

Abstract
Cytomegalovirus (CMV) infection leads to the development of adaptive and humoral immune responses that are among the largest for any pathogen, and intriguingly, the magnitude of the immune response increases with age, a phenomenon termed “memory inflation.” Elevated CMV-specific immunity has been correlated with an increased mortality rate in elderly individuals and with impaired vaccination responses. The latent phase of CMV infection is characterized by intermittent episodes of subclinical viral reactivation and the production of immunogenic transcripts that may maintain memory inflation of virus-specific cytotoxic lymphocytes. However, the relative importance of CMV reactivation in the development of memory inflation is uncertain, as is the potential for antiviral treatment to reverse this effect. Here, we administered valaciclovir for up to 12 months in mice with established murine CMV (MCMV) infection. Treatment reduced the magnitude of the MCMV-specific CD8+ T-lymphocyte response by 80%, and the residual MCMV tetramer-specific lymphocytes exhibited a less differentiated phenotype. In addition, latent MCMV infection suppressed the proportion of naïve CD8+ T cells by 60% compared to antiviral-treated mice or MCMV-negative animals. Furthermore, treatment led to a reduction in influenza A viral loads following a challenge in elderly MCMV-infected animals and also reduced the differentiation of influenza virus-specific cytotoxic lymphocytes. These observations demonstrate that MCMV-specific memory inflation is maintained by viral replication and that therapeutic intervention could lead to improved immune function.
INTRODUCTION
Cytomegalovirus (CMV) is a highly prevalent betaherpesvirus that rarely elicits symptomatic infection in healthy individuals, but it is associated with a range of clinical complications in immunocompromised individuals. CMV is never eradicated from the host and is believed to undergo intermittent episodes of subclinical reactivation that may be triggered by episodes of stress and immune suppression.
CMV-infected humans develop large populations of CMV-specific CD8+ and CD4+ T lymphocytes, which typically demonstrate a late-differentiated phenotype that is believed to reflect a strong replicative history. Interestingly, these virus-specific T-cell populations increase with age and come to dominate the memory compartments in elderly CMV-seropositive individuals (1, 2); they also have the capacity to impair host immunity to other coresident infections (3). This “memory inflation” of CMV-specific cytotoxic lymphocytes has been implicated as a contributory factor in the development of immune senescence (4). Epidemiological studies have demonstrated that CMV-seropositive octogenarians are at an elevated risk of mortality, and those individuals with a higher proportion of CD8+ CD28− memory T cells and inverted CD4/CD8 T-cell ratios are almost exclusively CMV seropositive in a profile that has been termed the immune risk phenotype (5). In addition, epidemiological studies have confirmed that elderly individuals with the highest quartile of anti-CMV-specific serum immunoglobulin G (IgG) were at the highest risk of all-cause mortality (6). The CMV-specific antibody titer is correlated with the magnitude of cellular immunity; therefore, there are potentially considerable benefits from treatments that can suppress the CMV-specific adaptive immune response.
Murine CMV (MCMV) infection also leads to the expansion of virus-specific cytotoxic CD8+ T lymphocytes that demonstrate memory inflation. MCMV can infect a variety of cell types (7); however, the anatomical reservoirs of latent MCMV may be limited to sites such as the salivary glands, liver sinusoidal endothelium (8), and nonhematopoietic cells within the lymph nodes (9, 10). The degree of T-cell inflation against individual viral peptides is governed by their patterns of expression and the nature of the infection model (11–14). Inflation of the MCMV-specific CD8+ T cells is known to occur against a series of immunodominant T-cell epitopes, such as M38316-323, m139419-426, and IE3416-426, within chronically infected C57BL/6 mice (13). Indeed, the frequency of cytotoxic lymphocytes that come against each antigen can exceed 10% of the CD8+ T-cell pool. These inflationary populations also exhibit a highly differentiated membrane phenotype indicative of repeated antigen exposures (14). The adoptive transfer of inflationary MCMV-specific CD8+ T cells into MCMV-infected animals demonstrated these T cells to have a short half-life (45 to 60 days), with their frequency maintained at a high level through the recruitment of naïve and less differentiated T cells into memory through intermittent antigen exposures (14, 15). However, the importance of antigen persistence in the generation and maintenance of CMV-specific immunity remains uncertain.
Recently, it was shown that prior/latent MCMV infection can result in significantly reduced CD8+ T-cell responses to challenging infections (16) and elevated viral loads (17), thus suggesting that the presence of reactivated MCMV could impair immunity in older animals. Therapeutic intervention using an antiviral drug that has the capacity to suppress MCMV reactivation has the potential to block the repopulation of MCMV-specific CD8+ T cells from the naïve pool while permitting the natural decay of the preexisting immune response. Such a feature is seen in the decline of the HIV-specific CD8+ T-cell population following the introduction of highly active antiretroviral therapy (HAART) (18). It might be anticipated that any such reduction in the MCMV-specific immune response would enable regeneration of the peripheral naïve T-cell compartment with subsequent improvement in the immune responses to heterologous pathogens. Here, we show that long-term administration of a drug that inhibits CMV replication, valaciclovir, leads to profound suppression of MCMV-specific T-cell memory formation, regeneration of the naïve CD8+ T-cell compartment, and reduction of viral load following an influenza virus challenge.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the Animals (Scientific Procedures) Act 1986. The protocol was approved (project license 40/2998) by the Home Office, and the experiments were performed at the Biomedical Services Unit at the University of Birmingham. All efforts were made to minimize suffering, and where appropriate, procedures were performed under anesthesia.
Microplaque reduction assay.
Mouse embryo fibroblasts were incubated (37°C at 5% CO2) with MCMV strain Smith at a multiplicity of infection (MOI) of ≈0.01 for 4 days in increasing concentrations of aciclovir (Sigma) or valaciclovir hydrochloride (GlaxoSmithKline). The cells were fixed with 10% formaldehyde and stained with 0.5% crystal violet, and the plaques were counted at ×10 magnification.
Mice and antiviral treatment.
Female C57BL/6 mice were obtained from Harlan UK Ltd. and kept at the Biomedical Service Unit at the University of Birmingham. The animals were housed within a negative-pressure incubator to prevent the possible spread of infection to other mouse colonies.
The animals received the antiviral valaciclovir hydrochloride orally at a concentration of 1 mg/ml in their drinking water. This was replaced with a fresh solution once a week during the course of the experiment. The dosages were calculated based on the average consumption of drinking water.
Virus strains and infections.
MCMV strain Smith (ATCC) was cultured, and the titer was calculated by a limited dilution of viral stock onto mouse embryo fibroblasts. The mice were inoculated by intraperitoneal injection (i.p.) with 1 × 106 PFU of MCMV strain Smith (19).
The influenza A virus (IAV) A/Puerto Rico/8/34/England/939/69 clone 7a (H3N2) was cultured and titrated in fertilized hen's eggs (20). The mice were inoculated by intranasal injection (i.n.) with a 1 × 106 50% egg infective dose (EID50) of IAV.
Tetramer staining, antibodies, and intracellular cytokine stimulation assay.
