

Abstract
The adeno-associated virus (AAV) encodes four regulatory proteins called Rep. The large AAV Rep proteins Rep68 and Rep78 are essential factors required in almost every step of the viral life cycle. Structurally, they share two domains: a modified version of the AAA+ domain that characterizes the SF3 family of helicases and an N-terminal domain that binds DNA specifically. The combination of these two domains imparts extraordinary multifunctionality to work as initiators of DNA replication and regulators of transcription, in addition to their essential role during site-specific integration. Although most members of the SF3 family form hexameric rings in vitro, the oligomeric nature of Rep68 is unclear due to its propensity to aggregate in solution. We report here a comprehensive study to determine the oligomeric character of Rep68 using a combination of methods that includes sedimentation velocity ultracentrifugation, electron microscopy, and hydrodynamic modeling. We have determined that residue Cys151 induces Rep68 to aggregate in vitro. We show that Rep68 displays a concentration-dependent dynamic oligomeric behavior characterized by the presence of two populations: one with monomers and dimers in slow equilibrium and a second one consisting of a mixture of multiple-ring structures of seven and eight members. The presence of either ATP or ADP induces formation of larger complexes formed by the stacking of multiple rings. Taken together, our results support the idea of a Rep68 molecule that exhibits the flexible oligomeric behavior needed to perform the wide range of functions occurring during the AAV life cycle.
INTRODUCTION
The adeno-associated virus (AAV) nonstructural protein Rep68 belongs to the superfamily 3 of helicases (SF3), whose main characteristic is their multifunctionality (1). Whereas the role of most helicases is to unwind DNA ahead of the replication fork, SF3 helicases also work as initiators of DNA replication, transcriptional regulators, and motor pumps to pack DNA into empty capsids (2). This multifunctionality is due in part to the presence of a specialized AAA+ domain that is at the core of all DNA transactions performed by these proteins. Their catalytic activities are dependent on the formation of oligomeric complexes, where the ATP binding and hydrolysis occurring at the interface of neighboring subunits drive conformational changes that promote translocation or remodeling of target substrates (3). Functional diversity is achieved through formation of different number of oligomers and the presence of specialized associated domains (4). For instance, the AAA+ core of AAV Rep proteins contains a β-hairpin insertion that is involved in the coupling of ATP hydrolysis to DNA translocation/unwinding, and Rep68 and Rep78 have an origin binding domain (OBD) at the N terminus (5–7). Combination of these structural features allows AAV Rep proteins to play a central role in virtually every step of the viral life cycle, such as DNA replication, transcription regulation of the p5 promoter, and site-specific integration (8–11). The AAV Rep proteins have distinct characteristics that position them apart from other SF3 family members, such as simian virus 40 large T antigen (SV40-LTag) and papillomavirus E1 protein (PV-E1). A case in point is the OBD that contains an endonuclease activity that is required for DNA replication and site-specific integration and is similar to domains from other nonstructural proteins from geminiviruses and bacteriophages that use rolling-circle replication (RCR) to replicate their genome (12). Moreover, the minimal AAA+ helicase domain of Rep proteins is monomeric, in contrast to the equivalent domains of SV40-LTag and PV-E1, which form hexameric rings (13–15). This important structural difference may be a reflection of the diversity of DNA binding sites that Rep proteins recognize and the special mechanisms used to replicate the AAV genome. The AAV single-stranded genome contains palindromic sequences at both ends that fold into three-way junction structures called inverted terminal repeats (ITRs). The start of replication initiates at the 3′ end, which is used as a primer for leading strand synthesis. This initial cycle of replication, which requires unwinding of the 5′-end ITR, leaves a DNA molecule with an ITR hairpin end that needs to be replicated to generate a double-stranded DNA (dsDNA) molecule (16, 17). To complete replication, Rep68/Rep78 bind to a specific site called the Rep binding site (RBS) within the ITR and in a reaction that is ATP dependent unwind and nick DNA, generating a new 3′ end (18). The nature of the Rep-DNA complex formed during this process remains unknown, but because of the variety of DNA substrates generated—from double-stranded DNA during the initial binding to single-stranded DNA after the melting reaction—it may require the assembly and disassembly of Rep-DNA complexes with different stoichiometries (19). For instance, binding to both AAV ori and AAV S1 sites may be a stepwise process where initial binding to the RBS site is followed by additional recruitment of more Rep molecules, as has been reported for similar initiator proteins such as SV40-Tag and PV-E1. Therein lies the importance of determining the peculiar oligomeric nature of the apo-Rep68/Rep78 proteins in order to understand their DNA-directed assembly. Recently, we and others have determined that the linker that connects the OBD to the AAA+ domain is critical for the oligomerization of Rep proteins and may be important for DNA complex formation (20, 21). Our current knowledge of Rep68/Rep68 oligomerization derives mostly from studies of these proteins bound to the AAV ori or other DNA targets showing a variety of oligomers from hexameric to double octameric (19, 22). In this study, we have used a multifaceted approach involving size-exclusion chromatography, analytical ultracentrifugation, modeling, plus electron microscopy, and we show below that Rep68 has a dynamic behavior in solution and forms a mixture of monomer, dimers, tetramers, and multiple-ring structures.
