla luciferase under the control of a minimal herpesvirus TK promoter was used as an internal control. The results are the average of four independent experiments, and the error bars denote the standard deviation. An asterisk denotes significant differences (P < 0.05) in HES5 promoter activity in Neuro-2A cells following cotransfection with the ORF2 reporter and a Notch1 or Notch3 expression plasmid relative to HES5 promoter activity following transfection with a plasmid expressing Notch1 or Notch3 plus the empty vector, as determined by the Student t test.
Neuro-2A cells were cotransfected with plasmids expressing ORF2, Notch1, or Notch3; the HES5 promoter and luciferase activities were measured to test whether ORF2 influenced trans activation of the HES5 promoter. ORF2 consistently inhibited Notch1-mediated trans activation of the HES5 promoter by 4-fold, which was significantly different from the empty-vector control (Fig. 5C). ORF2 also significantly reduced Notch3-mediated trans activation of HES5 promoter activity in a dose-dependent fashion (Fig. 5D).
Additional studies were performed to identify mutant ORF2 constructs that do not interfere with Notch-mediated trans activation of the HES5 promoter. All of the transposon mutant constructs inhibited Notch1 (Fig. 6A)- or Notch3 (Fig. 6B)-mediated trans activation of the HES5 promoter similarly to WT ORF2. In contrast, ORF2-ΔNLS, ORF2-P, and ORF2-AP were unable to significantly interfere with Notch1- or Notch3-mediated trans activation of the HES5 promoter compared to the empty vector. In summary, specific ORF2 sequences were necessary for interference with Notch1- or Notch3-mediated trans activation of the HES5 promoter.
Fig 6.
Localization of ORF2 sequences important for Notch-mediated trans activation of the HES5 promoter. Neuro-2A cells were cotransfected with a plasmid expressing the Notch1 or Notch3 ICD, a plasmid expressing WT or mutant ORF2, and a plasmid containing the firefly luciferase gene downstream of the HES5 promoter. Luciferase assay was performed as described in the legend to Fig. 5. The results are the average of three independent experiments, and the error bar denotes the standard deviation. Asterisks denote significant differences (P < 0.05) in HES5 promoter activity in Neuro-2A cells following cotransfection with the ORF2 reporter and a Notch1 or Notch3 expression plasmid relative to HES5 promoter activity following transfection with a plasmid expressing Notch1 or Notch3 plus the empty vector, as determined by the Student t test.
ORF2 promotes Notch3 degradation.
Studies were subsequently performed to test whether ORF2 influences the steady-state levels of Notch. The rationale for this study was that the assembly of a Notch-CSL complex at a promoter leads to recruitment of the cyclin-dependent kinase 8 (CDK8)-cyclin C complex (48). CDK8-cyclin C phosphorylates Notch, and phosphorylated Notch is a substrate for the nuclear E3 ubiquitin ligase Sel10. Sel10 interacts with the C-terminal PEST region of Notch, and this is required for proteasome-dependent degradation. When Notch is degraded, the coactivator complex dissociates from CSL, leading to the recruitment of a corepressor complex and repression of gene expression.
To examine whether ORF2 has an effect on steady-state Notch3 levels, cotransfection studies were performed and Notch3 protein levels were examined by Western blot analysis. Cells were also treated with CHX to block de novo protein synthesis to examine the turnover of steady-state Notch3 protein levels. In the absence of ORF2, Notch3 levels were reduced within 2 h after CHX treatment unless the proteasome inhibitor lactacystin was added to cultures (Fig. 7A). In the presence of ORF2, Notch3 protein levels were consistently lower than those of the empty-vector control, even before CHX treatment (Fig. 7A). One hour after CHX treatment, the levels of Notch3 were also lower in cells cotransfected with ORF2 than in the empty-vector control. An ORF2 frameshift mutant construct (2A/ORF2) that expresses mRNA (25) but does not express a protein recognized by the Flag monoclonal antibody or the ORF2-specific antibody had no effect on Notch3 stability (Fig. 7B). Even prolonged exposure of the Western blot that was probed with the ORF2-specific antibody did not reveal a specific band in cells transfected with the 2A/ORF2 construct. In contrast to Notch3, ORF2 had no obvious effect on steady-state Notch1 protein levels (data not shown).