The MCMV tetramer (IE3) and IAV tetramers (nucleoproteins NP366 to NP374) were synthesized by the NIH Tetramer Core Facility at Emory University. Where possible, the tetramer staining and peptide stimulations were performed on 1 × 106 lymphocytes. The following fluorescently conjugated antibodies were used to label the cells for flow cytometry: CD8α (Ly-2), CD27 (LG.3A10), CD44 (IM7), CD62L (Ly-22), CD122 (TM-b1), and CD127 (A7R34). For intracellular cytokine staining, splenocytes were stimulated for 6 h with 5 μg/ml of peptide in the presence of brefeldin A. The cells were then washed and surface stained for CD8α and CD4 before fixation, permeabilization, and staining for gamma interferon (IFN-γ) (clone XMG1.2) and tumor necrosis factor alpha (TNF-α) (clone MP6-XT22). The samples were acquired on an LSR II flow cytometer and analyzed using FACSDiva software. Multivariant statistical analyses were performed using GraphPad Prism5.
In vitro influenza A virus quantitation assay.
Following perfusion, right-hand lung lobes were extracted from mice and placed immediately into 1 ml of serum-free RPMI medium on ice. The lung samples were homogenized and stored at −80°C. The lung viral load was determined using the previously described virus titration assay (21). Briefly, serial 2-fold dilutions of lung homogenates were incubated with MDCK cells with a methylcellulose overlay in a 24-well plate for 48 h. The cells were fixed, permeabilized, and stained with a monoclonal antibody specific for the PR8 hemagglutinin. The cells were incubated with a peroxidase-labeled secondary antibody, and the plate was developed using aminoethylcarbazole (AEC) reagents. Each stained cell was counted, and the total number of PFU per lung sample was calculated.
RESULTS
Replication of MCMV was inhibited by valaciclovir in vitro.
The acycloguanosine class of antivirals has a broad spectrum of activity against herpesviruses. To generate a guanine nucleotide analogue capable of inhibiting the viral DNA polymerase, these drugs undergo three phosphorylation reactions, the first of which is mediated by viral thymidine kinase (TK). Although CMVs do not encode typical TKs, they are able to phosphorylate acycloguanosines through the phosphotransferase enzyme M97 (22, 23). Here, the MCMV strain Smith was tested for its sensitivity to aciclovir or valaciclovir in a microplaque reduction assay (Fig. 1A and B). Plaque formation was completely inhibited with both antiviral drugs at concentrations exceeding 1.25 μM. The 50% effective concentrations (EC50) of aciclovir and valaciclovir toward MCMV were calculated to be 0.5 μM, demonstrating that MCMV has a level of sensitivity to acycloguanosines equivalent to that of herpes simplex virus type 1 (HSV-1) and herpes simplex virus type 2 (HSV-2) (24). Due to this similarity in antiviral efficacy and improved bioavailability over aciclovir, valaciclovir was selected as the optimal candidate for suppressing MCMV replication in vivo (25). A 1-mg/ml valaciclovir hydrochloride suspension was added to drinking water and made available ad libitum to the mice. On average, the animals consumed 3.7 mg of antiviral medication per day (Fig. 1C). The dosage achieved was equivalent to approximately 10 g/day for a 75-kg adult and is comparable to the doses used to suppress CMV following solid organ transplantations (26).
Fig 1.
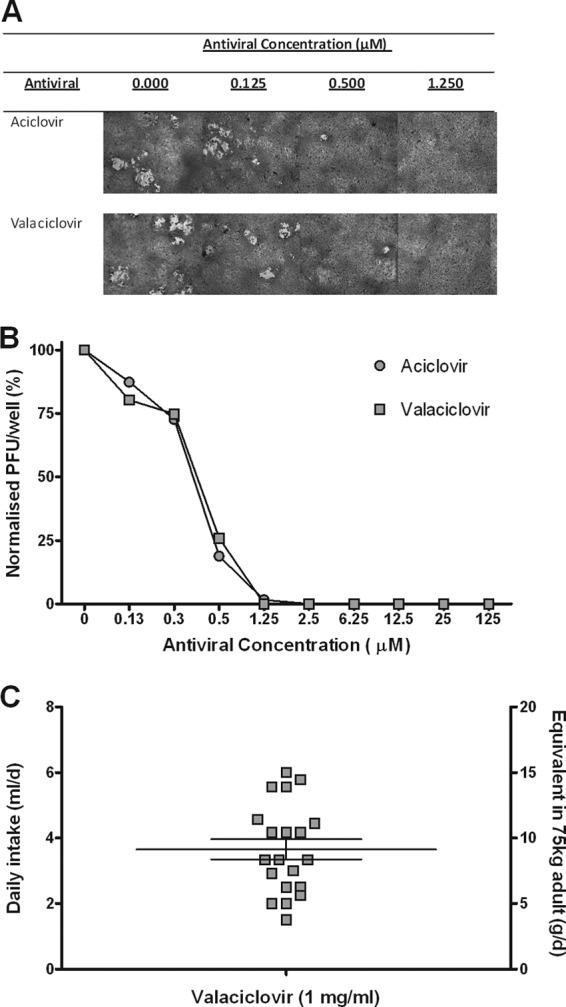
Replication of MCMV strain Smith is inhibited by valaciclovir in vitro. (A) Images of primary mouse embryo fibroblasts cocultured with MCMV strain Smith (MOI, ≈0.01) in media containing aciclovir or valaciclovir (0 to 125 μM). (B) Mean numbers of PFU were determined at each antiviral concentration following 4-day incubation at 37°C, 5% CO2. The graph shows antiviral concentration (μM) versus the normalized number of PFU/well (% maximum PFU). (C) The average daily fluid intake of valaciclovir suspension (1 mg/ml) was recorded per mouse each day (ml/day), and the approximate equivalent dose was calculated for a 75-kg individual (equivalent dose [g/d] = daily intake [ml/day] × antiviral [mg/ml] × [1,000/mass of the mouse] × 75).
Prolonged antiviral treatment led to profound suppression of the MCMV-specific CD8+ T-cell response in aged mice.
C57BL/6 mice were inoculated with MCMV at 6 to 8 weeks of age and kept for 12 months to facilitate the development of memory inflation in an aged population. At this time, antiviral treatment was administered to the animals for either 3 or 6 months in order to determine the potential impact on T-cell immunity (Fig. 2A). Splenocytes were isolated from groups of mice immediately before antiviral treatment and then at 3 and 6 months after treatment had started. The magnitudes of the individual MCMV-specific CD8+ T-cell responses prior to treatment ranged from 4 to 10% of the total CD8+ T-cell pool against the epitopes M38, M45, M57, m139, and IE3 (Fig. 2B to F). Mice that had received antiviral treatment for 3 months showed no significant reductions in the frequency of MCMV-specific T cells in comparison to the MCMV-untreated animals. This finding is comparable to that of Snyder et al., who studied the effects of 3 months of antiviral treatment in younger mice exposed to a mutant famciclovir-sensitive MCMV viral strain (27). However, analyses of individual peptide responses after 6 months of antiviral treatment revealed nonsignificant reductions of between 30 and 40% for all epitopes except IE3 (Fig. 2B to F).
Fig 2.