MATERIALS AND METHODS
Protein expression and purification.
Rep68 wild type (wt), as well as the other mutants proteins used here, was expressed and purified as described before (19). Briefly, the His6-PreScission protease (PP) cleavage site-Rep68 fusion protein was expressed in Escherichia coli BL21(DE3)/pLysS bacteria at 37°C for 3 h in LB medium containing 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Cell pellets were lysed in Ni-buffer A (20 mM Tris-HCl [pH 7.9 at 4°C], 500 mM NaCl, 5 mM imidazole, 10% glycerol, 0.2% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 1 mM TCEP [Tris(2-carboxyethyl)phosphine]). After five 10-s cycles of sonication, the fusion protein was purified using an Ni column preequilibrated in Ni-buffer A. The protein that eluted was desalted using buffer A and a HiPrep 26/10 desalting column (GE Healthcare). The His-PP tag was removed by PreScission protease treatment using 150 μg PP/mg His-PP-Rep68. After overnight incubation at 4°C, buffer was exchanged using the same desalting column and Ni-buffer A. Subsequent Ni column chromatography using buffer B (the same as buffer A but with 1 M imidazole) was performed to remove the uncleaved fusion protein, and untagged Rep68 was eluted with 30 mM imidazole. Rep68 was finally purified by gel filtration chromatography using a HiLoad Superdex 200 16/60 column (GE Healthcare) and size-exclusion buffer (25 mM Tris-HCl [pH 8.0], 200 mM NaCl, 2 mM TCEP). Rep68 wt and mutant proteins were concentrated (1.0 mg/ml for wt and the Cys mutants and 10 mg/ml for the double mutant of Cys). All proteins were flash-frozen in liquid N2 and stored at −80°C.
Analytical gel filtration chromatography.
Samples of the different versions of Rep68 (200 μl) at 4-mg/ml concentrations were chromatographed on a Superose 6 10/300 GL column (GE Healthcare) with a flow rate of 0.5 ml/min. Size-exclusion buffer was used for all chromatographic analyses. Protein elution was detected from its absorbance at 280 nm. A standard curve for protein molecular masses was generated using the proteins carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), alcohol dehydrogenase (ADH; 150 kDa), β-amylase (210 kDa), apoferritin (443 kDa), and thyroglobulin (669 kDa) (Sigma-Aldrich).
Cross-linking of Rep68*.
The cross-linking reactions for the Rep68 Cys151Ser mutant (Rep68*) were done according to a protocol adapted from that of Packman and Perham (23). The protein concentration was 82 μM (5 mg/ml) in 20 mM phosphate buffer, added with 300 mM NaCl, pH 8.0. A 30-fold molar excess of 100 mM dimethyl pimelimidate dihydrochloride (DMP; MP Biomedicals, LLC) was added to the reaction mixture, and the mixture was incubated at room temperature. The reaction was quenched by addition of Tris (pH 7.5) to a final concentration of 50 mM after 1 h.
Analytical ultracentrifugation.
Sedimentation velocity experiments were carried out using a Beckman Optima XL-I analytical ultracentrifuge (Beckman Coulter Inc.) equipped with both four- and eight-hole rotors. Samples were loaded in the cells, using in all cases size-exclusion buffer. Samples were centrifuged in 2-sector carbon-filled Epon centerpieces typically at 25,000 rpm and 20°C. Sectors were loaded with a 420-μl sample volume. Typically, 200 or more scans were collected at 5-min intervals at 25,000 rpm. Concentration profiles were collected using both UV absorption (280 nm) and Rayleigh interference optical systems. Results were analyzed using both the SEDFIT and SEDPHAT programs (24, 25).
DNA binding.
Fluorescence anisotropy experiments were performed on an ISS PC1 fluorimeter (ISS, Champaign, IL). The DNA substrate used was a single-stranded oligo(dT)38 oligonucleotide modified with a 5′ 6-carboxyfluorescein molecule (Integrated DNA Technologies). Reactions were performed in a volume of 500 μl with a buffer containing 25 mM HEPES (pH 7.0), 100 mM NaCl, and a final concentration of 5 nM DNA. Protein concentrations were in the range of 0 to 2.0 μM. Samples were incubated at 20°C for 30 min prior to measurement. Binding activity was followed at an excitation of 492 nm and an emission of 520 nm. Data analysis was performed as described by Yoon-Robarts et al. (7).
Helicase assay.
