

Abstract
The major inducible 70-kDa heat shock protein (hsp70) is host protective in a mouse model of measles virus (MeV) brain infection. Transgenic constitutive expression of hsp70 in neurons, the primary target of MeV infection, abrogates neurovirulence in neonatal H-2d congenic C57BL/6 mice. A significant level of protection is retained after depletion of T lymphocytes, implicating innate immune mechanisms. The focus of the present work was to elucidate the basis for hsp70-dependent innate immunity using this model. Transcriptome analysis of brains from transgenic (TG) and nontransgenic (NT) mice 5 days after infection identified type I interferon (IFN) signaling, macrophage activation, and antigen presentation as the main differences linked to survival. The pivotal role of type I IFN in hsp70-mediated protection was demonstrated in mice with a genetically disrupted type I IFN receptor (IFNAR−/−), where IFNAR−/− eliminated the difference in survival between TG and NT mice. Brain macrophages, not neurons, are the predominant source of type I IFN in the virus-infected brain, and in vitro studies provided a mechanistic basis by which MeV-infected neurons can induce IFN-β in uninfected microglia in an hsp70-dependent manner. MeV infection induced extracellular release of hsp70 from mouse neuronal cells that constitutively express hsp70, and extracellular hsp70 induced IFN-β transcription in mouse microglial cells through Toll-like receptors 2 and 4. Collectively, our results support a novel axis of type I IFN-dependent antiviral immunity in the virus-infected brain that is driven by hsp70.
INTRODUCTION
The major inducible 70-kDa heat shock protein (hsp70) is highly induced by fever (1), a frequent accompaniment of virus infection (2), and the phylogenetic conservation of the febrile response suggests that hsp70 induction plays a host protective role (reviewed in references 3 and 4). hsp70 is expressed in the major histocompatibility complex (MHC) III region of the human genome (5). It supports MHC I antigen expression (6) and can be released into the extracellular environment by secretion or following cell death to activate macrophages and stimulate release of proinflammatory cytokines (7, 8). These characteristics illustrate the potential of hsp70 to stimulate innate immunity, although the mechanism by which hsp70 might contribute to innate antiviral responses and the relative significance remain to be shown.
The ability of hsp70 to mediate protective antiviral immunity was first established in the mouse model of measles virus (MeV) brain infection. Neonatal C57BL/6 mice are susceptible to infection following intracranial inoculation with Edmonston MeV (Ed MeV), where neurons are the primary target of productive viral replication (9–11). Mice lack the constitutive expression of hsp70 that is observed in primates and other mammals (12–14). Therefore, transgenic (TG) mice that express human hsp70 through a neuron-specific enolase promoter can be used to evaluate the impact of virus-hsp70 interactions within infected cells on uninfected immune cells mediating both innate and adaptive responses. Constitutive expression of hsp70 in neurons of H-2d congenic C57BL/6 mice results in complete protection against intracranial inoculation with 4 × 104 50% tissue culture infective doses (TCID50) of Ed MeV (9). In contrast, nontransgenic (NT) mice exhibit approximately 30% mortality, which is correlated to elevated brain viral burden and cytopathic effects. T cell depletion increases mortality in both MeV-infected TG and NT mice, although mortality is significantly lower in the TG groups, indicating an innate immune component to hsp70-dependent protection. This is supported by increased levels of transcripts for CD68, MHC II, Toll-like receptor 2 (TLR2), and TLR4 within total brain RNA of infected TG mice relative to infected NT mice at 5 days postinfection (p.i.). Five days p.i. is prior to the onset of virus-induced mortality and measurable adaptive immune responses observed at ≥10 days p.i. (9, 12). CD68 and MHC II are markers of macrophage activation, and TLR2 and TLR4 mediate innate immune responses of brain macrophages, responding to ligands that may include extracellular hsp70 (15, 16). The mediators of hsp70-dependent innate antiviral immunity and their contribution to host protection are unknown.
The focus of the present work was to define innate host responses to infection that are the basis for hsp70-mediated protection against viral infection. The first step was to analyze patterns of host gene expression within infected tissues at 5 days p.i., with results supporting type I interferon (IFN) signaling as a major difference in the hsp70-dependent host response to infection. Subsequent in vivo studies with IFNAR−/− mice established proof of the central role of type I IFN in hsp70-mediated host protection, and in vitro modeling provided a mechanism that directly links hsp70 expression to IFN-β induction.
MATERIALS AND METHODS
The Institutional Animal Care and Use Committee for The Ohio State University provided supervision of animal care for all aspects of the study, including review and approval of the experimental protocols in accordance with the Public Health Service Act of the United States of America. Animal care facilities are AAALAC accredited.
Infection of hsp70 TG and NT mice.
hsp70-mediated protection is H-2 dependent, being observed in H-2d resistant mice (9, 10). Accordingly, H-2d congenic mice were the focus of the present work. Characterization of H-2d congenic C57BL/6 mice that constitutively overexpress human hsp70 (from the HSPA1A gene) under the control of the neuron-specific enolase promoter has been described previously; these mice are referred to here as hsp70-transgenic (TG) mice (9, 10). Male and female TG and nontransgenic (NT) mice, approximately 42 h of age, were inoculated intracranially in the left cerebral hemisphere with 4 × 104 TCID50 Ed of MeV in 1× phosphate-buffered saline (PBS), for a total volume of 10 μl (9, 10, 12). Control mock-infected mice were inoculated with 1× PBS alone. Brains were harvested at 5 and 10 days p.i. for global transcript analysis, routine light microscopic evaluation of formalin-fixed tissue sections, and immunohistochemical staining of paraformaldehyde-fixed tissues. Brains were also harvested at 1, 2, and 3 days p.i. for transcript analysis by reverse transcription-PCR (RT-PCR). For transcript analyses, the right half of the brain (contralateral to the inoculation hemisphere) was processed for total RNA isolation using the RNeasy lipid tissue minikit (Qiagen), followed by DNase I treatment (TURBO DNA-free; Ambion).
Global transcript analysis.
The integrity and quantity of isolated total RNA were determined using an Agilent Bioanalyzer 2100 in the MicroArray Shared Resource (MASR), The Ohio State University Comprehensive Cancer Center (OSUCCC). The MASR performed sample preparation and labeling, chip hybridization, chip scanning, and initial image analysis by following Affymetrix protocols. cRNA was synthesized using the GeneChip T7-oligo(dT) promoter primer kit (17) and analyzed using mouse genome 430 arrays (Affymetrix). Background correction and normalization were performed to correct technical bias, and gene expression levels for individual mice (n = 4 per group) were summarized using the robust multiarray average (RMA) method (18). Filtering methods based on the percentage of samples with expression values above noise level removed genes from the analyses that were not expressed or were expressed at low levels (log2 expression level < 6), resulting in the analysis of 27,000 genes in the 43,000-probe set. For remaining genes, linear models were performed to detect differentially expressed genes between groups of interest (e.g., transgenic versus nontransgenic at the 5th day postinfection). In order to improve the estimates of variability and statistical tests for differential expression, variance smoothing methods were employed (19). A moderated 2-sample t test was used to compare samples between groups, establishing P values for use in genome-wide discovery. Significance level for genome-wide discovery was determined by controlling the number of false positives (20). Statistical software programs SAS 9.2 and R were used for this analysis.
Data for differentially expressed genes were analyzed through the use of Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA), where the association between genes exhibiting differences in expression between groups and canonical pathways in the Ingenuity Knowledge Base were determined. The significance of the association between genes (data sets) and canonical pathways was based upon Fisher's exact test, which calculates a P value determining the probability that the association between genes in the data set and the canonical pathway is explained by chance alone.
Histochemical and immunohistochemical tissue analyses.
Analyses focused on coronal sections at −2.0 bregma, providing good representation of cortices, diencephalon, and hippocampus. For formalin-fixed paraffin-embedded tissue sections, mice were euthanized and whole brains were harvested at 5 days p.i. for immersion fixation in 10% formalin. Paraffin-embedded tissue sections were evaluated for viral cytopathic effects and inflammatory responses following routine hematoxylin and eosin (H&E) staining. Infiltration of blood origin monocytes was measured by CD45 immunohistochemistry as previously described (21, 22). Analysis of microglial activation was also based upon immunohistochemical staining for MHC II in paraformaldehyde-fixed frozen sections as previously described (23). Staining employed rat anti-mouse I-A/I-E (M5/114.15.2, catalog number 556999; BD Pharmingen). The signal from MHC II staining was digitized and quantified by using the positive pixel counter algorithm in Aperio Imagescope (Aperio). Within each section, positivity of immunoreactivity in the hippocampus contralateral to the inoculation hemisphere was calculated as the total number of positive pixels divided by the total number of pixels.
IFN-β transcript analysis in brain.
