

Abstract
Hantavirus pulmonary syndrome (HPS) is a severe respiratory disease characterized by pulmonary edema, with fatality rates of 35 to 45%. Disease occurs following infection with pathogenic New World hantaviruses, such as Andes virus (ANDV), which targets lung microvascular endothelial cells. During replication, the virus scavenges 5′-m7G caps from cellular mRNA to ensure efficient translation of viral proteins by the host cell cap-dependent translation machinery. In cells, the mammalian target of rapamycin (mTOR) regulates the activity of host cap-dependent translation by integrating amino acid, energy, and oxygen availability signals. Since there is no approved pharmacological treatment for HPS, we investigated whether inhibitors of the mTOR pathway could reduce hantavirus infection. Here, we demonstrate that treatment with the FDA-approved rapamycin analogue temsirolimus (CCI-779) blocks ANDV protein expression and virion release but not entry into primary human microvascular endothelial cells. This effect was specific to viral proteins, as temsirolimus treatment did not block host protein synthesis. We confirmed that temsirolimus targeted host mTOR complex 1 (mTORC1) and not a viral protein, as knockdown of mTORC1 and mTORC1 activators but not mTOR complex 2 components reduced ANDV replication. Additionally, primary fibroblasts from a patient with tuberous sclerosis exhibited increased mTORC1 activity and increased ANDV protein expression, which were blocked following temsirolimus treatment. Finally, we show that ANDV glycoprotein Gn colocalized with mTOR and lysosomes in infected cells. Together, these data demonstrate that mTORC1 signaling regulates ANDV replication and suggest that the hantavirus Gn protein may modulate mTOR and lysosomal signaling during infection, thus bypassing the cellular regulation of translation.
INTRODUCTION
Hantaviruses (family Bunyaviridae) are negative-sense RNA viruses harbored by small mammals and have worldwide distribution (1–5). Despite global spread, hantavirus strains are confined to the geographic range of the host species, and human infections occur following zoonotic transmission from asymptomatic rodent carriers (6). Hantaviruses are the etiologic agent for two distinct human diseases; Old World hantaviruses are endemic in Europe, Russia, and China and cause hemorrhagic fever with renal syndrome, whereas New World hantaviruses are endemic in the Americas and cause hantavirus pulmonary syndrome (HPS) (6, 7). HPS is a severe respiratory disease that manifests as a flu-like illness, progressing to pulmonary edema and shock (6, 7). In the Americas, most HPS infections are caused by the Sin Nombre virus (SNV) or the Andes virus (ANDV), and these infections have resulted in an average fatality rate of 39% (8–10). Extracorporeal membrane oxygenation support has been shown to improve the survival rates for patients with severe HPS (11, 12); however, no HPS-specific vaccines or antivirals currently exist.
Efforts to understand HPS have focused on how the virus usurps cellular signaling. Disease symptoms are likely due to a combination of dysregulated host immune responses and viral replication in the host cells (7). ANDV preferentially targets the microvascular endothelium, and viral replication results in increased endothelial permeability (13–17). These data demonstrate that viral replication actively disrupts endothelial signaling to cause disease pathogenesis.
To promote viral replication, hantaviruses scavenge 5′-m7G caps from cellular mRNA through endonuclease activity found in the viral polymerase (18–20). The viral polymerase requires the presence of the scavenged caps to initiate viral transcription in a manner similar to that of the La Crosse virus (18, 20, 21). After transcription, the 5′-m7G caps might help recruit the host cell cap-dependent translation machinery to viral mRNA. For instance, the viral nucleocapsid protein can bind to both the cap and ribosomal protein S19, potentially recruiting the ribosomal preinitiation complex (22–24). The nucleocapsid protein can also replace components of the host eukaryotic translation initiation factor 4F (eIF4F) complex (24). Together, these data suggest that hantavirus protein translation is dependent on the host cap-dependent translation machinery.
In host cells, the protein kinase mammalian target of rapamycin (mTOR) acts as an environmental sensor to activate progrowth pathways while inhibiting catabolic processes in the cells (25). By integrating amino acid, energy, and oxygen availability signals, mTOR regulates the activity of host cap-dependent translation (26). The mTOR protein exists in two distinct complexes, mTOR complex 1 (mTORC1) and mTORC2, which are composed of different subunits that phosphorylate and regulate distinct cellular signaling cascades (25, 26). Much is known about the signaling controlled by mTORC1, but little is known about the activities of mTORC2. Recent evidence suggests that mTORC2 phosphorylates and activates Akt (S473) and regulates the actin cytoskeleton and cell migration (27, 28). Rapamycin, an allosteric inhibitor of mTORC1 but not mTORC2, has been useful for defining the activities of mTOR complex 1 (29).
mTORC1 signaling is activated following growth factor stimulation at the plasma membrane, leading to the activation of phosphatidylinositol kinase and Akt and inhibition of the tuberous sclerosis complex (TSC1, hamartin, and TSC2, tuberin) (Fig. 1) (30–33). Inhibition of the TSC1-TSC2 complex relieves its GTPase-activating protein (GAP) function, increasing the amount of GTP-bound Rheb in the cell (34, 35). GTP-bound Rheb, the RagA/B-RagC/D heterodimer, and the ragulator complex (composed of LAMTOR1 [p18, c11orf59], LAMTOR2 [p14, ROBLD3], and LAMTOR3 [MP1, MAPKSP1]) help recruit and activate mTORC1 on the lysosomal membrane (36, 37). Upon activation, mTORC1 phosphorylates multiple substrates that regulate translation, cell size, RNA splicing, DNA replication, and vesicle transport (38, 39). The first and best-characterized mTORC1 substrates include eIF4E-binding protein 1 (4E-BP1) and p70/85 S6 kinases (S6K) (31, 40–42). Phosphorylation and activation of S6K lead to phosphorylation of ribosomal protein S6, which promotes cap-dependent translation by increasing the translation of mRNAs containing polypyrimidine tracts (5′TOP) in the 5′ untranslated regions (42–44). These 5′TOP-containing mRNAs encode components of the translation machinery, such as ribosomal proteins and translation elongation factors (43).
Fig 1.
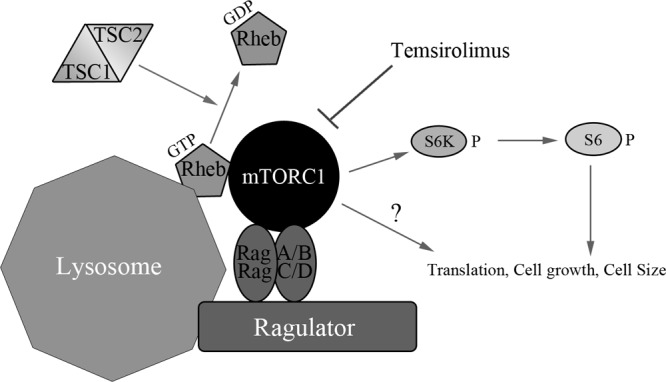
Outline of the general mTOR signaling pathway illustrating upstream mTOR activators and downstream mTOR substrates. The canonical signaling pathway senses growth factor stimulation at the plasma membrane, leading to the activation of phosphatidylinositol kinase and Akt and inhibition of the TSC1-TSC2 complex. Inhibition of the TSC1-TSC2 complex relieves its GTPase-activating protein (GAP) function, increasing the amount of GTP-bound Rheb in the cell. GTP-bound Rheb, the RagA/B-RagC/D heterodimer, and the ragulator complex(composed of LAMTOR1 [p18, c11orf59], LAMTOR2 [p14, ROBLD3], and LAMTOR3 [MP1, MAPKSP1]) help recruit and activate mTORC1 to the lysosomal membrane. Upon activation, mTORC1 phosphorylates and activates the p70/85 S6 kinases (S6K). Temsirolimus allosterically inhibits mTORC1 activity and reduces mTORC1 substrate phosphorylation. Active S6K phosphorylates ribosomal protein S6, promoting cap-dependent translation. In addition to S6K, mTORC1 also phosphorylates multiple substrates that regulate translation, cell growth, and cell size.