DNA unwinding was measured using a fluorescence resonance energy transfer (FRET)-based fluorometric assay based on the protocol of Bjornson et al., with some modifications (26). The assay uses a DNA molecule labeled at each strand with a FRET pair. The DNA site consists of an 18-bp duplex region and a 10-nucleotide 3′ tail in the bottom strand. The top strand has been labeled with Iowa-Black Dark quencher (IB) at the 3′ end, and the bottom strand has been labeled with cyanine 5 (Cy5) at the 5′ end. IB quenches the fluorescent intensity of Cy5, and upon DNA unwinding, the increase in fluorescent intensity of Cy5 is measured. Fluorescent stopped-flow kinetics studies were carried out using an ISS PC1 fluorimeter (ISS, Champaign, IL) equipped with a Hi-Tech SFA-20 rapid kinetics stopped-flow mixer (TgK Scientific Limited, United Kingdom). The Rep68 protein and the ATP were mixed in a buffer containing 25 mM HEPES, 50 mM NaCl, pH 7.0. A reaction mix of protein, fluorescent DNA (IB/Cy5-dsDNA), and MgCl2 was placed in one syringe of the device, and the ATP mix was placed in a second. Samples were equilibrated at 20°C for 5 min before mixing. Helicase activities were followed using 30 nM fluorescent DNA and 1 μM protein in each case for 900 s. ATP was kept at a 1 mM final concentration. An excitation of 645 nm (Cy5) and an emission of 670 nm (IB) were used for these experiments with a slit of 1 mm. Data results were fit by nonlinear least-squares, using the Origin (version 5.0) program, to Ft = F0[1 − exp(−a · k)], where a is the amplitude of the fluorescence, k is the first order rate constant, and Ft and F0 are the total and initial fluorescent signals, respectively.
ATPase assay.
The ATPase assay was performed using a protocol modified from that of Baginski et al. (27). The conditions of the reactions were as follows: 1 mM ATP and 1 μM protein in the presence of 1 mM MgCl2 were mixed in a final volume of 125 μl in reaction buffer A (25 mM Tris-HCl, pH 8.0, 200 mM NaCl, 2 mM TCEP). The reaction mixture was incubated at 25°C, and aliquots were removed every 15 s. This reaction mixture was stopped with 250 μl of solution MA (0.5% ammonium molybdate, 3% ascorbic acid–0.5 N HCl). After 20 min of incubation in ice water, solution CA (2% sodium citrate tribasic dihydrate, 2% sodium arsenite, 2% acetic acid) was added to the samples. After 20 min of incubation at room temperature, samples were read at 840 nm in a spectrophotometer (Agilent Technologies) with a multiple-cell thermoregulated compartment. The formation of inorganic phosphate was calculated from phosphate standard curves.
Virus production and purification.
HEK293T cells maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, United Kingdom) supplemented with 10% fetal calf serum (FCS; Invitrogen, United Kingdom) at 37°C and in 5% CO2 were transfected using polyethylenimine (PEI) with 45 μg of pAV2 plasmid (containing the AAV type 2 [AAV2] genome) or its C151S-mutated version, pMB2, and 135 μg of helper plasmid pXX6 (University of North Carolina Vector Core Facility; see http://genetherapy.unc.edu/mta.htm or a map and the sequence). Cells were incubated in DMEM containing 2% fetal bovine serum, 100 units/ml of penicillin, 100 μg/ml streptomycin, and 25 mM HEPES. After 72 h, the cells were harvested and lysed in 150 mM NaCl, 50 mM Tris at pH 8.5, followed by three freeze (−80°C)-thaw (37°C water bath) cycles. The crude lysate was treated for 30 min at 37°C with 150 units/ml of Benzonase (Sigma) and cleared by centrifugation. The virus was purified on an iodixanol step gradient as previously described (28). The gradient was formed in clear ultracentrifugation tubes (Beckman) by first adding 2.8 ml of 15% iodixanol (Optiprep density gradient medium; Sigma) in 1 M sodium chloride, 1× TD buffer (1× phosphate-buffered saline, 1 mM MgCl2, 2.5 mM KCl) and then underlying, in succession, 1.88 ml 25% iodixanol in 1× TD buffer containing 12.5 μg/ml phenol red (Gibco, Grand Island, NY), 1.55 ml 40% iodixanol in 1× TD buffer, and 1.55 ml 60% iodixanol containing 12.5 μg/ml phenol red. The cell lysate was then applied on top of the gradient, which was centrifuged at 40,000 rpm for 3 h at 18°C in a Sorvall Discovery 90SE ultracentrifuge using a TH641 rotor. The virus was extracted with an 18-gauge needle from the 40%-60% iodixanol interphase as well as the majority of the 40% iodixanol phase. A Vivaspin20, 100-kDa-cutoff concentrator (Sartorius Stedim, Goettingen, Germany) was used to concentrate the virus in a final volume of 700 μl as well as to exchange the buffer to lactated Ringer's solution (Baxter, Deerfield, IL).
