

Abstract
Alpha interferon (IFN-α) production is triggered when influenza virus RNA is detected by appropriate pattern recognition receptors in the host cell. IFN-α induces the expression of more than 300 interferon-stimulated genes (ISGs), and this blunts influenza virus replication. The human ISG MxA can inhibit influenza A virus replication in mouse cells by interfering with a step in the virus replication cycle after primary transcription of the negative-strand RNA genome to mRNA (J. Pavlovic, O. Haller, and P. Staeheli, J. Virol. 66:2564–2569, 1992). To determine the role of MxA in blocking human influenza A virus replication in primate cells, we manipulated MxA expression in rhesus kidney epithelial cells (LLC-MK2) and human lung carcinoma cells (A549). We found that IFN-α treatment prior to influenza virus infection suppressed virus replication and induced the expression of many ISGs, including MxA. However, IFN-α-mediated suppression of virus replication was abolished by small interfering RNA (siRNA) knockdown of MxA expression in IFN-treated cells. In addition, influenza virus replication was suppressed in Vero cells stably transfected with MxA. A strand-specific reverse transcription-PCR (RT-PCR) assay showed that positive-strand influenza virus mRNA and negative-strand genomic RNA (gRNA) accumulated to high levels at 8 h after infection in control Vero cells containing the empty vector. However, in Vero cells stably transfected with MxA positive-strand influenza virus mRNA, complementary positive-strand influenza virus genome RNA (cRNA) and influenza virus gRNA were drastically suppressed. Thus, in primate cells, MxA inhibits human seasonal influenza virus replication at a step prior to primary transcription of gRNA into mRNA. Taken together, these results demonstrate that MxA mediates control of influenza virus replication in primate cells treated with IFN-α.
INTRODUCTION
Influenza A viruses are enveloped viruses with a segmented negative-strand RNA genome and a complex replication cycle. After attachment, the virus enters the cell by receptor-mediated endocytosis (1) and the viral nucleocapsids (vRNPs), which contain the RNA genome bound to nucleoprotein (NP) and the viral RNA-dependent RNA polymerase (2, 3), are released into the cytoplasm and transported to the nucleus (4). In the nucleus, the negative-strand viral RNA genome segments (genomic RNA [gRNA]) are transcribed to produce viral mRNAs, a process referred to as primary transcription. Viral mRNAs are translocated to the cytoplasm and use the host cell translational machinery to produce new viral proteins. When sufficient levels of NP traffic to, and accumulate in, the nucleus, the viral polymerase switches from producing mRNA to producing full-length positive-strand complementary genome RNA (cRNA) (5, 6). Production of cRNA from gRNA is referred to as secondary transcription. These cRNAs are in turn used as the template for de novo synthesis of new negative-strand gRNAs. Newly made gRNA is then complexed with the viral polymerase and encapsidated by NP to form new vRNPs, which are then exported from the nucleus (7) and transported to the cell membrane for packaging into daughter virions that bud from the cell (8).
Understanding the cell biology of influenza virus replication is important because seasonal influenza A viruses cause highly contagious upper respiratory tract infections in humans that result in high morbidity and mortality each year, mostly in the young, old, and immunocompromised (9). Thus, seasonal influenza A viruses have both public health and economic impacts that make identifying new interventions for controlling virus replication in humans an important area of research. Type I interferons (alpha and beta interferons [IFN-α and -β, respectively]) elaborated as part of the innate immune response suppress influenza A virus replication and reduce disease in mice, ferrets, and nonhuman primates (10–12). Infected cells express type I IFNs when the single-stranded negative-strand RNA influenza virus genome is recognized by pattern recognition receptors in the cytoplasm and endosomes. Type I IFN expression in turn induces the expression of hundreds of interferon-stimulated genes (ISGs), such as myxovirus resistance genes (MxA in primates and Mx1 in mice), RNA-dependent protein kinase (PKR), oligoadenylate synthetase (OAS), interferon-stimulated gene 15 (ISG15), and interferon-inducible transmembrane protein 3 (IFITM3) (13). ISGs are effective against a variety of viruses, with a range of antiviral activities that can include blocking protein synthesis, degrading RNA genomes, or misdirecting viral components away from the cellular sites of virus replication (14).
Mx1 is the ISG that blocks influenza A virus replication in mouse cells. In the A2G mouse line, which is resistant to infection with mouse-adapted influenza virus strains, influenza virus resistance was mapped to Mx1 (15, 16). In contrast to the functional Mx1 gene present in A2G mice and most wild mice (17, 18), the majority of inbred laboratory strains have a deletion or nonsense mutation in the Mx1 gene rendering it nonfunctional (19). Functional Mx1 is located in the nucleus of mouse cells, and it blocks primary transcription of the influenza virus genome by directing the viral gene segments to degradation pathways (20–22). However, the mechanism by which type I IFNs inhibit influenza virus replication in primate cells has not been determined. In contrast to mouse cells, where Mx1 is located in the nucleus (20–22), MxA, the primate homologue of Mx1, is located in the cytoplasm of primate cells. While MxA can block influenza virus replication in murine cells (38), the role of MxA in suppressing human influenza virus replication in primate cells has not been determined.
In our previous studies, we noted that while the level of influenza virus replication correlates with the mRNA levels of IFN-α and most ISGs in the respiratory tract of rhesus macaques (RM), MxA expression was suppressed by uncontrolled virus replication (23). When influenza virus replication was moderated by administering oseltamivir phosphate (Tamiflu), MxA expression was greatly increased while expression of IFN and other ISGs was decreased (23). Because MxA expression is tightly regulated by type I IFN (24), these observations suggest that influenza virus actively suppresses MxA expression. The purpose of this study was to determine the role of MxA in IFN-α-induced control of influenza A virus replication in primate cells.
MATERIALS AND METHODS
Cells and viruses.