Real-time quantitative RT-PCR (qRT-PCR) analysis of total brain RNA was used to quantify IFN-β transcripts at 1 day p.i. in the brain hemisphere opposite the virus inoculation site, using 5 animals per group. Two micrograms of total RNA was denatured at 70°C for 5 min and annealed to 0.4 μg of random primers. Primer-annealed RNA was added to reverse transcriptase reaction mixtures that included 25 nmol of dNTP and 1 μl of AffinityScript multiple-temperature reverse transcriptase (Stratagene) in a final volume of 20 μl. The cDNA reaction mixtures were incubated for 90 min at 48°C. Levels of IFN-β transcript cDNAs were quantified by SYBR green real-time qPCR using a Roche 480 LightCycler. Commercially available primers were employed (Real Time Primers, Elkins Park, PA) that recognize a conserved portion of the mouse IFN-β gene (GenBank accession number NM_010510). Plasmid containing mouse IFN-β cDNA (GenBank accession number NM_010510) was obtained from GeneCopoeia (Rockville, MD) and used to establish a standard curve. The qPCR was performed using SYBR green LightCycler 480 SYBR green I master mix (Roche). The reaction program included preincubation at 95°C for 10 min and 45 cycles of amplification at 95°C for 15 s, at 58°C for 10 s, and at 72°C for 15 s, followed by a 2-s incubation at 81°C. Total SYBR green fluorescence was measured at 81°C. Quantifications were based upon fit point analysis with arithmetic baseline adjustment. Melting peak and melting curve analyses were performed using the polynomial calculation method. At the conclusion of the reactions, amplicons were further analyzed by 1.5% agarose gel electrophoresis.
Generation of hsp70-TG and NT H-2d IFNAR−/−mice.
C57BL/6 (H-2b) mice carrying a disrupted type I IFN receptor gene (IFNAR−/−) were obtained from the Paul-Ehrlich-Institute, Langen, Germany (24). hsp70-TG H-2d mice carrying the mutant type I IFN receptor gene were generated by backcrossing hsp70-TG H-2d mice with C57BL/6 IFNAR−/− (H-2b) mice for two generations. Genetic screening was performed in F2 and F3 generations in order to obtain animals homozygous for H-2d, mutant allele of the IFNAR, and hsp70 transgene. Peripheral blood mononuclear cells, isolated from whole blood, were used to monitor the H-2 complex by flow cytometry using monoclonal antibodies specific for H-2b (AF6-88.5, fluorescein isothiocyanate conjugated; BD Pharmingen) and H-2d (SF1-1.1, R-phycoerythrin conjugated; BD Pharmingen) alloantigens. The presence and level of the hsp70 construct were confirmed by real-time qPCR analysis of genomic DNA isolated from tail biopsy specimens using Gentra Puregene kits (Qiagen). DNAs from established heterozygotes and homozygotes were used as standards (9, 25). The expression of wild-type and mutant alleles of type I IFN receptor were confirmed by PCR analysis of the genomic DNA using two sets of primers. For the wild-type allele, a 150-bp fragment was amplified using 5′-AAGATGTGCTGTTCCCTTCCTCTGCTCTGA-3′ and 5′-ATTATTAAAAGAAAAGACGAGGCGAAGTGG-3′ sense and antisense primers, respectively. For the mutant allele, a 340-bp fragment was amplified using 5′-TCAGCGCAGGGGCGCCCGGTTCTTT-3′ and 5′-ATCGACAAGACCGGCTTCCATCCGA-3′ sense and antisense primers, respectively. Mice were maintained as homozygotes for the H-2d allele, hsp70 transgene, and type I IFN receptor mutant allele.
Survival analysis of infected mice.
Male and female hsp70-TG and NT H-2d IFNAR−/− neonatal mice (approximately 42 h of age), referred to here as TG and NT IFNAR−/−, were inoculated intracranially in the left cerebral hemisphere with 4 × 104 TCID50 of Ed MeV diluted to 10 μl in PBS (9, 10, 12). Control mice were inoculated with PBS. Removal criteria included a weight of <30 to 50% of the mean litter body weight (typically associated with dehydration), social withdrawal, and seizure activity. Kaplan-Meier survival curves were constructed, and Wilcoxon and log rank tests were used to define differences in survival between treatment groups. Results were compared to differences in mortality between infected TG and NT mice.
In vitro modeling.
The ability of Ed MeV-infected neurons to induce IFN-β in uninfected microglia was modeled using the mouse neuroblastoma cell line N2a and the mouse microglial cell line BV-2. N2a cells have been stably transfected to constitutively express hsp70 (N2a-HSP) and together with vector-transfected controls (N2a-V) have been used to characterize the MeV transcriptional response to hsp70 and the impact upon viral fitness (26). N2a-HSP and N2a-V cells were infected at a multiplicity of infection (MOI) of 1.0 with Ed MeV and monitored for cell death and hsp70 release between 0 and 60 h p.i. Triplicate cell populations in 6-well plates were analyzed, using uninfected N2a-HSP and N2a-V cells as controls. Tissue culture medium (Dulbecco modified Eagle medium [DMEM] containing 5% fetal calf serum) was harvested at defined intervals and clarified by centrifugation (1,000 × g, 15 min, 4°C), and fresh medium was applied to cultures. Cell death was quantified based upon release of lactate dehydrogenase (LDH) into clarified culture supernatants, using a commercially available assay (Biovision, Mountain View, CA). For this purpose, 10 μl of culture medium is used in an enzyme coupling reaction that generates a yellow product, where color intensity measured at 450 nm correlates with the number of cells lysed. Maximal LDH release was defined by cells treated with 1% Triton X-100 for 30 min. hsp70 concentrations in culture supernatants were measured using a commercially available enzyme-linked immunosorbent assay (ELISA) (hsp70 high-sensitivity enzyme immunometric assay; Enzo Life Sciences, Farmingdale, NY). The standard curve was defined using serial dilutions of a purified human recombinant, hsp70-1 (product of the HSPA1A gene). The primary antibody used for detection is a rabbit polyclonal antibody recognizing human, rat, and mouse hsp70 and which does not cross-react with other hsp70 family members (e.g., hsc70).
Effects of extracellular hsp70 were tested on BV-2 cells (a mouse microglial cell line) using culture supernatants from infected and uninfected N2a cells and a highly purified form of hsp70-1 that has been used to study structural interactions with the C terminus of the MeV N protein (27, 28). BV-2 cells were seeded in replicate wells of six-well plates (0.2 × 106 cells/well), and the medium was replaced with culture supernatant from N2a cells or purified hsp70 added to BV-2 culture medium at 12 h postseeding. Culture supernatants from N2a cells, representing 1 to 20 h p.i., were treated with short-wave UV light (254 nm) and titrated to rule out the presence of infectious virus. hsp70 contained in tissue culture supernatants from infected cells either was left untreated or was neutralized by incubation (1 h, 37°C) with rabbit polyclonal IgG against hsp70 (15 μg/ml) (SPA-812; Enzo Life Sciences). Normal rabbit total IgG was used as a neutralization negative control. Addition of purified recombinant hsp70 to BV-2 cells included control treatments: addition of 30 μg/ml of polymyxin B in combination with hsp70 and use of heat-denatured hsp70 (100°C, 10 min). Total BV-2 cell RNA was harvested at 6 h posttreatment (Qiagen RNeasy minikit). Samples were treated with DNase I (TURBO DNA free; Ambion), and 1.0 μg of RNA was reverse transcribed using 0.4 μg of oligo(dT)15 primers and multiple-temperature reverse transcriptase (Affinity Script). Real-time qRT-PCR analysis was used to quantify IFN-β cDNA levels as described above. Confirmatory experiments examined the potential of hsp70 to induce IFN-β transcription in a second mouse microglial cell line (NR-9460), and also in primary cultures of bone marrow-derived macrophages, using established procedures for isolation and culture (29).
Antibody blockade of TLR2 and TLR4 signaling employed mouse monoclonal antibody against mouse and human TLR2 (eBioscience; 16-9024), mouse IgG1 isotype control, rat monoclonal antibody against TLR4 (Imgenex; IMG-428E), and rat IgG2a isotype control. Antibodies were added to culture media 1 h prior to the addition of hsp70, at a concentration of 20 μg/ml.
Statistical analysis for in vitro studies included two-tailed unpaired t test (t test) and one-way analysis of variance (ANOVA). Bonferroni's multiple-comparison test was used to compare pairs of groups as the posttest of ANOVA.
Microarray data accession number. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE4226.
RESULTS
hsp70 enhances expression of type I interferon and interferon-responsive genes following viral infection.
Transcriptome analysis was performed on whole brain tissues to obtain an unbiased picture of how hsp70 alters host innate responses to viral infection. Gene expression patterns were determined in hsp70 TG and NT mice infected with Ed MeV. Total brain mRNA (n = 4 per challenge group) was analyzed on mouse genome microarrays, sampling the hemisphere opposite the side of viral inoculation. We discovered 457 upregulated genes and 543 downregulated genes in TG relative to NT mice on day 5 p.i. using a P value cutoff of 9.3 × 10−4.