Slot blot.
HEK293T cells were transfected with pAV2 or pMB2 with Lipofectamine 2000 (Sigma) and after 24 h infected with adenovirus at a multiplicity of infection of 10. After an additional 48 h, the cells were harvested, lysed in 350 μl of 0.2 M NaOH–10 mM EDTA, and boiled for 15 min. Each sample was loaded in triplicate onto a nylon hybridization membrane (Amersham Biosciences). The membranes were rinsed in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), dried, UV cross-linked, and prehybridized in 0.75× nylon wash solution (NW; 40.6 g Na2HPO4, 18.65 g EDTA, 500 g SDS in 3.58 liters of H2O, pH 7.2) buffer at 65°C. The membranes were hybridized overnight in 0.75 × NW buffer to a radiolabeled probe (labeled using a Prime-It RmT random primer labeling kit from Stratagene, [32P]dCTPs from PerkinElmer) consisting of either a rep sequence or an ampicillin sequence. Primers used to amplify the rep probe were 5′-AACTGGACCAATGAGAACTTTCC-3′ and 5′-AAAAAGTCTTTGACTTCCTGCTT-3′. To generate the ampicillin probe, a 587-bp fragment of the mini-pDG plasmid was amplified by PCR, using the primers ND44 5′-AATCAGTGAGGCACCTATCTCAGC-3′ and 5′-AACTCGGTCGCCGCATACACTATT-3′. The membranes were washed twice with 0.5× NW buffer, followed by an additional wash in 0.1× NW buffer. Finally, the membranes were exposed to a phosphorimager screen for 2 h, and the image was analyzed with ImageQuant software (GE Healthcare Life Sciences).
Determination of infectious particle production.
HEK293T cells (105) were seeded and triple transfected 2 days later with plasmids encoding the recombinant AAV2 (rAAV2)-green fluorescent protein (GFP) (pTRUF11), AAV Rep (wt or C151S mutant), and Cap (pDG or pMB1) constructs and adenovirus helper functions (pXX6). Cells were harvested 3 days after transfection and treated as described above for virus production. Increasing volumes of crude lysate were used to infect HeLa cells. At 3 days postinfection, HeLa cells were harvested and the percentage of GFP-positive cells was determined by fluorescence-activated cell sorter (FACS) analysis (FACSCalibur; BD).
Sequencing of viral preparations.
PCR was used to prepare a fragment of rep containing the C151S mutation. The primers used were ND140 (forward, 5′-GTTTCCTGAGTCAGATTCGCG-3′) and ND45 (reverse, 5′-AAAAAGTCTTTGACTTCCTGCTT-3′), both at 0.5pmol/μl. Deoxynucleoside triphosphates (0.2 mM; NEB) and Go Taq polymerase enzyme (0.05 U/μl; Promega) were added to the reaction mixture. The cycling parameters were 2 min at 94°C, followed by 35 cycles of 30 s at 94°C, 40 s at 64°C, and 1 min at 72°C, with a final 10 min of incubation at 72°C. The resulting fragment was cloned into the pCR2.1 TOPO vector (Invitrogen) and sequenced using primers 5′-AAAAAGTCTTTGACTTCCTGCTT-3′ and MB3 5′-GTCAGGCTCATAATCTTTCCCGCA-3′ at Eurofins MWG Operon. The results were analyzed using the BLAST (NCBI website) program to confirm the expected viral sequence.
Transmission electron microscopy (TEM) analysis.
Protein samples at 0.1 mg/ml were adsorbed directly onto carbon-coated copper grids. Following negative staining with 0.75% (wt/vol) uranyl formate, samples were visualized in an electron microscope (Tecnai F20) operated at 200 kV, and images were collected at a magnification of ×50,000 under low-dose conditions on a Gatan 4k × 4k charge-coupled-device camera. Particle windowing, two-dimensional (2D) alignment, and classification reconstruction were carried out with EMAN2 software. During the entire process, the default settings of this image-processing software were followed for eight iterative alignments. The 2D averages were obtained from a final set of 560 particles.
Generation of Rep68 models.
An initial model of AAV2 OBD was built using the crystal structure of AAV5 OBD (Protein Data Bank [PDB] accession number 1RZ9) encompassing residues 1 to 98. The structure of AAV2 Rep40 (PDB accession number 1S9H) contains residues 225 to 490. Using the program MODELLER, we generated the last 46 residues known to be unstructured as an extended polypeptide chain (29). We generated two linker models; the first was modeled as an extended polypeptide. The second model was generated using the Robetta server with the sequence from residues 205 to 230 (30). The three regions (OBD, linker, and helicase domain) were put together using the program COOT (31).