Madin-Darby canine kidney (MDCK) cells were maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine (Invitrogen Life Technologies, Grand Island, NY). RM monkey kidney cells (LLC-MK2) were maintained in MEM containing 0.1 mM MEM nonessential amino acids (NEAA; Invitrogen Life Technologies, Grand Island, NY), and human lung carcinoma cells (A549) were maintained in Ham's F-12K medium supplemented with 10% FBS. These cells produce IFN-α and the complete type I IFN-stimulated gene response after influenza virus infection. Thus, with these cells we can determine if MxA plays a role in controlling influenza A virus replication in the context of the global type I IFN response to virus infection. A Vero monkey kidney cell line (Vero) constitutively expressing MxA (VA9 cells) and a MxA-negative Vero cell line (VN36 cells) were generously provided by Georg Kochs at the University of Freiburg, Freiburg, Germany (25). Vero cells are incapable of producing type I IFN in response to influenza virus infection; thus, they were used to determine the effect of MxA expression alone on influenza virus replication without the confounding effects of the global type I IFN response.
VA9 and VN36 cells were maintained in Dulbecco's modified Eagle's medium containing l-glutamine (DMEM) supplemented with 10% fetal bovine serum (FBS) and 2 mg/ml Geneticin (Invitrogen Life Technologies, Grand Island, NY). MDCK, LLC-MK2, and A549 cells were obtained from the ATCC (Manassas, VA).
The minimally passaged human isolate influenza virus strains A/Memphis/7/01 (generously provided by R. Webby, St. Jude Children's Research Hospital, Memphis, TN) and A/Wyoming/3/03 (Influenza Reagent Resource, Manassas, VA) were grown on MDCK cells to 106.5 50% tissue culture infective doses (TCID50)/ml and 104.5 TCID50/ml, respectively. Titers were determined by TCID50 assays on MDCK cells as previously described (23).
Influenza virus infections.
For all influenza A virus infections, cells were inoculated at a multiplicity of infection (MOI) of 0.01 for 1 h at 37°C. Infected cells were then maintained in infection medium consisting of MEM supplemented with 0.3% bovine serum albumin (BSA) fraction V (Invitrogen Life Technologies, Grand Island, NY), 2 mM l-glutamine, 0.1 mM NEAA, and 2 μg/ml of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin (Sigma-Aldrich, St. Louis, MO). For TCID50 assays, supernatants were collected at 36 h after infection and frozen at −80°C. For strand-specific PCR, cells were trypsinized with trypsin-EDTA (Cellgro, Manassas, VA) and pelleted. Cell pellets were then lysed with TRIzol reagent (Invitrogen Life Technologies, Grand Island, NY) and stored at −80°C.
siRNA knockdown experiments.
A high-performance purity (HPP) small interfering RNA (siRNA) directed against rhesus macaque MxA was generated against the full-length mRNA sequence cloned from rhesus macaque peripheral blood mononuclear cells (PBMCs) and submitted to GenBank. The MxA-specific siRNA (siMxA) was 5′-ATGGGAATCAGTCATGAGCTA-3′ (rhesus macaque MxA GenBank accession no. EF101561) with DNA overhangs and was unmodified. siRNA experiments were carried out in 6-well flat-bottom cell culture plates containing LLC-MK2 rhesus kidney epithelial cells starting at 2.5 × 105 cells/well. siRNAs for siMxA or the negative-control siRNA (tagged with Alexa 488) were transfected at a concentration of 100 nM into cells using Oligofectamine reagent (Invitrogen Life Technologies, Grand Island, NY) according to the manufacturer's instructions. Six hours later, the transfection solution was aspirated and 20,000 IU of Pegasys IFN-α-2a in complete medium was added to the cultures. Cells were then incubated for 24 h, washed with phosphate-buffered saline (PBS) again, and infected with A/Memphis/7/01 influenza A virus as described above. Supernatants were frozen at −80°C and later used to determine the levels of infectious virus by determining TCID50 in MDCK cells.
Viral ssRT-QPCR assays.
A strand-specific reverse transcription-quantitative PCR (ssRT-QPCR) assay based on a published method for quantifying all three influenza virus RNA species (11) was modified for A/Wyoming/3/03 H3N2 and A/Memphis/7/01 H1N1. Cell pellets were lysed using TRIzol reagent per the manufacturer's instructions (Invitrogen Life Technologies, Grand Island, NY) and stored at −80°C. To perform the assay, the samples stored in TRIzol were thawed and total RNA was extracted using Qiagen's RNeasy microkit per the manufacturer's instructions. cDNA synthesis was performed as described below with one exception. cDNA synthesis from the cRNA template was performed with the addition of 6.5 μl of saturated trehalose (using trehalose dihydrate; Life Sciences Advanced Technologies, St. Petersburg, FL) in place of nuclease-free water to reduce nonspecific amplification of mRNA by cRNA primers (11).
To construct plasmids for standard curves, total RNA was extracted from 1 × 106 A549 cells at 6 h post-influenza virus infection with either A/Wyoming/3/03 H3N2 or A/Memphis/7/01 H1N1 (MOI, 0.01) using an RNeasy minikit per the manufacturer's instructions (Qiagen Inc., Valencia, CA). cDNA was made from 5 μl of total RNA using tagged cDNA synthesis oligonucleotides in segment 5 for each specific strand of interest. For A/Memphis/7/01, cDNA was synthesized using oligonucleotides specific for gRNA, A/Mem H1N1 seg5 vtagsynth (GGC CGT CAT GGT GGC GAA TGA ATG GAA GGA AAA CAA GGA TTG C); mRNA, A/Mem H1N1 seg5 mtagsynth (CCA GAT CGT TCG AGT CGT TTT TTT TTT TTT TTT TCA TTA ATT GTC); and cRNA, A/Mem H1N1 seg5 ctagsynth (GCT AGC TTC AGC TAG GCA TCA GTA GAA ACA AGG GTA TTT TTC ATT). A/Wyoming/3/03 cDNA was synthesized using primers specific for gRNA, A/Wyo H3N2 seg5 vtagsynth (GGC CGT CAT GGT GGC GAA TGA ATG GGC GGA AAA CAA GAA GTG C); mRNA, A/Wyo H3N2 seg5 mtagsynth (CCA GAT CGT TCG AGT CGT TTT TTT TTT TTT TTT TCC TTA ATT GTC); and cRNA, A/Wyo H3N2 seg5 ctagsynth (GCT AGC TTC AGC TAG GCA TCA GTA GAA ACA AGG GTA TTT TTC CTT). cDNA synthesis was performed using the Quantitect reverse transcription kit (Qiagen) as follows.
