

Abstract
Infectious bursal disease (IBD) is an acute, highly contagious, and immunosuppressive avian disease caused by IBD virus (IBDV). Although IBDV-induced immunosuppression has been well established, the underlying exact molecular mechanism for such induction is not very clear. We report here the identification of IBDV VP4 as an interferon suppressor by interaction with the glucocorticoid-induced leucine zipper (GILZ) in host cells. We found that VP4 suppressed the expression of type I interferon in HEK293T cells after tumor necrosis factor alpha (TNF-α) treatment or Sendai virus (SeV) infection and in DF-1 cells after poly(I·C) stimulation. In addition, the VP4-induced suppression of type I interferon could be completely abolished by knockdown of GILZ by small interfering RNA (siRNA). Furthermore, knockdown of GILZ significantly inhibited IBDV growth in host cells, and this inhibition could be markedly mitigated by anti-alpha/beta interferon antibodies in the cell cultures (P < 0.001). Thus, VP4-induced suppression of type I interferon is mediated by interaction with GILZ, a protein that appears to inhibit cell responses to viral infection.
INTRODUCTION
Infectious bursal disease (IBD), also called Gumboro disease, is an acute, highly contagious disease in young chickens that occurs across the world (1). Its causative agent, IBD virus (IBDV), destroys its target cells, the B-lymphocyte precursors. The diseased chickens suffer from a severe immunosuppression which leads to an increased susceptibility to other pathogens (2).
IBDV is an Avibirnavirus belonging to Birnaviridae family, which is composed of nonenveloped viruses containing two segments of double-stranded RNAs (A and B) (3). Whereas the short RNA, segment B (2.8 kb), encodes VP1, a RNA-dependent RNA polymerase (RdRp) (4, 5), segment A, the large molecule (3.17 kb), contains two partially overlapping open reading frames (ORFs) (2, 6). The first ORF encodes the nonstructural viral protein 5 (VP5), and the second one encodes a 110-kDa pVP2-VP4-VP3 precursor that can be cleaved by the proteolytic activity of VP4 to form viral proteins VP2, VP3, and VP4 (3, 6–8). VP2 and VP3 are the major structural proteins, constituting 51% and 40% of the virion, respectively (9). VP4, a viral protease, is able to cleave in trans and is responsible for the interdomain proteolytic autoprocessing of the pVP2-VP4-VP3 polyprotein encoded by RNA segment A into pVP2 precursor (48 kDa) as well as VP4 (28 kDa) and VP3 (32 kDa) (2, 10), and pVP2 is further processed at its C-terminal domain by VP4 to generate the mature capsid protein VP2 (41 kDa) and four small peptides (11). VP4 self-assembles and forms tubes with a diameter of about 25 nm (12). VP5, a highly basic, cysteine-rich nonstructural (NS) protein (17 kDa), is not present in the virion and can be detected only in IBDV-infected cells (13). Several lines of evidence suggest that it may play a role in the induction of apoptosis during IBDV infection (13–16).
Viruses have refined various strategies to suppress the host response against viral dissemination (17). IBDV infection induces altered expression of multiple genes that are related to T- and B-cell activation and differentiation, as well as activation of genes involved in Toll-like receptor (TLR)- and interferon (IFN)-mediated antiviral responses (18). Recently, it has been reported that IFN-α has strong antiviral activity in IBDV-infected cells (19), suggesting that type I interferon of host cells may play a critical role in combating IBDV.
Although VP4 has been known as a viral protease to cleave polyprotein pVP2-VP4-VP3 (20, 21), it may also be involved in viral pathogenesis (22). Thus, the overall functions of VP4 remain to be elucidated. In this study, we found that VP4 acts as a major IBDV component responsible for suppressing type I interferon expression via interaction with the glucocorticoid-induced leucine zipper (GILZ) of host cells. In support of a role for GILZ in cytokine response induced by VP4, knockdown of GILZ by small interfering RNA (siRNA) abolished VP4-induced suppression of type I interferon expression, accompanied by inhibition of IBDV replication.
MATERIALS AND METHODS
Cells and virus.
Both DF-1 cells (immortal chicken embryo fibroblasts [CEF]) and HEK293T cells were obtained from the ATCC. All cells were cultured in Dulbecco modified Eagle medium (DMEM) (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS) in a 5% CO2 incubator. Lx, a cell culture-adapted IBDV strain, was kindly provided by Jue Liu (Beijing Academy of Agriculture and Forestry, Beijing, China).
Reagents.
Endotoxin-free plasmid preparation kits were purchased from Aidlab (Beijing, China). All restriction enzymes were purchased from NEB (USA). pCMV-Myc, pDsRed-monomer-N1, pRK5-FLAG, and pEGFP-N1 vectors were obtained from Clontech (USA). Anti-c-Myc (sc-40), anti-green fluorescent protein (anti-GFP; sc-9996), anti-GILZ (sc-33780), and anti-β-actin (sc-1616-R) antibodies were obtained from Santa Cruz Biotechnology (USA). Anti-human IFN-α1 (ab11408) and anti-human IFN-β (ab6979) were purchased from Abcam (United Kingdom). Poly(I·C) and anti-FLAG monoclonal antibody (F1804) were purchased from Sigma. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG, tetramethyl rhodamine isocyanate (TRITC)-conjugated goat anti-rabbit IgG, and horseradish peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit IgG antibodies were purchased from DingGuo (China). OPTI-MEM I, RNAiMAX, and Lipofectamine LTX were purchased from Invitrogen. 4′,6-Diamino-2-phenylindole (DAPI) was purchased from Beytime (China). Recombinant human tumor necrosis factor alpha (TNF-α) was purchased from PeproTech (USA). An enhanced chemiluminescence (ECL) kit was purchased from Kangwei Biological Company (China).
Constructs.
IFN-α4, IFN-β, and NF-κB promoter luciferase reporter plasmids (pGL3-IFN-α4, pGL3-IFN-β, and pGL3-NF-κB) were kindly provided by Hongbin Shu (23, 24). pCMV-Myc, pDsRed-monomer-N1, and pEGFP-N1 vectors were purchased from Clontech. IBDV vp4 was cloned from IBDV strain Lx using the following specific primers: sense, 5′-AGGATAGCTGTGCCGGTGGTCTCCACAT-3′; antisense, 5′-TTTGATGAACGTTGCCCAGTT-3′ (GenBank accession no. 6539893). Human gilz was cloned from HEK293T cells using the specific primers 5′-ATGGCCCAGTCCAAGCTCGA-3′ (sense) and 5′-TTACACCGCAGAACCACCA-3′ (antisense) according to the sequence in GenBank (accession no. 62865623). Chicken gilz was cloned from DF-1 cells using the specific primers 5′-ATGAGCACCGGCGTGTACCA-3′ (sense) and 5′-TTACACCGCAGAACCACCA-3′ (antisense) with reference to the sequence in GenBank (accession no. NM_001077234). All the primers were synthesized by Sangon Company (China).
Immunofluorescence antibody assay (IFA).
HEK293T cells were infected with IBDV Lx at a multiplicity of infection (MOI) of 10 and incubated for 3 h at 37°C. Uninfected HEK293T cells were used as negative controls. Three hours after incubation, the medium was changed for fresh DMEM with 2% FBS and further incubated for 24 h. After incubation, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2%Triton X-100, blocked with 1% bovine serum albumin (BSA), and incubated with chicken anti-IBDV antiserum, followed by FITC-conjugated goat anti-chicken IgG antibodies, and were visualized by a fluorescence microscope. After infection with IBDV at an MOI of 10, HEK293T cells were fixed with 4% paraformaldehyde, permeabilized with 0.2%Triton X-100, blocked with 1% bovine serum albumin, and incubated with mouse anti-IBDV VP4 antiserum and rabbit anti-GILZ antibodies, followed by FITC-conjugated goat anti-mouse IgG (green) and TRITC-conjugated goat anti-rabbit IgG (red), and were visualized with a fluorescence microscope.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR) analysis.