RESULTS
AAV2 Rep68 forms multiple oligomeric species in solution with a tendency to aggregate.
An initial characterization of Rep68 in solution was carried out using gel filtration chromatography and sedimentation velocity. Gel filtration profiles on a Superose 6 column clearly showed the presence of three major distributions. A fraction of the protein, P1, eluted at the void volume (V0), thus indicating the presence of aggregates with molecular masses larger than the exclusion limit of the column, i.e., ∼40 MDa (Fig. 1A). The two other species, P2 and P3, eluted at 13.3 and 15.9 ml, which correspond to molecular masses of ∼550 kDa and ∼110 kDa, respectively. These molecular masses should be considered approximations due to the elongated nature of the Rep68 molecule. At higher concentrations (>1 mg/ml), the fraction of total protein in the aggregate peak increased substantially and constituted the majority of the protein population. This behavior was also observed in sedimentation velocity studies using concentrations ranging from 2 μM to 16 μM (0.125 to 1 mg/ml), indicating that even at the lowest concentration, significant formation of aggregates with an apparent s20,w value of >40S occurs (Fig. 1B). The two other major species have apparent sedimentation coefficients of ∼4S and ∼13S. The width of the peaks suggests that each of these species may be composed of more than one component. In order to assess this possibility, we analyzed the data using the van Holde-Weischet (VHW) method implemented by the program UltraScan-SOMO (32). This method removes any effect from diffusion on the calculation of s20,w, and any spread of these values reflects the presence of multiple species. Figure 1C shows the VHW analysis of the same data, showing significant drifts in both 4S and 13S peaks, indicative of a heterogeneous mixture of multiple oligomeric species.
Fig 1.
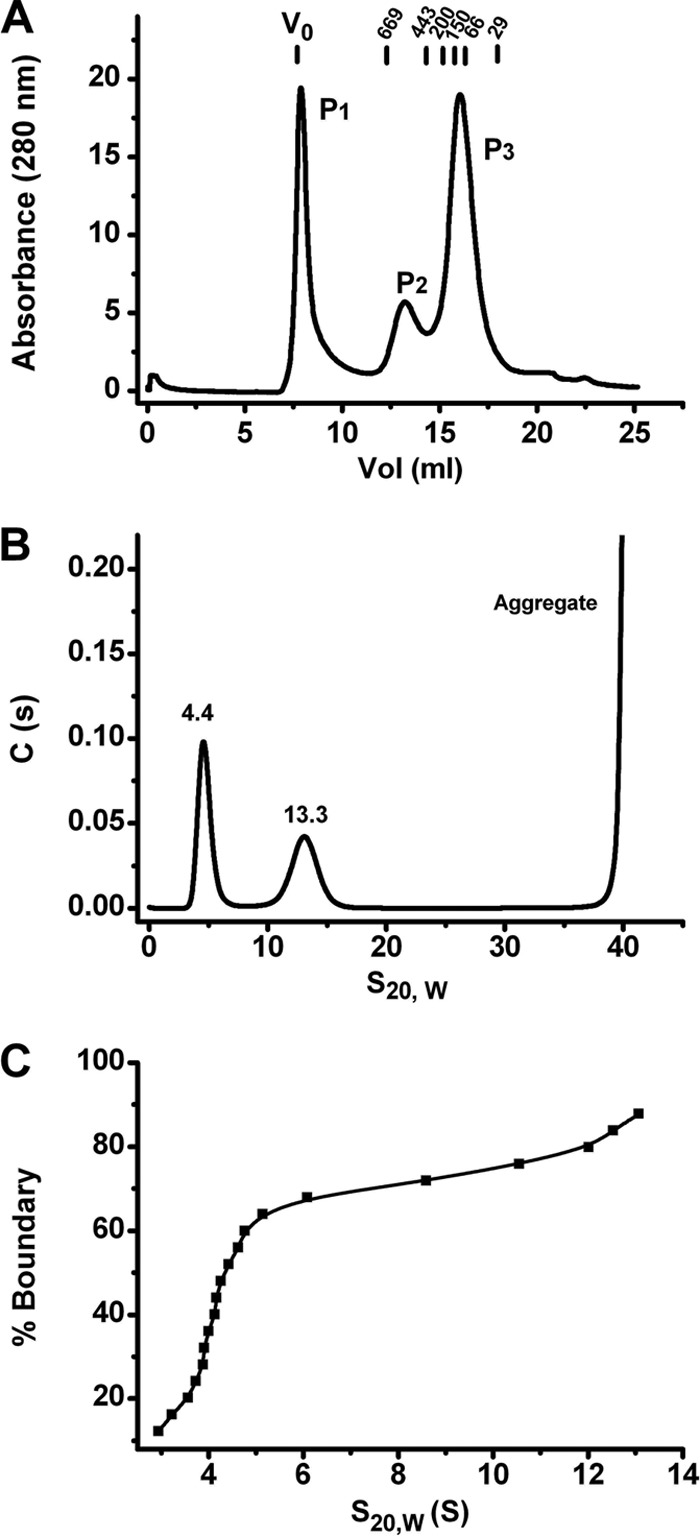
Rep68 wt oligomerization profile. (A) Rep68 wt (2 mg/ml) was injected in a Superose 6 10/300 GL column with a flow rate of 0.5 ml/min. Protein elution was followed by UV detection at 280 nm. The x axis represents the elution volume. Molecular mass standards are shown above the plot. (B) Sedimentation velocity profiles of Rep68 wt at a concentration of a 0.5 optical density (0.33 mg/ml). The experiment was run at 40,000 rpm and 20°C, as described in Materials and Methods. (C) van Holde-Weischet analysis of the sedimentation velocity data shown in panel B. The spread from the vertical at both 4S and 13S indicates the presence of more than one species in each population.