On ice, 5 μl of total RNA, 1 μl of nuclease-free water, and 1 μl of 7× gDNA Wipeout buffer were mixed. The mixture was then incubated at 42°C in a preheated thermal cycler for 3 min and immediately placed back on ice for 5 min. While still on ice, the following master mix was added: 4 μl 5× Quantiscript reverse transcriptase (RT) buffer, 1 μl Quantiscript RT, 1 μl 20 μM tagged strand-specific primer, and 7 μl nuclease-free water (or in the case of cRNA, 6.5 μl saturated trehalose and 0.5 μl nuclease-free water). The resulting mixture was vortexed briefly, spun down, and placed back on ice. The cDNA synthesis reaction mixtures were then placed in a preheated thermal cycler at 42°C for 30 min and heated to 95°C for 3 min, with a final dwell at 4°C. The cDNA reaction mixture was then diluted 1:10 with Tris-EDTA (TE), pH 8.0, and 5 μl was used as the template for PCR.
PCR was done with 5 μl strand-specific cDNA template, and PCR was performed with HotStarTaq Plus (Qiagen Inc., Valencia, CA) in a 25-μl total volume per the manufacturer's instructions. PCR primers were as follows: A/Memphis/7/01 gRNA amplicon, forward primer vRNAtag (GGC CGT CAT GGT GGC GAA T) and reverse primer A/Memseg5 vRNA_R (CTT AAT ATG AGT GCA GAC CGT GCC); A/Memphis/7/01 mRNA amplicon, forward primer A/Memseg5 cRNAmRNA_F (CGA TCG TGC CCT CCT TTG) and reverse primer mRNAtag (CCA GAT CGT TCG AGT CGT); A/Memphis/7/01 cRNA amplicon, forward primer A/Memseg5 cRNAmRNA_F (CGA TCG TGC CCT CCT TTG) and reverse primer cRNAtag (GCT AGC TTC AGC TAG GCA TC); A/Wyoming/3/03 gRNA amplicon, forward primer vRNAtag and reverse primer A/Wyoseg5 vRNA_R (CCT CTC AAT ATC AAT GCA GAT CTT GCC); A/Wyoming/3/03 mRNA amplicon, forward primer A/Wyoseg5 cRNAmRNA_F (CCG ATC GTG CCC TCT TTT G) and reverse primer mRNAtag; A/Wyoming/3/03 cRNA amplicon, forward primer A/Wyoseg5 cRNAmRNA_F (CCG ATC GTG CCC TCT TTT G) and reverse primer cRNAtag. PCR was done in an Eppendorf Mastercycler (Eppendorf AG. Hamburg, Germany). The cycling conditions were as follows: 95°C for 5 min, followed by 8 cycles of 94°C for 20 s, 63°C for 20 s [−1°C/cycle], and 72°C for 20 s; followed by 35 cycles of 94°C for 20 s, 56°C for 20 s, and 72°C for 20 s; and then 72°C for 10 min, with a final dwell at 4°C. One microliter of the PCR product was cloned into pCR-4 TOPO per the manufacturer's instructions. Plasmids were then purified using Qiagen's plasmid minikit per the manufacturer's instructions and sequenced, and plasmid copy numbers were calculated based on Nanodrop 2000 (Thermo Scientific, Wilmington, DE) spectrophotometer readings. To calculate the number of strand-specific copies, standard curves were made using plasmids containing 101 to 107 copies per well in at least 12 wells per dilution.
The ssRT-QPCR assay was performed using Qiagen's QuantiTect PCR probe kit (Qiagen, Valencia, CA) as follows. Five microliters of 10-fold-diluted species-specific cDNA was added to a master mix containing 12.5 μl of QuantiTect Probe PCR master mix, 0.5 μl of 10 μM probe, 1.0 μl each of 10 μM forward and reverse primers, and 5.0 μl of nuclease-free water. All primers and probes were ordered through Integrated DNA Technologies, Inc., Coralville, IA. A universal probe was used to detect gRNA species in both viruses: universal vRNA_P1 (5,6-carboxyfluorescein [5,6-FAM]–TGA GAT CTT-ZEN-CGA WCT CAG CAT TTC CTG GRT TCC-3IABkFQ); the universal probe to detect mRNA or cRNA species was universal cRNAmRNA_P1 (5,6-FAM–AGT AAT GAA-ZEN-GGA TCT TAT TTC TTC GGA GAC AAT GC-3IABkFQ). Other primers and probes were as follows: to detect A/Memphis/7/01 gRNA, forward primer vRNAtag (GGC CGT CAT GGT GGC GAA T), reverse primer A/Memseg5 vRNA_R (CTT AAT ATG AGT GCA GAC CGT GCC), and probe univ vRNA_P1; for A/Memphis/7/01 mRNA, forward primer A/Memseg5 cRNAmRNA_F (CGA TCG TGC CCT CCT TTG), reverse primer mRNAtag (CCA GAT CGT TCG AGT CGT), and probe univ cRNAmRNA_P1; for A/Memphis/7/01 cRNA, forward primer A/Memseg5 cRNAmRNA_F, reverse primer cRNAtag (GCT AGC TTC AGC TAG GCA TC), and probe univ cRNAmRNA_P1; for A/Wyoming/3/03 gRNA, forward primer vRNAtag, reverse primer A/Wyoseg5 vRNA_R (CCT CTC AAT ATC AAT GCA GAT CTT GCC), and probe univ vRNA_P1; for A/Wyoming/3/03 mRNA detection, forward primer A/Wyoseg5 cRNAmRNA_F (CCG ATC GTG CCC TCT TTT G), reverse primer mRNAtag, and probe univ cRNAmRNA_P1; for A/Wyoming/3/03 cRNA, forward primer A/Wyoseg5 cRNAmRNA_F, reverse primer cRNAtag, and probe univ cRNAmRNA_P1. ssRT-QPCRs were run on an ABI 7900 system heated at 95°C for 15 min, followed by 45 cycles of 95°C for 15 s and 60°C for 1 min.
Immunocytochemistry.