Six months of antiviral treatment leads to partial suppression of the MCMV-specific CD8+ T-cell response. (A) Mice were infected with MCMV strain Smith (1 × 106 PFU) at 6 to 8 weeks of age. Twelve months after MCMV infection, groups remained untreated or were treated with valaciclovir for 3 months or 6 months. The frequencies of MCMV-specific IFN-γ+ CD8+ T cells from the spleen samples analyzed by ex vivo stimulation with immunodominant peptides from M45 (B), M57 (C), M38 (D), m139 (E), or IE3 (F) were then determined. The results are shown as percentages of the total CD8+ T-cell pool. The number of mice was 3 or 4 at each time point (mean ± standard error of the mean [SEM]).
These observations were then extended to assess the influence of a longer period of antiviral treatment in a new group of animals. In order to allow for a comparable assessment in mice at the age of 18 months, antiviral treatment was started in the animals at 6 months of age and valaciclovir was continued for 12 months (Fig. 3A). Mice in this group were also infected with MCMV at 6 to 8 weeks of age.
Fig 3.

Twelve months of antiviral treatment leads to profound suppression of the MCMV-specific CD8+ T-cell response in aged mice. (A) Mice were infected with MCMV strain Smith (1 × 106 PFU) at 6 to 8 weeks of age. Six months after MCMV infection, the groups remained untreated (MCMV−AV) (n = 9) or treated with valaciclovir (MCMV+AV) (n = 11) for 12 months. In addition, an uninfected age-matched control group was included (MCMV-neg) (n = 5). (B) Frequencies of IFN-γ-positive MCMV-specific CD8+ T cells against individual MCMV peptides at 18 months of age from the spleen samples of MCMV−AV (n = 9) and MCMV+AV (n = 11) mice. (C) The magnitude of the total MCMV-specific T-cell immune response is shown as the sum of the five immunodominant IFN-γ-specific CD8+ T-cell responses (data in panels A and B were combined from two independent experiments) (mean ± SEM). The data were analyzed by the two-way Mann-Whitney U test (not significant [ns], P > 0.05; *, P < 0.05; **, P < 0.01; and ***, P < 0.001). (D) The rate of decline in the CD8+ T-cell immune response against each MCMV peptide following 3, 6, and 12 months of antiviral treatment. The relative changes to the MCMV immune response between untreated and treated MCMV infection are shown; the data from a single experiment are shown; the numbers of mice range from 3 to 4 per time point.
Following 12 months of antiviral treatment, the animals that received the valaciclovir (MCMV+AV) had profound suppression in the magnitude of splenic MCMV-specific T cells in comparison to mice in the infected control group (MCMV−AV). This pattern was seen against every MCMV peptide, with the degree of suppression ranging from 70 to 85% of that seen in the untreated MCMV−AV group (Fig. 3B; Table 1). The most profound effect was seen in relation to immunity to the IE3 epitope, where IE3-specific CD8+ T cells accumulated to 10% of the entire CD8+ T-cell pool in the MCMV−AV group, compared to 1.2% following antiviral treatment; this represents a relative reduction of 84% (P < 0.001) (Fig. 3B; Table 1). The M38 and M45 T-cell frequencies were also significantly reduced following the longer period of treatment. The global MCMV-specific IFN-γ+ lymphocyte response from these mice averaged 24% of the CD8+ T-cell pool in the untreated group compared to 5% in those treated with the antiviral for 12 months (Fig. 3C); this indicates that valaciclovir can suppress the overall MCMV-specific CD8+ T-cell response by almost 80%.
Table 1.
Magnitude of the MCMV-specific CD8+ T-cell response in mice aged 18 months with or without 12 months of valaciclovir treatment
| Epitope | IFN-γ (% CD8)a |
% reductionb | |
|---|---|---|---|
| MCMV−AV | MCMV+AV | ||
| M38 | 4.87 (±1.45) | 1.21 (±0.39) | 75.15 |
| M45 | 4.88 (±1.46) | 1.35 (±0.41) | 72.40 |
| M57 | 1.59 (±1.15) | 0.32 (±0.13) | 80.03 |
| m139 | 2.84 (±1.24) | 0.74 (±0.37) | 73.05 |
| IE3 | 9.87 (±1.88) | 1.62 (±0.43) | 83.61 |
| Total | 23.95 (±4.48) | 5.23 (±1.11) | 78.16 |
Results are shown as the mean of the percentage of IFN-γ-secreting CD8+ T cells against MCMV-immunodominant peptides (±SEM).
Percent reduction observed between MCMV−AV–treated (n = 9) and MCMV+AV–treated (n = 11) mice. Data are combined from two independent experiments.
We then went on to examine the declines in the frequency of the individual MCMV peptide-specific T-cell responses, relative to those in the untreated control groups, over time in order to estimate the half-life of decay following the introduction of antiviral treatment. The individual peptide-specific responses showed a broadly similar percentage reduction over 12 months, and the average half-life of decay (t1/2) over this period ranged from 200 to 250 days (Fig. 3D). However, differences were observed in the kinetics of the declines of CD8+ T-cell immunity against individual peptides. In particular, the immune responses to IE3 did not start to decline until after 6 months of treatment but then fell rapidly, with an estimated t1/2 of approximately 45 days, which was the same rate seen by Snyder et al. (2008) following adoptive transfer (14). This could suggest that the antiviral treatment reduced MCMV loads to an absent level.
In order to determine if reductions in the frequency of cytokine-responsive MCMV-specific cells in the MCMV+AV group were simply due to the loss of functional response, we then went on to examine the numbers of tetramer-binding cells. Splenocytes were stained with a major histocompatibility complex class I (MHC-I)-peptide tetramer containing immunodominant IE3 (Fig. 4A and B). In keeping with the results obtained by cytokine secretion analysis, the proportion of IE3 tetramer-binding cells was profoundly reduced from 15% in the MCMV−AV group to approximately 1% of CD8+ T cells in the MCMV+AV group (Fig. 4B). Yet, the frequency of m139 tetramer-binding cells was not reduced by the treatment (data not shown).
Fig 4.

Valaciclovir reduces the degree of differentiation of IE3- but not m139-specific CD8+ T cells. (A) Representative fluorescence-activated cell sorter (FACS) plots of IE3 tetramer-specific CD8+ T cells from spleen samples 18 months post-MCMV infection plus or minus 12 months of antiviral treatment. (B) Mean frequency of IE3 tetramer-specific CD8+ T cells measured by tetramer staining in MCMV−AV (n = 3) and MCMV+AV (n = 3) mice. The graphs show the phenotype of IE3- (C) and m139- (D) specific CD8+ T cells in MCMV−AV mice (n = 3) in relation to MCMV+AV–treated (n = 3) mice. The results are shown as the percentages of CD27+, CD44+, CD62L+, CD122+, and CD127+ expression on tetramer-positive cells (% Tet) (mean ± SEM). The data were analyzed by 2-way analysis of variance (ANOVA) plus Bonferroni's posttest correction (*, P < 0.05).
Antiviral treatment reduced the degree of differentiation of MCMV IE3-specific T cells.