Total RNA was prepared from HEK293T or DF-1 cells using a Qiagen RNeasy kit per the manufacturer's instructions. The total RNA was treated with DNase I, and 1 μg of total RNA was used for cDNA synthesis by reverse transcription using an RT-PCR kit (TaKaRa). The specific primers for human IFN-α1 (5′-CAATATCTACGATGGCCTCGC-3′ and 5′-AGAGATGGCTGGAGCCTTCTG-3′), IFN-β (5′-GATTCATCTAGCACTGGCTGG-3′ and 5′-CTTCAGGTAATGCAGAATCC-3′), p50 (5′-GCGAGAGGAGCACAGATACC-3′ and 5′-CTGATAGCCTGCTCCAGGTC-3′), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 5′-CAACTACATGGTTTACATGTTCC-3′ and 5′-GGACTGTGGTCATGAGTCCT-3′) were designed with reference to previous publications (25–27) and synthesized by Sangon Company. Specific primers for chicken IFN-α1 (5′-CCAGCACCTCGAGCAAT-3′ and 5′-GGCGCTGTAATCGTTGTCT-3′), IFN-β (5′-GCCTCCAGCTCCTTCAGAATACG-3′ and 5′-CTGGATCTGGTTGAGGAGGCTGT-3′), p65 (5′-CCACAACACAATGCGCTCTG-3′ and 5′-AACTCAGCGGCGTCGATG-3′), and GAPDH (5′-TGCCATCACAGCCACACAGAAG-3′ and 5′-ACTTTCCCCACAGCCTTAGCAG-3′) were designed with reference to previous publications (28–30). The analysis of real-time PCR was carried out with a Light Cycler 480 (Roche, USA). The PCR was performed in a 20-μl volume containing 1 μl of cDNA, 10 μl of 2× SYBR green Premix Ex Taq (TaKaRa), and a 0.4 μM concentration of each gene-specific primer. Thermal cycling parameters were as follows: 94°C for 2 min; 40 cycles of 94°C for 20 s, 55°C for 20 s, and 72°C for 20 s; and 1 cycle of 95°C for 30 s, 60°C for 30 s, and 95°C for 30 s. The final step was to obtain a melt curve for the PCR products to determine the specificity of the amplification. All samples were carried out in triplicate on the same plate, and the GAPDH gene was utilized as the reference gene. Expression levels of genes were calculated relative to the expression of the GAPDH gene and expressed as fold increase or decrease relative to the control samples.
Coimmunoprecipitation and Western blot analysis.
For immunoprecipitation, HEK293T or DF-1 cells (6 × 105) were seeded on 6-well plates and cultured for 24 h before cotransfection with pCMV-Myc-GILZ and pEGFP-VP4 or pRK5-flag-VP4 or empty vectors as controls by standard calcium phosphate precipitation. Twenty-four hours after transfection, cell lysates were prepared using a nondenaturing lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1% NP-40; 5 mM EDTA; 10% glycerol; 10 mM dithiothreitol [DTT]; 1× complete cocktail protease inhibitor). The cell lysates were incubated with 2 μg of anti-c-Myc or anti-FLAG antibody at 4°C for 2 h and then mixed with 20 μl of a 50% slurry of protein A/G plus agarose and incubated for another 2 h. Beads were washed three times with the lysis buffer and boiled with 2× SDS loading buffer for 10 min. The samples were fractionated by electrophoresis on 12% SDS-PAGE gels, and resolved proteins were transferred onto polyvinylidene difluoride (PVDF) membranes. After blocking with 5% skim milk, the membranes were incubated with either anti-Myc, anti-FLAG, or anti-GFP antibody, followed by an appropriate HRP-conjugated secondary antibody. Blots were developed using an ECL kit. For endogenous pulldown assay, HEK293T cells or DF-1 cells were transfected with pEGFP-VP4 or pRK5-VP4 or with empty vector. Thirty-six hours after transfection, the cell lysates were subjected to immunoprecipitation with anti-GFP antibody or anti-FLAG antibody and immunoblotted with anti-GILZ, anti-GFP, or anti-FLAG antibody.
Confocal laser scanning microscopy assays.
HEK293T cells (2 × 105) or DF-1 cells (1 × 105) were seeded on coverslips in 24-well plates and were cultured overnight before transfection with pEGFP-VP4 and pDsRed-GILZ. Twenty-four hours after transfection, the cells were fixed with 1% paraformaldehyde and the nuclei were stained with DAPI. For endogenous protein staining, IBDV- or mock-infected cells were fixed with 1% paraformaldehyde and permeabilized with 0.2% Triton X-100 for 15 min, blocked with 1% bovine serum albumin, and then probed with mouse anti-IBDV VP4 antiserum and rabbit anti-GILZ antibodies, followed by FITC-conjugated goat anti-mouse IgG (green) and TRITC-conjugated goat anti-rabbit IgG (red). After three washes with PBS, the cells were stained for nuclei with DAPI. The samples were analyzed with a laser confocal scanning microscope (C1 standard detector; Nikon, Japan).
Luciferase reporter gene assays.
HEK293T cells (2.0 × 105) were seeded on 24-well plates and transfected with reporter gene plasmids (pGL3-IFN-α4, pGL3-IFN-β, or pGL3-NF-κB) by standard calcium phosphate precipitation. To normalize for transfection efficiency, we added 0.01 μg of pRL-TK Renilla luciferase reporter plasmid to each transfection. Six hours after transfection, cells were mock infected or infected with IBDV at an MOI of 20. Twelve hours after infection, cells were treated with TNF-α at a final concentration of 20 ng/ml or with medium as a control. Twelve hours after TNF-α treatment, luciferase reporter gene assays were performed. For the measurement of TNF-α- or Sendai virus (SeV)-induced activation of type I interferon and NF-κB promoters, cells were transfected with reporter gene plasmids (pGL3-IFN-α4, pGL3-IFN-β, or pGL3-NF-κB), and 18 h after transfection, cells were treated with TNF-α at a final concentration of 20 ng/ml or infected with SeV at an MOI of 10, with treatment with medium and mock infection as controls. Twelve hours after TNF-α treatment or 24 h after SeV infection, luciferase reporter gene assays were performed with a dual-specific luciferase assay kit (Promega, USA). Firefly luciferase activities were normalized on the basis of Renilla luciferase activities. All reporter assays were repeated at least three times. Data shown are average values ± standard deviations (SD) from one representative experiment.
Nuclear protein extraction and EMSA.
Crude nuclear proteins were extracted from HEK293T cells using a nuclear-cytoplasmic extraction kit (Thermo Fisher) with a protease inhibitor mixture. 5′-Biotin-labeled NF-κB consensus double-stranded oligonucleotides (5′-AGTTGAGGGGACTTTCCCAGG-3′) were synthesized by AuGCT Biotechnology (31). Detection of the NF-κB-oligonucleotide complex was performed using a LightShift chemiluminescent electrophoretic mobility shift assay (EMSA) kit (Thermo Fisher) per the manufacturer's instructions. Briefly, nuclear protein (3 to 5 μg) was incubated with 20 fmol of biotin-labeled oligonucleotides for 20 min at room temperature in a 20-μl reaction volume containing 10 mM HEPES-KOH (pH 7.9), 50 mM KCl, 2.5 mM MgCl2, 1 mM DTT, 10% glycerol, 1 μg of DNase-free BSA, and 2.5 μg of polydeoxyinosinic-deoxycytidylic acid. The resulting products were resolved by electrophoresis on a 6% polyacrylamide gel using 0.5× Tris-borate EDTA (TBE) buffer. NF-κB-oligonucleotide complex was electroblotted to a nylon membrane (Millipore). After incubation in blocking buffer for 15 min at room temperature, the membrane was incubated with streptavidin-HRP conjugate for 15 min at room temperature. Color was developed using Light Shift chemiluminescence detection reagents (Thermo Fisher).
RNA interference (RNAi) knockdown of GILZ.
The siRNA was designed by Genechem Company (Shanghai, China) and used to knock down GILZ in HEK293T cells. The siRNAs for targeting GILZ in HEK293T cells included the following: RNAi#1 (sense, 5′-GUGAGAACACCCUGUUGAAtt-3′; antisense, 5′-UUCAACAGGGUGUUCUCACtt-3′), RNAi#2 (sense, 5′-GAAGAAUCAUCUGAUGUAUtt-3′; antisense, 5′-AUACAUCAGAUGAUUCUUCtt-3′), RNAi#3 (sense, 5′-UCUGGUGAAGAAUCAUCUGtt-3′; antisense, 5′-CAGAUGAUUCUUCACCAGAtt-3′), and negative control (sense, 5′-UUCUCCGAACGUGUCACGUtt-3′; antisense, 5′-ACGUGACACGUUCGGAGAAtt-3′). The sequences of siRNA for targeting GILZ in DF-1 cells included the following: RNAi#1 (sense, 5′-GCGUGGUGGCCAUUGACAAtt-3′; antisense, 5′-UUGUCAAUGGCCACCACGCtt-3′), RNAi#2 (sense, 5′-AGGAACUGUUGGAGAAGAAtt-3′; antisense, 5′-UUCUUCUCCAACAGUUCCUtt-3′), RNAi#3 (sense, 5′-GCCAGCGUGGUGGCCAUUGtt-3′; antisense, 5′-CAAUGGCCACCACGCUGGCtt-3′), and negative control (sense, 5′-UUCUCCGAACGUGUCACGUtt-3′; antisense, 5′-ACGUGACACGUUCGGAGAAtt-3′). To transfect cells with the interference RNAs against GILZ, we seeded HEK293T or DF-1 cells (4 × 105) cells on 6-well plates and cultured them for at least 20 h prior to transfection. The cells were transfected with siRNA using RNAiMAX according to the manufacturer's instructions (Invitrogen). Double transfection was performed at a 24-h interval. Forty-eight hours after the second transfection, cells were harvested for further analysis.