Analysis of the association between genes exhibiting differences in expression between groups and canonical pathways in the Ingenuity Knowledge Base showed that differences in gene expression at day 5 p.i. were primarily attributed to greater induction of interferon-responsive genes and genes associated with macrophage activation in TG mice (Tables 1 to 4). The significance of the association was P = 9.2 × 10−3 for interferon induction and antiviral responses. Macrophage activation was represented by both macrophage maturation (P = 1.7 × 10−4) and antigen presentation (P = 1.0 × 10−2) pathways.
Table 1.
Increased expression of genes related to IFN signaling in MeV-infected TG relative to NT mice, 5 days p.i.a
| Gene product | GenBank accession no. | Description | (TGi − NTi) − (TGu − NTu) |
TGi − TGu |
NTi − NTu |
|||
|---|---|---|---|---|---|---|---|---|
| Fold change | P | Fold change | P | Fold change | P | |||
| CHUK | AU045682 | Conserved helix-loop-helix ubiquitous ligase; IκB kinase alpha | 1.3 | 9.9E−4 | 1.2 | 4.5E−4 | — | — |
| STAT-1 | AW214029 | Signal transducer and activator of transcription | 1.9 | 3.1E−3 | 6.9 | 1.3E−12 | 3.5 | 1.5E−9 |
| USP18 | NM_011909 | Ubiquitin-specific peptidase 18 | 1.9 | 2.3E−2 | 21.9 | 3.8E−14 | 11.3 | 6.6E−12 |
| TGTP | NM_011579 | T cell-specific GTPase (inducible in T cells and macrophages) | 2.3 | 1.2E−2 | 5.1 | 1.5E−7 | 2.2 | 1.9E−3 |
| IFIT1L | NM_008331 | IFN-induced protein with tetratricopeptide repeats 1-like | 1.9 | 2.4E−2 | 27.7 | 8.7E−15 | 14.3 | 1.1E−12 |
| IFI27 | AY090098 | IFN-α-inducible protein 27 | 1.6 | 4.4E−2 | 6.7 | 5.2E−12 | 4.3 | 1.2E−9 |
| RIG-I | BB401061 | Retinoic acid-inducible gene I | 1.6 | 2.7E−2 | 2.2 | 6.7E−6 | 1.4 | 2.3E−2 |
| IRF7 | NM_016850 | IFN regulatory factor 7 | 1.7 | 5.0E−2 | 4.7 | 1.8E−8 | 2.7 | 1.6E−5 |
| IRF9 | NM_008394 | IFN regulatory factor 9 | 1.4 | 2.2E−2 | 2.8 | 6.6E−11 | 2.0 | 6.7E−8 |
| RNase L | BF714880 | 2′,5′-oligoadenylate synthetase-dependent RNase L | 1.3 | 6.2E−4 | 1.4 | 7.4E−7 | — | — |
Results are based upon transcriptome analysis of total brain RNA. All changes represent induction, and the absence of a significant difference is indicated by a dash. Superscript “i” and “u” represent infected and uninfected mice, respectively.
Table 4.
Increased expression of genes reflecting macrophage activation/antigen presentation in MeV-infected TG relative to NT mice, 10 days p.i.a
| Gene product | GenBank accession no. | Description | (TGi − NTi) − (TGu − NTu) |
TGi − TGu |
NTi − NTu |
|||
|---|---|---|---|---|---|---|---|---|
| Fold change | P | Fold change | P | Fold change | P | |||
| H2-D1 | M83244 | MHC I | 11.9 | 1.8E−11 | 14.3 | 2.5E−15 | — | — |
| H2-K1 | BC018402 | MHC I | 4.2 | 5.7E−9 | 4.4 | 2.8E−12 | — | — |
| H2-EA | U13648 | MHC II | 1.7 | 1.3E−4 | 1.6 | 8.5E−6 | — | — |
| STAT-1 | AW214029 | Signal transducer and activator of transcription | — | — | 23.0 | 9.8E−18 | 23.8 | 7.7E−18 |
| TLR2 | NM_011905 | Toll-like receptor 2 | — | — | 4.8 | 6.2E−12 | 5.8 | 5.0E−13 |
| TLR4 | AF185285 | Toll-like receptor 4 | — | — | 1.4 | 3.6E−4 | 1.3 | 1.0E−3 |
Results are based upon transcriptome analysis of total brain RNA. All changes represent induction, and the absence of a significant difference is indicated by a dash. Superscript “i” and “u” represent infected and uninfected mice, respectively.
Gene lists associated with the pathways were expanded using a P value cutoff of 0.05, and the analysis was extended to include 10 days p.i. in order to define the temporal nature of any differences (Tables 1 to 4). The level of gene expression in sham-inoculated animals was subtracted from the expression profile in the infected groups: (infected TG − infected NT) − (uninfected TG − uninfected NT). Comparisons also included subtracting the value for uninfected TG mice from that for infected TG mice and subtracting the value for uninfected NT mice from that for infected NT mice to distinguish between differences in the magnitude of a response measured in both the TG and NT groups versus a response measured in only one group.
Infected TG mice showed enhanced expression of STAT-1, CHUK (IκB kinase alpha), and eight interferon-stimulated genes relative to infected NT mice at 5 days p.i. (Table 1). STAT-1 and CHUK are involved in macrophage activation as well as interferon signaling (30, 31). Seven of the eight interferon (IFN)-inducible genes (i.e., the genes for USP18, IFIT1L, IFI27, RIG-I, interferon regulatory factor 7 [IRF7], IRF9, and RNase L) are specific to type I IFN (31–35). By day 10, interferon-responsive genes were expressed at increased levels in both TG and NT mice, with no statistically significant differences between the infected groups (Table 2). The exceptions to this expression pattern were CHUK and RNase L, for which significant induction was observed only at 5 days p.i. and only in TG mice.
Table 2.
Increased expression of genes related to IFN signaling in MeV-infected TG relative to NT mice, 10 days p.i.a
| Gene product | GenBank accession no. | Description | (TGi − NTi) − (TGu − NTu) |
TGi − TGu |
NTi − NTu |
|||
|---|---|---|---|---|---|---|---|---|
| Fold change | P | Fold change | P | Fold change | P | |||
| CHUK | AU045682 | Conserved helix-loop-helix ubiquitous ligase; IκB kinase alpha | 1.2 | 1.8E−2 | — (1.1) | — (0.07) | — | — |
| STAT-1 | AW214029 | Signal transducer and activator of transcription | — | — | 23.0 | 9.8E−18 | 23.8 | 7.7E−18 |
| USP18 | NM_011909 | Ubiquitin-specific peptidase 18 | — | — | 71.2 | 8.9E−19 | 104.3 | 1.3E−18 |
| TGTP | NM_011579 | T cell-specific GTPase (inducible in T cells and macrophages) | — | — | 55.1 | 7.1E−16 | 31.8 | 1.8E−14 |
| IFIT1L | NM_008331 | Interferon-induced protein with tetratricopeptide repeats 1-like | — | — | 102.5 | 1.5E−18 | 148.5 | 2.6E−19 |
| IFI27 | AY090098 | IFN-α-inducible protein 27 | — | — | 22.4 | 3.5E−17 | 28.6 | 6.5E−18 |
| RIG-I | BB401061 | Retinoic acid-inducible gene I | — | — | 4.3 | 4.7E−11 | 4.8 | 1.1E−11 |
| IRF7 | NM_016850 | IFN regulatory factor 7 | — | — | 15.7 | 5.4E−14 | 18.4 | 1.6E−14 |
| IRF9 | NM_008394 | IFN regulatory factor 9 | — | — | 5.0 | 1.7E−15 | 6.1 | 1.2E−16 |
Results are based upon transcriptome analysis of total brain RNA. All changes represent induction, and the absence of a significant difference is indicated by a dash. Superscript “i” and “u” represent infected and uninfected mice, respectively.
The literature shows that IFN-β is the predominant type I IFN produced in the central nervous system of mice in response to neurotropic viral infection (36), and expression of IFN-inducible genes at 5 days p.i. is consistent with IFN-β expression at 1 day (37). Expression of IFN-β at 1 day p.i. was directly confirmed by real-time qRT-PCR of total brain RNA from the hemisphere opposite the site of viral inoculation, with transcript levels being significantly increased in infected relative to uninfected TG mice (P < 0.05; t test) (Fig. 1). In NT mice, an increase was observed in infected animals, but the increase relative to uninfected controls was not statistically significant. Collectively, these data indicate that hsp70 leads to increased IFN-β expression, which is correlated to increased protection of TG mice against infection.