Here we demonstrate that treatment with the rapamycin analogue temsirolimus (CCI-779), approved by the U.S. Food and Drug Administration (FDA) for the treatment of renal cell carcinoma, blocks ANDV protein expression and virion release but not viral entry in primary human microvascular endothelial cells. We confirmed that temsirolimus targeted the host mTORC1 and not a viral protein, as knockdown of mTORC1 and mTORC1 activators but not mTORC2 reduced ANDV replication. Furthermore, we show that the ANDV glycoprotein Gn colocalized with mTOR and lysosomes in infected cells. Together, these data demonstrate that mTORC1 plays a role in ANDV replication and suggest that the hantavirus Gn protein may regulate mTOR and lysosomal signaling during infection, thus bypassing the cellular regulation of translation.
MATERIALS AND METHODS
Cells, viruses, and reagents.
Primary human microvascular lung endothelial (HMVEC-L) cells (Lonza, Walkersville, MD) were grown in Lonza endothelial basal medium 2 (EBM-2) supplemented with EGM-2-MV SingleQuots of growth factors (Lonza). Experiments with HMVEC-L cells were performed using multiple donors. A549 cells were grown in Dulbecco's modified Eagle's medium (DMEM) (item no. 10313; Invitrogen, Carlsbad, CA) supplemented with 10% HyClone fetal bovine serum (FBS) (Thermo Scientific, Waltham, MA), 2 mM l-glutamine (Invitrogen), and 1× penicillin-streptomycin (P/S) (Invitrogen). Fibroblasts from a wild-type (WT) donor (submission no. GM00038) and a donor with clinically diagnosed tuberous sclerosis (submission no. GM06149) were purchased from the Coriell Institute (Camden, NJ) and grown in DMEM (Invitrogen) supplemented with 15% HyClone FBS (Thermo Scientific), 2 mM l-glutamine (Invitrogen), and 1× P/S (Invitrogen). Andes virus (strain Chile 9717869) was grown and propagated and titers were determined in Vero E6 cells. The mTOR inhibitor temsirolimus, obtained from LC Laboratories (Woburn, MA), was dissolved in 100% ethanol. Recombinant human insulin (Sigma, St. Louis, MO) was resuspended in sterile water, and the pH was adjusted to 2 to 3 with dilute HCl. The antibodies used for Western blotting, purchased from Cell Signaling Technology (Beverly, MA), were directed against p70 S6 kinase (1:500) (item no. 9202), phospho-p70/85 S6 kinase (1:500; p70, T389; p85, T412) (item no. 9206), S6 ribosomal protein (1:500) (item no. 2217), phospho-S6 ribosomal protein (1:500; S235/6) (item no. 2211), RagA (detects both RagA and RagB) (1:500) (item no. 4357), Rheb (1:500) (item no. 4935), and LAMTOR1 (1:500; p18, C11orf59) (item no. 8975). Antiraptor (1:500) (item no. 05-1470) was purchased from Millipore (Billerica, MA), and antirictor (1:1,000) (item no. A500-02A) was purchased from Bethyl Laboratories (Montgomery, TX).
Western analysis.
HMVEC-L cells or primary fibroblast cells were seeded in 12- or 6-well tissue culture plates. Monolayers were infected with ANDV at a multiplicity of infection (MOI) of 0.5. Experiments with ANDV were conducted under biosafety level 3 (BSL3) conditions at the Centers for Disease Control and Prevention. Gamma-irradiated virus was generated by exposing infectious virus to a high-energy 60Co source for a dose of 5 × 106 rads. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology, Beverly, MA) supplemented with 0.1% SDS, 10 mM sodium fluoride (Sigma, St. Louis, MO), and 10 mM sodium orthovanadate (Sigma), and protein concentrations were determined using the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein were separated on SDS-PAGE gels (4 to 12% NuPAGE bis-Tris precast gels; Invitrogen, Carlsbad, CA) or Tris-Tricine gels (10 to 20% Novex Tricine gels; Invitrogen); proteins were then transferred to nitrocellulose membranes. Membranes were blocked with 5% milk in Tris-buffered saline containing 0.05% Tween 20 (TBST) for 1 h and were probed with antibodies in blocking solution for 1 h. Bands were detected using anti-mouse (Thermo Scientific Pierce, Waltham, MA) or anti-rabbit (GE Healthcare, Chalfont St. Giles, United Kingdom) antibodies conjugated to horseradish peroxidase, and blots were developed using SuperSignal West Dura extended-duration substrate (Thermo Scientific Pierce).
Cellular toxicity.
To measure toxicity, HMVEC-L cells were seeded into 96-well plates at 4 × 103 cells/well and allowed to grow to confluence over 3 days. Cells were then treated with temsirolimus or an ethanol control for 3 days, and toxicity was measured using the PrestoBlue cell viability reagent (Invitrogen, Carlsbad, CA).
Metabolic labeling.
To measure the rates of total protein synthesis, cells were metabolically labeled with [35S]cysteine. To do this, HMVEC-L cells were infected with ANDV at an MOI of 0.5 for 1 h, and the inoculum was removed and replaced with fresh medium (EBM-2 supplemented with EGM-2-MV SingleQuots of growth factors; Lonza, Walkersville, MD) containing temsirolimus or ethanol. At 8 h postinfection, cells were washed for 30 min with RPMI medium lacking cysteine (containing temsirolimus or ethanol) and labeled for 16 h with prewarmed cysteine-free RPMI medium (Sigma, St. Louis, MO) containing 100 μCi [35S]cysteine (PerkinElmer, Waltham, MA) (containing temsirolimus or ethanol). Cells were lysed in RIPA buffer, proteins were separated on SDS-PAGE gels, and the gels were dried for autoradiography. Cell lysates were also separated on SDS-PAGE gels and transferred to nitrocellulose membranes for Western analysis.
ANDV pseudovirion entry assays.
An expression vector encoding the human codon-optimized ANDV GPC gene was synthesized (DNA2.0, Menlo Park, CA), and HIV pseudoparticles bearing ANDV GPC were prepared essentially as described previously (45). Briefly, 1 × 106 293T-LentiX cells (Clontech, Mountain View, CA) were cotransfected with plasmid DNA encoding the HIV genome containing the firefly luciferase gene (pNL4-3.Luc.R−E−) and an expression vector encoding the vesicular stomatitis virus (VSV) or ANDV glycoprotein or an empty vector. The ratio of HIV plasmid to glycoprotein expression vector was 8:1. Pseudoparticles were harvested from the medium 48 and 72 h posttransfection, and supernatants were combined and filtered to remove cellular debris. Pseudoparticles titers were determined on HMVEC-L cells using the LentiX provirus quantitation kit (Clontech). To measure ANDV glycoprotein-dependent entry, pseudoparticles were added to HMVEC-L cells and allowed to adsorb for 6 h. After this time, the inoculum was removed and replaced with phenol red-free medium (EBM [item no. CC-3129] or EGM-2-MV [item no. CC-4147]; Lonza, Walkersville, MD). At 3 days postinfection, firefly luciferase levels were measured using the luciferase assay system (Promega, Madison, WI). Neutralization of the ANDV or VSV glycoprotein was performed by incubating pseudoparticles for 30 min at 37°C with sera specific for ANDV glycoprotein (polyclonal rabbit anti-ANDV) or VSV glycoprotein (mouse anti-VSV-G) (item no. 8G5F11; KeraFast, Winston-Salem, NC) or control serum (mouse IgG [clone 1E2.2; Millipore, Billerica, MA] or rabbit IgG [item no. 2729; Cell Signaling Technology, Beverly, MA]). Neutralized particles were adsorbed onto HMVEC-L cells for 6 h, and luciferase levels were measured 3 days after infection. For drug treatments, cells were treated with temsirolimus or an equivalent amount of an ethanol vehicle control for 1 h, and pseudoparticles were adsorbed to cells in medium containing fresh drugs. After 6 h of adsorption, the inoculum was removed and replaced with fresh medium lacking compounds. Luciferase levels were measured 3 days after infection.