Measurement of IBDV growth in DF-1 cells and HEK293T cells.
Untreated DF-1 cells or HEK293T cells or cells receiving GILZ-specific siRNAs or control siRNA were infected with IBDV at an MOI of 10, and cell cultures were collected at different time points (12, 24, 48, and 72 h) after infection. The cell culture samples were freeze-thawed three times and centrifuged at 2,000 × g for 10 min, and the supernatants were saved at −80°C until use. To neutralize interferon's activity, 2 μg of anti-human IFN-α1 (ab11408; Abcam) and anti-human IFN-β (ab6979; Abcam) were added to GILZ RNAi cell culture or RNAi controls 3 h before IBDV infection. Forty-eight hours after IBDV infection, the cell culture samples were freeze-thawed three times and centrifuged at 2,000 × g for 10 min, and the supernatants were saved at −80°C until use. The viral contents in the supernatants were titrated using 50% tissue culture infective doses (TCID50) in DF-1 cells. Briefly, the viral solution was diluted 10-fold in DMEM. A 100-μl aliquot of each diluted sample was added to the wells of 96-well plates, followed by addition of 100 μl of DF-1 cells at a density of 5 × 105 cells/ml. Cells were cultured for 5 days at 37°C in 5% CO2. Tissue culture wells with cytopathic effect (CPE) were determined as positive. The titer was calculated based on a previously described method (32).
Statistical analysis.
The significance of the differences between GILZ RNAi cells and controls in gene expression, promoter activities, and viral growth was determined by the Mann-Whitney test and analysis of variance (ANOVA) accordingly.
RESULTS
Infection of HEK293T cells with IBDV inhibits TNF-induced expression of type I interferon.
IBDV is capable of replicating in multiple types of cells, including chicken B cells (33), CEF and DF-1 cells (34), and Vero and HEK293T cells (35). To determine if the IBDV Lx strain could replicate in HEK293T cells, we infected this cell line with the virus at an MOI of 10 and examined the viral growth with immunofluorescence antibody assay (IFA). Twenty-four hours after IBDV infection, a large number of immunofluorescent cells could be detected after IFA staining using chicken anti-IBDV antiserum (Fig. 1A to D). In addition, kinetics of virus production was also examined in IBDV-infected cells (Fig. 1E). These data indicate that the IBDV Lx strain could replicate in HEK293T cells. As type I interferon plays a critical role in the host response against IBDV infection (19) and HEK293T cells do not produce detectable TLR3 activity (36), we examined the expression of type I interferon in TNF-α-stimulated HEK293T cells with or without IBDV infection using qRT-PCR assay (37). Because TNF induces activation of NF-κB (38, 39), and NF-κB regulates type I IFN expression (37), we employed TNF-α as an inducer in our assay to examine the effect of IBDV infection on TNF-induced type I interferon expression. We found that TNF-induced expression of IFN-α and IFN-β was significantly reduced in IBDV-infected cells compared to that of controls (P < 0.01) (Fig. 1F and G), suggesting an inhibitory effect of IBDV infection on type I interferon expression in host cells. Since the expression of type I IFN is regulated by transcriptional regulator NF-κB (37), we next examined the mRNA expression of NF-κB in IBDV-infected cells stimulated with TNF-α. Consistent with the above observation, infection of HEK293T cells with the IBDV Lx strain inhibited TNF-induced expression of NF-κB (Fig. 1H) (P < 0.001), suggesting that IBDV-induced suppression of type I IFN may be associated with transcriptional regulator NF-κB.
Fig 1.
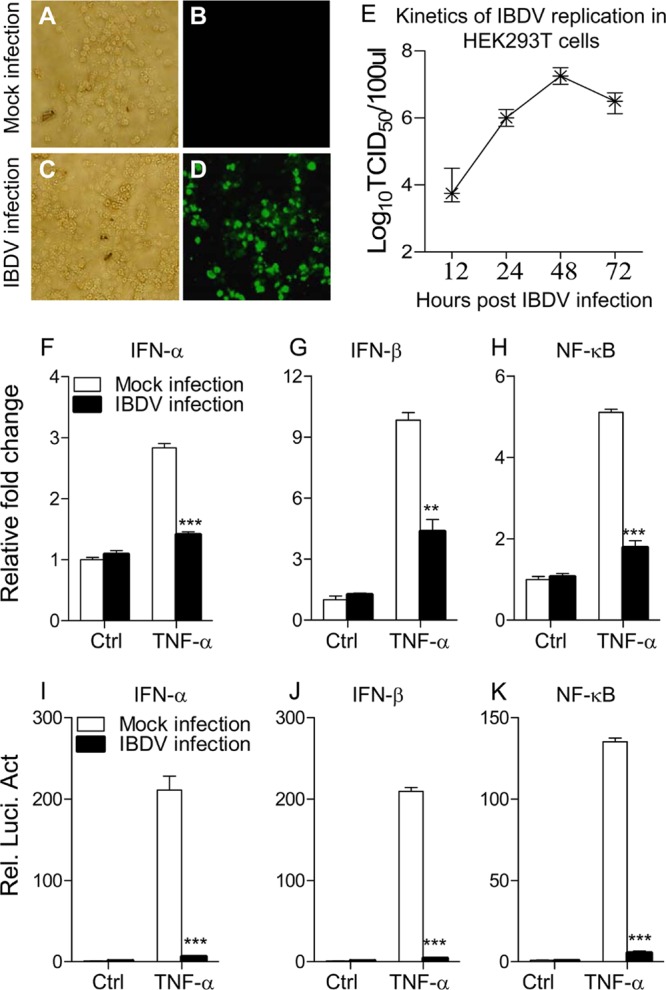
Infection of HEK293T cells with IBDV strain Lx inhibits TNF-induced expression of IFN-α, IFN-β, and NF-κB. (A to D) Replication of IBDV strain Lx in HEK293T cells. HEK293T cells were mock infected (A and B) or infected with IBDV Lx at an MOI of 10 (C and D) and incubated for 3 h at 37°C. Twenty-four hours after IBDV infection, cells were subjected to IFA staining using chicken anti-IBDV antiserum followed by FITC-conjugated rabbit anti-chicken IgG antibodies and were visualized under a fluorescence microscope. (E) Kinetics of IBDV replication in HEK293T cells. HEK293T cells were infected with IBDV at an MOI of 10. At different time points (12, 24, 48, and 72 h) after IBDV infection, the viral titers in the cell cultures were determined as TCID50 using 96-well plates. (F to H) Effects of IBDV infection on TNF-induced mRNA expression of IFN-α, IFN-β, and NF-κB. HEK293T cells (2 × 105) were mock infected or infected with IBDV at an MOI of 20. Twelve hours after IBDV infection, cells were treated with TNF-α at a final concentration of 20 ng/ml. Twelve hours after TNF treatment, mRNA expression of IFN-α, IFN-β, and NF-κB was measured by qRT-PCR using specific primers. The relative level of gene expression is calculated as follows: gene expression of IBDV-infected cells or TNF-treated cells (mock- or IBDV-infected cells)/gene expression of mock-infected controls. (I to K) Effects of IBDV infection on TNF-induced activation of IFN-α, IFN-β, and NF-κB promoters. HEK293T cells (2.0 × 105) were seeded on 24-well plates and transfected with the indicated reporter gene plasmids. Six hours after transfection, cells were mock infected or infected with IBDV at an MOI of 20. Twelve hours after IBDV infection, cells were treated with TNF-α at a final concentration of 20 ng/ml. Twelve hours after TNF-α treatment, luciferase reporter gene assays were performed. The data are normalized as in panels F to H. Results are representative of three independent experiments. Data are represented as means ± SD; n = 3. ***, P < 0.001; **, P < 0.01. Ctrl, control.