Fig 1.
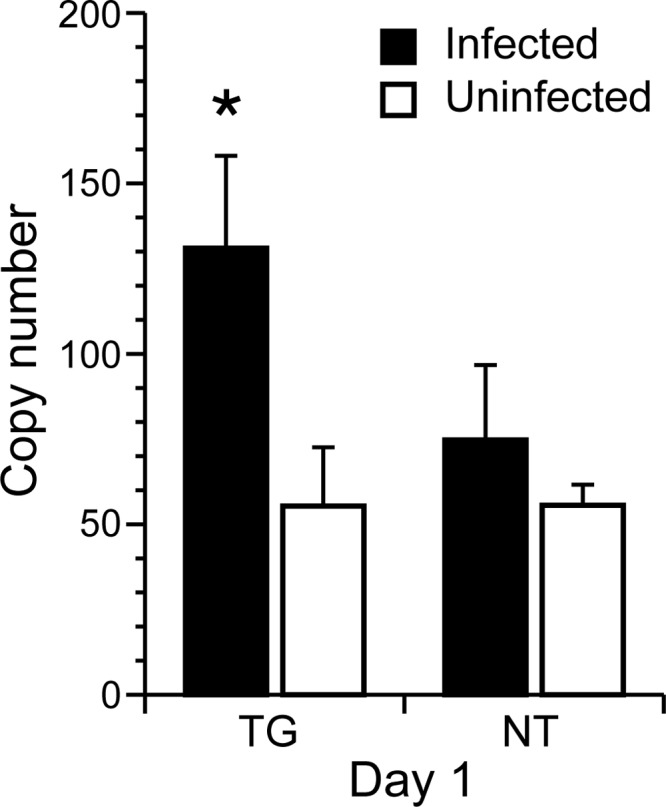
Transcript levels of IFN-β in total brain RNA of TG and NT mice at 1 day p.i. TG and NT mice (n = 5 per group) received intracranial inoculations of 4 × 104 TCID50 of Ed MeV. Total brain RNA was isolated from the hemisphere opposite the inoculation site. Mice inoculated with PBS served as uninfected controls. Significant elevation of IFN-β transcript levels following infection was observed only in hsp70-TG mice (P < 0.05; t test).
In order to establish the causal relationship between enhanced type I IFN expression and hsp70-mediated protection, we used mice in which the gene for the type I IFN receptor was disrupted (IFNAR−/−) and measured the impact upon hsp70-mediated host protection against intracranial viral challenge. TG, TG IFNAR−/−, NT, and NT IFNAR−/− mice were infected with Ed MeV and monitored over a 28-day time course for survival. As observed in previous studies (9), NT mice suffered 35% mortality (65% survival), whereas nearly all TG mice survived (95%), a significant difference between groups (P < 0.05) (Fig. 2). Disruption of the type I IFN receptor significantly increased mortality in both TG and NT mice (P value of 0.0002 for the difference between TG and TG IFNAR−/− mice and P value of 0.0035 for the difference between NT and NT IFNAR−/− mice). Now, however, there was no statistically significant difference in survival between the TG IFNAR−/− and NT IFNAR−/− groups (P = 0.51). These data clearly establish the causal relationship between hsp70-dependent IFN-β induction and protection against infection.
Fig 2.

Disruption of type I IFN signaling results in loss of hsp70-mediated host protection. Mice were inoculated intracranially in the left cerebral hemisphere with 4 × 104 TCID50 of Ed MeV. Sham-inoculated controls received PBS. Kaplan-Meier statistics were used to generate survival curves, and all mice were censored on day 28 p.i. The Wilcoxon and log rank tests of survival confirmed the significant increase in survival in infected TG mice (n = 14) relative to NT H-2d mice (n = 10) that has been previously reported (P < 0.05) (9). In contrast, disruption of the type I IFN receptor significantly decreased survival in NT IFNAR−/− (n = 14) relative to NT mice, and in TG IFNAR−/− (n = 15) relative to TG mice, eliminating a significant survival advantage in TG IFNAR−/− relative to NT IFNAR−/− mice (P = 0.51). Results support the pivotal role of type I IFN in hsp70-mediated host protection.
hsp70 enhances macrophage activation in response to viral infection.
Canonical pathway analysis shows that brain macrophage activation is enhanced by hsp70 in response to infection. Increased levels of STAT-1 expression were found at 5 and 10 days p.i. and of CHUK expression at 5 days p.i. (Tables 3 and 4). CHUK is part of the NF-κB signaling pathway and is thus essential for TLR-mediated microglial activation through NF-κB as well as for type I IFN induction (30). TLR2 and TLR4 are primarily expressed on cells of myeloid origin in the mouse brain (i.e., microglia), and both are specific markers for macrophage activation in the brain after infection, with TL2 expression being affected to a greater degree than TLR4 (38). TLR2 was upregulated by infection at 5 and 10 days and TLR4 at 10 days p.i., although no significant differences were observed between TG and NT mice (Tables 3 and 4). Expression of other TLRs (TLR2 to -5 and TLR7, -9, -12, and -13) was not affected by infection. Additional markers of macrophage activation are the induction of MHC I and II molecules, and infected hsp70-TG mice demonstrated a significant increase in expression of H2-D1, H2-K1, and H2-EA at 5 days p.i. The genes for two of these (H-2D1 and H2-K1) exhibited the overall greatest induction, and levels of expression were further increased at 10 days. Induction of genes for antigen-presenting molecules was not detected in NT mice at 5 or 10 days p.i.
Table 3.
Increased expression of genes reflecting macrophage activation and antigen presentation in MeV-infected TG relative to NT mice, 5 days p.i.a
| Gene product | GenBank accession no. | Description | (TGi − NTi) − (TGu − NTu) |
TGi − TGu |
NTi − NTu |
|||
|---|---|---|---|---|---|---|---|---|
| Fold change | P | Fold change | P | Fold change | P | |||
| H2-D1 | M83244 | MHC I | 7.1 | 4.7E−9 | 7.5 | 3.0E−12 | — | — |
| H2-K1 | BC018402 | MHC I | 2.5 | 1.7E−5 | 2.5 | 8.2E−8 | — | — |
| H2-EA | U13648 | MHC II | 1.9 | 2.1E−2 | 2.0 | 1.0E−3 | — | — |
| CHUK | AU045682 | Conserved helix-loop-helix ubiquitous ligase; IκB kinase alpha | 1.3 | 9.9E−4 | 1.2 | 4.5E−4 | — | — |
| STAT-1 | AW214029 | Signal transducer and activator of transcription | 1.9 | 3.1E−3 | 6.9 | 1.3E−12 | 3.5 | 1.5E−9 |
| TLR2 | NM_011905 | Toll-like receptor 2 | — | — | 2.0 | 2.8E−5 | 1.7 | 6.8E−4 |
Results are based upon transcriptome analysis of total brain RNA. All changes represent induction, and the absence of a significant difference is indicated by a dash. Superscript “i” and “u” represent infected and uninfected mice, respectively.
In order to verify macrophage activation in situ, we performed immunohistochemical staining of MHC II in brain tissue sections harvested at 5 days p.i. Constitutive MHC II expression is not observed in normal brains of humans and rodents; however, expression is highly inducible in activated microglia and inducible to very low levels in endothelial cells and astrocytes (39, 40). The analysis was focused on the hippocampus, since virus-induced cytopathic effects in the hippocampus have been previously correlated to mortality (9). Specific staining was readily detected in infected TG mice, in cells with a morphology and localization characteristic of perivascular and parenchymal microglia (Fig. 3A). Using the pixel counter algorithm (Aperio Imagescope) to quantify signal on digitized slides, the level of positivity was significantly greater for TG mice than for NT mice (P < 0.05; t test) (Fig. 3B). The level of MHC II signal in infected NT mice was not significantly different from the signal obtained from uninfected control tissues. Macrophages originating from blood are characterized by high-level expression of CD45 (common leukocyte antigen), in contrast to a low level of expression in resident microglia (41). CD45 was not detected in these regions (data not shown), indicating that infiltrating monocytes are not a significant contributor to the brain macrophage population at 5 days p.i.
Fig 3.

Expression of MHC II in hippocampus of infected TG relative to infected NT mice. Signal was evaluated in the hemisphere opposite the inoculation site. (A) Representative expression of MHC II in infected NT and TG mice (NT and TG, respectively) and the uninfected controls (NT and TG Uninf) at 5 days p.i. (n = 4/group). Shown are paraformaldehyde-fixed frozen sections, with immunoperoxidase staining and hematoxylin counterstain, at magnifications of ×4 (left four panels) and ×40 (right). Virus-induced cytopathic effects or cellular inflammatory infiltrates affecting the granular cell neurons of the dentate gyrus (GrDG) and pyramidal cell neurons of the CA1, CA2, and CA3 regions of the hippocampus are not present at this early p.i. interval, although induced MHC II expression on parivascular and parenchymal microglia is evident in TG mice. The signal levels between uninfected TG and NT mice and infected NT mice were equivalent. (B) Quantification of pixel counts from digitized images (number of positive pixels/total number of pixels) shows that the increased staining in infected TG relative to infected NT mice is statistically significant (P < 0.05, t test).