Cytospin slides were fixed with 100% acetone at 4°C for 15 min and then air dried and stored at −20°C. Before applying the first antibody, slides were blocked with 10% normal goat serum at room temperature for 20 min. MxA antibody (clone M143; a gift from Georg Kochs, Institute for Medical Microbiology and Hygiene, Freiburg, Germany) was diluted 1:100 in Tris-buffered saline (TBS)–5% bovine serum albumin (BSA)–2% normal monkey serum and placed onto the slides, and the slides were incubated at 4°C overnight. Binding of the MxA primary antibody was detected using Alexa Fluor 568-labeled polyclonal goat anti-mouse IgG (Life Technologies, Grand Island, NY), diluted in 1:400 TBS-5% BSA-2% normal monkey serum at room temperature for 1 h. Subsequently, an influenza virus nucleoprotein (NP) monoclonal antibody (fluorescein isothiocyanate [FITC]) (clone M2110169; Fitzgerald Industries International, North Acton, MA) was diluted 1:50, and a rabbit anti-FITC antibody (Invitrogen, Carlsbad, CA) was added to the slide and incubated at room temperature for 1 h. Binding of the rabbit anti-FITC antibodies was detected using Alexa Fluor 488-labeled goat anti-rabbit IgG (Life Technologies, Grand Island, NY). Slides were washed 3 times for 5 min in Tris-buffered saline (TBS) containing 0.5% Tween 20. Slides were briefly incubated in a 4′,6-diamidino-2-phenylindole (DAPI) solution to stain nuclei. Primary antibodies were replaced by mouse IgG (Dako, Carpinteria, CA) and included with each staining series as the negative control. All slides were coverslipped using Prolong Gold with 4′,6-diamidino-2-phenylindole dihydrochloride hydrate (DAPI) (Life Technologies, Grand Island, NY) to stain nuclei. Slides were visualized with epifluorescent illumination using a Zeiss Imager Z1 microscope (Carl Zeiss Inc., Thornwood NY) and appropriate filters. Digital images were captured using a Zeiss AxioCam system.
Statistical analysis.
Statistical analyses were performed using Prism 5.0 software (GraphPad Software). A two-tailed t test was used to compare two groups. For comparisons of more than 2 groups, one-way analysis of variance (ANOVA) with Tukey's multiple comparison test was used.
RESULTS
IFN-α-induced MxA is necessary to control influenza virus replication in primate cells.
To determine if IFN-α-induced MxA expression correlated with suppressed influenza A virus replication, LLC-MK2 cells were treated with recombinant human pegylated IFN-α (Pegasys; Roche Pharmaceuticals) 24 h prior to A/Memphis/7/01 inoculation (MOI, 0.01) (Fig. 1A). We found that IFN-α induced expression of MxA and suppressed influenza A virus replication in a dose-dependent manner (Fig. 1A and B). In fact, the highest concentration of IFN-α tested (2 × 104 IU/ml) induced a 102.5-fold increase in MxA mRNA levels in LLC-MK2 cells and reduced influenza virus matrix gene RNA levels 101.5-fold compared to control cultures. Thus, administration of exogenous IFN-α to LLC-MK2 cells induced MxA expression and suppressed influenza A virus replication.
Fig 1.

MxA is necessary for control of human seasonal influenza virus replication in IFN-α-stimulated LLC-MK2 cells. (A) Effect of IFN-α administration on MxA mRNA expression 24 h after treatment. Bars represent an average of three replicate cultures (mean ± standard error). (B) Effect of IFN-α administration on A/Memphis/7/01 virus replication. Values represent the log10 copies of matrix RNA/ml of culture supernatant. Bars are an average of three replicate cultures (mean ± standard error). (C) Knockdown of rhesus macaque MxA. Cell cultures were transfected with siRNAs against MxA (siMxA) or with AllStar negative-control siRNA (siNeg), and 24 h later, IFN-α was added. Bars represent the average of two experiments run in triplicate (mean ± standard error). (D) Immunofluorescence staining of cytospin slides of cells transfected with siMxA or siNeg and inoculated with IFN-α for MxA (red) and DAPI (blue). (E) Infectious influenza virus titers (TCID50/ml) in supernatant of cultures transfected with siMxA or siNeg. Six hours later, IFN-α was added, and 24 h after that, the cells were infected with A/Memphis/7/01 and compared to control cultures infected with A/Memphis/7/01 alone or treated with IFN-α and then infected with A/Memphis/7/01. Bars represent the average of four experiments run in triplicate (mean ± standard error). The P value was generated using an ANOVA, and the results of Tukey's post hoc pairwise comparisons are shown only if differences were significant.
To determine if MxA expression is necessary to control influenza virus replication, MxA mRNA was knocked down in interferon-treated LLC-MK2 cells prior to infection. LLC-MK2 cells were transfected with MxA siRNA designed for rhesus macaque MxA (siMxA) 6 h prior to administration of 2 × 104 IU/ml of IFN-α. There was a 90% reduction in MxA mRNA and protein levels in LLC-MK2 cells transfected with MxA siRNA compared to those in IFN-α-treated LLC-MK2 cells transfected with a negative-control siRNA (Qiagen's AllStars) (Fig. 1C and D). The level of influenza virus replication in cells administered IFN-α 24 h prior to A/Memphis/7/01 inoculation was similar to the level in IFN-α-treated cells that were transfected with an irrelevant siRNA but lower than that in control cells infected with A/Memphis/7/01 (Fig. 1E). Thus, IFN-α-treated cells produced 101.3-fold less infectious virus than did untreated LLC-MK2 cultures infected with A/Memphis/7/01 (ANOVA, P = 0.0029; Tukey's test, P < 0.05) (Fig. 1E). However, when MxA expression was knocked down in IFN-α-treated cells by transfection with siMxA, LLC-MK2 cells produced infectious virus in amounts similar to those of untreated control cells (Fig. 1E). This siRNA effect was MxA specific, as the levels of influenza virus produced by IFN-α-treated cells transfected with the negative-control siRNA (siNeg) were similar to those in IFN-α-treated cells and significantly lower than those in untreated cells (ANOVA, P = 0.0029; Tukey's test, P < 0.01) (Fig. 1E). When the experiment was repeated in human A549 cells, only 70 to 80% knockdown of MxA mRNA could be achieved. As expected, this level of MxA knockdown blunted, but did not completely abrogate, the anti-influenza virus effect of IFN-α (data not shown). Thus, knocking down >90% of IFN-induced MxA expression was required to abrogate the anti-influenza virus effect of interferon as was achieved in LLC-MK2 cells. This result confirms that MxA expression is necessary for IFN-α-mediated suppression of human seasonal influenza virus replication in human and nonhuman primate cells.