To determine whether the inhibitory effect of IBDV on type I IFN expression occurs at or upstream of the transcriptional level, we employed luciferase reporter gene assays to examine the cytokine gene promoter activities (37, 40). HEK293T cells were transiently transfected with type I interferon or NF-κB reporter gene plasmids, and then cells were mock infected or infected with IBDV at an MOI of 20. Twelve hours after IBDV infection, cells were stimulated with TNF-α for 12 h and their luciferase activities were measured. All luciferase activities were normalized to a cotransfected Renilla luciferase plasmid control. As shown in Fig. 1I to K, IBDV infection significantly inhibited the reporter activities of IFN-α, IFN-β, and NF-κB promoters following stimulation with TNF-α (P < 0.001). Thus, the inhibitory effect of IBDV on the transactivation of these promoters suggests that IBDV suppresses type I interferon expression at or upstream of the transcriptional level, which may eventually lead to a suppressed immune response in host cells.
VP4 is mainly responsible for IBDV-induced suppression of type I interferon expression.
Since IBDV infection suppressed type I interferon expression in host cells, we proposed that one or more components of IBDV affect type I interferon expression by engaging host proteins involved in regulating this cellular response. To test this hypothesis, we cloned the vp1, vp2, vp3, vp4, and vp5 genes from the IBDV Lx strain, made a GFP fusion for each of these proteins, and expressed them in HEK293T cells by transfection. All five protein fusions were expressed well in this cell line when transfected with 5 μg of each individual plasmid (Fig. 2A). As SeV infection causes typical type I interferon expression (41), we used this strategy to examine the effect of each protein fusion on type I interferon expression. We found that transient expression of GFP-VP4 significantly suppressed SeV-induced type I interferon expression in host cells compared to the GFP controls (P < 0.05) (Fig. 2B and C). In comparison, expression of GFP-VP1, -VP2, -VP3, or -VP5 did not display marked downregulated expression of type I interferon in SeV-infected cells. In contrast, GFP-VP3 and -VP5 enhanced SeV-induced type I interferon expression. Similar results were also obtained from the examination of mRNA expression of NF-κB (Fig. 2D). As overexpression of GFP-VP4 does not affect cell viability (14), these results suggest that VP4 is the major viral component responsible for IBDV-induced suppression of type I interferon expression, leading to suppressed immune responses in host cells.
Fig 2.

The VP4 protein is mainly responsible for IBDV-induced suppression of type I interferon expression in HEK293T cells. (A) Expression of GFP-VP1, -VP2, -VP3, -VP4, or -VP5 fusion proteins in HEK293T cells. HEK293T cells were transfected with 5 μg of pEGFP-N1, pEGFP-VP1, pEGFP-VP2, pEGFP-VP3, pEGFP-VP4, or pEGFP-VP5 plasmid. Twenty-four hours after transfection, cell lysates were prepared and examined by Western blotting (WB) using anti-GFP antibodies. (B to D) Effects of viral components on mRNA expressions of the type I IFN and NF-κB in host cells after SeV infection. HEK293T cells were left untransfected (NT) or were transfected with 5 μg of pEGFP-N1, pEGFP-VP1, pEGFP-VP2, pEGFP-VP3, pEGFP-VP4, or pEGFP-VP5 as described for panel A. Six hours after transfection, cells were mock infected or infected with SeV at an MOI of 10. Twenty-four hours after SeV infection, mRNA expression of IFN-α, IFN-β, and NF-κB was measured by quantitative RT-PCR using specific primers. The relative level of mRNA expression is calculated as follows: gene expression of plasmid-transfected cells or SeV-infected cells/gene expression of mock-infected nontransfected controls. Results are representative of three independent experiments. Data are represented as means ± SD; n = 3. **, P < 0.01; *, P < 0.05.
VP4 suppresses TNF-induced activation of type I interferon and NF-κB.
The fact that VP4 is the major IBDV component responsible for suppressing type I interferon expression prompted us to investigate the role of VP4 in this suppression. We examined the type I interferon response in pEGFP-vp4-transfected cells by measuring TNF-α-induced activation of type I IFN and NF-κB promoters (37). Consistent with the above observation, transient expression of VP4 markedly inhibited TNF-induced activation of type I IFN and NF-κB promoters compared to that of controls (Fig. 3A to C) (P < 0.01). Similar results were also obtained from the examination of reporter activities using SeV infection (Fig. 3D to F). These results indicate that IBDV VP4 suppresses type I interferon expressions at or upstream of the transcriptional level in host cells.
Fig 3.
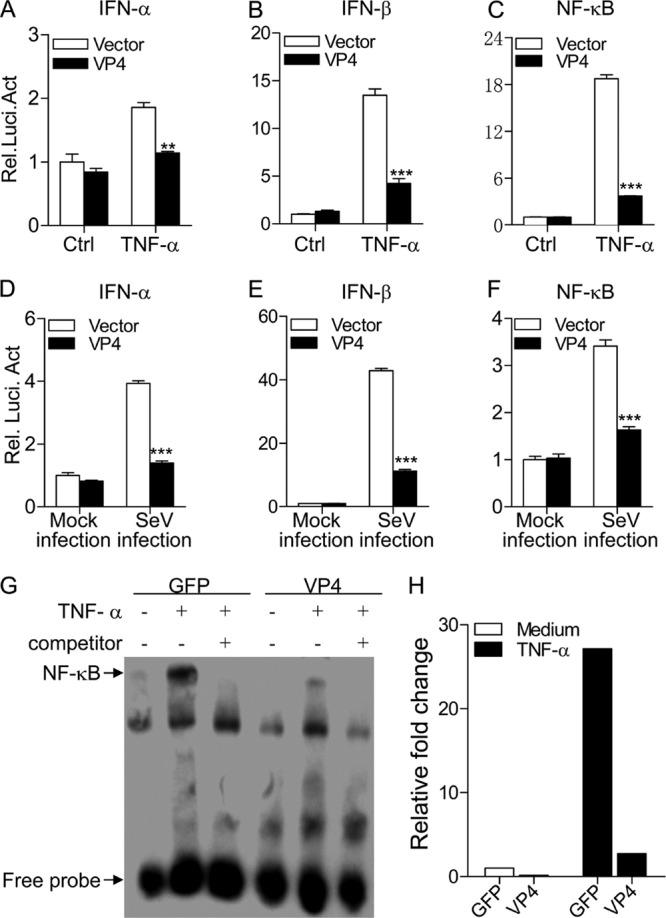
Expression of VP4 inhibits TNF-α- or SeV-induced activation of IFN-α, IFN-β, and NF-κB promoters. (A to C) Effects of VP4 on TNF-induced activation of IFN-α, IFN-β, and NF-κB promoters. HEK293T cells (2.0 × 105) were transfected with pEGFP-vp4 or empty vector (pEGFP-N1) as a control together with the indicated reporter plasmids. Eighteen hours after transfection, cells were treated with 20 ng/ml of TNF-α or medium as a control for 12 h before reporter activities were examined with a dual-specific luciferase assay kit. (D to F) Effects of VP4 on SeV-induced activation of IFN-α, IFN-β, and NF-κB promoters. HEK293T cells (2.0 × 105) were transfected with pEGFP-vp4 or empty vector as controls together with the indicated reporter plasmids. Eighteen hours after transfection, cells were mock infected or infected with SeV at an MOI of 10 for 24 h before reporter activities were examined with a dual-specific luciferase assay kit. Results are representative of three independent experiments. Data are represented as means ± SD; n = 3. ***, P < 0.001; **, P < 0.01; *, P < 0.05. (G) Impact of VP4 on TNF-induced nuclear translocation of NF-κB p65. HEK293T cells (5.0 × 105) were transfected with pEGFP-vp4 or empty vector as a control. Twenty hours after transfection, cells were treated with 100 ng/ml of TNF-α or medium as controls for 45 min before crude nuclear proteins were extracted. The nuclear proteins were then subjected to EMSA for determining the nuclear translocation of NF-κB p65. The detection of the NF-κB-oligonucleotide complex was performed using a Light Shift chemiluminescence EMSA kit. (H) The density of relative nuclear translocated NF-κB p65 bands in panel G was quantitated by densitometry and normalized to that of vector control. Results are representative of three independent experiments.
To further determine the inhibitory effects of VP4 on NF-κB-mediated signaling, we extracted nuclear proteins from pEGFP-vp4- or pEGFP-transfected cells with or without TNF-α treatment and performed EMSA to examine the impact of VP4 on TNF-induced nuclear translocation of NF-κBp65, a master regulator of all TLR-induced responses (42). As shown in Fig. 3G and H, the nuclear translocation of NF-κBp65 remarkably increased in pEGFP-N (empty vector)-transfected cells after TNF-α treatment. In contrast, the TNF-induced nuclear translocation of NF-κBp65 was markedly reduced in cells transfected with pEGFP-vp4, indicating that VP4 plays an inhibitory role in the NF-κB-mediated cell response.