Virion release and viral RNA quantification.
To measure the effects of temsirolimus on virus release, HMVEC-L cells were infected with ANDV at an MOI of 1. After 1 h of adsorption, the inoculum was removed, cells were washed twice with Hanks' balanced salt solution (HBSS) (Invitrogen, Carlsbad, CA), and the medium was replaced with DMEM supplemented with 10% FBS (with or without temsirolimus or ethanol). Three days postinfection, the medium was removed and stored at −80°C. Cells were lysed with TriPure isolation reagent (Roche, Indianapolis, IN), and total RNA was extracted using RNeasy columns (Qiagen, Valencia, CA). To quantify virion release, supernatants were diluted 2-fold and added to monolayers of naïve Vero E6 cells. After 1 h of adsorption, the inoculum was removed, cells were washed with HBSS, and the medium was replaced with an agar overlay containing 1× basal Eagle medium (BME) (Invitrogen), 20 mM HEPES (Invitrogen), 4% FBS, P/S, and 1% SeaKem ME agarose (Lonza, Walkersville, MD). At 5 to 6 days postinfection, wells were treated for 1 h with 3.7% formaldehyde, the agarose was removed, cells were fixed for 15 min with 2% formaldehyde, and plaques were visualized by immunohistochemistry. Cells were incubated with monoclonal mouse antinucleoprotein antibody (1:200) or polyclonal rabbit anti-ANDV antibody (1:200) and Alexa Fluor 488-conjugated anti-mouse or anti-rabbit secondary antibodies (Invitrogen). Viral RNA was quantified through TaqMan analysis conducted with an Applied Biosciences 7500 fast real-time PCR system (Invitrogen). Total viral RNA was quantified with a Superscript III Platinum one-step quantitative reverse transcription (qRT)-PCR system (Invitrogen) using primers and probes specific to the viral S segment (AAAACATCACAGCACACGAACAA [forward primer] and CTGCCTTCTCGGCATCCTTA [reverse primer] and the probe 6-FAM-AGCTCGTGACTGCTCGGCA-BHQ-1 [where 6-FAM is 6-carboxyfluorescein and BHQ-1 is black hole quencher 1]) or the viral M segment (CCAGCCAATAAAGAAGAGTCTATT [forward primer] and ACCATAACAATGAAATGCAGTCT [reverse primer] and the probe 6-FAM-TCGCATCCATCCAATGACCCAGGGG-BHQ-1). Data were normalized to values for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) controls (item no. 4310884E; Invitrogen).
RNA interference.
To knock down host proteins, HMVEC-L cells were transfected with 50 nM small interfering RNA (siRNA) using 0.05% DharmaFECT 1 transfection reagent (Thermo Scientific Dharmacon, Waltham, MA). After 16 h, the medium was removed and replaced with complete medium. After a 48-h knockdown, cells were infected with ANDV at an MOI of 0.5. After 2 days, cells were lysed and processed for Western analysis. RNAs complementary to human proteins, including raptor (item no. L-004107-00), rictor (L-016984-00), Rheb (L-009692-00), RagA (L-016070-00), RagB (L-012189-01), and LAMTOR1 (p18) (L-020916-02), were purchased from Dharmacon. To confirm losses of mTORC1 activity following mTORC1, mTOC2, and mTORC1 lysosomal activator knockdown, HMVEC-L cells were transfected with siRNA for 2 days and serum starved overnight. After 16 h, cells were deprived of amino acids with HBSS (Invitrogen, Carlsbad, CA) for 1 h and stimulated with 100 nM insulin in complete medium (EBM-2 with EGM-2-MV SingleQuots of growth factors; Lonza, Walkersville, MD) for 30 min. Cells were lysed, and mTOR activity was measured through Western analysis.
Microscopy.
To visualize host and viral protein localization, HMVEC-L cells were seeded onto gelatin-coated glass coverslips and infected with ANDV (MOI of approximately 0.25) or mock infected for 3 days. After 3 days, cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 15 min, permeabilized with 0.1% Triton X-100 in PBS, and stained with primary and secondary antibodies in 1% BSA in PBS for 1 h. Slips were mounted on glass slides with ProLong Gold antifade reagent containing DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen, Carlsbad, CA). The following antibodies were used for microscopy: anti-lysosome-associated membrane protein 2 (LAMP2) (1:50, clone H4B4) (Developmental Studies Hybridoma Bank, University of Iowa), anti-mTOR (1:50) (Cell Signaling Technology, Beverly, MA), and anti-Gn (1:50) (SNV-28). Human Fab SNV-28 was engineered by using a phage display library from RNA extracted from the bone marrow of an HPS-convalescent patient and was provided by Paul Parren and Dennis Burton (46). SNV-28 specificity was determined through radioimmunoprecipitation assays (data not shown). Secondary antibodies used for microscopy included Alexa Fluor 633-conjugated goat anti-mouse IgG (1:300) (Invitrogen), Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:300) (Invitrogen), and DyLight 549-conjugated goat anti-human F(ab′)2 fragment (Jackson ImmunoResearch, West Grove, PA).
RESULTS
mTOR activity regulates ANDV protein expression.
To investigate whether mTOR signaling plays a role during ANDV replication, we treated primary human lung microvascular endothelial (HMVEC-L) cells with the FDA-approved rapamycin analogue temsirolimus (in an ethanol carrier) for 1 h and then infected the cells with ANDV at an MOI of 0.5. At 3 days postinfection, we observed that temsirolimus but not the ethanol carrier alone reduced viral Gn, Gc, and nucleocapsid protein levels (Fig. 2A). When values were normalized to actin levels, the levels of the viral glycoproteins Gn and Gc were reduced 72% and 53%, respectively, and appeared more sensitive to 0.1 μM temsirolimus than the viral nucleocapsid levels (reduced 32%). We hypothesize that the rapid Gn turnover observed by Hussein et al. contributes to increased Gn sensitivity (47). The inhibitory effects on viral protein expression were observed at a concentration 100-fold below the concentration for 50% cellular toxicity (CC50) (10.9 ± 1.9 μM) measured over the same time period (see Fig. S1 in the supplemental material). To confirm inhibition of mTOR catalytic activity following temsirolimus treatment, we probed for phosphorylation of the direct mTOR substrate p70 S6K (Fig. 2B). Temsirolimus treatment effectively reduced mTOR activity, as indicated by a reduction in p70 S6K phosphorylation (at T389) relative to untreated and ethanol-treated cells (Fig. 2B).
Fig 2.

Temsirolimus reduces ANDV protein levels but not total cellular protein levels. (A) HMVEC-L cells were treated with temsirolimus or ethanol for 1 h, virus was adsorbed in drug-containing medium (MOI of 0.5), and the inoculum was removed and replaced with fresh medium (containing temsirolimus or an equivalent amount of the ethanol vehicle control) after adsorption. At 72 h postinfection, monolayers were lysed and proteins were separated by SDS-PAGE. The expressions of viral nucleocapsid (N) and glycoproteins (Gn and Gc) were observed through Western analysis. The bar graph indicates the densitometric analysis of Gn, Gc, and nucleocapsid protein expressions following treatment with 0.1 μM temsirolimus. Viral protein expression was normalized to actin levels and compared with values for the untreated Gn, Gc, and nucleocapsid protein samples. (B) To confirm the loss of mTOR activity following temsirolimus treatment, HMVEC-L cells were infected with ANDV (MOI of 0.5) and treated with temsirolimus or a carrier. Cells were lysed at 16 h postinfection, proteins were separated by SDS-PAGE, and mTOR activity was assessed through S6K phosphorylation with Western analysis. (C) To measure the effects of temsirolimus on total protein synthesis, HMVEC-L cells were infected as described for panel B. At 8 h postinfection, cells were washed for 30 min with medium lacking cysteine (with or without temsirolimus) and labeled for 16 h with prewarmed cysteine-free RPMI medium containing 100 μCi [35S]cysteine (with or without temsirolimus). Duplicate protein samples were transferred to membranes for Western analysis of viral protein Gc levels. The bar graph indicates densitometric analysis of Gc protein expression normalized to actin levels. Values for control samples treated with ethanol were set at 100%.