Fig 7.

ORF2 reduces the steady-state levels of Notch3. (A) Neuro-2A cells were transfected with the designated plasmids and then collected and processed for Western blot analysis. Cultures were treated with 100 μM CHX for 1 or 2 h at 40 h after transfection. To inhibit proteasome activity, cells were treated with lactacystin (15 μM) 10 h prior to CHX treatment. (B) The 2A/ORF2 plasmid contains ORF2 cloned such that there is a one-nucleotide frameshift downstream of the N-terminal Flag epitope. The 2B/ORF2 plasmid contains ORF2 in frame with the N-terminal Flag epitope. The Western blots were probed with a Flag-specific monoclonal antibody or an ORF2-specific peptide antibody. (C, D) The effects of the respective mutant ORF2 constructs on steady-state Notch3 protein levels were examined. Neuro-2A cells were cotransfected with ORF2 or the designated mutant ORF2 constructs and a plasmid expressing Notch3. Cultures were treated with CHX as described above. A mouse anti-Flag antibody was used to detect WT and mutant ORF2, while Notch3 was detected by using a rabbit anti-Notch3 antibody. β-Actin protein levels were determined to confirm that equal amounts of protein were loaded in each lane. The molecular weights (103) of the respective bands are shown to the left of each Western blot.
To identify ORF2 sequences that influence Notch3 stability, Neuro-2A cells were cotransfected with the Notch3 expression plasmid plus the designated mutant ORF2 constructs and Western blot assays were performed to measure Notch protein levels. Three transposon mutant constructs that did not stimulate neurite formation (ORF2-134, ORF2-240, and ORF2-271) and one that behaved like the WT (ORF2-529) were used for these studies. ORF2-240, but not the other transposon mutant constructs, had slightly higher levels of Notch3 than WT ORF2 (Fig. 7C). The ORF2-NLS mutant construct, but not the two phosphorylation mutant constructs, had little or no effect on Notch3 protein stability (Fig. 7D). These studies suggested that ORF2 reduced the half-life of Notch3 and that nuclear localization of ORF2 was necessary for this function.
DISCUSSION
In this study, we demonstrated that the expression of ORF2 in Neuro-2A cells allowed neurite formation to occur in the presence of activated Notch1 or Notch3. As expected, Notch1 or Notch3 interferes with neurite formation in serum-arrested Neuro-2A cells (31–34). Although previous studies demonstrated that ORF2 interfered with the ability of Notch1 and Notch3 to activate productive infection and trans activate certain viral promoters (26), they did not address whether ORF2 influences Notch-mediated signaling in the context of known cellular functions. It now seems clear that, in general, ORF2 interferes with Notch-mediated signaling. Activated Notch is required for the growth of certain human tumors (49–51), suggesting that ORF2 may have therapeutic value with respect to inhibition of the growth of tumors that are dependent on Notch for proliferation.
Many other genes, in addition to Notch, negatively regulate neurite sprouting. These include A1 adenosine receptor, merlin, adenomatous polyposis coli protein, β-catenin, Cdc42-interacting protein 4, harmine, HES-1, HES-5, proline/serine-rich coiled-coil protein 1 (also known as DDA3), and Van Gogh 1 (47, 52–59). Conversely, a number of genes promote neurite sprouting. For example, a complex containing diacylglycerol kinase ζ, Rac1, and syntrophin promotes neurite outgrowth (60, 61). In addition, the Brn-3a transcription factor, CD47 (also referred to as integrin-associated protein), degenerin/epithelial Na+ channel protein, Dickkopf-1, insulin-like growth factors I and II, NF-κB, plasticity-related gene 5, the Prickle 1 or 2 gene, protruding, retina-derived growth factor, and Wnt-3a promote neurite outgrowth (62–71). Several proteins that positively or negatively regulate neurite outgrowth belong to the Wnt signaling pathway (adenomatous polyposis coli protein, β-catenin, Wnt-3a, and Dickkopf-1). The Wnt and Notch signaling pathways have considerable cross talk, and thus, activation or repression of the Notch signaling pathway impacts Wnt signaling (72, 73). It is not clear whether ORF2 influences the Wnt signaling pathway and/or other genes that regulate neurite outgrowth.