VP4 interacts with GILZ.
After identification of VP4 as a suppressor of the antiviral response in host cells, we furthered our investigation to study the mechanism of such suppression by searching for its cellular targets. To this end, we used VP4 as bait in the yeast two-hybrid system to screen a cDNA library generated from the chicken bursa of Fabricius. The positive clones were tested for β-galactosidase activity in two additional rounds of selection (turning blue) (Fig. 4A to C), and the plasmids from these clones were rescued, sequenced, and subjected to a BLAST search against the NCBI database. Among 56 positive clones, 25 GILZ clones were identified. In addition, we found that during the course of IBDV infection, endogenous levels of GILZ in DF-1 cells markedly increased (data not shown). This protein might be relevant to VP4 function because it inhibits the immune response (43, 44). Thus, we constructed a plasmid that allows the expression of Myc-GILZ for analyzing its interaction with VP4 in HEK293T cells. When lysates of cells expressing both EGFP-VP4 and Myc-GILZ were immunoprecipitated with Myc antibody, EGFP-VP4 was detected in the precipitate, indicating that VP4 interacted with ectopically expressed GILZ in HEK293T cells (Fig. 4D). Similar results were obtained in an experiment using the DF-1 cells (Fig. 4E), indicating that the observed interaction between these two proteins is not cell type specific. To further substantiate the binding of VP4 to GILZ, we expressed VP4 in HEK293T or DF-1 cells and examined its interaction with endogenous GILZ using a pulldown assay. The binding of GFP-VP4 or FLAG-VP4 with endogenous GILZ was readily detectable in cells expressing the viral protein VP4 (Fig. 4F and G). These results demonstrate that VP4 interacts with GILZ in host cells.
Fig 4.

Interaction of IBDV VP4 with host cellular protein GILZ. (A to C) Yeast two-hybrid screening of IBDV VP4 binding proteins. Yeast colonies cotransformed with pGBKT7-VP4 and pGADT7-GILZ (A), cotransformed with pGBKT7-Lam and pGADT7-T antigen as a negative control (B), or cotransformed with pGBKT7-p53 and pGADT7-T antigen as a positive control (C) were incubated at 30°C and checked periodically for positivity (turning blue). (D to E) Interaction of VP4 with exogenous GILZ. HEK293T (D) or DF-1 (E) cells were transfected with 2.5 μg of the indicated plasmids. Twenty-four hours after transfection, cell lysates were prepared and immunoprecipitated (IP) with anti-Myc(D) or anti-FLAG (E) antibodies. VP4 and GILZ in the immune complex were immunoblotted with anti-GFP or anti-Myc or anti-FLAG antibodies. (F and G) Interaction of VP4 with endogenous GILZ. HEK293T (F) or DF-1 (G) cells were transfected with 5 μg of pEGFP-vp4 (F), pRK5-vp4 (G), pRK5-vp5 (G), or empty vectors as controls, and immunoprecipitation assays were performed with anti-GFP or anti-FLAG antibodies. GILZ in the immune complex was examined by Western blotting using anti-GILZ antibody. Data are representative of three experiments with similar results.
VP4 colocalizes with GILZ in host cells.
To determine the subcelluar localization of VP4 and GILZ, we performed a confocal microscopy assay with HEK293T cells transfected to express DsRed-GILZ and GFP-VP4. Transfection of HEK293T cells with pEGFP-vp4 or pDsRed-gilz indicated that both VP4 and GILZ were located in cytoplasm (Fig. 5A and B). When cells were transfected with both plasmids, we found colocalization of VP4 with GILZ in the transfected cells (Fig. 5C to E). To determine whether endogenous GILZ colocalizes with VP4 in IBDV-infected cells, we infected HEK293T or DF-1 cells and performed an IFA to examine the interaction of VP4 with endogenous GILZ. Consistent with the above observation, the endogenous GILZ also colocalized with VP4 in the cytoplasm of IBDV-infected cells (Fig. 5F to Q). These results clearly demonstrate that VP4 interacts with GILZ in the cytoplasm of host cells.
Fig 5.
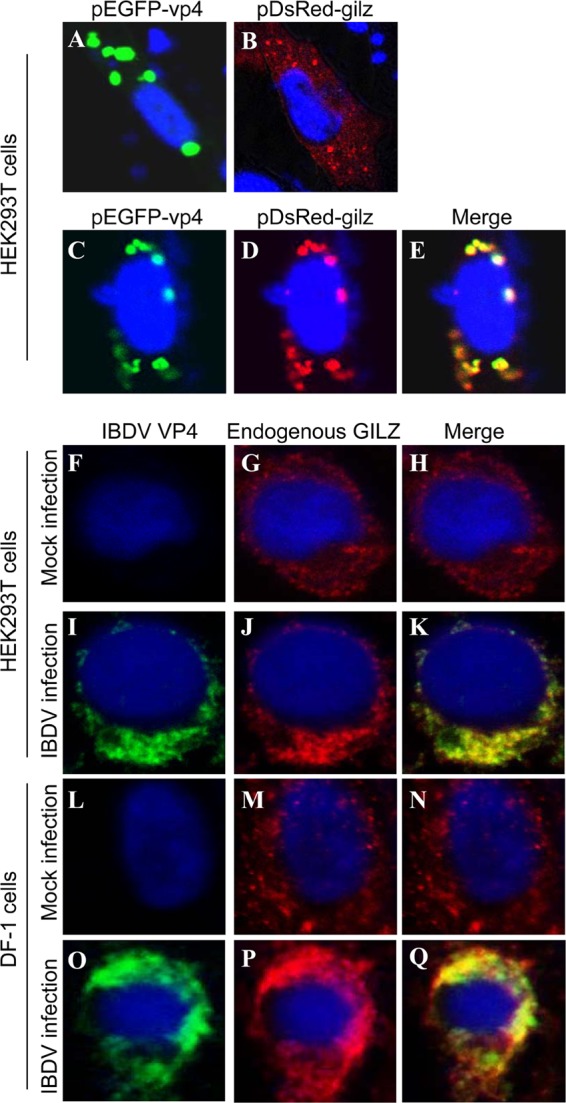
Colocalization of VP4 with GILZ in the cell. (A and B) Expression of exogenous VP4 (A) or GILZ (B) in HEK293T cells. HEK293T cells (2 × 105) were seeded on 24-well plates with coverslips in the wells and cultured overnight. Cells were transfected with pEGFP-vp4 (A) or pDsRed-gilz (B). Twenty-four hours after transfection, cells were fixed and observed with a laser confocal scanning microscope. (C to E) Colocalization of VP4 with exogenous GILZ. HEK293T cells (2 × 105) were seeded on 24-well plates with coverslips in the wells and cultured overnight. Cells were transfected with both pEGFP-vp4 and pDsRed-gilz. Twenty-four hours after transfection, cells were fixed and observed with a laser confocal scanning microscope. (F to Q) Colocalization of IBDV VP4 with endogenous GILZ in IBDV-infected cells. HEK293T (F to K) or DF-1 (L to Q) cells were mock infected (F to H and L to N) or infected with IBDV Lx (I to K and O to Q) at an MOI of 10 and incubated for 3 h at 37°C. Twenty-four hours after IBDV infection, cells were fixed and probed with mouse anti-VP4 antibodies and rabbit anti-GILZ antibodies, followed by the FITC-conjugated goat anti-mouse antibody (green) and TRITC-conjugated goat anti-rabbit antibody (red). Nuclei were counterstained with DAPI (blue) and were visualized under a fluorescence microscope.
GILZ is required for VP4-induced suppression of type I interferon expression.
The facts that VP4 inhibits type I interferon expression and interacts with GILZ suggest that GILZ might play a critical role in VP4-induced suppression of the cytokine response and that knockdown of GILZ would therefore affect cell responses. To test this hypothesis, we made three GILZ RNAi constructs, and we found that one could effectively lower the cellular level of GILZ without causing discernible changes in cell morphology (Fig. 6A and B). We then transfected HEK293T cells receiving this siRNA or control siRNA with pEGFP-vp4 plasmid and examined the activation of type I interferon and NF-κB promoters in these cells after stimulation with TNF-α or infection with SeV. Knockdown of GILZ completely abolished VP4-induced suppression of TNF-induced activation of type I interferon and NF-κB promoters (Fig. 6C to E). Consistently, knockdown of GILZ also abolished VP4-induced suppression of SeV-induced activation of type I interferon and NF-κB promoters (Fig. 6F to H). Furthermore, we knocked down GILZ expression in DF-1 cells (immortal chicken embryo fibroblasts) by siRNA (Fig. 7A and B) and examined the expression of type I interferon and NF-κB in these cells after stimulation with poly(I·C). Consistent with the above observation, VP4 suppressed poly(I·C)-induced expression of type I interferon and NF-κB in DF-1 cells, and this suppression could be abolished by knockdown of GILZ (Fig. 7C to E). These data clearly demonstrate that GILZ is required for the VP4-induced suppression of type I interferon expression at or upstream of the transcriptional level in host cells.