To determine if the effect of temsirolimus on viral protein levels was due to inhibition of total cellular protein synthesis, we metabolically labeled cells following temsirolimus treatment and/or ANDV infection. To do this, HMVEC-L cells were infected with ANDV at an MOI of 0.5 for 1 h, and the inoculum was removed and replaced with fresh medium or medium containing temsirolimus. At 8 h postinfection, cells were washed for 30 min with medium lacking cysteine (with or without temsirolimus) and labeled for 16 h with prewarmed cysteine-free RPMI medium containing 100 μCi [35S]cysteine (with or without temsirolimus). Under these conditions, we observed that ANDV replication alone did not reduce total protein levels relative to the uninfected cells (Fig. 2C, top, left lanes). Treatment of HMVEC-L cells with 1 μM temsirolimus reduced Gc protein levels (Fig. 2C, bottom) but had no effect on total cellular protein levels (Fig. 2C, top, right lanes). These data confirm that the effect of temsirolimus is specific to viral protein expression and not expression of cellular proteins.
mTOR activity is not necessary for Andes glycoprotein-mediated entry.
MacGurn et al. recently demonstrated that yeast TORC1 activity regulates the endocytosis of amino acid transporters (48). To investigate whether mTOR participates in ANDV entry, we infected cells with luciferase-expressing lentiviruses containing the ANDV glycoprotein (GPC) or VSV GPC. To characterize pseudovirion specificity, we neutralized pseudovirions with sera specific to the viral glycoproteins. VSV pseudovirions were effectively neutralized with monoclonal VSV glycoprotein-neutralizing antibody but not polyclonal anti-ANDV sera (Fig. 3A). In contrast, ANDV pseudovirions were effectively neutralized with polyclonal anti-ANDV sera but not with VSV glycoprotein-specific antibody (Fig. 3A). Neither ANDV nor VSV pseudoparticles were neutralized with isotype control sera (data not shown). To test whether mTOR functions during viral entry, we pretreated cells with temsirolimus for 1 h and adsorbed ANDV and VSV pseudovirions for 6 h in drug-containing medium. Compound and inoculum were removed after adsorption, and luciferase levels were measured at 3 days postinfection (Fig. 3B). Pretreatment with temsirolimus had no effect on ANDV or VSV pseudovirion entry (Fig. 3B). These data indicate that mTOR activity is necessary for efficient ANDV protein expression but not viral entry.
Fig 3.
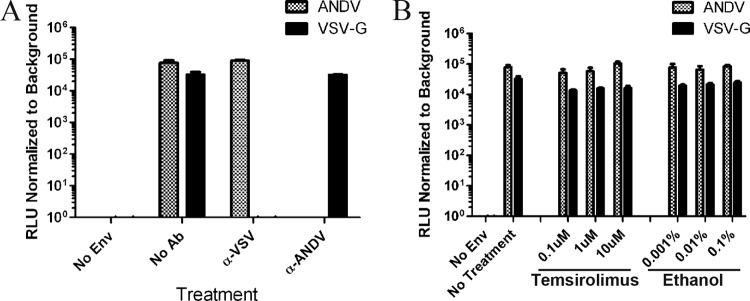
mTOR activity is not necessary for Andes virus glycoprotein-mediated entry. (A) To confirm pseudoparticle specificity, virions were neutralized with antibodies specific for the ANDV or the VSV glycoprotein. Virions were adsorbed to HMVEC-L cells (MOI of 14) for 6 h, and the inoculum was removed and replaced with fresh medium. Luciferase expression was measured at 3 days postinfection. RLU, relative light units. (B) Temsirolimus did not block ANDV or VSV pseudoparticle entry into HMVEC-L cells. Cells were pretreated for 1 h with temsirolimus or an equivalent amount of ethanol and infected for 6 h with pseudoparticles in drug-containing medium, and the inoculum was removed and replaced with fresh medium lacking temsirolimus and ethanol. Luciferase expression was measured at 3 days postinfection. Results were reproduced in two separate experiments performed in triplicate. Env, envelope protein; Ab, antibody.
Temsirolimus reduces ANDV release but not viral RNA synthesis.
The observation that mTOR activity was necessary for efficient ANDV protein production in HMVEC-L cells indicated that temsirolimus might reduce subsequent virion formation. To test this, we infected HMVEC-L cells with ANDV at an MOI of 1 for 1 h and then treated cells with temsirolimus for 3 days. After that time, the titer of virus in the culture medium was determined, and total RNA was isolated from the infected cells. The amount of virus released from HMVEC-L cells was reduced by 50% with temsirolimus treatment but not with ethanol treatment (Fig. 4A). Intriguingly, the levels of viral S segment RNA were increased following temsirolimus treatment (Fig. 4B). However, no significant differences in the levels of viral M segment RNA were observed following temsirolimus treatment (see Fig. S2 in the supplemental material). These data suggest that mTOR primarily regulates ANDV protein expression. Furthermore, a reduction in viral protein expression appears to also reduce infectious virion packaging and release, resulting in increases in the accumulation and levels of viral S segment RNA. However, we cannot rule out the possibility that S segment turnover might be disrupted (through either increased transcription or decreased RNA degradation) under these conditions.
Fig 4.
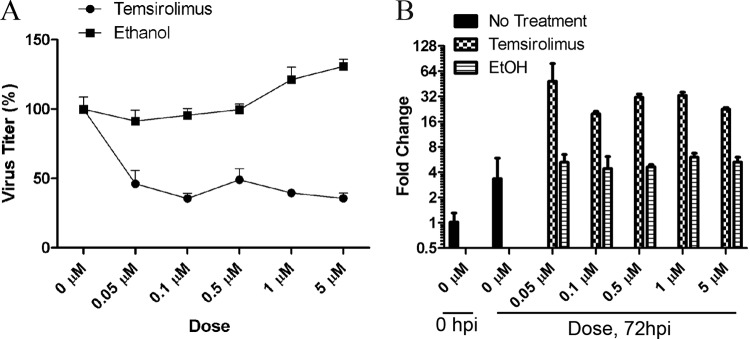
Temsirolimus reduces ANDV release but not viral RNA synthesis. (A) Temsirolimus reduces ANDV release from HMVEC-L cells. HMVEC-L cells were infected with ANDV (MOI of 1), and medium containing temsirolimus or an equivalent amount of the ethanol vehicle control was added to the cells. At 72 h postinfection, the supernatant was removed, and viral titers were determined. Temsirolimus reduced ANDV release by approximately 50% at all concentrations tested. (B) Temsirolimus does not inhibit ANDV RNA production. Total RNA was extracted from HMVEC-L cells treated as described for panel A and subjected to TaqMan analysis of ANDV S segment RNA. Viral transcript levels were normalized to cellular glyceraldehyde-3-phosphate dehydrogenase transcript levels, and data are presented as fold change relative to that of the input virus at 1 h postadsorption. Results were reproduced in two separate experiments performed in quadruplicate. EtOH, ethanol; hpi, hours postinfection.
ANDV does not increase S6K phosphorylation or activity during infection.