The phenotype of inflationary MCMV-specific CD8+ T cells has been characterized as a late-differentiated profile (CD44+ CD62Llo CD27lo CD122lo CD127lo, where lo indicates low expression) (14). In order to investigate if antiviral treatment had any influence on T-cell differentiation, we then studied the membrane phenotype of MCMV-specific cells following antiviral treatment. Here, the IE3-specific responses appeared to be most affected by the antiviral treatment. Expression of the lymph node homing molecule CD62L is downregulated to enable the lymphocyte to exit the lymph node; therefore, it can be used as an indirect measure of activation. CD62L expression was retained on 20.2% of the IE3 tetramer-binding cells in the MCMV+AV–treated group compared to only 1.6% in the untreated MCMV−AV group (P < 0.05), showing that treatment reduces the levels of persistent activation. Likewise, the expression of CD122 (the high-affinity interleukin-2 beta receptor [IL-βR]), usually absent in MCMV-specific T cells (28), was significantly higher in the antiviral-treated (4.8%) group than in the untreated mice (0.2%) (P < 0.05). The expressions of CD27 and CD127 (the interleukin-7 receptor alpha [IL-7Rα]) were also increased on IE3 tetramer-positive cells following the antiviral treatment (Fig. 4C). However, these patterns were much less pronounced on the m139 tetramer-binding CD8+ T cells (Fig. 4D). These data indicate that antiviral treatment is able to limit the degrees of differentiation of the MCMV-specific immune response in specific populations that are more sensitive to the exposure of valaciclovir.
Antiviral therapy reduced the accumulation of effector memory T cells during MCMV infection and restored the proportion of naïve T cells.
CMV infection leads to both a substantial increase in the memory T-lymphocyte pool and an associated reduction in the proportion of naïve T cells (29, 30); similarly, murine T cells can be categorized into memory subsets based on CD44 and CD62L coexpression (Fig. 5A). There is concern that a decline in naïve T-cell frequencies may influence immune competence in CMV-seropositive individuals. After the resolution of primary MCMV infection, there is also a reduction in the proportion of naïve T cells (TN) (CD8+ CD44− CD62L+) and an accumulation of cytotoxic effector memory cells (TEM) (CD8+ CD44+ CD62L−). Indeed, in our studies, we observed gradual accumulations of TEM cells over time, and these became the most prominent of the CD8+ T-cell subsets 40 weeks post-MCMV infection (Fig. 5B).
Fig 5.

Antiviral treatment restores the naïve and memory CD8+ T-cell pools to those seen in MCMV-neg mice. (A) Representative FACS plots of CD8+ T-cell memory subsets: naïve (TN) (CD44− CD62L+), central memory (TCM) (CD44+ CD62L+), effector memory (TEM) (CD44+ CD62L+), and double negative (TDN) (CD44− CD62L−) phenotypes. (B) Frequencies of naïve, central memory, effector memory, and intermediate CD8+ T cells following MCMV infection, where the numbers of mice range from 3 to 4 per time point (mean ± SEM). (C) The numbers of live splenocytes isolated from MCMV−AV (n = 6) mice 18 months postinfection and following 12 months of antiviral treatment (MCMV+AV) (n = 8). The data were analyzed by the two-way Mann-Whitney U test (ns, P > 0.05). (D) Frequencies of TN, TCM, TEM, and TDN CD8+ T cells are shown from spleen samples of MCMV-neg (n = 3), MCMV−AV (n = 3), and MCMV+AV (n = 4) groups of mice (mean ± SEM). The data were analyzed by 2-way ANOVA plus Bonferroni's posttest correction (*, P < 0.05; **, P < 0.01).
In order to investigate if the antiviral treatment was able to modulate the accumulation of memory T cells during MCMV infection, we then tried to determine the T-cell memory subsets in mice aged 18 months of age depending on whether they had received 12 months of antiviral therapy. The MCMV+AV mice had numbers of T cells in the spleen equivalent to those in the control MCMV−AV mice (Fig. 5C), yet clear differences were observed in the memory phenotype of the CD8+ T-cell pool. The TEM population was the most dominant CD8+ T-cell subset detected in the untreated MCMV−AV mice and comprised 68% of the entire splenic CD8+ T-cell pool. However, the TEM population was only 40% in the antiviral-treated group (P < 0.001) (Fig. 5D).
The TN CD8+ T-cell subset remained the most abundant T-cell population in the spleens of MCMV-uninfected mice (MCMV-neg) at 18 months of age and comprised 37% of the CD8+ T-cell pool. In comparison, the TN CD8+ T-cell pool was depleted by 60% in the MCMV−AV group, such that it represented only 15% of CD8+ T cells in this group. Importantly, valaciclovir therapy completely restored the proportion of TN CD8+ T cells to a value of 45% of the CD8+ T-cell pool (P < 0.05) (Fig. 5D). The frequencies of central memory T cells (TCM) (CD8+ CD44+ CD62L+) were also comparable between the MCMV-negative (MCMV-neg) and the MCMV+AV groups, but they were reduced in the MCMV−AV group. This likely reflects sporadic MCMV reactivation in the untreated animals, which drives the differentiation of central memory cells into TEM CD8+ T cells. These data demonstrate that the typical “senescent” profile of CD8+ memory T-cell accumulation associated with CMV infection can be reversed with the use of antiviral therapy.
Prolonged antiviral treatment reversed the elevated influenza viral load associated with viral challenge of MCMV-infected mice.
CMV infection has emerged as an important determinant of the efficacy of influenza vaccination in clinical studies (31). Influenza A virus is a good model for testing MCMV-induced immune senescence, as infection with this virus is generally nonlethal in younger adult mice, whereas elderly mice suffer from increased complications associated with impaired IAV-specific adaptive immunity (32, 33). Because we had observed that antiviral therapy was able to restore the size of the naïve lymphocyte pool in elderly MCMV-infected mice (Fig. 5D), we then investigated if such treatment was able to improve the immunological and clinical responses to IAV infection. All three groups of 18-month-old mice were challenged with A/Puerto Rico/8/34/England/939/69 (H3N2) IAV, and the subsequent immune responses were determined 10 days postinfection (Fig. 3A).
No acute deaths were seen in the MCMV-neg or MCMV+AV groups, whereas 25% of the control MCMV−AV animals succumbed to the IAV challenge within the first 5 days (Fig. 6A); however, this finding was not statistically significant. Furthermore, the MCMV−AV animals displayed the most severe signs of illness following the IAV challenge, as demonstrated by decreased activity, hunched posture, and weight loss, most likely due to the highest viral burden measured at approximately 5,500 PFU/lung sample. In comparison, the viral burden was 1,200 PFU/lung sample in the MCMV-neg mice (P < 0.05) and 1,900 PFU/lung sample in the MCMV+AV group, demonstrating that valaciclovir therapy indirectly helped to prevent the development of elevated influenza viral loads (Fig. 6B).
Fig 6.