Collectively, the in vivo data establish a correlation between increased type I IFN production and enhanced macrophage activation in infected hsp70-TG mice relative to those in infected NT mice. This finding is of potential significance in that activated uninfected brain macrophages, not infected neurons, are the predominant source of IFN-β in the virus-infected brain (42). MeV infection does not induce significant levels of IFN-β in neurons, a phenomenon that may reflect a more general restriction of type I IFN production in this cell type (36, 43, 44). In vitro studies were subsequently designed to establish an hsp70-dependent link between infected neurons and the induction of IFN-β in uninfected macrophages. The hypothesis tested was that virus infection causes extracellular release of hsp70 from neurons, and the extracellular hsp70 binds TLR2 and -4 on macrophages to induce IFN-β.
hsp70 release by MeV-infected neuronal cells.
Mouse neuroblastoma cells (N2a) were used to address the potential release of hsp70 upon infection. These cells faithfully reproduce neuronal patterns of gene expression and support a noncytolytic mode of MeV replication that is characteristic of MeV in vivo neuronal infection (9, 10). Like cortical neurons, N2a cells support release of exosomes (45), an established mechanism for hsp70 release from viable cells. Stably transfected N2a cell lines have been generated that either lack basal hsp70 (N2a-V) or constitutively express hsp70 (N2a-HSP). These lines reproduce the pattern of hsp70 expression observed in NT and hsp70-TG mice, and Ed MeV replication parameters have been thoroughly characterized in these cells (26). The cell lines offer a significant advantage over the use of primary cultures in studying virus-hsp70 interaction. Published work comparing in vitro and in vivo expression of hsp70 in rodents shows that primary neuronal (and glial) cultures exhibit a stress-conditioned phenotype (i.e., a phenotype mediated by elevated basal levels of endogenous heat shock proteins, including hsp70) (46, 47). Primary cultures could thus obscure differences between infected neurons derived from hsp70-transgenic and nontransgenic mice, whereas baseline and/or virus-induced endogenous hsp70 levels are stable and defined in the N2a cells modeling neuronal infection.
N2a-HSP and N2a-V cells were infected with Ed MeV (MOI = 1) and monitored for hsp70 release into culture supernatant by ELISA and for cell viability based upon LDH release. Previous work shows that under these infection conditions, significant viral transcript production (i.e., secondary transcription) begins after 12 h p.i., with antigen expression detectable in approximately 18% of N2a-V and N2a-HSP cells by immunocytochemistry at 24 h (26). Peak viral transcript production occurs at 60 h p.i., which corresponds to peak infectious viral progeny release (26). Significant increases in extracellular hsp70 levels were observed between 0 and 10 h p.i. in both infected N2a-HSP cells and N2a-V cells, with the level of release being significantly greater in the infected N2a-HSP cells. Levels of extracellular hsp70 were an average of 3.3 ng/ml above background for N2a-HSP cells, compared to 0.4 ng/ml for N2a-V cells. The source of hsp70 in the N2a-V cells is likely virus-induced endogenous hsp70, previously reported for Ed MeV in this cell line (48). Infection did not induce hsp70 release in subsequent postinfection intervals (Fig. 4). No significant differences between LDH levels in culture supernatants between infected and uninfected cell controls were found between 0 and 60 h p.i. (Fig. 4), consistent with the noncytolytic nature of infection that is detected by light microscopy.
Fig 4.

MeV infection induces early extracellular release of hsp70 from viable neuronal cells. Mouse neuroblastoma (N2a) cells were infected with Ed MeV at an MOI of 1.0 and monitored for 60 h p.i. The N2a cells were stably transfected to constitutively express hsp70 (N2a-HSP) or contained empty vector (N2a-V). Virus-induced cell death was not observed, based upon lack of a significant difference in LDH release between infected and uninfected cells (−). Extracellular hsp70 levels were measured by ELISA. Results reflect levels measured over infected (Inf) and uninfected (Uninf) cells at discrete postinfection intervals. The shaded region represents background measured over uninfected cells, and asterisks denote significant increases over background (ANOVA; P < 0.05). Detergent-lysed cells served as positive controls for the LDH and hsp70 analyses.
hsp70 induces IFN-β transcription in a mouse microglial cell line.
The mouse microglial cell line BV-2 was used to establish the potential of extracellular hsp70 to induce IFN-β through TLR2 and -4. BV-2 cells are known for their ability to model inflammatory responses observed in primary microglial culture (49).
Initial experiments used tissue culture supernatants containing hsp70 released from Ed MeV-infected N2a-HSP and N2a-V cells. BV-2 medium was replaced with the supernatant, cells were incubated for 6 h, and total cell RNA was analyzed by real-time RT-PCR for IFN-β transcripts. The supernatants from N2a cells were harvested at 20 h p.i., reflecting the early postinfection interval in which IFN-β induction is observed in vivo. Significant production of cell-free infectious viral progeny does not occur until 36 h p.i. in N2a cells, although we included UV treatment to inactivate any low level of infectious virus that might be present. Viral titrations showed that no virus is detectable in media recovered from N2a-V cells at 20 h, whereas an average of 370 TCID50/ml is recovered from N2a-HSP cells and this infectivity is eliminated by UV treatment. We could not demonstrate IFN-β induction in BV-2 by these low levels of inactivated virus, using dilutions of UV-treated stock virus added to the BV-2 culture media (data not shown). Significant induction of IFN-β transcripts was observed in hsp70-containing media from infected N2a-HSP cells relative to uninfected N2a-HSP cells (Fig. 5A). Significant induction was not observed using media from infected N2a-V cells. The specific role of hsp70 in mediating IFN-β induction was shown using antibody neutralization of hsp70 in supernatants from infected N2a-HSP cells (Fig. 5B).
Fig 5.

hsp70 released from Ed MeV-infected N2a-HSP cells can induce IFN-β transcription in a mouse microglial cell line (BV-2). (A) Supernatants harvested from infected (Inf) and uninfected (Uninf) N2a-HSP and N2a-V cells were cocultured with BV-2 cells for 6 h, followed by quantification of IFN-β transcripts in the BV-2 cells by real-time RT-PCR analysis of total cell RNA. Significant induction of IFN-β transcripts was observed in BV-2 cells treated with supernatant recovered from infected N2a-HSP, relative to cells treated with supernatant from uninfected N2a-HSP cells and infected and uninfected N2a-V cells (ANOVA; P < 0.05). (B) Polyclonal rabbit anti-hsp70 IgG (αHSP70), added to culture supernatants from infected N2a-HSP cells, suppressed induction of IFN-β transcripts in BV-2 cells to levels observed in cells cocultured with supernatants derived from uninfected N2a-HSP cells. Significant differences are noted by an asterisk (ANOVA; P < 0.05). Negative-control rabbit total IgG caused a partial nonspecific suppression of IFN induction, although the level of suppression was not statistically significant.
To confirm these results, we examined the potential of purified recombinant hsp70 to induce IFN-β transcription in BV-2 cells. hsp70-1 was added to culture media for 6 h, followed by total cell RNA isolation for quantification of IFN-β transcripts by real-time qRT-PCR. A range of concentrations was employed (i.e., 2.2 to 142 nM), bracketing the concentration found in culture supernatants from infected N2a-HSP cells (i.e., approximately 95 nM). hsp70 induced IFN-β transcripts in BV-2 cells in a dose-dependent manner (Fig. 6A). Induction was not suppressed by the addition of polymyxin B, whereas induction was abrogated when the hsp70 was heat denatured (Fig. 6B). This is proof that induction was mediated by hsp70 and not by lipopolysaccharide (LPS) contamination. Induction of IFN-β through hsp70 was confirmed in a second mouse microglial cell line, NR-9460 (50), and also in primary cultures of mouse bone marrow-derived macrophages (Fig. 6C).
Fig 6.