To determine the localization of MxA and influenza virus nucleoprotein (NP) in LLC-MK2 cells, IFN-α (2 × 104 IU/ml) was added to cultures 24 h prior to A/Memphis/7/01 inoculation (MOI, 0.01). Twenty-four hours after infection, cytospins of the infected and uninfected cells were labeled to detect MxA and NP and examined by fluorescence microscopy. In the IFN-α-stimulated, uninfected cells (Fig. 2A), MxA was diffusely distributed throughout the cytoplasm, and in addition, most cells had a perinuclear space devoid of MxA labeling. In the influenza virus-infected cells (Fig. 2B), the distribution of MxA was no longer diffuse. In some infected cells, MxA was found concentrated in perinuclear aggregates, while in other cells, MxA colocalized with NP in the cytoplasm. In some cells, both types of MxA localization were apparent.
Fig 2.
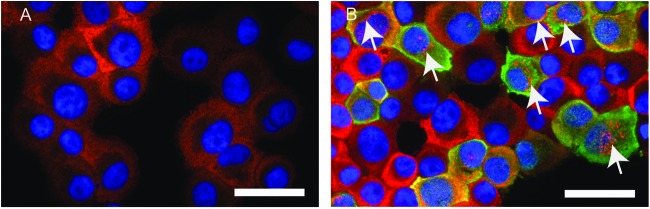
MxA localizes in perinuclear aggregates within the cytoplasm of influenza virus-infected, IFN-α-stimulated LLC-MK2 cells. Cytospins of IFN-α-stimulated LLC-MK2 cells were immunofluorescent antibody labeled to detect MxA protein (red), influenza virus nucleoprotein (green), and nuclei (blue). (A) Uninfected cells. (B) Influenza A virus-infected cells 24 h after inoculation. Arrows denote perinuclear aggregates of MxA. Yellow indicates colocalization of MxA and NP. Bars, 50 μm.
MxA expression is sufficient to control human seasonal influenza virus replication.
To determine whether MxA expression is sufficient to control seasonal human influenza virus replication in primate cells, we used Vero cells stably transfected with human MxA (VA9 cells) or a negative-control Vero cell line transfected with an empty vector (VN36) (Fig. 3A and B); both cell lines were generously provided by Georg Kochs (25). Vero cells are unable to produce type I IFN in response to viral infection (26, 27), but they respond normally to exogenous type I IFN. The absence of an endogenous interferon response to infection by influenza virus makes Vero cells uniquely appropriate for assessing the effect of exogenous MxA expression on influenza A virus replication. Note that VA9 cells express about 101-fold more MxA mRNA than do IFN-α-stimulated VN36 cells (P < 0.0001) (Fig. 3A). VA9 or VN36 cells were infected with A/Memphis/7/01 (MOI of 0.01), and strand-specific RT-PCR was used to determine the levels of matrix and NP gRNA (28). Eight hours after infection, influenza virus matrix and NP gRNA levels were approximately 101.5-fold lower in VA9 cells than in VN36 cells (P < 0.0001) (Fig. 3C and D). Because influenza virus NP and matrix gRNA levels were similarly affected by MxA expression, we used NP RNA levels as a marker for influenza virus replication. This demonstrates that expression of human MxA in Vero cells is sufficient to control human seasonal influenza virus replication in primate cells.
Fig 3.

A/Memphis/7/01 replication is inhibited in Vero cells stably transfected with human MxA. (A) MxA mRNA levels were assessed by TAQman PCR in untreated MxA-transfected Vero cells (MxA+; VA9) and empty-plasmid-transfected control Vero cells (MxA−; VN36) treated with IFN-α. The levels of MxA mRNA in these cultures relative to those in untreated VN36 cell cultures are shown. Bars represent the average of three samples (mean ± standard error). (B) Immunofluorescence was performed on cytospins of VA9 and VN36 cells for MxA (red) and nuclei (blue). (C) VA9 and VN36 cells were infected with A/Memphis/7/01, and the levels of nucleoprotein (NP) gRNA were assessed 8 h later. (D) VA9 and VN36 cells were infected with A/Memphis/7/01, and the levels of matrix gRNA were assessed 8 h later. Values presented are negative-strand gRNA copies per cell. Bars represent data from two experiments run in triplicate (mean ± standard error). (P values were generated with a two-tailed Student t test.)
MxA blocks influenza virus replication at, or before, primary transcription of the RNA genome.
To determine at what point MxA inhibits the replication cycle of influenza A virus in primate cells, ssRT-PCR was used to monitor the levels of positive-stranded NP mRNA, positive-stranded NP cRNA, and negative-stranded NP gRNA in cells infected with A/Memphis/7/01 (Fig. 4). Replication of human influenza virus isolates is hindered at 41°C (29, 30) during transcription of cRNA to gRNA. This block in secondary transcription is due to the unstable interaction between cRNA and the virus polymerase above 37°C. Thus, when human influenza virus-infected cells are cultured at 41°C, influenza virus gRNA levels are suppressed but influenza virus mRNA levels are high (31). To confirm that the ssRT-PCR method produced similar results, the levels of influenza virus mRNA, cRNA, and gRNA in A549 cells infected with A/Memphis/7/01 (MOI, 0.01) and maintained at 41°C or treated with IFN-α were determined and compared to those in control A549 cultures (37°C and no IFN-α). In cultures infected and maintained at 41°C, we found that gRNA levels were suppressed but mRNA and cRNA levels were high, similarly to 37°C control cultures (Fig. 4A to C). This result is consistent with the published description of the negative effect of high temperature on influenza virus RNA synthesis (31) and suggests that the ssRT-PCR method can be used to detect blocks at specific steps in the influenza virus replication cycle.
Fig 4.