Oxidation of Rep68 cysteine residues is the major contributor to Rep68 aggregation in vitro.
Aggregation of Rep68 hinders not only the further characterization of its oligomeric properties but also future functional and structural work. To determine the cause of Rep68 aggregation, we pursued a systematic study on the influence of conditions such as pH, salt, detergents, as well as chaotropic agents and reducing agents. We determined that only reducing agents and specifically TCEP significantly reduced the fraction of Rep68 aggregates present (Fig. 2). Protein samples at a concentration of 2 mg/ml were analyzed on a Superose 6 column equilibrated with buffer A containing different TCEP concentrations ranging from 3 to 5 mM. These data clearly show that the aggregate peak eluting at the void volume of the column is reduced as the concentration of the reducing agent TCEP is increased. This indicates that aggregation is caused by oxidation, which is perhaps caused by the formation of intermolecular disulfide bonds.
Fig 2.
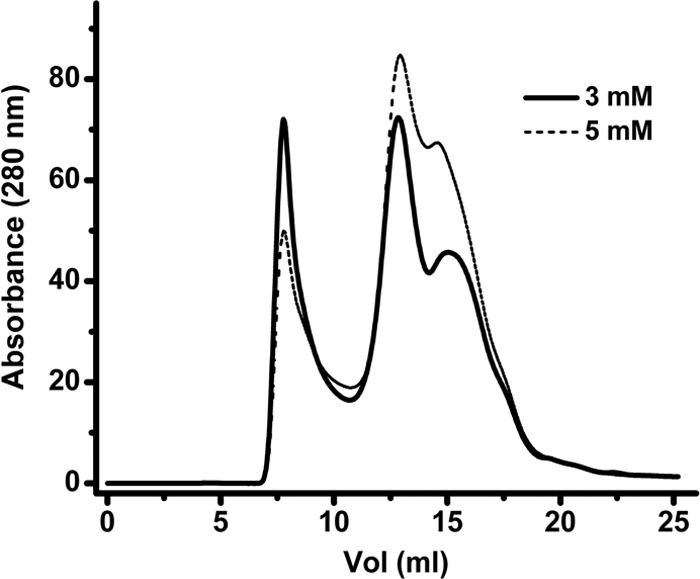
Effect of TCEP on aggregation of Rep68 wt. Rep68 wt (2 mg/ml) was injected with different concentrations of TCEP (3 and 5 mM). A Superose 6 10/300 GL column was used with a flow rate of 0.5 ml/min. Protein elution was followed by UV detection at 280 nm. The aggregation peak of the protein at an elution volume of ∼7 ml decreases as the TCEP concentration increases.
Mutation of Cys151 prevents Rep68 aggregation.
To identify cysteine resides with the potential to form intermolecular disulfide bonds, we analyzed the crystal structure of AAV2 Rep40 and an AAV2 OBD model based on the crystal structure of the AAV5 OBD AB (33). The structures show that of the six cysteine residues, four are buried or semiburied in the protein and only two, Cys151 and Cys405, are fully exposed to the solvent. Cys151 resides in the OBD and Cys405 is located in β hairpin 1 of the helicase domain. Interestingly, Cys405 is conserved in other AAV serotypes, while Cys151 is not. We thus proceeded to make single Cys mutants and an additional mutant in which both residues were replaced by serine/alanine. The mutant proteins were purified and analyzed on a Superose 6 column. Figure 3 shows that mutation of Cys151 to alanine or serine abolished the formation of aggregates at all concentrations tested. In contrast, the Cys405Ser mutant protein still showed significant formation of aggregate eluting at the void volume (Fig. 3B). As expected, the double mutant behaved like the single Cys151Ser mutant (Fig. 3C).
Fig 3.