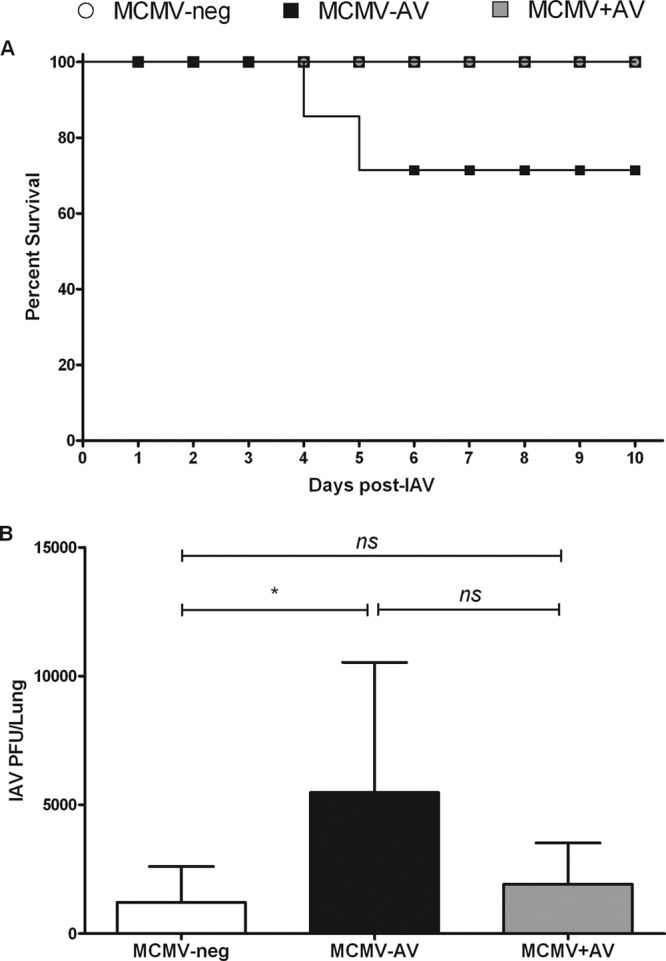
Chronic antiviral treatment modulates the clinical outcome of MCMV-infected elderly mice following influenza virus infection. (A) Kaplan-Meier survival curve of MCMV-neg (n = 5), MCMV−AV (n = 8), and MCMV+AV (n = 4) mice following IAV infection. (B) Viral titers in lung samples from MCMV-neg (n = 5), MCMV−AV (n = 6), and MCMV+AV (n = 4) mice were determined 10 days post-IAV infection. The bars represent the plaque assay (mean ± SEM). The data were analyzed by the Mann-Whitney U test (ns, P > 0.05; *, P < 0.05).
Valaciclovir treatment reduced the degree of differentiation of the IAV-specific immune response in MCMV-infected mice following influenza virus infection.
The magnitude and phenotype of the NP-specific immune response were then measured using an MHC-peptide tetramer specific for an immunodominant epitope from the influenza virus nucleoprotein NP366-374. Frequencies of tetramer-binding cells were measured in bronchoalveolar lavage fluid (BAL), caudal mediastinal lymph nodes (CMLN), and the spleen in order to study adaptive immunity in local tissues, the draining lymph node, and a systemic organ, respectively.
Very recently (in 2012), Cicin-Sain et al. showed that the magnitude of the IAV-specific CD8+ T-cell response was reduced in latently MCMV-infected mice (16); however, we did not observe significant alterations to the frequency of NP-specific CD8+ T cells. The magnitude of the NP-specific immune response was greatest in BAL fluid, where it represented 6% of the CD8+ T-cell pool and was comparable between all groups. NP-specific tetramer staining was seen on 1 to 2% of CD8+ T cells in both the CMLN and spleen samples, and this value again was similar in all groups (Fig. 7B). Consequently, the elevated IAV titers in the MCMV−AV group were unlikely to be direct results of the magnitude of the NP-specific immune response and could relate to either an impaired function of the cytotoxic lymphocytes (34) or failings in other immune cells, such as CD4+ T cells (35).
Fig 7.
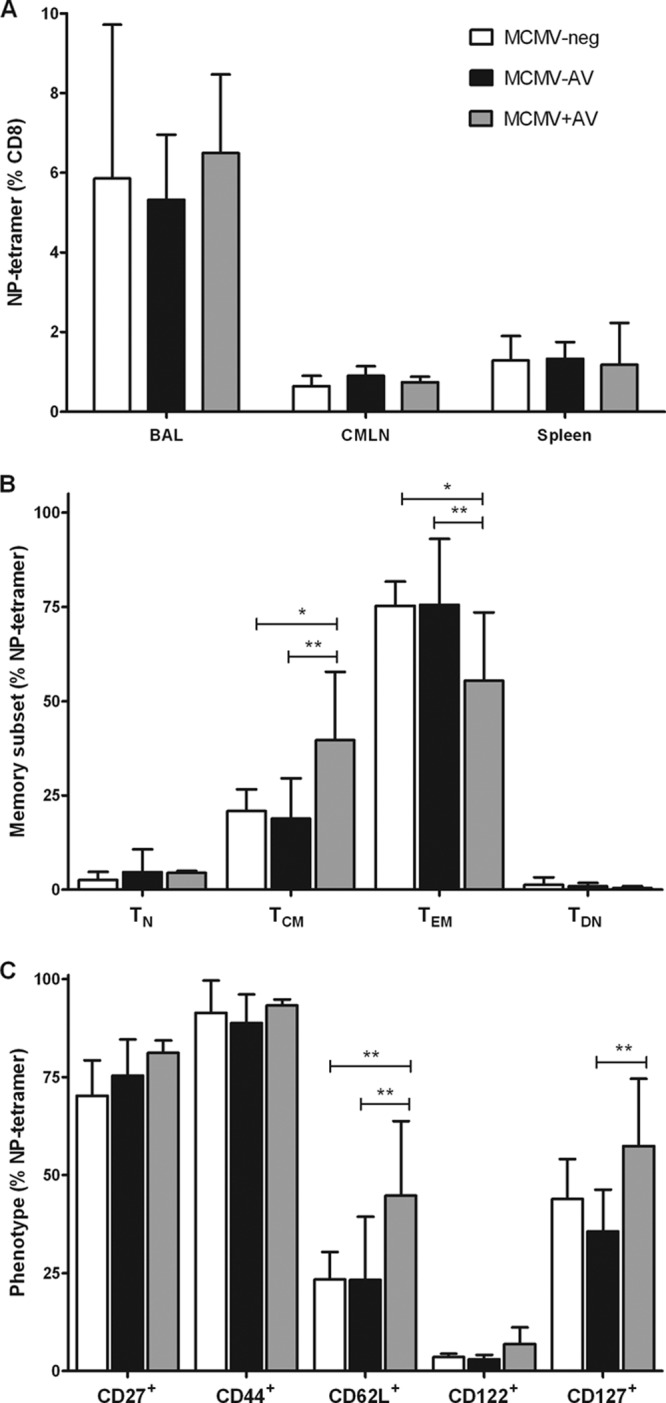
Valaciclovir treatment leads to a less differentiated IAV-specific CD8+ T-cell phenotype following influenza challenge. (A) The percentage of NP tetramer-specific CD8+ T cells within the BAL (n = 3 to 6), CMLN (n = 4 to 5), and spleen (n = 3 to 6). Shown also are results of analyses of memory subsets (B) and surface phenotypes (C) of NP tetramer-specific CD8+ T cells in CMLN (mean ± SEM). The data were analyzed by 2-way ANOVA plus Bonferroni's posttest correction (*, P < 0.05; **, P < 0.01).