Interferon mediates a block to influenza virus replication at, or before, primary transcription of the negative-strand RNA genome to mRNA. A549 cells were infected with A/Memphis/7/01 and incubated at 37°C or 41°C or treated with IFN-α and incubated at 37°C (37°C + IFN-α). (A to C) ssRT-PCR was used to determine the levels of NP negative-stranded viral genome (gRNA) copies/cell (A), positive-stranded complementary genome (cRNA) copies/cell (B), and positive-stranded mRNA copies/cell (C). (D) Ratio of mRNA/gRNA copies/cell. (E) Area under the curve of mRNA/gRNA ratios. (F) Number of cells in the influenza virus-infected cultures. Total pg of RNA/culture was converted to cell number based on the assumption that there are 10 pg RNA/cell. The ANOVA P value is indicated, and the results of Tukey's post hoc pairwise comparisons are shown only if differences were significant. All data points represent the average of two experiments run in triplicate (mean ± standard error).
To determine at what step MxA blocks influenza virus replication, IFN-α was administered to A549 cells 24 h prior to infection with A/Memphis/7/01. The levels of gRNA, mRNA, and cRNA were low from 0 to 12 h postinfection (p.i.) compared to high levels of all 3 RNAs in control cultures (Fig. 4A to C). The ratio of mRNA to gRNA is a measure of the efficiency of gRNA production. This ratio was much higher in control cells or IFN-α-treated cells incubated at 37°C than in 41°C cultures, a finding that is consistent with a block in gRNA production (Fig. 4D). In fact when the mRNA/gRNA ratios from 0 to 12 h were converted to an area under the curve (AUC) value and the mRNA/gRNA AUC values between all three culture conditions were compared (Fig. 4E), IFN-α-treated cultures had 3-fold-lower mRNA/gRNA AUC values than did 41°C cultures (ANOVA, P < 0.0001; Tukey's test, P < 0.05) (Fig. 4E) and 1.5-fold-lower mRNA/gRNA AUC values than did 37°C control cultures (ANOVA, P < 0.0001; Tukey's test, P < 0.05) (Fig. 4E).
To rule out the possibility that the decrease in influenza virus replication in 41°C cultures was due to increased cell death at high temperature, we determined the level of total (cellular plus viral) RNA in infected cells incubated at 37°C, IFN-α-treated and infected cells incubated at 37°C, and infected cells incubated at 41°C (Fig. 4F). We found that there was no decrease in the level of total RNA in any of the cultures from 1 to 12 h after inoculation (Fig. 4F). Thus, cell death does not contribute to the decrease in influenza virus replication in 41°C cultures. Taken together, these data support the conclusion that primary transcription is the step at which MxA suppresses human influenza virus replication. Thus, in primate cells, IFN-α, and by extension MxA, blocks the influenza A virus replication cycle at, or before, primary transcription, when the negative-strand gRNA is transcribed into mRNA by the viral RNA polymerase.
To confirm that primary transcription is the step at which MxA suppresses human influenza virus replication, MxA-transfected Vero (VA9) and empty-vector control (VN36) cells were infected with A/Memphis/7/01 (MOI of 0.01) and influenza virus RNA levels were analyzed 8 h later (Fig. 5A). This time point was selected because gRNA and mRNA levels are high in control cells at 8 h p.i. (Fig. 4A and C). At 8 h p.i., all 3 viral RNA species (gRNA, mRNA, and cRNA) were 102-fold lower in MxA+ VA9 cells than in control VN36 cells infected at 37°C (Fig. 5A to C). Further, compared to VN36 cells incubated at 41°C, VA9 cells had 102-fold-lower influenza virus mRNA and cRNA levels (Fig. 5B and C) and 101-fold lower influenza virus gRNA levels (Fig. 5A). Although influenza virus mRNA and cRNA levels in VN36 cells at 41°C and 37°C were similar at 8 h p.i., influenza virus gRNA levels were lower in 41°C VN36 cells than in 37°C VN36 cells (Fig. 5A). Finally, the influenza virus mRNA-to-gRNA ratios in VA9 cells and VN36 cells incubated at 37°C were similar and were significantly lower than those in 41°C VN36 cells (ANOVA, P < 0.0001; Tukey's test, P < 0.05) (Fig. 5D). These results are consistent with the conclusion that in Vero cells, as in A549 cells, MxA blocks influenza A virus replication at, or before, primary transcription of the negative-strand RNA genome to mRNA.
Fig 5.

MxA blocks replication of human seasonal influenza virus at, or before, primary transcription of genome RNA to mRNA. (A to C) VA9 (MxA+), VN36 (MxA−; Cntrl), or VN36 cells incubated at 41°C (MxA−; Cntrl 41°C) were infected with A/Memphis/7/01 for 8 h, and the numbers of gRNA (A), mRNA (B), and cRNA (C) copies/cell were determined by ssRT-QPCR. (D) Further, the ratio of mRNA/gRNA copies/cell was calculated. (E to G) VA9 (MxA+), VN36 (MxA−; Cntrl), or VN36 cells incubated at 41°C (MxA−; Cntrl 41°C) were infected with A/Wyoming/3/03 for 8 h, and the numbers of gRNA (E), mRNA (F), and cRNA (G) copies/cell were determined by ssRT-QPCR. (H) Further, the ratio of mRNA/gRNA copies/cell was calculated. All data points represent the averages of two experiments run in triplicate (bars represent means ± standard errors). P values, generated using a Kruskal-Wallis (KW) test or 1-way ANOVA (as indicated), are shown if there is a significant difference among the groups, and the results of Dunn's (KW test) or Tukey's (ANOVA) post hoc pairwise comparisons are shown if differences between any 2 groups were significant.