In general, there was a correlation between the abilities of the respective mutant ORF2 constructs to inhibit neurite formation, the bICP0 early promoter (44), BHV-1 productive infection (44), and Notch-mediated trans activation of the HES-5 promoter (Table 1 contains a summary of the results for the respective mutant constructs). The ability of four transposon mutant ORF2 constructs (ORF2-95, ORF2-134, ORF2-240, and ORF2-271) to enhance the degradation of Notch3 did not correlate with neurite formation, activation of productive BHV-1 infection, and activation of BHV-1 promoters, suggesting that sufficient levels of Notch3 were present to perform these functions. Because of the complexity of the regulation of neurite formation, it is also possible that ORF2 inhibits other cellular proteins that interfere with neurite formation. Interestingly, two phosphorylation mutant constructs (ORF2-P and ORF2-AP) did not effectively inhibit Notch-mediated trans activation of the HES5 promoter, whereas the same mutant constructs promoted neurite formation and inhibited Notch1-mediated trans activation of the bICP0 early promoter with WT efficiency (44). For activated Notch to stimulate transcription, Notch, CSL, and MAML must form a complex at a consensus CSL binding site (reviewed in references 27 and 29). Consequently, ORF2 may (i) interfere with the formation of the Notch-CSL-MAML complex, (ii) prevent the Notch-CSL-MAML complex from interacting with certain CSL consensus binding sites, or (iii) interfere with certain coactivators recruited to the Notch-CSL-MAML complex (27, 29). Reduction of the steady-state levels of Notch3 by ORF2 would also interfere with the trans activation potential of Notch3. We speculate that ORF2 phosphorylation influences trans activation of the bICP0 early promoter but not the HES5 promoter because Notch-mediated trans activation requires the formation of different complexes on these two promoters. The ORF2 phosphorylation mutant constructs are more stable than ORF2 (44), suggesting that the differential effects of inhibiting Notch-mediated trans activation were not merely due to lower levels of mutant ORF2 in transfected cells. Considering the complexity of Notch-mediated transcriptional activation and neurite sprouting, it is not surprising that different mutant ORF2 constructs may affect one process but not others. On the basis of our present studies, we believe that ORF2 interference with Notch3 functions is mediated by the ability of ORF2 to interact with Notch3, sequester it to the rim of the nucleus (26), and enhance Notch3 degradation.
Table 1.
Summary of the Notch-inhibiting functions of ORF2 and a panel of mutant ORF2 constructsa
| Construct | Inhibition of: |
||||
|---|---|---|---|---|---|
| Activation of BHV-1 promoters | Activation of productive BHV-1 infection | Neurite formation | HES5 activation | Notch3 degradation | |
| ORF2 | +++ | +++ | +++ | +++ | +++ |
| ORF2-95 | − | − | − | +++ | +++ |
| ORF2-134 | − | − | − | +++ | +++ |
| ORF2-240 | − | − | − | +++ | +++ |
| ORF2-271 | − | − | − | +++ | +++ |
| ORF2-310 | +++ | +++ | +++ | +++ | +++ |
| ORF2-333 | +++ | +++ | +++ | +++ | +++ |
| ORF2-469 | +++ | +++ | +++ | +++ | +++ |
| ORF2-529 | +++ | +++ | +++ | +++ | +++ |
| ORF2-NLS | − | − | − | − | − |
| ORF2-P | +++ | +++ | +++ | − | +++ |
| ORF2-AP | +++ | +++ | +++ | − | +++ |
Studies describing the effects of ORF2 on the activation of BHV-1 promoters and productive infection were previously published (26, 44). The abilities of ORF2 to inhibit neurite formation, HES5 promoter activation, and Notch3 degradation are presented in this study. +++, inhibition; −, lack of inhibition.