The most significant observations were related to the degrees of differentiation of the IAV-specific T cells in the different groups. Twice as many NP-specific cells from the CMLN of MCMV+AV mice than from the other two groups displayed a TCM phenotype with a corresponding 40% reduction in the proportion of the TEM subset (Fig. 7C). The reduced degree of differentiation of the NP-specific pool was also apparent, as there were elevated frequencies of cells that retained the expression of CD62L and CD127 in the MCMV+AV group (Fig. 7D). The absolute numbers of cells within the CMLN were not affected by treatment (data not shown).
DISCUSSION
Several epidemiological studies of older individuals have demonstrated an association between an elevated CMV-specific immune response and a relative reduction in overall survival. As such, an understanding of the immunological mechanisms that drive memory inflation is important for the development of potential therapeutic interventions that may enhance healthy aging.
CMV-specific memory inflation is seen in both the cellular and humoral arms of the immune response, but there is debate as to the importance of viral antigen in driving this process. The M38, m139, and IE3 epitopes that undergo the most profound inflation exhibit a differentiated effector phenotype and show impaired production of interleukin-2 (IL-2), both of which reflect a history of repeated antigen stimulations (13, 14). As such, the presence of a peptide antigen might be expected to play an important role. Previous attempts to suppress herpesvirus-specific T-cell immunity using antiviral drugs have shown limited success. A model of systemic murine HSV infection has been used to generate an immune profile that resembles memory inflation, and antiviral therapy suppressed immunity by approximately 30% following 7 months of therapy when it was started soon after infection (36). The role of antigen dependence has also been tested in MCMV models in which antivirals were administered to animals infected with a famciclovir-sensitive MCMV mutant strain, and suppression of cellular immunity was not evident after 12 weeks of treatment (27). We chose to use valaciclovir in our studies, as it has demonstrated excellent efficacy against MCMV in vitro and is also well tolerated in human subjects for periods of prolonged treatment. We did not select a more potent inhibitor, such as ganciclovir, as valaciclovir has markedly fewer side effects and does not have further toxic bystander effects on hematopoietic progenitor cells (37–39).
Memory inflation in humans is seen most markedly in elderly individuals, and we therefore chose to study mice aged 18 months in our studies. There has been very little previous analysis of this group, possibly due to the technical and financial requirements of maintaining virally infected animals for prolonged periods of time. Recently, den Braber et al. (2012) showed that naïve T-cell frequencies in elderly mice are almost exclusively sustained by thymic output, whereas the naïve T-cell pool of humans in this age group relies largely on homeostatic proliferation (40). Therefore, it might be anticipated that the effect of antiviral treatment is less pronounced in CMV-infected humans than in mice, although in this regard, it is of interest that MCMV-specific memory inflation does still occur in thymectomized mice (41). It has been suggested that both the TN and TCM pools proliferate in response to antigen stimulation to maintain CD8+ T-cell memory inflation (42); therefore, future studies should consider how age and the extent of thymic output may contribute to inflationary processes.
Sophisticated adoptive transfer studies of MCMV-specific T cells into naïve and infected mice have estimated that the half-life of inflationary MCMV-specific T cells is around 45 to 60 days (14). However, we found that 3 months of antiviral treatment was insufficient for reducing T-cell inflation, with reductions in peptide-specific immunity starting to become evident after 6 months and profound suppression of the MCMV CD8+ T-cell response after 12 months across a range of both the inflationary and noninflationary MCMV peptides. Furthermore, treatment led to an almost 80% reduction in the combined CD8+ T-cell response to five MCMV-immunodominant CD8+ T-cell epitopes in elderly mice. It should be noted that the mice had been infected for at least 6 months prior to starting antiviral therapy, so the model therefore begins to resemble the situation in the human population, where individuals will have been infected for many years prior to potential consideration of antiviral therapy. In addition, the MCMV-specific CD8+ T-cell responses were not completely abrogated, certain responses (e.g., m139) were not affected by antiviral treatment, and factors such as homeostatic proliferation are likely to retain a residual role. An interesting feature that we observed was that the degree of differentiation of the remaining population of the IE3-specific lymphocytes was markedly reduced (Fig. 4C), indicating that reactivation is required to both promote the differentiation and maintain the majority of the MCMV-specific CD8+ T-cell pool.
It was of interest to measure the rate of decay of the MCMV-specific immune response following the introduction of antiviral therapy. The half-lives for the five immunodominant MCMV-specific CD8+ T-cell populations were between 200 and 250 days when taken over the 12-month period (Fig. 3D). This is considerably longer than has previously been estimated for MCMV-specific responses (14), although it was of interest to note that the kinetics of T-cell decline were not linear. There was a tendency for the t1/2 of decay to shorten with time; the IE3-specific response was the most marked in this regard, with cells declining to a t1/2 of 45 days in the last 6 months of treatment, which we hypothesize was due to a more complete suppression of the IE3 antigen.
Factors that may influence the degree of memory inflation are the nature and origin of the target antigen. The levels of suppression of CD8+ T-cell immunity against individual peptide epitopes were comparable in our study. However, the most profound reduction was seen in the response to IE3, which is a highly inflationary epitope and has demonstrated some unique biological features in the C57BL/6 model. In particular, the response develops late after infection and is highly dependent on the presence of a CD4+ T-cell response (43). It has been suggested that priming of the IE3-specific immune response may require CD4-mediated licensing of dendritic cells (43). As such, it is possible that the uptake of viral protein by dendritic cells is suppressed during valaciclovir treatment and leads to the dramatic suppression of this response. Snyder et al. (2011) have also shown that IE3-specific CD8+ T cells do not accumulate if famciclovir is administered both prior to and immediately following MCMV infection (27). We also observed partial decreases in the frequency of M45- and M57-specific lymphocytes during the most prolonged course of treatment. These reductions were statistically significant for only M45; as such, we believe that the classification of M45 and M57 as noninflationary populations of MCMV-specific T cells may not be definitive, as these cells were also lost following adoptive transfer (14), indicating some requirement for antigen stimulation.
Our study has shown that valaciclovir needs to be taken for prolonged periods in order to suppress the accumulation of CMV-specific T cells, and this raises the question of the potential toxicity of long-term administration of the drug. Indeed, it has been reported that aciclovir can cause apoptosis of murine CD8+ T lymphocytes at high doses in vitro and in vivo (37–39). Here, we used the antiviral drug at half the dose others have used and did not observe any reductions in the actual numbers of viable cells from the spleen (Fig. 5C). In addition, if valaciclovir was lymphotoxic specifically toward CD8+ T cells, one would expect an increase in the CD4/CD8 T-cell ratio; however, this was not the case, and MCMV+AV-treated mice had CD4/CD8 T-cell ratios that were equivalent to those of MCMV-neg mice (data not shown). It is notable that the drug has been in clinical use for over 20 years with no reports of aciclovir-induced lymphopenia. The loss of MCMV-specific cells is therefore unlikely to be a result of direct CD8+ T-cell lymphotoxicity, and this is reinforced by the observation that the magnitude of NP-specific CD8+ lymphocytes was not affected by antiviral treatment.