To determine if the effects of MxA are applicable to other human influenza virus isolates, we repeated the infections of VA9 and VN36 cells with the human seasonal isolate A/Wyoming/3/03 (Fig. 5E to H). We chose this isolate because A/Wyoming/3/03 is insensitive to Mx1 overexpression in mouse cells (32). At 8 h after infection, all three species of A/Wyoming/3/03 RNA were 101- to 102-fold lower in VA9 cells than in 37°C VN36 cells or 41°C VN36 cells (Fig. 5E to G). Influenza virus gRNA in VA9 cells was 101-fold lower than that in 37°C VN36 cells (Fig. 5E). Influenza virus mRNA and cRNA levels in VA9 cells were nearly 100-fold lower than those in 37°C VN36 and 41°C VN36 cells (Fig. 5F and G). The ratio of mRNA to gRNA in VA9 cells was much lower than that in 41°C VN36 cells (ANOVA, P < 0.0001; Tukey's test, P < 0.05) (Fig. 5H). In contrast to A/Memphis/7/01, the mRNA/gRNA ratio in A/Wyoming/3/03-infected 37°C VN36 cells was similar to that in 41°C VN36 cells (Fig. 5D and H). This slight difference in results with the two virus strains could be due to a difference in the replication kinetics of the two strains. In summary, MxA expression in primate cells reduces the levels of all three viral RNA species to similar extents after infection with two human seasonal influenza A virus strains and this effect is due to a block in viral replication at, or before, primary transcription of gRNA to mRNA.
DISCUSSION
Administration of exogenous IFN-α to primate cell cultures increased MxA expression and suppressed influenza virus replication, reducing infectious virus titers more than 10-fold. Among the many ISGs induced by IFN-α treatment of primate cells, we found that MxA expression was necessary and sufficient to produce the type I IFN-mediated suppression of human seasonal influenza virus replication. This conclusion is based on three separate lines of evidence. First, the dose of IFN-α added to primate cell cultures correlates with the level of MxA expression and the extent to which influenza virus replication is suppressed. Second, when siRNA is used to knock down IFN-α-induced MxA expression, leaving the levels of all other ISGs high, influenza A virus replication is not suppressed. Third, MxA overexpression in Vero cells by itself suppresses human seasonal influenza virus replication 102-fold compared to that in control Vero cells transfected with the empty vector. Finally, experiments with a second strain of human seasonal influenza A virus produced similar results. Thus, A/Wyoming/3/03 replication was blunted in MxA-expressing Vero (VA9) cells. These results demonstrate that MxA is the IFN-α-stimulated effector molecule that is responsible for suppressing human seasonal influenza A virus replication in primate cells.
In addition, we found that MxA blocks the influenza virus life cycle in primate cells before primary transcription of the negative-strand virus genome to mRNA. This conclusion is based on the fact that MxA expression suppressed production of all three influenza virus RNA species, mRNA, cRNA, and gRNA, equally. This general effect on influenza virus RNA production can be explained only if MxA blocks a step upstream of, or during, primary gRNA transcription to mRNA. However, it remains to be determined how MxA exerts its effect so early in the virus replication cycle. MxA is a GTPase protein that localizes to distinct compartments of the smooth endoplasmic reticulum (21, 33–35). In the case of La Crosse virus, which replicates in the cytoplasm, MxA molecules are released from the smooth endoplasmic reticulum to bind newly translated La Crosse virus NP, an interaction that prevents the switch from mRNA to gRNA production (34). Thus, as we describe above for influenza virus, production of all three La Crosse virus RNA species is dramatically suppressed (36). However, because influenza A virus replicates in the nucleus, this cannot explain the effect of MxA on influenza virus replication. Further, the low-level accumulation of all three species of influenza viral RNA in the presence of MxA that we observed is consistent with a small fraction of influenza virus RNPs eluding MxA in the cytoplasm and translocating to the nucleus, where production of mRNA, cRNA, and gRNA could proceed unimpeded by MxA. This hypothesis could form the basis of additional experiments to define the detailed mechanism behind MxA-mediated early suppression of influenza virus replication in primate cells.
This study makes clear that the ability of Mx1 to suppress virus replication in mouse cells does not predict the extent of MxA activity against the virus in primate cells. The human seasonal isolate A/Wyoming/3/03 is insensitive to Mx1 in mouse cells (32), but as we show here, it is very sensitive to MxA in primate cells (Fig. 4). In addition to the differences in the abilities of Mx1 and MxA to control influenza virus replication in mouse and human cells, the source of virus also likely affects its susceptibility to MxA. Passaging influenza viruses in laboratory mice that lack a functional Mx gene selects for viruses that are acutely sensitive to MxA expression in mouse cells (32, 37). In contrast, human influenza virus isolates are selected for the ability to replicate, at least to some extent, in the presence of MxA. Thus, MxA may work in very different ways to suppress replication of a mouse-adapted influenza A virus in mouse cells or a minimally passaged human influenza virus in human cells.
We also found that the intracellular distribution of MxA in IFN-α-stimulated primate cells was altered by influenza virus infection, with NP and MxA colocalizing to the cytoplasm and MxA aggregating in yet-to-be-defined perinuclear structures. There was no evidence that MxA translocated to the nucleus of influenza A virus-infected primate cells. Thus, while Mx1 suppresses influenza virus replication in the nucleus of mouse cells (20–22), it seems that MxA blocks influenza virus replication in the cytoplasm of primate cells.
We previously showed that IFN-α can control human influenza virus replication in the respiratory tract of primates (11, 23), and the results reported here show that MxA is the specific ISG that inhibits influenza virus replication in primate cells. We also found that MxA blocks human seasonal influenza virus replication at a step prior to, or at, primary transcription of the RNA genome to mRNA. In addition to expanding and clarifying the critical role of the primate innate immune response in effective influenza virus immunity, the data reported here suggest novel therapeutic approaches to treating influenza virus infections of humans.
ACKNOWLEDGMENTS
We thank Richard Webby and David Walker at St. Jude Children's Research Hospital for the influenza A virus and tissue culture protocols; Georg Kochs at the University of Freiburg, Freiburg, Germany, for the VA9 and VN36 cell lines; and the Primate Services Unit at the CNPRC and Tracy Rourke for excellent technical assistance.