Fig 6.

GILZ mediates the inhibitory effect of VP4 on TNF-α or SeV-induced activation of IFN-α, IFN-β, and NF-κB promoters. (A and B) Effects of GILZ RNAi on the expression of endogenous GILZ. HEK293T cells (2.0 × 105) were transfected with siRNA (RNA#1 to RNA#3) or controls as described in Materials and Methods. Forty-eight hours after the second transfection, cell lysates were prepared and examined by Western blotting with anti-GILZ antibody. Endogenous β-actin expression was used as internal controls. The band density of GILZ in GILZ RNAi-treated cells (A) was quantitated by densitometry, and the relative levels of GILZ (B) were calculated as follows: band density of GILZ/band density of β-actin. (C to H) Knockdown of GILZ abolished the inhibitory effect of VP4 on TNF-α- or SeV-induced activation of IFN-α, IFN-β, and NF-κB promoters. HEK293T cells (2.0 × 105) were transfected with the GILZ RNAi#1 construct or RNAi control. Twenty-four hours after the first transfection, cells were cotransfected with an RNAi construct together with pEGFP-vp4 and the indicated reporter plasmids. Eighteen hours after transfection, cells were treated with 20 ng/ml of TNF-α (C to E) or infected with SeV (F to H) at an MOI of 10. Medium or mock-infected cells were used as controls. Twelve hours after TNF-α treatment or 24 h after SeV infection, cells were collected and the reporter activities of IFN-α, IFN-β, and NF-κB were determined by luciferase reporter gene assays using a dual-specific luciferase assay kit. Data are presented as means ± SD; n = 3. ***, P < 0.001; **, P < 0.01.
Fig 7.

GILZ mediates the inhibitory effect of VP4 on poly(I·C)-induced expressions of IFN-α, IFN-β, and NF-κB in DF-1 cells. (A and B) Effect of GILZ RNAi on the expression of endogenous GILZ in DF-1 cells. DF-1 cells (2.0 × 105) were transfected with siRNA constructs (RNA#1 to RNA#3) or controls. Double transfection was performed at a 24-h interval. Forty-eight hours after the second transfection, cell lysates were prepared and examined by Western blotting with anti-GILZ antibody. Endogenous β-actin expression was used as internal controls (A). The band density of GILZ in GILZ RNAi-treated cells (A) was quantitated by densitometry, and the relative levels of GILZ (B) were calculated as follows: band density of GILZ/band density of β-actin. (C to E) Knockdown of GILZ abolished the inhibitory effect of VP4 on poly(I·C)-induced expression of IFN-α, IFN-β, and NF-κB. DF-1 cells (2.0 × 105) were cotransfected with RNAi constructs together with pEGFP-vp4 expression plasmids or pEGFP-N1 (vector) controls. Eighteen hours after transfection, cells were treated with 0.8 μg of poly(I·C). Twenty-four hours after poly(I·C) treatment, mRNA expression of IFN-α, IFN-β, and NF-κB was measured by qRT-PCR using specific primers. The expression levels of mRNA were calculated in relation to the expression level of GAPDH. Results are representative of three independent experiments. Data are represented as means ± SD; n = 3. ***, P < 0.001; **, P < 0.01.
IBDV growth is inhibited by type I interferon or GILZ knockdown.
To determine the role of GILZ in type I interferon expression in IBDV-infected cells, we knocked down GILZ expression in HEK293T cells by siRNA, infected these cells with the IBDV Lx strain at an MOI of 10, and examined type I interferon expression by qRT-PCR assay. As expected, knockdown of GILZ markedly enhanced mRNA expression of type I interferon in IBDV-infected cells (P < 0.001) (Fig. 8A and B). These results suggest that GILZ is involved in suppressing type I interferon expression in IBDV-infected cells.
Fig 8.

Knockdown of GILZ inhibits IBDV growth by type I interferon. (A and B) GILZ is required for IBDV-induced suppression of type I interferon expression in host cells after TNF-α treatment. HEK293T cells were treated with GILZ-RNAi or RNAi-control (Ctrl-RNAi) constructs or medium only as an untreated control and were mock infected or infected with IBDV at an MOI of 10. Twelve hours after IBDV infection, cells were treated with TNF-α at a final concentration of 20 ng/ml. Twelve hours after TNF-α treatment, mRNA expression of IFN-α and IFN-β were measured by qRT-PCR using specific primers. The expression levels of mRNA were calculated in relation to that of GAPDH. Results are representative of three independent experiments. Data are represented as means ± SD; n = 3. ***, P < 0.001. (C and D) Knockdown of GILZ inhibits IBDV growth. DF-1 (C) or HEK293T (D) cells were treated with GILZ-RNAi or control-RNAi constructs or medium only as controls and were infected with IBDV at an MOI of 10. At different time points (12, 24, 48, and 72 h) after IBDV infection, the viral titers in the cell cultures were determined as TCID50 using 96-well plates. The significance of the differences between GILZ-RNAi and controls was determined by ANOVA (P < 0.01). (E) Type I interferon mediates the inhibitory effect of GILZ-RNAi on IBDV growth. Anti-human IFN-α1 (4 × 104 neutralizing units/ml) and anti-human IFN-β (5 × 104 neutralizing units/ml) were added to GILZ knockdown cells or RNAi controls 3 h ahead of IBDV infection. Forty-eight hours after IBDV infection, the culture samples were freeze-thawed three times and centrifuged at 2,000 × g for 10 min. The viral titers were titrated using TCID50 in DF-1 cells. Results are representative of three independent experiments. Data are represented as means ± SD; n = 3. ***, P < 0.001.
Since type I interferon plays a critical role in host response to IBDV infection (19) and GILZ suppresses type I interferon expression, we hypothesized that IBDV might take advantage of GILZ to suppress the host response for its own benefit and that knockdown of GILZ would therefore inhibit IBDV growth in host cells. To test this hypothesis, we examined the viral replication in GILZ knockdown cells by measuring viral loads in IBDV-infected DF-1 or HEK293T cell cultures at different time points postinfection. Consistent with its postulated role in suppression of antiviral response, cells with lower GILZ levels markedly inhibited IBDV growth (P < 0.01) (Fig. 8C and D), and this inhibition could be effectively blocked by specific antibodies against type I IFN but not by its isotype-matched IgG controls (P < 0.001) (Fig. 8E), suggesting that GILZ suppresses the immune response via inhibiting type I IFN expression, which might be employed by IBDV as an important strategy to evade the host antiviral response.
DISCUSSION
IBD is an acute, highly contagious viral disease causing damage in lymphoid organs in birds, especially the bursa of Fabricius (45). Importantly, IBDV-infected chickens suffer from immunosuppression with compromised humoral and cellular immune responses (46, 47), leading to susceptibility of chickens to other diseases. Thus, IBD remains a threat to the poultry industry worldwide.
Although IBDV-induced immunosuppression has been well established (46–48), the exact molecular mechanism for such induction is unclear. IBDV-induced immunosuppression in the host may involve multiple factors, such as those involved in proinflammatory response and apoptosis, cytokine regulation, and the cellular immune response (29). It was reported that IBDV infection interferes with the transcription of chicken type I and II interferon mRNA (49), suggesting that the immunosuppressive effects of IBDV might be attributed, at least in part, to the suppression of chicken interferon by IBDV infection. In addition, type I interferon inhibits IBDV growth in chicken embryo fibroblast cultures (19), indicating that type I interferon plays an important role in the host anti-IBDV response. Our data show that IBDV infection inhibits TNF-induced expressions of IFN-α, IFN-β, and NF-κB at or upstream of the transcriptional level in host cells, and transfection of HEK293T cells with pEGFP-vp4 inhibits TNF-induced activation of IFN-α and -β promoters. Using an SeV infection system (41, 50), we also found that VP4 inhibited SeV-induced activation of type I interferon and NF-κB promoters. As type I interferon expression is regulated by transcriptional regulator NF-κB (37), VP4-induced suppression of the type I interferon response might result from the inhibitory effect of VP4 on the activation of NF-κB. Consistent with these observations, transfection with pEGFP-vp4 inhibits TNF or SeV-induced activation of NF-κB reporter as demonstrated by luciferase reporter gene assay and EMSA (Fig. 3). Thus, VP4-induced suppression of type I interferon expression is not specific to IBDV-infected cells but a general signaling inhibition for type I interferon expression in host cells.