ORF2, in the absence of other viral gene products, inhibits apoptosis (25, 70) in transiently transfected cells. The antiapoptosis functions of ORF2 are believed to be crucial for the latency-reactivation cycle because an LR mutant virus that contains stop codons at the amino terminus of ORF2 induces higher levels of apoptosis in TG neurons during the establishment of latency (22) and is not reactivated from latency after DEX treatment (6). ORF2 promotion of neurite formation in the presence of Notch may maintain normal neuronal functions, including neuronal survival, after infection. Productive BHV-1 infection induces Notch1 protein levels (26), suggesting that during the establishment of latency, ORF2 maintains axonal projections in infected neurons by promoting neurite sprouting in the presence of activated Notch. In the absence of ORF2 (LR mutant virus, for example), we predict that certain infected neurons are more susceptible to loss of axonal projections because activated Notch is present. It is well established that neurons with damaged or cut axons can undergo Wallerian degeneration, a slow form of neuronal death (36, 74). The fact that Notch stimulates productive infection (26) also favors virus-induced neuronal cell death. In neuronal progenitor cells, Notch activation induces apoptosis by a p53-dependent pathway (38). Collectively, these observations suggest that the ability of ORF2 to restrain Notch functions complements the antiapoptosis functions of ORF2 to promote the establishment and life-long maintenance of latency.
During DEX-induced reactivation from latency, Notch3 RNA (26), as well as protein, levels are increased in TG, and HES6 RNA levels are increased during reactivation from latency (43). Examination of DEX-induced transcription in TG of calves latently infected with BHV-1 revealed that many genes activated by Notch signaling are induced (data not shown), suggesting that the Notch signaling pathway may enhance reactivation from latency. Since ORF2 protein expression is reduced during DEX-induced reactivation, we predict that activated Notch family members would not be restrained by ORF2, suggesting that activated Notch destabilizes normal neuronal functions and may induce neuronal cell death, as well as reactivation from latency.
ACKNOWLEDGMENTS
This research was supported by a grant from the USDA National Institute of Food and Agriculture Competitive Grants Program (09-01653). A grant to the Nebraska Center for Virology (1P20RR15635) has also supported certain aspects of these studies. Devis Sinani was partially supported by a fellowship from Ruth L. Kirschstein National Research Service Award 1 T32 AIO60547 (National Institute of Allergy and Infectious Diseases).
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Turin L, Russo S, Poli G. 1999. BHV-1: new molecular approaches to control a common and widespread infection. Mol. Med. 5:261–284 [PMC free article] [PubMed] [Google Scholar]
- 2. Jones C. 2009. Regulation of innate immune responses by bovine herpesvirus 1 and infected cell protein 0. Viruses 1:255–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones C, Chowdhury S. 2010. Bovine herpesvirus type 1 (BHV-1) is an important cofactor in the bovine respiratory disease complex. Vet. Clin. North Am. Food Anim. Pract. 26:303–321 [DOI] [PubMed] [Google Scholar]
- 4. Jones C, Chowdhury S. 2007. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex, and development of improved vaccines. Anim. Health Res. Rev. 8:187–205 [DOI] [PubMed] [Google Scholar]
- 5. Schang L, Jones C. 1997. Analysis of bovine herpesvirus 1 transcripts during a primary infection of trigeminal ganglia of cattle. J. Virol. 71:6786–6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inman M, Lovato L, Doster A, Jones C. 2002. A mutation in the latency related gene of bovine herpesvirus 1 interferes with the latency-reactivation cycle of latency in calves. J. Virol. 76:6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv. Virus Res. 51:81–133 [DOI] [PubMed] [Google Scholar]
- 8. Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin. Microbiol. Rev. 16:79–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones C, Newby TJ, Holt T, Doster A, Stone M, Ciacci-Zanella J, Webster CJ, Jackwood MW. 2000. Analysis of latency in cattle after inoculation with a temperature sensitive mutant of bovine herpesvirus 1 (RLB106). Vaccine 18:3185–3195 [DOI] [PubMed] [Google Scholar]