This work was supported by Public Health Service grant P51RR00169 from the National Center for Research Resources and grants U01AI57624 and U01AI5726 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Matlin KS, Reggio H, Helenius A, Simons K. 1981. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 91:601–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Compans RW, Content J, Duesberg PH. 1972. Structure of the ribonucleoprotein of influenza virus. J. Virol. 10:795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murti KG, Webster RG, Jones IM. 1988. Localization of RNA polymerases on influenza viral ribonucleoproteins by immunogold labeling. Virology 164:562–566 [DOI] [PubMed] [Google Scholar]
- 4. Martin K, Helenius A. 1991. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 65:232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beaton AR, Krug RM. 1986. Transcription antitermination during influenza viral template RNA synthesis requires the nucleocapsid protein and the absence of a 5′ capped end. Proc. Natl. Acad. Sci. U. S. A. 83:6282–6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vreede FT, Brownlee GG. 2007. Influenza virion-derived viral ribonucleoproteins synthesize both mRNA and cRNA in vitro. J. Virol. 81:2196–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shapiro GI, Gurney T, Jr, Krug RM. 1987. Influenza virus gene expression: control mechanisms at early and late times of infection and nuclear-cytoplasmic transport of virus-specific RNAs. J. Virol. 61:764–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. 2003. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. U. S. A. 100:2002–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brydon EW, Morris SJ, Sweet C. 2005. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 29:837–850 [DOI] [PubMed] [Google Scholar]
- 10. Kugel D, Kochs G, Obojes K, Roth J, Kobinger GP, Kobasa D, Haller O, Staeheli P, von Messling V. 2009. Intranasal administration of alpha interferon reduces seasonal influenza A virus morbidity in ferrets. J. Virol. 83:3843–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matzinger SR, Carroll TD, Fritts L, McChesney MB, Miller CJ. 2011. Exogenous IFN-alpha administration reduces influenza A virus replication in the lower respiratory tract of rhesus macaques. PLoS One 6:e29255 doi:10.1371/journal.pone.0029255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tumpey TM, Szretter KJ, Van Hoeven N, Katz JM, Kochs G, Haller O, Garcia-Sastre A, Staeheli P. 2007. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J. Virol. 81:10818–10821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeuchi O, Akira S. 2007. Recognition of viruses by innate immunity. Immunol. Rev. 220:214–224 [DOI] [PubMed] [Google Scholar]
- 14. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lindenmann J. 1964. Inheritance of resistance to influenza virus in mice. Proc. Soc. Exp. Biol. Med. 116:506–509 [DOI] [PubMed] [Google Scholar]
- 16. Lindenmann J. 1962. Resistance of mice to mouse-adapted influenza A virus. Virology 16:203–204 [DOI] [PubMed] [Google Scholar]
- 17. Haller O, Acklin M, Staeheli P. 1987. Influenza virus resistance of wild mice: wild-type and mutant Mx alleles occur at comparable frequencies. J. Interferon Res. 7:647–656 [DOI] [PubMed] [Google Scholar]
- 18. Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. 1986. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 44:147–158 [DOI] [PubMed] [Google Scholar]
- 19. Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O. 1988. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 8:4518–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelhardt OG, Sirma H, Pandolfi PP, Haller O. 2004. Mx1 GTPase accumulates in distinct nuclear domains and inhibits influenza A virus in cells that lack promyelocytic leukaemia protein nuclear bodies. J. Gen. Virol. 85:2315–2326 [DOI] [PubMed] [Google Scholar]
- 21. Haller O, Kochs G. 2002. Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic 3:710–717 [DOI] [PubMed] [Google Scholar]
- 22. Zurcher T, Pavlovic J, Staeheli P. 1992. Nuclear localization of mouse Mx1 protein is necessary for inhibition of influenza virus. J. Virol. 66:5059–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carroll TD, Matzinger SR, Genesca M, Fritts L, Colon R, McChesney MB, Miller CJ. 2008. Interferon-induced expression of MxA in the respiratory tract of rhesus macaques is suppressed by influenza virus replication. J. Immunol. 180:2385–2395 [DOI] [PubMed] [Google Scholar]
- 24. Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J. Virol. 81:7776–7785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frese M, Kochs G, Meier-Dieter U, Siebler J, Haller O. 1995. Human MxA protein inhibits tick-borne Thogoto virus but not Dhori virus. J. Virol. 69:3904–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. 1988. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. U. S. A. 85:5259–5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43:247–252 [DOI] [PubMed] [Google Scholar]
- 28. Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods 173:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baigent SJ, McCauley JW. 2003. Influenza type A in humans, mammals and birds: determinants of virus virulence, host-range and interspecies transmission. Bioessays 25:657–671 [DOI] [PubMed] [Google Scholar]
- 30. Massin P, van der Werf S, Naffakh N. 2001. Residue 627 of PB2 is a determinant of cold sensitivity in RNA replication of avian influenza viruses. J. Virol. 75:5398–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dalton RM, Mullin AE, Amorim MJ, Medcalf E, Tiley LS, Digard P. 2006. Temperature sensitive influenza A virus genome replication results from low thermal stability of polymerase-cRNA complexes. Virol. J. 3:58 doi:10.1186/1743-422X-3-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dittmann J, Stertz S, Grimm D, Steel J, Garcia-Sastre A, Haller O, Kochs G. 2008. Influenza A virus strains differ in sensitivity to the antiviral action of the Mx-GTPase. J. Virol. 82:3624–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Accola MA, Huang B, Al Masri A, McNiven MA. 2002. The antiviral dynamin family member, MxA, tubulates lipids and localizes to the smooth endoplasmic reticulum. J. Biol. Chem. 277:21829–21835 [DOI] [PubMed] [Google Scholar]
- 34. Reichelt M, Stertz S, Krijnse-Locker J, Haller O, Kochs G. 2004. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic 5:772–784 [DOI] [PubMed] [Google Scholar]
- 35. Stertz S, Reichelt M, Krijnse-Locker J, Mackenzie J, Simpson JC, Haller O, Kochs G. 2006. Interferon-induced, antiviral human MxA protein localizes to a distinct subcompartment of the smooth endoplasmic reticulum. J. Interferon Cytokine Res. 26:650–660 [DOI] [PubMed] [Google Scholar]
- 36. Frese M, Kochs G, Feldmann H, Hertkorn C, Haller O. 1996. Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J. Virol. 70:915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zimmermann P, Manz B, Haller O, Schwemmle M, Kochs G. 2011. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J. Virol. 85:8133–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pavlovic J, Haller O, Staeheli P. 1992. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 66:2564–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]