Innate immunity is the first line of host defense against pathogenic infection. TNF-induced NF-κB signaling is an essential portion of the innate immune response in hosts to viral infection (38, 39). It was reported that proinflammatory cytokine TNF-α had been detected in the tissues of IBDV-infected chickens (51), suggesting that TNF may induce the inflammatory response in an IBDV-infected host. In the present study, we first found that TNF-induced type I interferon expression in host cells was inhibited by IBDV infection (Fig. 1F to K). Second, among the viral components, VP4 markedly suppressed the expression of type I interferon in host cells after SeV infection, and the suppressive effect of VP4 on the expression of I interferon and NF-κB occurs at or upstream of the transcriptional level in host cells, as demonstrated by reporter gene assay and EMSA (Fig. 3). Third, VP4 specifically interacts with GILZ under all tested conditions (Fig. 4 and 5). Fourth, the abolishment of VP4-induced suppression of type I interferon could be achieved by the knockdown of GILZ expression (Fig. 6 and 7). Finally, knockdown of GILZ inhibited viral growth, and this inhibition could be effectively abolished by anti-type I IFN antibodies in the cell culture (Fig. 8). Clearly, VP4 suppresses the type I interferon response of the host by interacting with GILZ. As type I interferon is a critical anti-IBDV cytokine (19, 49), these results provide strong evidence that VP4, via engagement with GILZ, suppresses the innate immune response.
GILZ, also known as TSC22 domain family protein 3, is a glucocorticoid-responsive molecule (43, 52). The GILZ protein consists of three major domains: the N-terminal, LZ, and C-terminal domains (43). A proline-rich region in the C-terminal domain of GILZ is necessary for direct binding to the p65 subunit of NF-κB (53). Currently, three isoforms of the GILZ protein in humans (GenBank accession numbers NM_198057.2 for the longest one, NM_004089.3 for the medium one, and NM_001015881.1 for the shortest), four in mice (54), and one in chickens (GenBank accession no. DQ917420.1) have been identified. The similarity of chicken GILZ with human GILZ ranges from 55.5% to 80%, while with mice it ranges from 30% to 90%. Interestingly, the amino acid sequence of the C-terminal domain of chicken GILZ is identical to that of the three isoforms of human GILZ, indicating that this portion (containing an NF-κB binding site) is highly conservative. GILZ plays an important role in the regulation of the immune response, such as inhibiting inflammation (43, 55) and preventing the T cell response (44). It has been reported that GILZ inhibits activities NF-κB (53). Our data show that VP4 markedly suppressed TNF-α-, SeV-, or poly(I·C)-induced activation of NF-κB (Fig. 3), and this suppression can be abolished by the knockdown of GILZ expression (Fig. 6 and 7), suggesting that VP4, by interacting with GILZ, inhibits NF-κB signaling, thus leading to the immunosuppression of the host. In this regard, it can be proposed that knockdown of GILZ may enhance the antiviral response in IBDV-infected cells. The fact that cells with lower GILZ levels markedly inhibited IBDV growth and that this inhibition could be effectively inhibited by specific antibodies against type I interferon provides strong evidence in support of this hypothesis (Fig. 8).
Of note, the mechanism underlying the immunosuppression induced by pathogenic infection may vary. Suppression of cytokine expression in host cells in such a case might be only one of the tricks exploited by the pathogens to evade immune response. Therefore, several questions are raised. For example, how does VP4 activate GILZ—by phosphorylation, acetylation, enzymatic cleavage, or something else? Similarly, what molecular features of VP4 interact with GILZ, leading to a suppressed cytokine response? And is GILZ taken advantage of by any other pathogens to suppress the immune response in host cells?
In summary, our results reveal that IBDV VP4 interacts with GILZ to suppress the innate immune response in host cells. The observations that knockdown of GILZ abolished VP4-induced suppression of type I interferon expression and reduced IBDV growth in host cells suggest that GILZ plays a critical role in IBDV-induced immunosuppression. These findings have provided insights for further studies of the molecular mechanism of IBDV infection.
ACKNOWLEDGMENTS
We thank Zhao-Qing Luo for critical reading of the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (no. 31272543 and 31072117) and Earmarked Fund for Modern Agro-Industry Technology Research System (no. NYCYTX-41).
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Pitcovski J, Gutter B, Gallili G, Goldway M, Perelman B, Gross G, Krispel S, Barbakov M, Michael A. 2003. Development and large-scale use of recombinant VP2 vaccine for the prevention of infectious bursal disease of chickens. Vaccine 21:4736–4743 [DOI] [PubMed] [Google Scholar]
- 2. Stricker RL, Behrens SE, Mundt E. 2010. Nuclear factor NF45 interacts with viral proteins of infectious bursal disease virus and inhibits viral replication. J. Virol. 84:10592–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Azad AA, Barrett SA, Fahey KJ. 1985. The characterization and molecular cloning of the double-stranded RNA genome of an Australian strain of infectious bursal disease virus. Virology 143:35–44 [DOI] [PubMed] [Google Scholar]
- 4. Pan J, Lin L, Tao YJ. 2009. Self-guanylylation of birnavirus VP1 does not require an intact polymerase activity site. Virology 395:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. von Einem UI, Gorbalenya AE, Schirrmeier H, Behrens SE, Letzel T, Mundt E. 2004. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J. Gen. Virol. 85:2221–2229 [DOI] [PubMed] [Google Scholar]
- 6. Kibenge FS, McKenna PK, Dybing JK. 1991. Genome cloning and analysis of the large RNA segment (segment A) of a naturally avirulent serotype 2 infectious bursal disease virus. Virology 184:437–440 [DOI] [PubMed] [Google Scholar]
- 7. Hudson PJ, McKern NM, Power BE, Azad AA. 1986. Genomic structure of the large RNA segment of infectious bursal disease virus. Nucleic Acids Res. 14:5001–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jagadish MN, Staton VJ, Hudson PJ, Azad AA. 1988. Birnavirus precursor polyprotein is processed in Escherichia coli by its own virus-encoded polypeptide. J. Virol. 62:1084–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dobos P, Hill BJ, Hallett R, Kells DT, Becht H, Teninges D. 1979. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J. Virol. 32:593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Birghan C, Mundt E, Gorbalenya AE. 2000. A non-canonical lon proteinase lacking the ATPase domain employs the ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus. EMBO J. 19:114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Da Costa B, Chevalier C, Henry C, Huet JC, Petit S, Lepault J, Boot H, Delmas B. 2002. The capsid of infectious bursal disease virus contains several small peptides arising from the maturation process of pVP2. J. Virol. 76:2393–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Granzow H, Birghan C, Mettenleiter TC, Beyer J, Kollner B, Mundt E. 1997. A second form of infectious bursal disease virus-associated tubule contains VP4. J. Virol. 71:8879–8885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mundt E, Kollner B, Kretzschmar D. 1997. VP5 of infectious bursal disease virus is not essential for viral replication in cell culture. J. Virol. 71:5647–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z, Wang Y, Xue Y, Li X, Cao H, Zheng SJ. 2012. Critical role for voltage-dependent anion channel 2 in infectious bursal disease virus-induced apoptosis in host cells via interaction with VP5. J. Virol. 86:1328–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yao K, Goodwin MA, Vakharia VN. 1998. Generation of a mutant infectious bursal disease virus that does not cause bursal lesions. J. Virol. 72:2647–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yao K, Vakharia VN. 2001. Induction of apoptosis in vitro by the 17-kDa nonstructural protein of infectious bursal disease virus: possible role in viral pathogenesis. Virology 285:50–58 [DOI] [PubMed] [Google Scholar]