- 10. Jones C, Geiser V, Henderson G, Jiang Y, Meyer F, Perez S, Zhang Y. 2006. Functional analysis of bovine herpesvirus 1 (BHV-1) genes expressed during latency. Vet. Microbiol. 113:199–210 [DOI] [PubMed] [Google Scholar]
- 11. Rock D, Lokensgard J, Lewis T, Kutish G. 1992. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J. Virol. 66:2484–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Winkler MT, Doster A, Sur JH, Jones C. 2002. Analysis of bovine trigeminal ganglia following infection with bovine herpesvirus 1. Vet. Microbiol. 86:139–155 [DOI] [PubMed] [Google Scholar]
- 13. Workman A, Perez S, Doster A, Jones C. 2009. Dexamethasone treatment of calves latently infected with bovine herpesvirus 1 (BHV-1) leads to activation of the bICP0 early promoter, in part by the cellular transcription factor C/EBP-alpha. J. Virol. 83:8800–8809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol. 61:3820–3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rock DL, Beam SL, Mayfield JE. 1987. Mapping bovine herpesvirus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits. J. Virol. 61:3827–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kutish G, Mainprize T, Rock D. 1990. Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. J. Virol. 64:5730–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jaber T, Workman A, Jones C. 2010. Small noncoding RNAs encoded within the bovine herpesvirus 1 latency-related gene can reduce steady-state levels of infected cell protein 0 (bICP0). J. Virol. 84:6297–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Silva LF, Jones C. 2012. Two microRNAs encoded within the bovine herpesvirus 1 latency-related gene promote cell survival by interacting with RIG-I and stimulating NF-κB-dependent transcription and beta interferon signaling pathways. J. Virol. 86:1670–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang Y, Inman M, Zhang Y, Posadas NA, Jones C. 2004. A mutation in the latency-related gene of bovine herpesvirus 1 inhibits protein expression from open reading frame 2 and an adjacent reading frame during productive infection. J. Virol. 78:3184–3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meyer F, Perez S, Jiang Y, Zhou Y, Henderson G, Jones C. 2007. Identification of a novel protein encoded by the latency-related gene of bovine herpesvirus 1. J. Neurovirol. 13:569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inman M, Lovato L, Doster A, Jones C. 2001. A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. J. Virol. 75:8507–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lovato L, Inman M, Henderson G, Doster A, Jones C. 2003. Infection of cattle with a bovine herpesvirus 1 strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J. Virol. 77:4848–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ciacci-Zanella J, Stone M, Henderson G, Jones C. 1999. The latency-related gene of bovine herpesvirus 1 inhibits programmed cell death. J. Virol. 73:9734–9740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Henderson G, Perng G-C, Nesburn A, Wechsler S, Jones C. 2004. The latency related gene of bovine herpesvirus 1 can suppress caspase 3 and caspase 9 during productive infection. J. Neurovirol. 10:64–70 [DOI] [PubMed] [Google Scholar]
- 25. Shen W, Jones C. 2008. Open reading frame 2, encoded by the latency-related gene of bovine herpesvirus 1, has antiapoptotic activity in transiently transfected neuroblastoma cells. J. Virol. 82:10940–10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Workman A, Sinani D, Pittayakhajonwut D, Jones C. 2011. A protein (ORF2) encoded by the latency-related gene of bovine herpesvirus 1 interacts with Notch1 and Notch3. J. Virol. 85:2536–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bray SJ. 2006. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7:678–689 [DOI] [PubMed] [Google Scholar]
- 28. Cornell R, Eisen JS. 2005. Notch in the pathway: the roles of Notch signalling in neural crest development. Semin. Cell Dev. Biol. 16:663–672 [DOI] [PubMed] [Google Scholar]
- 29. Ehebauer M, Penelope P, Arias AM. 2006. Notch, a universal arbiter of cell fate decisions. Science 314:1414–1415 [DOI] [PubMed] [Google Scholar]
- 30. Justice NJ, Jan YN. 2002. Variations on the Notch pathway in neural development. Curr. Opin. Neurobiol. 12:64–70 [DOI] [PubMed] [Google Scholar]