Purified hsp70 induces IFN-β transcripts in mouse microglial cell lines and primary cultures of bone marrow-derived macrophages. Purified recombinant hsp70-1 was incubated for 6 h with the mouse microglial cell lines BV-2 and NR-9460 and primary cultures of bone marrow-derived macrophages. Total RNA was then isolated for real-time RT-PCR quantification of IFN-β transcripts. (A) hsp70 induced IFN-β transcripts in a dose-dependent manner in BV-2 cells. Asterisks denote significant increases over untreated cells (ANOVA; P < 0.05). (B) Induction of IFN-β transcripts by 142 nM hsp70 was not diminished by the addition of 30 μg/ml of polymyxin B, whereas induction was abrogated when the hsp70 was heat denatured. Asterisks denote significant increases over untreated cells and cells treated with denatured hsp70 (ANOVA; P < 0.05). (C) Addition of 142 nM hsp70 also resulted in significant induction of IFN-β transcripts in the mouse microglial cell line NR-9460 and primary cultures of macrophages derived from NT mice (t test; P < 0.05).
hsp70 is a known ligand of TLR2 and TLR4, where extracellular hsp70 activates microglia and stimulates proinflammatory cytokine gene expression through NF-κB (16, 51). The ability of hsp70 to induce IFN-β has not previously been addressed, although other TLR4 ligands (e.g., LPS) are capable of IFN-β induction, requiring signaling through IRF3 (44). The role of TLR2 and TLR4 in hsp70-mediated induction of IFN-β transcription was shown in this study by inhibiting the response with TLR-specific antibodies. Immunocytochemical staining of BV-2 cells and RT-PCR analysis of total cell RNA showed that BV-2 cells express both TLR2 and -4 (data not shown). Antibody against TLR2, applied 1 h prior to the addition of 142 nM hsp70, suppressed induction of IFN-β by approximately 68%, whereas isotype control antibody had no significant effect (Fig. 7A). Similarly, antibody against TLR4 suppressed induction of IFN-β by approximately 79%, and the isotype control antibody had no inhibitory effect (Fig. 7B). These data indicate that hsp70 interaction with both TLR2 and TLR4 results in the induction of IFN-β in microglial cells.
Fig 7.

Antibody against TLR2 and TLR4 inhibits hsp70-mediated induction of IFN-β. BV-2 cells were treated with 142 nM hsp70 for 6 h, followed by quantitation of IFN-β transcripts by real-time RT-PCR. Induction was significantly inhibited (*) (ANOVA; P < 0.05) by the addition of 20 μg/ml of monoclonal antibody specific to TLR2 (A) or TLR4 (B), applied 1 h prior to the addition of hsp70. Identical concentration of isotype control antibodies had no effect on induction.
DISCUSSION
Induction of type I IFN was a key difference in the response to infection between hsp70-TG and NT mice, being correlated to the increased survival that was associated with constitutive hsp70 expression in neurons. IFN-β is the principal type I IFN produced in brain and a key mediator of protection against neurovirulence for multiple viruses. The central role of type I IFN in hsp70-mediated host protection in the current study was proven by the loss of any significant difference in mortality between infected TG and NT mice when type I IFN signaling was disrupted. Results in TG mice are relevant to humans in that constitutive hsp70 levels are a human pattern of expression not observed in NT mice (14). MeV infection does not induce IFN-β in neurons (36, 43, 52), with activated microglia considered the predominant source of IFN-β in this system (42). Our results support a model explaining how virus-infected neuronal cells can induce IFN-β in uninfected brain macrophages, namely, through the extracellular release of hsp70, which can, in turn, engage microglial TLR2 and -4 (Fig. 8). In this capacity, extracellular hsp70 acts as a danger signal to the presence of virus infection, inducing innate immune responses of microglia. Although the concept of extracellular hsp70 as a danger signal to the immune system has previously been suggested by models of bacterial infection and tissue injury (53, 54), our results are the first to show that hsp70 is released from viable virus-infected neuronal cells and that hsp70 interacts with TLR2 and -4 of a brain macrophage line to induce IFN-β.
Fig 8.

Model for IFN-β induction in uninfected microglia in response to MeV neuronal infection. A neuron is pictured at the left, containing single-stranded viral genomes of MeV. Infection triggers extracellular release of hsp70, which then acts as a damage-associated molecular pattern (DAMP), interacting with TLR2 and TLR4 of uninfected brain macrophages (right). TLR signaling is known to induce IFN-β transcription through IRF3, AP-1, and NF-κB, and MHC expression is mediated by NF-κB.
Host protective roles of IFN-β span both innate and adaptive immunity, including the induction of antiviral IFN-stimulated gene products, induction of macrophage activation and antigen presentation, and support of virus-specific T cell activation to produce IFN-γ, which is ultimately responsible for noncytolytic viral clearance in the mouse model of MeV brain infection (37, 55, 56). Although neurons are limited in their capacity to produce IFN-β in response to MeV infection, they do maintain the capacity to respond to IFN-β by producing antiviral IFN-stimulated gene products (36). Eight IFN-stimulated genes were significantly increased in infected TG relative to NT mice, with the magnitude of increase ranging from 1.3- to 2.3-fold. Expression of combinations of interferon-stimulated genes has an additive inhibitory effect on viral replication, such that the individual fold changes underestimate the cumulative level of host protection (57). In fact, expression of several IFN-stimulated genes at low levels is viewed as a means of achieving a global antiviral effect with minimal impact on normal cell function (58). A less stringent analysis using the comparison involving subtraction of expression in infected NT mice from that in infected TG mice identified additional type I IFN-inducible genes that were significantly induced in both infected NT and TG mice at 5 days p.i. (P < 0.05), with greater induction in TG animals. These included 2′5′-oligoadenylate synthetase, eukaryotic translation initiation factor 2α (PKR), and MDA5. The P value approached but did not achieve significance in the higher-stringency comparison obtained by the equation (infected TG − infected NT) − (uninfected TG − uninfected NT), even though there was no significant difference in basal expression of these genes in uninfected TG and NT groups. Here, the differences between infected TG and NT mice were 1.5-fold for 2′,5′-oligoadenylate synthetase (P = 0.07), 1.4-fold for PKR (P = 0.09), and 1.5-fold for MDA5 (P = 0.06).
The hsp70-dependent increase in macrophage activation and antigen presentation may be a direct effect of extracellular hsp70 or an indirect consequence of hsp70-mediated IFN induction. IFN-β drives macrophage activation through the type I IFN receptor (IFNAR), resulting in the induction of antigen-presenting complexes and IFN-β expression by these activated cells, thus setting into motion an autocrine feedback loop (59). We were unable to establish the contribution of IFNAR to macrophage activation using infected IFNAR−/− mice. Deletion of type I IFN signaling resulted in a dramatic increase in virus-induced necrosis (data not shown), and necrotic tissue releases numerous ligands capable of macrophage activation that are in addition to IFN-β.
Enhanced expression of antigen-presenting complexes in microglia of hsp70-TG mice at 5 days p.i. may contribute to host protection by adaptive immune responses at 10 days p.i. Activation of virus-specific T cells and production of IFN-γ requires an encounter with an antigen-presenting cell in the presence of type I IFN (37, 56). The activation would rely on the well-established capacity of murine microglia to cross-present exogenous antigen (in this instance, viral antigen from infected neurons) on MHC I (60). In vitro studies with human neuronal and glial cell lines and in vivo work in mouse models show that acute MeV infection is a potent stimulus for MHC I expression in glia but not neurons (43, 61).
Increased mortality in the infected NT IFNAR−/− mice relative to that in infected NT mice indicates that type I IFNs do play a role in host protection despite the absence of constitutive hsp70 expression. This conclusion is supported by the transcriptome analysis of NT mice at 5 days p.i., where genes in the IFN signaling pathways are significantly induced by infection compared to uninfected NT mice. At 10 days p.i., levels of IFN-inducible genes are greater than observed at 5 days p.i., and the levels are now comparable between infected TG and NT mice. hsp70, in the context of viral infection, can thus be seen to enhance the early type I IFN response that is otherwise observed in NT mice, providing a greater margin for host protection against virus-induced mortality that begins as early as 11 days p.i.