- 17. Galluzzi L, Kepp O, Morselli E, Vitale I, Senovilla L, Pinti M, Zitvogel L, Kroemer G. 2010. Viral strategies for the evasion of immunogenic cell death. J. Intern. Med. 267:526–542 [DOI] [PubMed] [Google Scholar]
- 18. Wong RT, Hon CC, Zeng F, Leung FC. 2007. Screening of differentially expressed transcripts in infectious bursal disease virus-induced apoptotic chicken embryonic fibroblasts by using cDNA microarrays. J. Gen. Virol. 88:1785–1796 [DOI] [PubMed] [Google Scholar]
- 19. O'Neill AM, Livant EJ, Ewald SJ. 2010. Interferon alpha-induced inhibition of infectious bursal disease virus in chicken embryo fibroblast cultures differing in Mx genotype. Avian Dis. 54:802–806 [DOI] [PubMed] [Google Scholar]
- 20. Lejal N, Da Costa B, Huet JC, Delmas B. 2000. Role of Ser-652 and Lys-692 in the protease activity of infectious bursal disease virus VP4 and identification of its substrate cleavage sites. J. Gen. Virol. 81:983–992 [DOI] [PubMed] [Google Scholar]
- 21. Rodríguez-Lecompte JC, Kibenge FS. 2002. Site-directed mutagenesis of Avibirnavirus VP4 gene. Virology 292:241–246 [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Wu X, Li H, Wu Y, Shi L, Zheng X, Luo M, Yan Y, Zhou J. 2009. Antibody to VP4 protein is an indicator discriminating pathogenic and nonpathogenic IBDV infection. Mol. Immunol. 46:1964–1969 [DOI] [PubMed] [Google Scholar]
- 23. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740 [DOI] [PubMed] [Google Scholar]
- 24. Zhang M, Tian Y, Wang RP, Gao D, Zhang Y, Diao FC, Chen DY, Zhai ZH, Shu HB. 2008. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res. 18:1096–1104 [DOI] [PubMed] [Google Scholar]
- 25. Laderach D, Compagno D, Danos O, Vainchenker W, Galy A. 2003. RNA interference shows critical requirement for NF-kappa B p50 in the production of IL-12 by human dendritic cells. J. Immunol. 171:1750–1757 [DOI] [PubMed] [Google Scholar]
- 26. Payvandi F, Amrute S, Fitzgerald-Bocarsly P. 1998. Exogenous and endogenous IL-10 regulate IFN-alpha production by peripheral blood mononuclear cells in response to viral stimulation. J. Immunol. 160:5861–5868 [PubMed] [Google Scholar]
- 27. Sharma P, Kumar S, Kundu GC. 2010. Transcriptional regulation of human osteopontin promoter by histone deacetylase inhibitor, trichostatin A in cervical cancer cells. Mol. Cancer 9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abdul-Careem MF, Hunter BD, Lee LF, Fairbrother JH, Haghighi HR, Read L, Parvizi P, Heidari M, Sharif S. 2008. Host responses in the bursa of Fabricius of chickens infected with virulent Marek's disease virus. Virology 379:256–265 [DOI] [PubMed] [Google Scholar]
- 29. Li YP, Handberg KJ, Juul-Madsen HR, Zhang MF, Jorgensen PH. 2007. Transcriptional profiles of chicken embryo cell cultures following infection with infectious bursal disease virus. Arch. Virol. 152:463–478 [DOI] [PubMed] [Google Scholar]
- 30. Liu H, Zhang M, Han H, Yuan J, Li Z. 2010. Comparison of the expression of cytokine genes in the bursal tissues of the chickens following challenge with infectious bursal disease viruses of varying virulence. Virol. J. 7:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. 2007. Negative regulation of Toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science 317:675–678 [DOI] [PubMed] [Google Scholar]
- 32. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 33. Rodríguez-Lecompte JC, Nino-Fong R, Lopez A, Frederick Markham RJ, Kibenge FS. 2005. Infectious bursal disease virus (IBDV) induces apoptosis in chicken B cells. Comp. Immunol. Microbiol. Infect. Dis. 28:321–337 [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Qi X, Gao H, Gao Y, Lin H, Song X, Pei L, Wang X. 2009. Comparative study of the replication of infectious bursal disease virus in DF-1 cell line and chicken embryo fibroblasts evaluated by a new real-time RT-PCR. J. Virol. Methods 157:205–210 [DOI] [PubMed] [Google Scholar]
- 35. Upadhyay C, Ammayappan A, Patel D, Kovesdi I, Vakharia VN. 2011. Recombinant infectious bursal disease virus carrying hepatitis C virus epitopes. J. Virol. 85:1408–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C. 2005. Recognition of double-stranded RNA by human Toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J. Biol. Chem. 280:38133–38145 [DOI] [PubMed] [Google Scholar]
- 37. Wang YY, Liu LJ, Zhong B, Liu TT, Li Y, Yang Y, Ran Y, Li S, Tien P, Shu HB. 2010. WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-kappaB activation. Proc. Natl. Acad. Sci. U. S. A. 107:815–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen D, Li Z, Yang Q, Zhang J, Zhai Z, Shu HB. 2003. Identification of a nuclear protein that promotes NF-kappaB activation. Biochem. Biophys. Res. Commun. 310:720–724 [DOI] [PubMed] [Google Scholar]
- 39. Huang J, Teng L, Liu T, Li L, Chen D, Li F, Xu LG, Zhai Z, Shu HB. 2003. Identification of a novel serine/threonine kinase that inhibits TNF-induced NF-kappaB activation and p53-induced transcription. Biochem. Biophys. Res. Commun. 309:774–778 [DOI] [PubMed] [Google Scholar]
- 40. Carmody RJ, Maguschak K, Chen YH. 2006. A novel mechanism of nuclear factor-kappaB regulation by adenoviral protein 14.7K. Immunology 117:188–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. 2004. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J. Biol. Chem. 279:15652–15661 [DOI] [PubMed] [Google Scholar]
- 42. Carmody RJ, Chen YH. 2007. Nuclear factor-kappaB: activation and regulation during toll-like receptor signaling. Cell. Mol. Immunol. 4:31–41 [PubMed] [Google Scholar]
- 43. Beaulieu E, Morand EF. 2011. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat. Rev. Rheumatol. 7:340–348 [DOI] [PubMed] [Google Scholar]
- 44. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, Capel F, Balian A, Naveau S, Galanaud P, Lemoine FM, Emilie D. 2006. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood 107:2037–2044 [DOI] [PubMed] [Google Scholar]
- 45. Müller H, Islam MR, Raue R. 2003. Research on infectious bursal disease—the past, the present and the future. Vet. Microbiol. 97:153–165 [DOI] [PubMed] [Google Scholar]
- 46. Sharma JM, Kim IJ, Rautenschlein S, Yeh HY. 2000. Infectious bursal disease virus of chickens: pathogenesis and immunosuppression. Dev. Comp. Immunol. 24:223–235 [DOI] [PubMed] [Google Scholar]
- 47. van den Berg TP, Eterradossi N, Toquin D, Meulemans G. 2000. Infectious bursal disease (Gumboro disease). Rev. Sci. Tech. 19:509–543 [PubMed] [Google Scholar]
- 48. Peters MA, Lin TL, Wu CC. 2004. Infectious bursal disease virus polyprotein expression arrests growth and mitogenic stimulation of B lymphocytes. Arch. Virol. 149:2413–2426 [DOI] [PubMed] [Google Scholar]
- 49. Ragland WL, Novak R, El-Attrache J, Savic V, Ester K. 2002. Chicken anemia virus and infectious bursal disease virus interfere with transcription of chicken IFN-alpha and IFN-gamma mRNA. J. Interferon Cytokine Res. 22:437–441 [DOI] [PubMed] [Google Scholar]
- 50. Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, Feng D, Chung E, Pitha PM, Fitzgerald-Bocarsly P. 2003. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 74:1125–1138 [DOI] [PubMed] [Google Scholar]
- 51. Zhang L, Hu TJ, Liu HL, Shuai XH. 2011. Inhibitory effect of Sargassum polysaccharide on oxidative stress induced by infectious bursa disease virus in chicken bursal lymphocytes. Int. J. Biol. Macromol. 49:607–615 [DOI] [PubMed] [Google Scholar]
- 52. Wang Y, Lu Y, Yu D, Wang Y, Chen F, Yang H, Zheng SJ. 2008. Enhanced resistance of restraint-stressed mice to sepsis. J. Immunol. 181:3441–3448 [DOI] [PubMed] [Google Scholar]
- 53. Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, Donato V, Migliorati G, Riccardi C. 2007. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 35:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Soundararajan R, Wang J, Melters D, Pearce D. 2007. Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J. Biol. Chem. 282:36303–36313 [DOI] [PubMed] [Google Scholar]
- 55. Riccardi C. 2010. GILZ (glucocorticoid-induced leucine zipper), a mediator of the anti-inflammatory and immunosuppressive activity of glucocorticoids. Ann. Ig. 22:53–59 (In Italian.) [PubMed] [Google Scholar]