- 31. Berezovska O, McLean P, Knowles R, Frosh M, Lu FM, Lux SE, Hyman BT. 1999. Notch1 inhibits neurite outgrowth in postmitotic primary neurons. Neuroscience 93:433–439 [DOI] [PubMed] [Google Scholar]
- 32. Franklin JL, Berechid BE, Cutting FB, Presente A, Chambers CB, Folz DR, Ferreira A, Nye JS. 1999. Autonomous and non-autonomous regulation of mammalian neurite development by Notch1 and Delta1. Curr. Biol. 9:1448–1457 [DOI] [PubMed] [Google Scholar]
- 33. Levy OA, Lah JJ, Levy AI. 2002. Notch signaling inhibits PC12 cell neurite outgrowth via RBP-J-dependent and -independent mechanisms. Dev. Neurosci. 24:79–88 [DOI] [PubMed] [Google Scholar]
- 34. Sestan N, Artavanis-Tsakonas S, Rakic P. 1999. Contact-dependent inhibition of cortical neurite growth mediated by Notch signaling. Science 286:741–746 [DOI] [PubMed] [Google Scholar]
- 35. El Bejjani R, Hammerlund M. 2012. Notch signaling inhibits axon regeneration. Neuron 73:268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coleman MP, Freeman MR. 2010. Wallerian degeneration, wld(s), and nmnat. Annu. Rev. Neurosci. 33:245–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raff MC, Whitmore AV, Finn JT. 2002. Axonal self-destruction and neurodegeneration. Science 296:868–871 [DOI] [PubMed] [Google Scholar]
- 38. Yang Y, Klein R, Tian X, Cheng H-T, Kopan R, Shen J. 2004. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev. Biol. 269:81–94 [DOI] [PubMed] [Google Scholar]
- 39. Schuurmans C, Guillermot F. 2002. Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr. Opin. Neurobiol. 12:26–34 [DOI] [PubMed] [Google Scholar]
- 40. Conlon RA, Reaume AG, Rossant J. 1995. Notch1 is required for the coordinate segmentation of somites. Development 121:1533–1545 [DOI] [PubMed] [Google Scholar]
- 41. Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia A, Nye J, Conlon R, Mak T, Bernstein A, van der Kooy D. 2002. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 16:846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beatus P, Lundkvist J, Oberg C, Lendahl U. 1999. The Notch 3 intracellular domain represses Notch 1-mediated activation through Hairy/Enhancer of split (HES) promoters. Development 126:3925–3935 [DOI] [PubMed] [Google Scholar]
- 43. Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, Cook E, Doster A, Jones C. 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J. Virol. 86:2459–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sinani D, Jones C. 2011. Localization of sequences in a protein (ORF2) encoded by the latency-related gene of bovine herpesvirus 1 that inhibits apoptosis and interferes with Notch1-mediated trans-activation of the bICP0 promoter. J. Virol. 85:12124–12133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lassiter RN, Ball MK, Adams JS, Wright BT, Stark MR. 2010. Sensory neuron differentiation is regulated by Notch signaling in the trigeminal placode. Dev. Biol. 344:836–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fischer A, Gessler M. 2007. Delta-Notch—and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 35:4583–4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. 1999. Hes1 and Hes5 effectors in mammalian differentiation. EMBO J. 18:2196–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fryer CJ, White JB, Jones KA. 2004. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16:509–520 [DOI] [PubMed] [Google Scholar]
- 49. Brakenhoff RH. 2011. Another NOTCH for cancer. Science 333:1102–1103 [DOI] [PubMed] [Google Scholar]
- 50. Koch U, Radtke F. 2011. Notch in T-ALL: new players in a complex disease. Trends Immunol. 32:434–442 [DOI] [PubMed] [Google Scholar]
- 51. Sethi N, Kang Y. 2011. Notch signalling cancer progression and bone metastasis. Br. J. Cancer 105:1805–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Castella P, Sawai S, Nakao K, Wagner JA, Caudy M. 2000. HES-1 repression of differentiation and proliferation in PC12 cells: role for the helix 3-helix 4 domain in transcription repression. Mol. Cell. Biol. 20:6170–6183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Göckler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W. 2009. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 276:6324–6337 [DOI] [PubMed] [Google Scholar]
- 54. Hsieh P-C, Chiang M-L, Chang J-C, Yan Y-T, Wang F-F, Chou Y-C. 2012. DDA3 stabilizes microtubules and suppresses neurite formation. J. Cell Sci. 125:3402–3411 [DOI] [PubMed] [Google Scholar]