One consequence of signaling through mTOR involves the phosphorylation and activation of ribosomal protein S6, which promotes cap-dependent translation. This led us to investigate whether S6K and S6 phosphorylation are increased during ANDV infection. The mTOR substrate S6K is expressed through alternate translation to produce two nearly identical isoforms, p70 and p85 (41). The p70 S6K isoform is missing the first 23 amino acids found in the p85 isoform; however, both isoforms are direct mTOR substrates (41, 49). We observed no significant differences in S6K (Fig. 5A, p70S6K upper lanes, p85S6K; data not shown for p85 S6K) or S6 (Fig. 5A, lower lanes) phosphorylation or protein levels relative to mock- and inactivated virus-infected cells at the time points tested. Peng et al. and Clippinger et al. observed that serum withdrawal was necessary to observe virally induced increases in S6K phosphorylation during infection (50, 51). However, we did not observe any ANDV-induced differences in S6K phosphorylation following serum withdrawal (data not shown). These data indicate that S6K signaling downstream of mTOR is not upregulated during ANDV infection and suggest that other pathways downstream of mTOR are critical for viral replication.
Fig 5.
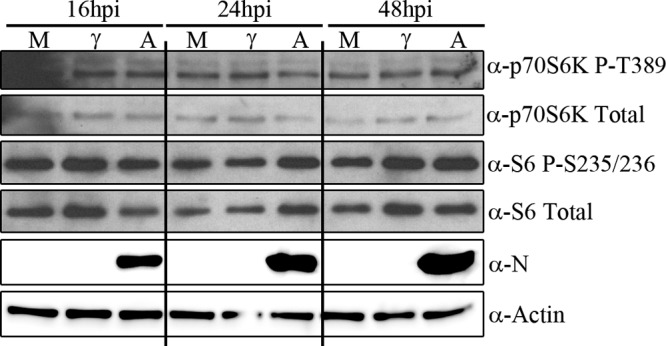
ANDV does not increase S6K phosphorylation or activity during infection. The time courses of S6K and S6 phosphorylation in HMVEC-L cells infected with ANDV (MOI of 3) are shown. Cells were infected with ANDV and lysed at different times postinfection. ANDV infection did not significantly increase p70 S6K, p85 S6K (data not shown), or S6 phosphorylation at all times tested (data for 0, 8, and 12 hpi, not shown). M, mock-infected cells; γ, gamma-irradiated inactivated virus; A, ANDV-infected cells. Results were reproduced in two separate experiments.
ANDV protein expression is increased in primary human fibroblasts derived from a donor with tuberous sclerosis.
Altogether, our data suggest that mTORC1 activity is necessary for efficient ANDV protein expression. To investigate whether mTORC1 activity is sufficient for ANDV protein expression, we infected primary human fibroblasts derived from a donor with tuberous sclerosis. Tuberous sclerosis manifests in patients as a multisystem disorder characterized by benign tumors and hamartomas and occurs following mutation of the TSC1-TSC2 complex (52). Loss or inhibition of the TSC1-TSC2 complex relieves its GTPase-activating protein (GAP) function, increasing the amount of GTP-bound Rheb, which increases mTOR activity (Fig. 1) (34, 35). Clinical samples derived from patients with tuberous sclerosis exhibit increased mTOR activity, and mTOR inhibitors have shown promise in the clinical setting (53). Thus, we hypothesized that mutations within TSC1 and TSC2 would increase mTORC1 activity while increasing ANDV protein expression. To investigate this, we infected primary human fibroblasts from an unaffected (WT) donor or from a donor diagnosed with tuberous sclerosis (TSC) with ANDV (MOI of 0.5) for 3 dpi. After that time, cell monolayers were lysed and proteins were separated by SDS-PAGE. We observed that ANDV nucleocapsid expression was increased in TSC fibroblasts relative to the control fibroblasts and that this effect was reduced following temsirolimus treatment (Fig. 6A). To confirm increased mTOR activity in the TSC fibroblasts, we probed for S6K phosphorylation. We observed that phosphorylation of the p85 S6K isoform (but not the p70 S6K isoform) was increased in TSC fibroblasts and that this phosphorylation was reduced following mTOR inhibition (Fig. 6B).
Fig 6.
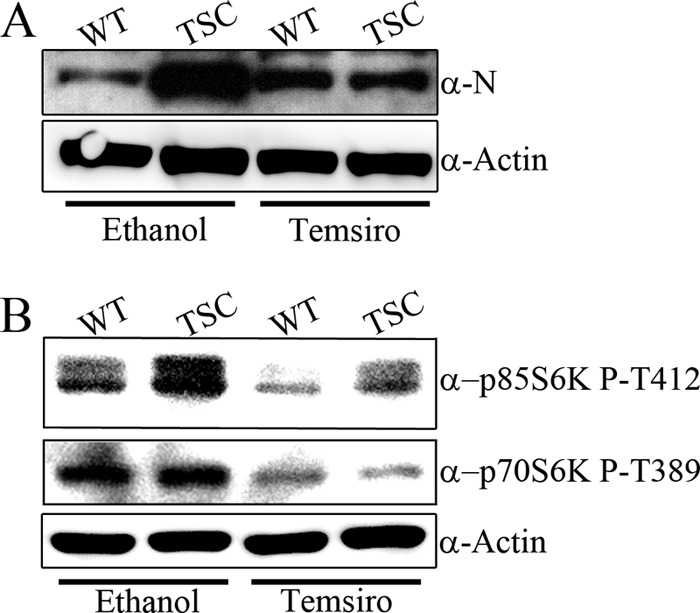
ANDV protein expression is increased in primary human fibroblasts derived from a patient with tuberous sclerosis disease. (A) Equal numbers of primary human fibroblasts from an unaffected (WT) donor or from a donor diagnosed with TSC were infected with ANDV (MOI of 0.5) for 72 hpi. After that time, monolayers were lysed and proteins were separated by SDS-PAGE. ANDV nucleocapsid expression was increased in TSC cells, relative to control levels, and reduced following 10 μM temsirolimus treatment. Equal protein loading was confirmed by probing for actin. Results were reproduced in two separate experiments. (B) WT and TSC cells are sensitive to temsirolimus treatment. WT and TSC cells were infected as described for panel A, and lysates were probed for phosphorylation of the mTOR substrates p70 S6K and p85 S6K. Increased mTOR activity in TSC cells, relative to WT cells, was confirmed by probing for p85 S6K phosphorylation (left lanes, upper panel). Phosphorylation of p70 S6K was unchanged in WT and TSC cells (left lanes, middle panel). Temsirolimus treatment reduced the phosphorylation of p70 S6K and p85 S6K (right lanes, upper and middle panels). Antibodies to detect total p85 S6K were nonspecific; therefore, actin was used as a loading control. Results were reproduced in two separate experiments.
Knockdown of mTORC1 and mTORC1 lysosomal activators but not mTORC2 reduces ANDV protein production.
To confirm that temsirolimus targets a cellular kinase and not a viral kinase, we knocked down components of mTORC1 or mTORC2 and infected cells with Andes virus. As a rapamycin analogue, temsirolimus allosterically inhibits mTOR signaling by binding to FK506-binding protein 12 (FKBP12), specifically disrupting mTORC1 but not mTORC2 activity (27, 54, 55). We observed that knockdown of raptor, a component specific to mTORC1 but not mTORC2, eliminated Gn levels and significantly reduced Gc and nucleocapsid levels (Fig. 7A). However, knockdown of rictor, a component specific to mTORC2 but not mTORC1, had no effect on viral protein production (Fig. 7A). We confirmed a loss of raptor or rictor expression through Western blot analysis (Fig. 7B). Knockdown of raptor (but not rictor) also reduced mTORC1 activity and p70 S6K phosphorylation (see Fig. S3 in the supplemental material).
Fig 7.