Results of the current work help to resolve the long-standing question of how infected neurons might trigger IFN-β induction and antigen cross-presentation in brain macrophages (55). Infected neurons may release both viral gene products (pathogen-associated molecular patterns [PAMPs]) and endogenous ligands (damage-associated molecular patterns [DAMPs]) capable of activating TLRs, and our work suggests a dominant role of hsp70 as a DAMP that engages TLR2 and -4 of macrophages, resulting in macrophage activation and induction of IFN-β (62, 63). Changes in TLR gene expression are a direct measure of pathway activation in response to TLR-specific ligands (64). Of the eight TLRs whose gene expression was detected in the transcriptome analysis, only TLR2 and -4 exhibited changes associated with infection. Inability to detect TLR4 induction at 5 days p.i. may reflect the limited sensitivity of the transcriptome analysis or the more complex transcriptional regulation of TLR4 in response to ligands, characterized by a biphasic response in which transcript levels are initially suppressed (64). The level of hsp70 release from MeV is proportionate to the intracellular hsp70 concentration, so the stimulus for IFN-β induction would be greater in TG relative to NT mice. hsp70 release from virus-infected cells has previously been described, but only in the context of lytic virus infection, namely, parvovirus infection of a tumor cell line (65). Release of hsp70 from viable cells that is documented in the current work is compatible with virus-induced exosomal secretion or microvesicular shedding (66). hsp70 is a major constituent of exosomes and exosomal release, and hsp70 content of exosomes has been shown to be increased following intracellular bacterial infection, in which it can induce proinflammatory responses of macrophages (8, 67). Neuronal cells, including mouse N2a and primary cortical neurons, release exosomes constitutively (45), which may explain the elevated baseline levels of hsp70 measured in culture supernatants of N2a-HSP relative to N2a-V (Fig. 4). Regulation of neuronal exosomal release is an active area of study (45), and work in progress will characterize the mechanism of virus-induced hsp70 release from viable cells. Extracellular release of hsp70 may also facilitate cross-presentation of viral antigen by brain macrophages (68). hsp70 binds the MeV N protein, specifically recognizing a domain containing an H-2d determinant of cell-mediated immunity (69, 70). These roles of extracellular hsp70 may be the basis for adjuvant effects observed following DNA vaccination, in which expression of a structural protein of Japanese encephalitis virus or herpes simplex virus type 1 (HSV-1) was protective against subsequent lethal viral challenge in neonatal mice when coexpressed with hsp70, an effect attributed to a stronger cellular immune response (71, 72). In addition to these direct effects of hsp70, we must also consider the potential contribution of hsp70-mediated stimulation of viral gene expression to antiviral immunity (9), where virus-hsp70 interaction may enhance extracellular release of DAMPS and/or PAMPS and increase levels of viral antigen available for cross-presentation.
In conclusion, we show that hsp70 may serve as a sensor of virus infection, enhancing the antiviral state through IFN-β. The model is relevant to MeV infection of humans, and the efficiency of this host response may explain the low incidence of MeV-induced neurological disease (reviewed in reference 13). Fever, a diagnostic feature of MeV infection in humans (1), is also a potent inducer of hsp70 and would thus drive an even more robust innate immune response. Supporting this view is our earlier work in mice, in which hyperthermic induction of hsp70 was correlated to enhanced viral clearance in brain (12). A role for hsp70 in eliciting innate immunity has potentially broad relevance. Viruses causing acute neurological disease include members of the herpesvirus, flavivirus, picornavirus, rhabdovirus, and bunyavirus families, and in each case there is evidence that pathogenicity is inversely related to type I IFN production (73–78). These observations warrant further investigation into mechanisms by which hsp70 is released from cells, particularly the relationship to virus gene expression, and how this novel axis of antiviral immunity might be exploited therapeutically.
ACKNOWLEDGMENTS
This work was supported by funds from the National Institute of Neurological Disorders and Stroke (R01NS31693).
We thank Phil Popovich for aid with the immunohistochemical staining.
Footnotes
Published ahead of print 7 November 2012
REFERENCES
- 1. Morrison-Bogorad M, Zimmerman AL, Pardue S. 1995. Heat-shock 70 messenger RNA levels in human brain: correlation with agonal fever. J. Neurochem. 64:235–246 [DOI] [PubMed] [Google Scholar]
- 2. Hutchins SS, Papania MJ, Amler R, Maes EF, Grabowsky M, Bromberg K, Glasglow V, Speed T, Bellini WJ, Orenstein WA. 2004. Evaluation of the measles clinical case definition. J. Infect. Dis. 189(Suppl 1):S153–S159 [DOI] [PubMed] [Google Scholar]
- 3. Mackowiak PA. 1994. Fever: blessing or curse? A unifying hypothesis. Ann. Intern. Med. 120:1037–1040 [DOI] [PubMed] [Google Scholar]
- 4. Mackowiak PA. 2000. Physiological rationale for suppression of fever. Clin. Infect. Dis. 31(Suppl 5):S185–S189 [DOI] [PubMed] [Google Scholar]
- 5. Milner CM, Campbell RD. 1990. Structure and expression of the three MHC-linked HSP70 genes. Immunogenetics 32:242–251 [DOI] [PubMed] [Google Scholar]
- 6. Wells AD, Rai SK, Salvato MS, Band H, Malkovsky M. 1998. Hsp72-mediated augmentation of MHC class I surface expression and endogenous antigen presentation. Int. Immunol. 10:609–617 [DOI] [PubMed] [Google Scholar]
- 7. Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. 2000. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 12:1539–1546 [DOI] [PubMed] [Google Scholar]
- 8. Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, Multhoff G, Arispe N, De Maio A. 2008. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 180:4299–4307 [DOI] [PubMed] [Google Scholar]
- 9. Carsillo T, Carsillo M, Traylor Z, Rajala-Schultz P, Popovich P, Niewiesk S, Oglesbee M. 2009. Major histocompatibility complex haplotype determines hsp70-dependent protection against measles virus neurovirulence. J. Virol. 83:5544–5555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carsillo T, Traylor Z, Choi C, Niewiesk S, Oglesbee M. 2006. hsp72, a host determinant of measles virus neurovirulence. J. Virol. 80:11031–11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakae H, Kohase M, Kurata T, Yoshikura H. 1997. Isolation of a measles virus variant: protection of newborn mice from measles encephalitis by 24 h prior intracerebral inoculation with the variant. Arch. Virol. 142:1937–1952 [DOI] [PubMed] [Google Scholar]
- 12. Carsillo T, Carsillo M, Niewiesk S, Vasconcelos D, Oglesbee M. 2004. Hyperthermic pre-conditioning promotes measles virus clearance from brain in a mouse model of persistent infection. Brain Res. 1004:73–82 [DOI] [PubMed] [Google Scholar]
- 13. Oglesbee M, Niewiesk S. 2011. Measles virus neurovirulence and host immunity. Future Virol. 6:85–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pardue S, Wang S, Miller MM, Morrison-Bogorad M. 2007. Elevated levels of inducible heat shock 70 proteins in human brain. Neurobiol. Aging 28:314–324 [DOI] [PubMed] [Google Scholar]
- 15. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. 2002. Novel signal transduction pathway utilized by extracellular HSP70: role of TLR2 and TLR4. J. Biol. Chem. 277:15028–15034 [DOI] [PubMed] [Google Scholar]
- 16. Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S. 2002. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 16:601–603 [DOI] [PubMed] [Google Scholar]
- 17. Beyer SJ, Zhang X, Jimenez RE, Lee ML, Richardson AL, Huang K, Jhiang SM. 2011. Microarray analysis of genes associated with cell surface NIS protein levels in breast cancer. BMC Res. Notes 4:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264 [DOI] [PubMed] [Google Scholar]
- 19. Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl. Genet. Mol. Biol. 3:Article3 [DOI] [PubMed] [Google Scholar]
- 20. Gordon A, Glazko G, Qiu X, Yakovlev A. 2007. Control of the mean number of false discoveries, Bonferroni and stability of multiple testing. Ann. Appl. Stat. 1:179–190 [Google Scholar]
- 21. Killedar S, DiRosario J, Divers E, Popovich PG, McCarty DM, Fu HY. 2010. Mucopolysaccharidosis IIIB, a lysosomal storage disease, triggers a pathogenic CNS autoimmune response. J. Neuroinflammation 7:39 doi:10.1186/1742-2094-7-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. 2003. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J. Comp. Neurol. 462:223–240 [DOI] [PubMed] [Google Scholar]
- 23. Kigerl KA, McGaughy VM, Popovich PG. 2006. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J. Comp. Neurol. 494:578–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerlach N, Schimmer S, Weiss S, Kalinke U, Dittmer U. 2006. Effects of type I interferons on Friend retrovirus infection. J. Virol. 80:3438–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921 [DOI] [PubMed] [Google Scholar]
- 26. Carsillo T, Zhang X, Vasconcelos D, Niewiesk S, Oglesbee M. 2006. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of Edmonston measles virus. J. Virol. 80:2904–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Couturier M, Buccellato M, Costanzo S, Bourhis JM, Shu Y, Nicaise M, Desmadril M, Flaudrops C, Longhi S, Oglesbee M. 2010. High affinity binding between Hsp70 and the C-terminal domain of the measles virus nucleoprotein requires an Hsp40 co-chaperone. J. Mol. Recognit. 23:301–315 [DOI] [PubMed] [Google Scholar]
- 28. Zhang X, Bourhis JM, Longhi S, Carsillo T, Buccellato M, Morin B, Canard B, Oglesbee M. 2005. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 337:162–174 [DOI] [PubMed] [Google Scholar]