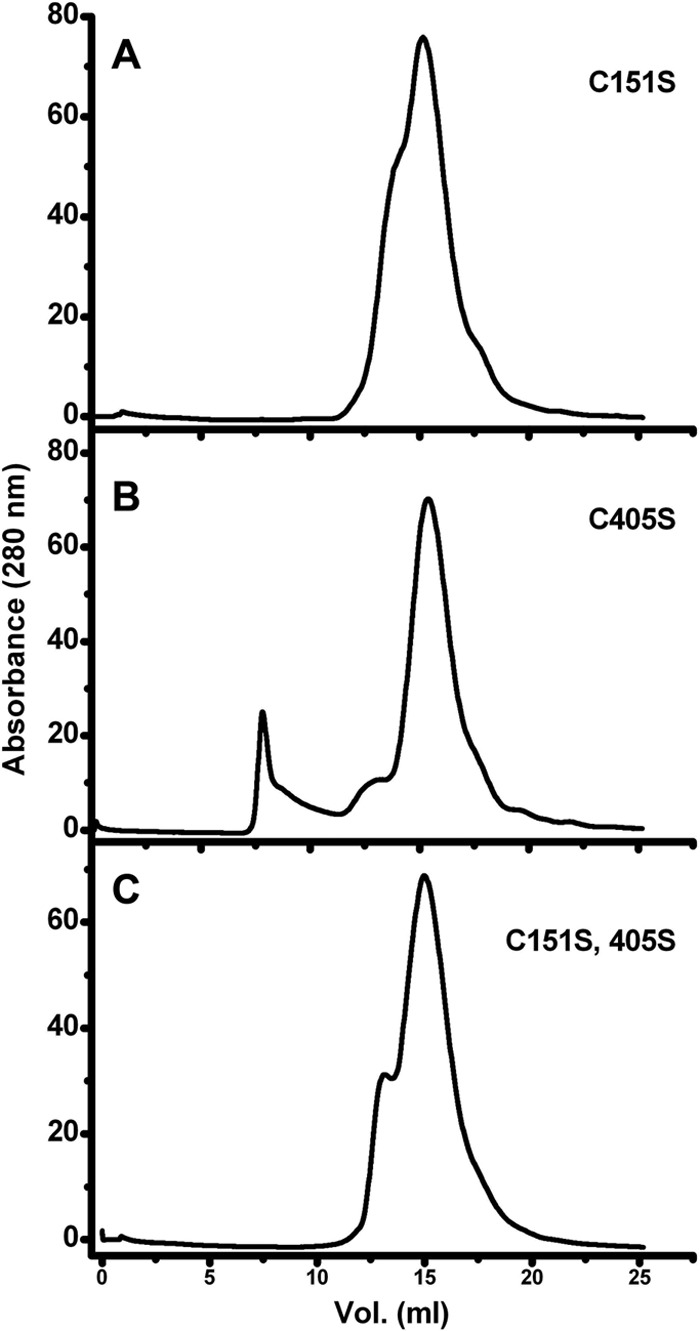
Effect of Cys mutations on Rep68 wt aggregation. Three Rep68 Cys mutants were evaluated by gel filtration chromatography: Rep68 C151S, Rep68 C405S, and a double Cys mutant, Rep68 C151A and C405S. A Superose 6 10/300 GL column was used with a flow rate of 0.5 ml/min. Protein elution was followed by UV detection at 280 nm. When the protein contains the Cys151S mutation (A), no aggregation was observed. In contrast, the C405S mutation still shows an aggregation peak eluting at the void volume of the column (B). (C) The double mutant behaves like the single C151S mutant. The concentration of the proteins was 32.65 μM (2 mg/ml).
The Rep68 C151S mutant is functionally equivalent to Rep68 wt.
In order to determine if the Cys151Ser mutation affects any of the biochemical activities of Rep68, we compared the biochemical activities of the C151S mutant with those of Rep68 wt. We performed ATPase (Fig. 4A), helicase (Fig. 4B), and DNA binding functional (Fig. 4C) assays in vitro. Results show that the C151S mutant protein performed as well as the wild-type protein in all these functional assays. In addition, we assessed the performance of the C151S mutant in a simple replication assay in HEK293T cells. Figure 4D shows comparable replication efficiencies by both the wt and mutant Rep68 proteins. To determine if the Rep C151S mutant was also able to support the production of infectious viral particles, we produced recombinant AAV2-GFP in the context of the mutant Rep protein and infected HeLa cells with increasing amounts of virus. Figure 4E shows that infectious rAAV2-GFP particles were efficiently formed in the presence of the C151S Rep, albeit at slightly lower titers than wt Rep. To investigate this small difference, we then purified both AAV2 wt and AAV2 containing the C151S mutation and assessed the fraction of empty particles in our preparations by transmission electron microscopy (Fig. 4F). The presence of the mutation in the purified viruses, confirmed by sequencing part of the viral DNA containing the mutation, did not affect the ratio of empty versus full particles.