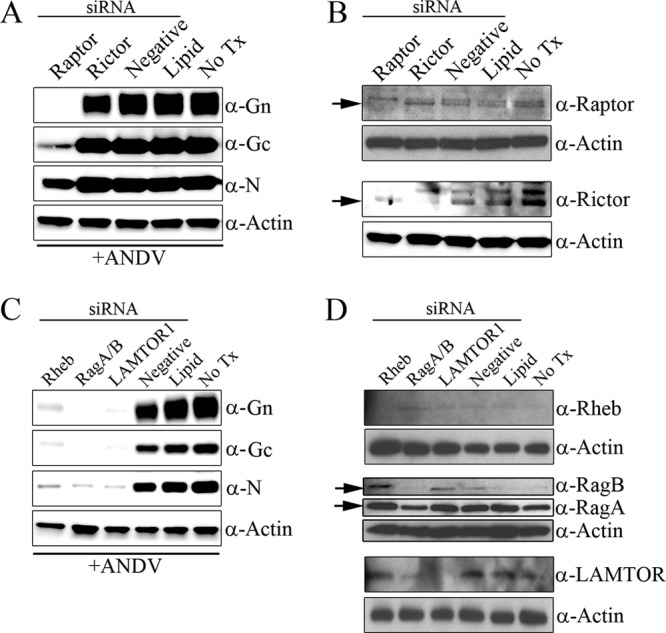
Knockdown of mTORC1 and mTORC1 lysosomal activators but not mTORC2 reduces ANDV protein production. (A and B) mTORC1 but not mTORC2 is necessary for ANDV protein production. HMVEC-L cells were transfected with 50 nM siRNA specific to mTORC1 (Raptor) or mTORC2 (Rictor), nonspecific RNA (Negative), or DharmaFECT 1 only (Lipid) or were not treated (No Tx) for 2 days. After that time, cells were infected with ANDV (MOI of 0.5). Monolayers were lysed at 48 hpi, and proteins were separated by SDS-PAGE. (A) Viral protein levels were reduced following knockdown of raptor but not rictor. (B) Knockdown of cellular proteins was confirmed by Western blot analysis using antibodies specific for raptor (the arrow indicates the lower raptor band; the upper band represents a nonspecific cross-reactive protein) or rictor (arrow). (C and D) mTORC1 lysosomal activators are necessary for ANDV protein production. HMVEC-L cells were transfected with 50 nM siRNA specific to Rheb, RagA/B, or LAMTOR1, nonspecific RNA (Negative), or DharmaFECT 1 only (Lipid) or were not treated (No Tx) for 2 days. Cells were then infected with ANDV (MOI of 0.5), and monolayers were lysed at 48 hpi. (C) Viral protein levels were reduced following knockdown of Rheb, RagA/B, and LAMTOR1 but not with treatment with nonspecific siRNA or DharmaFECT 1 only. (D) Knockdown of cellular proteins was confirmed by Western blot analysis using antibodies specific for Rheb, RagA/B, and LAMTOR1. Results were reproduced in two separate experiments.
Since knockdown of components of mTORC1 and treatment with the mTORC1 inhibitor were capable of reducing viral protein expression independently, we hypothesized that mTORC1 activity is necessary for viral replication. Recruitment of mTORC1 to the lysosomal surface is essential for amino acid-induced activation (36); therefore, we knocked down the expression of mTORC1 lysosomal recruiters/activators for 2 days and infected cells with ANDV for 2 dpi. We observed that knockdown of Rheb, RagA/B, and LAMTOR1 significantly reduced expression of the viral proteins Gn, Gc, and nucleocapsid (Fig. 7C). We confirmed losses of Rheb, RagA/B, and LAMTOR1 expression through Western blot analysis (Fig. 7D). We observed that knockdown of Rheb but not RagA/B or LAMTOR1 reduced mTORC1 activity and p70 S6K phosphorylation following insulin stimulation (see Fig. S3 in the supplemental material). These data are consistent with the observation that Rheb is required for mTORC1 activation but not its localization to lysosomes (36). In contrast, RagA/B expression and LAMTOR1 expression are required for targeting of mTOR to lysosomes but not for growth factor-mediated mTORC1 activity (36). Together, these data indicate that mTORC1 activation and localization on lysosomal membranes are necessary for ANDV replication.
ANDV glycoprotein Gn colocalizes with mTOR and LAMP2-positive vesicles.
Previously, we demonstrated that the SNV Gn protein colocalized with LAMP1 in lysosomes (46). Sancak et al. recently showed that mTOR, Rheb, RagA/B, and LAMTOR1 also colocalized with LAMP1 and LAMP2 in lysosomes (36, 37). These data suggest that ANDV Gn may also colocalize with mTOR in the lysosomal compartment. To investigate this, we infected HMVEC-L cells with ANDV for 3 days and stained for endogenous LAMP2, mTOR, and viral Gn proteins. In mock-infected cells, mTOR and LAMP2 colocalized in punctate vesicles (Fig. 8). The mTOR signal appeared strongest on the edges of the LAMP2-positive vesicles, whereas LAMP2 localized on the surface and lumen of mTOR-defined vesicles (Fig. 8, inset, yellow arrow and Mock). These data are consistent with the observations that mTOR localizes on the cytoplasmic side of lysosomes (36), while 90% of LAMP2 is membrane associated in the lysosomal lumen (56). During ANDV infection, we did not observe disruptions in LAMP2 and mTOR localization (Fig. 8). However, we could detect the viral protein Gn localized within the LAMP2- and mTOR-positive vesicles (Fig. 8). We occasionally observed Gn forming a doughnut-shaped structure within these vesicles, similar to the membrane-associated localization observed with mTOR (Fig. 8, +ANDV, inset arrowhead). Since Gn colocalizes with LAMP2 and mTOR, this proximity suggests that ANDV may directly modulate mTOR signaling through Gn.
Fig 8.

ANDV Gn protein colocalizes with LAMP2 and mTOR in infected cells. HMVEC-L cells were infected with ANDV for 3 days and stained with antibodies recognizing endogenous LAMP2, mTOR, and viral protein Gn. Nuclei are outlined in white. Note the doughnut-like mTOR- and LAMP2-positive vesicles in mock-transfected cells (mock, inset, yellow arrow) and ANDV-infected cells (inset, yellow arrow). Gn localizes within LAMP2- and mTOR-positive vesicles (inset, yellow arrow) and occasionally exhibits doughnut-like structures (inset, white arrowhead). Scale bar, 10 μm.
DISCUSSION
Here, we have explored the role of mTOR signaling during ANDV replication in primary human microvascular endothelial cells. We found that mTORC1 signaling is necessary for efficient ANDV protein expression and virus release but not virus entry. Inhibition of mTORC1 activity, either through allosteric mTOR inhibition, knockdown of raptor, or knockdown of mTOR lysosomal activators (Rheb, RagA/B, or LAMTOR1), reduced ANDV protein expression. Additionally, primary fibroblasts derived from a patient with tuberous sclerosis exhibited increased mTORC1 activity and increased ANDV protein expression relative to fibroblasts from an unaffected donor. Temsirolimus treatment of TSC cells reduced both mTOR activity and ANDV protein expression. Thus, mTORC1 plays an important role in ANDV replication by promoting viral protein expression.
During infection, viruses usurp the major cellular signaling pathways in order to replicate and survive. One of the main mechanisms through which viruses, such as polyomavirus, adenovirus, poxvirus, herpesvirus, hepatitis C virus, and Chikungunya virus, modulate mTOR signaling is by increasing the phosphorylation of S6K, S6, and 4E-BP1 (50, 57–61). In our study, ANDV infection did not increase the phosphorylation of any of the canonical mTOR substrates (data for S6K and S6 in Fig. 5; data for 4E-BP1 not shown). Despite this, we found that ANDV protein expression was regulated through mTORC1 and the mTORC1 lysosomal activators Rheb, RagA/B, and LAMTOR1 (a component of the ragulator complex). The involvement of the ragulator complex in viral protein expression is a novel finding and to our knowledge has not been reported for any other viruses. Other lysosomal mTORC1 effectors that regulate fatty acid, cholesterol, and lysosome biosynthesis (signaling pathways with potential importance during viral replication) have been described previously (62, 63).