- 29. Longbrake EE, Lai W, Ankeny DP, Popovich PG. 2007. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J. Neurochem. 102:1083–1094 [DOI] [PubMed] [Google Scholar]
- 30. Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, Ohara O, Akira S, Kaisho T. 2006. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440:949–953 [DOI] [PubMed] [Google Scholar]
- 31. Waddell SJ, Popper SJ, Rubins KH, Griffiths MJ, Brown PO, Levin M, Relman DA. 2010. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS One 5:e9753 doi:10.1371/journal.pone.0009753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlow DA, Teh SJ, Teh HS. 1998. Specific antiviral activity demonstrated by TGTP, a member of a new family of interferon-induced GTPases. J. Immunol. 161:2348–2355 [PubMed] [Google Scholar]
- 33. Fischer T, Aman J, van der Kuip H, Rudolf G, Peschel C, Aulitzky WE, Huber C. 1996. Induction of interferon regulatory factors 2′-5′ oligoadenylate synthetase, P68 kinase and RNase L in chronic myelogenous leukaemia cells and its relationship to clinical responsiveness. Br. J. Haematol. 92:595–603 [DOI] [PubMed] [Google Scholar]
- 34. Hui KP, Lee SM, Cheung CY, Mao H, Lai AK, Chan RW, Chan MC, Tu W, Guan Y, Lau YL, Peiris JS. 2011. H5N1 influenza virus-induced mediators upregulate RIG-I in uninfected cells by paracrine effects contributing to amplified cytokine cascades. J. Infect. Dis. 204:1866–1878 [DOI] [PubMed] [Google Scholar]
- 35. Schmeisser H, Mejido J, Balinsky CA, Morrow AN, Clark CR, Zhao T, Zoon KC. 2010. Identification of alpha interferon-induced genes associated with antiviral activity in Daudi cells and characterization of IFIT3 as a novel antiviral gene. J. Virol. 84:10671–10680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. 2006. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U. S. A. 103:7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pien GC, Nguyen KB, Malmgaard L, Satoskar AR, Biron CA. 2002. A unique mechanism for innate cytokine promotion of T cell responses to viral infections. J. Immunol. 169:5827–5837 [DOI] [PubMed] [Google Scholar]
- 38. Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. 2007. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem. Biophys. Res. Commun. 359:574–579 [DOI] [PubMed] [Google Scholar]
- 39. Kreutzberg GW. 1996. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19:312–318 [DOI] [PubMed] [Google Scholar]
- 40. Lemstra AW, Groen in't Woud JCM, Hoozemans JJM, van Haastert ES, Rozemuller AJM, Eikelenboom P, van Gool WA. 2007. Microglia activation in sepsis: a case-control study. J. Neuroinflammation 4:4 doi:10.1186/1742-2094-4-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. 1991. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl. Acad. Sci. U. S. A. 88:7438–7442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P. 2012. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J. Virol. 86:11223–11230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dhib-Jalbut S, Xia J, Rangaviggula H, Fang YY, Lee T. 1999. Failure of measles virus to activate nuclear factor-kappa B in neuronal cells: implications on the immune response to viral infections in the central nervous system. J. Immunol. 162:4024–4029 [PubMed] [Google Scholar]
- 44. Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. 2005. LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood 106:3114–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yuyama K, Sun H, Mitsutake S, Igarashi Y. 2012. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 287:10977–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marini AM, Kozuka M, Lipsky RH, Nowak TS., Jr 1990. 70-kilodalton heat shock protein induction in cerebellar astrocytes and cerebellar granule cells in vitro: comparison with immunocytochemical localization after hyperthermia in vivo. J. Neurochem. 54:1509–1516 [DOI] [PubMed] [Google Scholar]
- 47. Voisin PJ, Pardue S, Macouillard F, Yehia G, Labouesse J, Morrison-Bogorad M. 1996. Differential expression of heat shock 70 proteins in primary cultures from rat cerebellum. Brain Res. 739:215–234 [DOI] [PubMed] [Google Scholar]
- 48. Oglesbee MJ, Pratt M, Carsillo T. 2002. Role for heat shock proteins in the immune response to measles virus infection. Viral Immunol. 15:399–416 [DOI] [PubMed] [Google Scholar]
- 49. Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M. 2009. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26:83–94 [DOI] [PubMed] [Google Scholar]
- 50. Glaser KC, Hagos B, Molestina RE. 2011. Effects of Toxoplasma gondii genotype and absence of host MAL/Myd88 on the temporal regulation of gene expression in infected microglial cells. Exp. Parasitol. 129:409–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schell JB, Crane CA, Smith MF, Jr, Roberts MR. 2007. Differential ex vivo nitric oxide production by acutely isolated neonatal and adult microglia. J. Neuroimmunol. 189:75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robinzon S, Dafa-Berger A, Dyer MD, Paeper B, Proll SC, Teal TH, Rom S, Fishman D, Rager-Zisman B, Katze MG. 2009. Impaired cholesterol biosynthesis in a neuronal cell line persistently infected with measles virus. J. Virol. 83:5495–5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Awad H, Suntres Z, Heijmans J, Smeak D, Bergdall-Costell V, Christofi FL, Magro C, Oglesbee M. 2008. Intracellular and extracellular expression of the major inducible 70kDa heat shock protein in experimental ischemia-reperfusion injury of the spinal cord. Exp. Neurol. 212:275–284 [DOI] [PubMed] [Google Scholar]
- 54. Campisi J, Leem TH, Fleshner M. 2003. Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones 8:272–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Patterson CE, Lawrence DM, Echols LA, Rall GF. 2002. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J. Virol. 76:4497–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Slifka MK, Rodriguez F, Whitton JL. 1999. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature 401:76–79 [DOI] [PubMed] [Google Scholar]
- 57. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. U. S. A. 109:4239–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Faul EJ, Wanjalla CN, Suthar MS, Gale M, Wirblich C, Schnell MJ. 2010. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 6:e1001016 doi:10.1371/journal.ppat.1001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beauvillain C, Donnou S, Jarry U, Scotet M, Gascan H, Delneste Y, Guermonprez P, Jeannin P, Couez D. 2008. Neonatal and adult microglia cross-present exogenous antigens. Glia 56:69–77 [DOI] [PubMed] [Google Scholar]
- 61. Manchester M, Eto DS, Oldstone MB. 1999. Characterization of the inflammatory response during acute measles encephalitis in NSE-CD46 transgenic mice. J. Neuroimmunol. 96:207–217 [DOI] [PubMed] [Google Scholar]
- 62. Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511 [DOI] [PubMed] [Google Scholar]
- 63. Matzinger P. 2002. The danger model: a renewed sense of self. Science 296:301–305 [DOI] [PubMed] [Google Scholar]
- 64. Rehli M. 2002. Of mice and men: species variations of Toll-like receptor expression. Trends Immunol. 23:375–378 [DOI] [PubMed] [Google Scholar]
- 65. Moehler M, Zeidler M, Schede J, Rommelaere J, Galle PR, Cornelis JJ, Heike M. 2003. Oncolytic parvovirus H1 induces release of heat-shock protein HSP72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther. 10:477–480 [DOI] [PubMed] [Google Scholar]
- 66. Meckes DG, Jr, Raab-Traub N. 2011. Microvesicles and viral infection. J. Virol. 85:12844–12854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Anand PK, Anand E, Bleck CK, Anes E, Griffiths G. 2010. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS One 5:e10136 doi:10.1371/journal.pone.0010136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Basu S, Binder RJ, Ramalingam T, Srivastava PK. 2001. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14:303–313 [DOI] [PubMed] [Google Scholar]
- 69. Giraudon P, Buckland R, Wild TF. 1992. The immune response to measles virus in mice. T-helper response to the nucleoprotein and mapping of the T-helper epitopes. Virus Res. 22:41–54 [DOI] [PubMed] [Google Scholar]
- 70. Zhang X, Glendening C, Linke H, Parks CL, Brooks C, Udem SA, Oglesbee M. 2002. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 76:8737–8746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen W, Lin Y, Liao C, Hsieh S. 2000. Modulatory effects of the human heat shock protein 70 on DNA vaccination. J. Biomed. Sci. 7:412–419 [DOI] [PubMed] [Google Scholar]
- 72. Pack CD, Kumaraguru U, Suvas S, Rouse BT. 2005. Heat-shock protein 70 acts as an effective adjuvant in neonatal mice and confers protection against challenge with herpes simplex virus. Vaccine 23:3526–3534 [DOI] [PubMed] [Google Scholar]
- 73. Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. 2009. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 182:2297–2304 [DOI] [PubMed] [Google Scholar]
- 74. Faul EJ, Wanjalla CN, McGettigan JP, Schnell MJ. 2008. Interferon-beta expressed by a rabies virus-based HIV-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology 382:226–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hefti HP, Frese M, Landis H, Di Paolo C, Aguzzi A, Haller O, Pavlovic J. 1999. Human MxA protein protects mice lacking a functional alpha/beta interferon system against La Crosse virus and other lethal viral infections. J. Virol. 73:6984–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. 2005. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 79:4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sainz B, Jr, Halford WP. 2002. Alpha/beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 76:11541–11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 79:13350–13361 [DOI] [PMC free article] [PubMed] [Google Scholar]