- 55. Saengsawang W, Mitok K, Viesselmann C, Pietila L, Lumbard DC, Corey SJ, Dent EW. 2012. The F-BAR protein CIP4 inhibits neurite formation by producing lamellipodial protrusions. Curr. Biol. 22:494–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanchez-Alvarez L, Visanuvimol J, McEwan A, Su A, Imai JH, Colavita A. 2011. VANG-1 and PRKL-1 cooperate to negatively regulate neurite formation in Caenorhabditis elegans. PLoS Genet. 7:e1002257 doi:10.1371/journal.pgen.1002257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schulz A, Geissler KJ, Kumar S, Leichsenring G, Morrison H, Baader SL. 2010. Merlin inhibits neurite outgrowth in the CNS. J. Neurosci. 30:10177–10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thevananther S, Rivera A, Rivkees SA. 2001. A1 adenosine receptor activation inhibits neurite process formation by Rho kinase-mediated pathways. Neuroreport 12:3057–3063 [DOI] [PubMed] [Google Scholar]
- 59. Votin V, Nelson WJ, Barth AIM. 2005. Neurite outgrowth involves adenomatous polyposis coli protein and b-catenin. J. Cell Sci. 118:5699–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Woo S, Gomez TM. 2006. Rac1 and RhoA promote neurite outgrowth through formation and stabilization of growth cone point contacts. J. Neurosci. 26:1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yakubchyk Y, Abramovici H, Maillet J-C, Daher E, Obagi C, Parks RJ, Topham MK, Gee SH. 2005. Regulation of neurite outgrowth in N1E-115 cells through PDZ-mediated recruitment of diacylglycerol kinase zeta. Mol. Cell. Biol. 25:7289–7302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Broggini T, Nitsch R, Savaskan NE. 2010. Plasticity-related gene 5 (PRG5) induces filopodia and neurite growth and impedes lysophosphatidic acid- and nogo-A-mediated axonal retraction. Mol. Biol. Cell 21:521–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Drummond HA, Furtado MM, Meyers S, Grifoni S, Parker KA, Hoover A, Stec DE. 2006. ENaC proteins are required for NGF-induced neurite growth. Am. J. Physiol. Cell Physiol. 290:C404–C410 [DOI] [PubMed] [Google Scholar]
- 64. Endo Y, Beauchamp E, Woods D, Taylor WG, Toretsky JA, Uren A, Rubin JS. 2008. Wnt-3a and Dickkopf-1 stimulate neurite outgrowth in Ewing tumor cells via a Frizzled3 and c-jun N-terminal kinase-dependent mechanism. Mol. Cell. Biol. 28:2368–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC. 2000. NF-κB signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J. Neurosci. 20:7556–7563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Miyashita M, Ohnishi H, Okazawa H, Tomonaga H, Hayashi A, Fujimoto T-T, Furuya N, Matozaki T. 2004. Promotion of neurite and filopodium formation by CD47: roles of integrins, Rac, and Cdc42. Mol. Biol. Cell 15:3950–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mulholland MW, Romanchuk G, Simeone DM, Flowe K. 1992. Stimulation of myenteric plexus neurite outgrowth by insulin and insulin-like growth factors I and II. Life Sci. 51:1789–1796 [DOI] [PubMed] [Google Scholar]
- 68. Okuda H, Miyata S, Mori Y, Tohyama M. 2007. Mouse Prickle1 and Prickle2 are expressed in postmitotic neurons and promote neurite outgrowth. FEBS Lett. 581:4754–4760 [DOI] [PubMed] [Google Scholar]
- 69. Saita S, Shirane M, Natume T, Iemura S-I, Nakayama KI. 2009. Promotion of neurite extension by protrudin requires its interaction with vesicle-associated membrane protein-associated protein. J. Biol. Chem. 284:13766–13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith MD, Morris PJ, Dawson SJ, Schwartz ML, Schlaepfer WW, Latchman DS. 1997. Coordinate induction of the three neurofilament genes by the Brn-3a transcription factor. J. Biol. Chem. 272:21325–21333 [DOI] [PubMed] [Google Scholar]
- 71. Wagner JA, D'Amore PA. 1986. Neurite outgrowth induced by an endothelial cell mitogen isolated from retina. J. Cell Biol. 103:1363–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hayward P, Brennan K, Sanders P, Balayo T, DasGupta R, Perrimon N, Arlas AM. 2005. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 132:1819–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hayward P, Kalmar T, Arlas AM. 2008. Wnt/Notch signalling and information processing during development. Development 135:411–424 [DOI] [PubMed] [Google Scholar]
- 74. Wakatsuki S, Saitoh F, Araki T. 2011. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 13:1415–1423 [DOI] [PubMed] [Google Scholar]