During ANDV infection of HMVEC-L cells, we were intrigued that rapamycin had no effect on the translation of cellular proteins but specifically reduced the translation of viral proteins. Treatment of mammalian cells with rapamycin and rapamycin analogues does not completely block mTOR activity, and sensitivity varies by cell type (64–66). However, the loss of tumor suppressors can sensitize cells to temsirolimus treatment. Neshat et al. and Yu et al. found that cells and tumors that lack PTEN expression or have upregulated Akt signaling exhibit increased susceptibility to temsirolimus compared to wild-type cells (64, 67). Additionally, Thomas et al. found that cells deficient in the von Hippel-Lindau tumor suppressor (VHL) have increased hypoxia-inducible factor 1α (HIF-1α), HIF-2α, and vascular endothelial growth factor (VEGF) expression and exhibit increased sensitivity to temsirolimus treatment (68). ANDV replication is likely sensitized to mTORC1 inhibition by viral modulation of these cellular pathways.
Intriguingly, we found that while inhibition of mTORC1 activity appeared to regulate ANDV protein expression negatively, it had opposite effects on viral S segment RNA levels and did not change M segment RNA levels. These data suggest that mTOR primarily regulates ANDV protein expression. It is possible that reduced viral protein synthesis disrupts infectious virus packaging and release, yielding apparent increases in the levels of S segment RNA. However, we cannot rule out the possibility that S segment turnover might be disrupted (through either increased transcription or decreased RNA degradation) under these conditions. This observation is not without precedent, as rapamycin treatment of Sindbis virus infection in vitro has also increased levels of viral RNA through an unknown mechanism (69).
A previous study found that ANDV infection of lymphatic endothelial cells caused enhanced permeability in the presence of high levels of exogenously added VEGF and that this permeability was blocked following rapamycin treatment (70). However, it was unclear from that study whether the effects of rapamycin were due to inhibition of host VEGF receptor signaling or inhibition of viral replication. Here, we have shown that treatment with a rapamycin analogue (“rapalogue”) blocked ANDV replication through inhibition of host mTORC1 signaling. Therefore, the ability of rapamycin to block endothelial permeability during ANDV infection is likely due to two factors, i.e., reduced VEGF receptor host signaling and direct inhibition of viral replication (16).
We have shown that Gn colocalized with LAMP2 and mTOR during ANDV infection, which led us to predict that ANDV modulates mTOR signaling at lysosomes. A subset of Gn formed doughnut-like structures similar to the membrane localization observed for LAMP2 and mTOR, suggesting that a population of Gn could interact with mTORC1, RagA/B, or the ragulator complex. In this regard, we investigated whether ANDV infection disrupted mTOR or LAMP2 localization. Although Gn localized with mTOR and LAMP2, we did not observe differences in LAMP2 and mTOR localization in infected or uninfected cells, suggesting that while Gn localizes to lysosomes, it does not recruit mTOR to lysosomes.
Alternatively, Gn may not interact directly with mTORC1 or the mTORC1 activators but could maintain mTOR activity indirectly. Zoncu et al. recently proposed a model whereby mTORC1 activity is regulated by the accumulation of free amino acids within the lysosomal lumen (71). The concentrations of free amino acids are sensed by the vacuolar ATPase on the lysosomal membrane (71). The vacuolar ATPase interacts directly with the ragulator complex, in an amino acid-sensitive manner, to maintain mTORC1 activity (36, 71). Thus, autophagic turnover of Gn at lysosomes could maintain a pool of amino acids within the lysosomes that could activate mTORC1 indirectly. Previous work from our laboratory found that SNV Gn colocalized with LAMP1 within infected cells, and we hypothesized that the Gn-specific antibody recognized misfolded or degraded Gn within lysosomes (46). Also, Hussein et al. recently demonstrated that overexpressed Gn colocalized with the autophagosomal marker LC3, and they hypothesized that autophagic turnover of Gn was necessary for viral replication (47). Therefore, Gn turnover or direct Gn activity at lysosomes might maintain mTORC1 in an active state during viral replication.
We present here evidence that the FDA-approved mTOR inhibitor temsirolimus reduces hantavirus replication. These data are supported by the observation by Gavrilovskaya et al. that rapamycin blocks VEGF-induced permeability during ANDV infection (70). Thus, the FDA-approved mTOR inhibitors temsirolimus, sirolimus (rapamycin), and other rapalogues represent potential therapeutic agents for reducing hantavirus pathogenesis by inhibiting viral replication and thus, virus-induced permeability. These compounds provide new insights into the mechanisms of hantavirus replication and may represent new therapies for the treatment of hantavirus disease.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an ASM/CDC Program in Infectious Disease and Public Health Microbiology postdoctoral fellowship grant to S.M. Additional financial support was provided from CDC core funding.
We thank Eric Bergeron and Punya Shrivastava-Ranjan for insightful comments and discussions. The human Fab SNV-28 antibody was generously produced and provided by Paul Parren and Dennis Burton.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Footnotes
Published ahead of print 7 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02415-12.
REFERENCES
- 1. Bi Z, Formenty PB, Roth CE. 2008. Hantavirus infection: a review and global update. J. Infect. Dev. Ctries. 2:3–23 [DOI] [PubMed] [Google Scholar]
- 2. Luan VD, Yoshimatsu K, Endo R, Taruishi M, Huong VT, Dat DT, Tien PC, Shimizu K, Koma T, Yasuda SP, Nhi L, Huong VT, Arikawa J. 2012. Studies on hantavirus infection in small mammals captured in Southern and Central Highland area of Vietnam. J. Vet. Med. Sci. 74:1155–1162 [DOI] [PubMed] [Google Scholar]
- 3. Nichol ST, Spiropoulou CF, Morzunov S, Rollin PE, Ksiazek TG, Feldmann H, Sanchez A, Childs J, Zaki S, Peters CJ. 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262:914–917 [DOI] [PubMed] [Google Scholar]
- 4. Zeier M, Handermann M, Bahr U, Rensch B, Muller S, Kehm R, Muranyi W, Darai G. 2005. New ecological aspects of hantavirus infection: a change of a paradigm and a challenge of prevention: a review. Virus Genes 30:157–180 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y, Yuan J, Yang X, Zhou J, Yang W, Peng C, Zhang HL, Shi Z. 2011. A novel hantavirus detected in Yunnan red-backed vole (Eothenomys miletus) in China. J. Gen. Virol. 92:1454–1457 [DOI] [PubMed] [Google Scholar]
- 6. Schmaljohn CN, Nichol ST. 2007. Bunyaviridae, p 1741–1789 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 7. Macneil A, Nichol ST, Spiropoulou CF. 2011. Hantavirus pulmonary syndrome. Virus Res. 162:138–147 [DOI] [PubMed] [Google Scholar]
- 8. da Rosa Elkhoury M, da Silva Mendes W, Waldman EA, Dias JP, Carmo EH, Fernando da Costa Vasconcelos P. 2012. Hantavirus pulmonary syndrome: prognostic factors for death in reported cases in Brazil. Trans. R. Soc. Trop. Med. Hyg. 106:298–302 [DOI] [PubMed] [Google Scholar]
- 9. MacNeil A, Ksiazek TG, Rollin PE. 2011. Hantavirus pulmonary syndrome, United States, 1993-2009. Emerg. Infect. Dis. 17:1195–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martinez VP, Bellomo CM, Cacace ML, Suarez P, Bogni L, Padula PJ. 2010. Hantavirus pulmonary syndrome in Argentina, 1995-2008. Emerg. Infect. Dis. 16:1853–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dietl CA, Wernly JA, Pett SB, Yassin SF, Sterling JP, Dragan R, Milligan K, Crowley MR. 2008. Extracorporeal membrane oxygenation support improves survival of patients with severe hantavirus cardiopulmonary syndrome. J. Thorac. Cardiovasc. Surg. 135:579–584 [DOI] [PubMed] [Google Scholar]
- 12. Wernly JA, Dietl CA, Tabe CE, Pett SB, Crandall C, Milligan K, Crowley MR. 2011. Extracorporeal membrane oxygenation support improves survival of patients with hantavirus cardiopulmonary syndrome refractory to medical treatment. Eur. J. Cardiothorac. Surg. 40:1334–1340 [DOI] [PubMed] [Google Scholar]