Two features of CMV infection in humans are that it markedly increases the number of memory cells in the peripheral blood and reduces the naïve T pool. These factors could impair immune function in CMV-seropositive donors, and it is important to observe that antiviral treatment is able to correct this effect in the murine model. Naïve T cells comprised 37% of CD8+ T cells in the 18-month-old MCMV-neg mice, but this was reduced to only 15% in the MCMV-infected group. Remarkably, valaciclovir treatment was able to completely reverse this to a value of 42%. The effector memory CD8+ T-cell pool was almost doubled in frequency in the virally infected group, and this increase was completely suppressed with antiviral treatment. These observations seem particularly auspicious for a potential therapeutic role of valaciclovir in reversing the expansion of clonal CMV-specific CD8+ T cells within the peripheral blood of CMV-infected adult humans.
The use of an antiviral agent to suppress the CMV-specific immune response will only be of value if it improves the functional immune capacity of the host. In order to assess this, we used influenza as a pathogen challenge in our study groups. This is a relevant model, as influenza infection remains a major cause of death in elderly humans, and CMV-specific immunity has been shown to have been an important determinant of vaccine-dependent immunity in some studies (31, 44). It was of interest that the most severe clinical features following IAV infection were seen in the MCMV−AV group, and antiviral therapy was able to prevent these. The magnitude of the IAV-specific CD8+ immune response was comparable in all the study groups, but the degree of T-cell differentiation was suppressed in the antiviral group, suggesting a potential role of chronic MCMV infection in driving T-cell differentiation against heterologous pathogens. Indeed, this has been observed in relation to the CD4+ T-cell response in CMV-positive human subjects (45). One additional and intriguing observation was that some clinical and immunological differences following the IAV challenge were seen between valaciclovir-treated animals and MCMV-neg mice. MCMV infection has been shown to have beneficial effects against an unrelated pathogen challenge in young mice (46), and it is possible that antiviral treatment may unmask a potential benefit of an underlying MCMV infection that is otherwise lost in elderly animals due to uncontrolled memory inflation.
The data presented in this article have important implications for the potential treatment of patients with high levels of CMV-specific immunity that have been associated with immune senescence (47). Using the MCMV model, we have shown that memory inflation is reversible and is dependent on viral replication. Unfortunately, we were unable to show that antiviral treatment reduces the viral load, as MCMV titers were undetectable in latently infected mice. Valaciclovir is often given for periods of many years in order to suppress the reactivation of genital herpesvirus infection, and it is well tolerated. Viral resistance to valaciclovir is rare and is most commonly seen in the setting of immune suppression. The amount of valaciclovir used in our study was equivalent to the dose administered for the treatment of acute varicella zoster infection or control of CMV replication in patients undergoing immune suppression. However, the dose of valaciclovir that would be required to suppress CMV replication in immunocompetent subjects may be much less than this and is currently the subject of investigation. It may now be appropriate to consider the use of valaciclovir in CMV-seropositive people to determine if this can offer novel clinical opportunities to improve the health of the elderly population.
ACKNOWLEDGMENT
We thank the NIH Tetramer Core Facility at Emory University, Atlanta, Georgia, for producing the MHC-I tetramer reagents.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PA. 2002. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 169:1984–1992 [DOI] [PubMed] [Google Scholar]
- 2. Ouyang Q, Wagner WM, Voehringer D, Wikby A, Klatt T, Walter S, Muller CA, Pircher H, Pawelec G. 2003. Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp. Gerontol. 38:911–920 [DOI] [PubMed] [Google Scholar]
- 3. Khan N, Hislop A, Gudgeon N, Cobbold M, Khanna R, Nayak L, Rickinson AB, Moss PA. 2004. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J. Immunol. 173:7481–7489 [DOI] [PubMed] [Google Scholar]
- 4. Pawelec G, Derhovanessian E, Larbi A, Strindhall J, Wikby A. 2009. Cytomegalovirus and human immunosenescence. Rev. Med. Virol. 19:47–56 [DOI] [PubMed] [Google Scholar]
- 5. Wikby A, Johansson B, Olsson J, Lofgren S, Nilsson BO, Ferguson F. 2002. Expansions of peripheral blood CD8 T-lymphocyte subpopulations and an association with cytomegalovirus seropositivity in the elderly: the Swedish NONA immune study. Exp. Gerontol. 37:445–453 [DOI] [PubMed] [Google Scholar]
- 6. Roberts ET, Haan MN, Dowd JB, Aiello AE. 2010. Cytomegalovirus antibody levels, inflammation, and mortality among elderly Latinos over 9 years of follow-up. Am. J. Epidemiol. 172:363–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bevan IS, Sammons CC, Sweet C. 1996. Investigation of murine cytomegalovirus latency and reactivation in mice using viral mutants and the polymerase chain reaction. J. Med. Virol. 48:308–320 [DOI] [PubMed] [Google Scholar]
- 8. Seckert CK, Renzaho A, Tervo HM, Krause C, Deegen P, Kuhnapfel B, Reddehase MJ, Grzimek NK. 2009. Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J. Virol. 83:8869–8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seckert CK, Schader SI, Ebert S, Thomas D, Freitag K, Renzaho A, Podlech J, Reddehase MJ, Holtappels R. 2011. Antigen-presenting cells of haematopoietic origin prime cytomegalovirus-specific CD8 T-cells but are not sufficient for driving memory inflation during viral latency. J. Gen. Virol. 92:1994–2005 [DOI] [PubMed] [Google Scholar]
- 10. Walton SM, Torti N, Mandaric S, Oxenius A. 2011. T-cell help permits memory CD8(+) T-cell inflation during cytomegalovirus latency. Eur. J. Immunol. 41:2248–2259 [DOI] [PubMed] [Google Scholar]
- 11. Holtappels R, Pahl-Seibert MF, Thomas D, Reddehase MJ. 2000. Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62L(lo) memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J. Virol. 74:11495–11503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P. 2003. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 170:2022–2029 [DOI] [PubMed] [Google Scholar]
- 13. Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. 2006. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J. Immunol. 177:450–458 [DOI] [PubMed] [Google Scholar]
- 14. Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. 2008. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity 29:650–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klenerman P, Dunbar PR. 2008. CMV and the art of memory maintenance. Immunity 29:520–522 [DOI] [PubMed] [Google Scholar]