- 13. Gavrilovskaya IN, Gorbunova EE, Mackow NA, Mackow ER. 2008. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82:5797–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gorbunova E, Gavrilovskaya IN, Mackow ER. 2010. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 84:7405–7411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. 2011. VEGFR2 and Src kinase inhibitors suppress Andes virus-induced endothelial cell permeability. J. Virol. 85:2296–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shrivastava-Ranjan P, Rollin PE, Spiropoulou CF. 2010. Andes virus disrupts the endothelial cell barrier by induction of vascular endothelial growth factor and downregulation of VE-cadherin. J. Virol. 84:11227–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, Feddersen RM, Zumwalt RE, Miller GL, Khan AS, Rollin PE, Ksiazek TG, Nichol ST, Mahy BWJ, Peters CJ. 1995. Hantavirus pulmonary syndrome: pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–579 [PMC free article] [PubMed] [Google Scholar]
- 18. Garcin D, Lezzi M, Dobbs M, Elliott RM, Schmaljohn C, Kang CY, Kolakofsky D. 1995. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J. Virol. 69:5754–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hutchinson KL, Peters CJ, Nichol ST. 1996. Sin Nombre virus mRNA synthesis. Virology 224:139–149 [DOI] [PubMed] [Google Scholar]
- 20. Reguera J, Weber F, Cusack S. 2010. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 6:e1001101 doi:10.1371/journal.ppat.1001101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mir MA, Sheema S, Haseeb A, Haque A. 2010. Hantavirus nucleocapsid protein has distinct m7G cap- and RNA-binding sites. J. Biol. Chem. 285:11357–11368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng E, Haque A, Rimmer MA, Hussein IT, Sheema S, Little A, Mir MA. 2011. Characterization of the interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19). J. Biol. Chem. 286:11814–11824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haque A, Mir MA. 2010. Interaction of hantavirus nucleocapsid protein with ribosomal protein S19. J. Virol. 84:12450–12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mir MA, Panganiban AT. 2008. A protein that replaces the entire cellular eIF4F complex. EMBO J. 27:3129–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Magnuson B, Ekim B, Fingar DC. 2012. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441:1–21 [DOI] [PubMed] [Google Scholar]
- 26. Sengupta S, Peterson TR, Sabatini DM. 2010. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40:310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. 2004. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6:1122–1128 [DOI] [PubMed] [Google Scholar]
- 28. Oh WJ, Jacinto E. 2011. mTOR complex 2 signaling and functions. Cell Cycle 10:2305–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14:1296–1302 [DOI] [PubMed] [Google Scholar]
- 30. Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, Nellist M, Yeung RS, Halley DJ, Nicosia SV, Pledger WJ, Cheng JQ. 2002. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 277:35364–35370 [DOI] [PubMed] [Google Scholar]
- 31. Hayashi AA, Proud CG. 2007. The rapid activation of protein synthesis by growth hormone requires signaling through mTOR. Am. J. Physiol. Endocrinol. Metab. 292:E1647–E1655 [DOI] [PubMed] [Google Scholar]
- 32. Inoki K, Li Y, Zhu T, Wu J, Guan KL. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4:648–657 [DOI] [PubMed] [Google Scholar]
- 33. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. 2002. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 10:151–162 [DOI] [PubMed] [Google Scholar]
- 34. Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. 2003. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11:1457–1466 [DOI] [PubMed] [Google Scholar]
- 35. Inoki K, Li Y, Xu T, Guan KL. 2003. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17:1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. 2011. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332:1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. 2011. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332:1322–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE. 1992. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 257:973–977 [DOI] [PubMed] [Google Scholar]
- 41. Rosner M, Hengstschlager M. 2011. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene 30:4509–4522 [DOI] [PubMed] [Google Scholar]
- 42. von Manteuffel SR, Dennis PB, Pullen N, Gingras AC, Sonenberg N, Thomas G. 1997. The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70S6K. Mol. Cell. Biol. 17:5426–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jefferies HB, Reinhard C, Kozma SC, Thomas G. 1994. Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc. Natl. Acad. Sci. U. S. A. 91:4441–4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Flint M, Logvinoff C, Rice CM, McKeating JA. 2004. Characterization of infectious retroviral pseudotype particles bearing hepatitis C virus glycoproteins. J. Virol. 78:6875–6882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spiropoulou CF, Goldsmith CS, Shoemaker TR, Peters CJ, Compans RW. 2003. Sin Nombre virus glycoprotein trafficking. Virology 308:48–63 [DOI] [PubMed] [Google Scholar]
- 47. Hussein IT, Cheng E, Ganaie SS, Werle MJ, Sheema S, Haque A, Mir MA. 2012. Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J. Virol. 86:7520–7529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacGurn JA, Hsu PC, Smolka MB, Emr SD. 2011. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147:1104–1117 [DOI] [PubMed] [Google Scholar]
- 49. Meyuhas O, Dreazen A. 2009. Ribosomal protein S6 kinase from TOP mRNAs to cell size. Prog. Mol. Biol. Transl. Sci. 90:109–153 [DOI] [PubMed] [Google Scholar]
- 50. Clippinger AJ, Maguire TG, Alwine JC. 2011. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J. Virol. 85:9369–9376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peng L, Liang D, Tong W, Li J, Yuan Z. 2010. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J. Biol. Chem. 285:20870–20881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tomasoni R, Mondino A. 2011. The tuberous sclerosis complex: balancing proliferation and survival. Biochem. Soc. Trans. 39:466–471 [DOI] [PubMed] [Google Scholar]
- 53. Budde K, Gaedeke J. 2012. Tuberous sclerosis complex-associated angiomyolipomas: focus on mTOR inhibition. Am. J. Kidney Dis. 59:276–283 [DOI] [PubMed] [Google Scholar]
- 54. Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. 1994. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369:756–758 [DOI] [PubMed] [Google Scholar]
- 55. Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. 1994. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78:35–43 [DOI] [PubMed] [Google Scholar]
- 56. Lippincott-Schwartz J, Fambrough DM. 1987. Cycling of the integral membrane glycoprotein, LEP100, between plasma membrane and lysosomes: kinetic and morphological analysis. Cell 49:669–677 [DOI] [PubMed] [Google Scholar]
- 57. Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. 2012. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 86:6315–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 6:266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Joubert PE, Werneke SW, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow DJ, Albert ML. 2012. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 209:1029–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. 2011. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Invest. 121:3623–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zaborowska I, Walsh D. 2009. PI3K signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J. Virol. 83:3988–3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. 2011. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146:408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31:1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. 2001. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. U. S. A. 98:10314–10319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, Yu K. 2008. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 68:2934–2943 [DOI] [PubMed] [Google Scholar]
- 66. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284:8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yu K, Toral-Barza L, Discafani C, Zhang WG, Skotnicki J, Frost P, Gibbons JJ. 2001. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr. Relat. Cancer 8:249–258 [DOI] [PubMed] [Google Scholar]
- 68. Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. 2006. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 12:122–127 [DOI] [PubMed] [Google Scholar]
- 69. Mohankumar V, Dhanushkodi NR, Raju R. 2011. Sindbis virus replication, is insensitive to rapamycin and torin1, and suppresses Akt/mTOR pathway late during infection in HEK cells. Biochem. Biophys. Res. Commun. 406:262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gavrilovskaya IN, Gorbunova EE, Mackow ER. 2012. Andes virus infection of lymphatic endothelial cells causes giant cell and enhanced permeability responses that are rapamycin and vascular endothelial growth factor C sensitive. J. Virol. 86:8765–8772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. 2011. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334:678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.