- 16. Cicin-Sain L, Brien JD, Uhrlaub JL, Drabig A, Marandu TF, Nikolich-Zugich J. 2012. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PLoS Pathog. 8:e1002849 doi:10.1371/journal.ppat.1002849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mekker A, Tchang VS, Haeberli L, Oxenius A, Trkola A, Karrer U. 2012. Immune senescence: relative contributions of age and cytomegalovirus infection. PLoS Pathog. 8:e1002850 doi:10.1371/journal.ppat.1002850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weekes MP, Wills MR, Sissons JG, Carmichael AJ. 2006. Large HIV-specific CD8 cytotoxic T-lymphocyte (CTL) clones reduce their overall size but maintain high frequencies of memory CTL following highly active antiretroviral therapy. Immunology 118:25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sammons CC, Sweet C. 1989. Isolation and preliminary characterization of temperature-sensitive mutants of mouse cytomegalovirus of differing virulence for 1-week-old mice. J. Gen. Virol. 70:2373–2381 [DOI] [PubMed] [Google Scholar]
- 20. Collie MH, Sweet C, Smith H. 1980. Infection of neonatal and adult mice with non-passaged influenza viruses. Brief report. Arch. Virol. 65:77–81 [DOI] [PubMed] [Google Scholar]
- 21. Bachmann MF, Ecabert B, Kopf M. 1999. Influenza virus: a novel method to assess viral and neutralizing antibody titers in vitro. J. Immunol. Methods 225:105–111 [DOI] [PubMed] [Google Scholar]
- 22. Burns WH, Wingard JR, Bender WJ, Saral R. 1981. Thymidine kinase not required for antiviral activity of acyclovir against mouse cytomegalovirus. J. Virol. 39:889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glasgow LA, Richards JT, Kern ER. 1982. Effect of acyclovir treatment on acute and chronic murine cytomegalovirus infection. Am. J. Med. 73:132–137 [DOI] [PubMed] [Google Scholar]
- 24. Kern ER, Kushner NL, Hartline CB, Williams-Aziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. 2005. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother. 49:1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Miranda P, Burnette TC. 1994. Metabolic fate and pharmacokinetics of the acyclovir prodrug valaciclovir in cynomolgus monkeys. Drug Metab. Dispos. 22:55–59 [PubMed] [Google Scholar]
- 26. Winston DJ, Yeager AM, Chandrasekar PH, Snydman DR, Petersen FB, Territo MC, Valacyclovir Cytomegalovirus Study Group 2003. Randomized comparison of oral valacyclovir and intravenous ganciclovir for prevention of cytomegalovirus disease after allogeneic bone marrow transplantation. Clin. Infect. Dis. 36:749–758 [DOI] [PubMed] [Google Scholar]
- 27. Snyder CM, Cho KS, Bonnett EL, Allan JE, Hill AB. 2011. Sustained CD8+ T cell memory inflation after infection with a single-cycle cytomegalovirus. PLoS Pathog. 7:e1002295 doi:10.1371/journal.ppat.1002295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sierro S, Rothkopf R, Klenerman P. 2005. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. Eur. J. Immunol. 35:1113–1123 [DOI] [PubMed] [Google Scholar]
- 29. Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, Schonitzer D, Grubeck-Loebenstein B. 2005. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J. Virol. 79:3675–3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chidrawar S, Khan N, Wei W, McLarnon A, Smith N, Nayak L, Moss P. 2009. Cytomegalovirus-seropositivity has a profound influence on the magnitude of major lymphoid subsets within healthy individuals. Clin. Exp. Immunol. 155:423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trzonkowski P, Mysliwska J, Szmit E, Wieckiewicz J, Lukaszuk K, Brydak LB, Machala M, Mysliwski A. 2003. Association between cytomegalovirus infection, enhanced proinflammatory response and low level of anti-hemagglutinins during the anti-influenza vaccination—an impact of immunosenescence. Vaccine 21:3826–3836 [DOI] [PubMed] [Google Scholar]
- 32. Effros RB, Walford RL. 1983. The immune response of aged mice to influenza: diminished T-cell proliferation, interleukin 2 production and cytotoxicity. Cell Immunol. 81:298–305 [DOI] [PubMed] [Google Scholar]
- 33. Toapanta FR, Ross TM. 2009. Impaired immune responses in the lungs of aged mice following influenza infection. Respir. Res. 10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Humphreys IR, Clement M, Marsden M, Ladell K, McLaren JE, Smart K, Hindley JP, Bridgeman HM, van den Berg HA, Price DA, Ager A, Wooldridge L, Godkin A, Gallimore AM. 2012. Avidity of influenza-specific memory CD8(+) T-cell populations decays over time compromising antiviral immunity. Eur. J. Immunol. doi:10.1002/eji.201242575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, Swain SL. 2012. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J. Clin. Invest. 122:2847–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lang A, Brien JD, Nikolich-Zugich J. 2009. Inflation and long-term maintenance of CD8 T cells responding to a latent herpesvirus depend upon establishment of latency and presence of viral antigens. J. Immunol. 183:8077–8087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McGuffin RW, Shiota FM, Meyers JD. 1980. Lack of toxicity of acyclovir to granulocyte progenitor cells in vitro. Antimicrob. Agents Chemother. 18:471–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szczech GM. 1996. Preclinical development of antiviral drugs. Clin. Infect. Dis. 22:355–360 [DOI] [PubMed] [Google Scholar]
- 39. Wingard JR, Hess AD, Stuart RK, Saral R, Burns WH. 1983. Effect of several antiviral agents on human lymphocyte functions and marrow progenitor cell proliferation. Antimicrob. Agents Chemother. 23:593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mögling R, de Boer AB, Willems N, Schrijver EHR, Spierenburg G, Gaiser K, Mul E, Otto SA, Ruiter AFC, Ackermans MT, Miedema F, Borghans JAM, de Boer RJ, Tesselaar K. 2012. Maintenance of peripheral naïve T cells is sustained by thymus output in mice but not humans. Immunity 36:288–297 [DOI] [PubMed] [Google Scholar]
- 41. Loewendorf AI, Arens R, Purton JF, Surh CD, Benedict CA. 2011. Dissecting the requirements for maintenance of the CMV-specific memory T-cell pool. Viral Immunol. 24:351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Torti N, Walton SM, Brocker T, Rulicke T, Oxenius A. 2011. Non-hematopoietic cells in lymph nodes drive memory CD8 T cell inflation during murine cytomegalovirus infection. PLoS Pathog. 7:e1002313 doi:10.1371/journal.ppat.1002313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Snyder CM, Loewendorf A, Bonnett EL, Croft M, Benedict CA, Hill AB. 2009. CD4+ T cell help has an epitope-dependent impact on CD8+ T cell memory inflation during murine cytomegalovirus infection. J. Immunol. 183:3932–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. den Elzen WP, Vossen AC, Cools HJ, Westendorp RG, Kroes AC, Gussekloo J. 2011. Cytomegalovirus infection and responsiveness to influenza vaccination in elderly residents of long-term care facilities. Vaccine 29:4869–4874 [DOI] [PubMed] [Google Scholar]
- 45. Libri V, Azevedo RI, Jackson SE, Di Mitri D, Lachmann R, Fuhrmann S, Vukmanovic-Stejic M, Yong K, Battistini L, Kern F, Soares MV, Akbar AN. 2011. Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+CD45RA+CD27+ T cells: the potential involvement of interleukin-7 in this process. Immunology 132:326–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW., 4th 2007. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447:326–329 [DOI] [PubMed] [Google Scholar]
- 47. Wills M, Akbar A, Beswick M, Bosch JA, Caruso C, Colonna-Romano G, Dutta A, Franceschi C, Fulop T, Gkrania-Klotsas E, Goronzy J, Griffiths SJ, Henson S, Herndler-Brandstetter D, Hill A, Kern F, Klenerman P, Macallan D, Macualay R, Maier AB, Mason G, Melzer D, Morgan M, Moss P, Nikolich-Zugich J, Pachnio A, Riddell N, Roberts R, Sansoni P, Sauce D, Sinclair J, Solana R, Strindhall J, Trzonkowski P, van Lier R, Vescovini R, Wang G, Westendorp R, Pawelec G. 2011. Report from the second cytomegalovirus and immunosenescence workshop. Immun. Ageing 8:10. [DOI] [PMC free article] [PubMed] [Google Scholar]