Fig 4.

Functional comparison of Rep68 wt and the Rep68 C151S mutant. (A) ATPase activity showed no significant difference for the wt and C151S mutant proteins. (B) Helicase activity for WT and C151S proteins, using fluorescent stopped-flow kinetics). Helicase activities were followed using a 30 nM concentration. (C) Binding of DNA analysis of Rep C151S. A fluorescent probe (F-DNA-dsDNA) was used to monitor the binding of the protein. Binding was followed using 5 nM DNA and a range of protein concentrations from 50 to 2,000 nM. (D) Determination of viral replication in the presence of wt or C151S mutant Rep. The bar graph shows the quantification of three slot blot experiments. The signal obtained with the Rep-specific probe was normalized to that obtained with the ampicillin-specific probe, to normalize for input (transfected) DNA. The replication of the wt AAV2 genome in the presence of adenovirus was set as 100%. Data are presented as means ± SEMs. (E) Comparison of the production of rAAV2-GFP infectious particles in the presence of wt or C151S Rep. rAAV2-GFP particles were produced in HEK293T cells in the presence of adenovirus helper functions, AAV2 Cap, and wt or C151S Rep. Cells were harvested, lysed, freeze-thawed, and treated with the endonuclease Benzonase. Various volumes of crude lysate (in μl, x axis) were added to HeLa cells, and the percentage of GFP-positive infected cells was determined by FACS analysis. Data from four experiments are presented as means ± SEMs. (F) Visualization of wt AAV2 and AAV2-RepC151S viral particles by transmission electron microscopy at ×50,000 magnification. Empty viral particles appear as white rings. For each sample, 6 fields of approximately 200 particles were counted; the mean percentage of full particles and the standard deviation are indicated.
Rep68* forms multiple species in solution.
Similar to the results obtained for Rep68 wt (Fig. 1), Rep68* forms multiple oligomers at moderate protein concentrations. To further characterize the oligomeric behavior of Rep68*, an extensive sedimentation velocity study was undertaken at various Rep68* concentrations and in the presence and absence of either ATP or ADP. The results for apo-Rep68* show that the continuous sedimentation [c(S)] distributions change significantly as the total concentration of Rep68* is increased (Fig. 5A). At the lowest concentration (1 μM), two species are present, sedimenting at 4S and 13S. The number of species increases at higher concentrations, with as many as six species present at 18 μM and the appearance of a faster-sedimenting species at ∼27S. Figure 5B shows the effect of the Rep68* concentration on the weight (actually, the signal) average sedimentation coefficient (swt) for the overall reaction and the monomer-dimer equilibrium. These plots are considered a sensitive and powerful proof of species at equilibrium (34). The fact that swt increases significantly over the 18-fold concentration range indicates that dynamic protein-protein interactions are occurring. For the monomer-dimer reaction, swt was estimated by integrating over a somewhat variable range from 2.5S to 6.5S, which is necessary because at high Rep68* concentrations the larger particles (13S and 27S) interfere with the sedimentation of the monomer and dimer. The pattern exhibited is consistent with a strong monomer interaction that forms dimers (positive slope) and the well-known effect of an increasing concentration of a larger, more rapidly sedimenting species that interferes hydrodynamically. These results, although complex, are consistent with a kinetic process that seems to be near or at the boundary between a slow and an intermediate kinetic process (10−4 to 10−3/s and <10−5/s, respectively). This is illustrated in the behavior of the 4S peak present at low concentrations but at higher concentrations resolution into two peaks of monomer and dimers as a result of a better signal-to-noise ratio (Fig. 5A). In addition, calculation of the average molecular mass of the 4S species at the lowest concentration gives a molecular mass of ∼81 kDa, a value between that of a monomer and that of a dimer. Calculation of the molecular mass of the 13S species gives a range of values from 360 to 450 kDa at the lower concentrations. Together, these results indicate that in solution, Rep68 forms two major populations: a monomer-dimer population that is in slow equilibrium and sediments over the range of 3S to 5S and a 13S population consisting of a mixture of multiple oligomers of six or more Rep68* molecules. Interchange between the two populations occurs as the concentration increases and is mediated by an intermediate species that sediments with an s20,w value of between 5S and 9S, suggesting Rep68* trimers or tetramers. Sedimentation velocity data were also collected in the presence of both 1 mM ATP and ADP, and a representative set of these data at a 18 μM concentration is shown in Fig. 5C. These curves suggest that the presence of either ATP or ADP shifts the equilibrium in favor of higher-order oligomers. This effect can clearly be seen in Fig. 5D, which shows the amount of the 27S peak formed as a function of protein concentration in the presence or absence of nucleotides. The major difference with the apo-Rep68* is the formation of a species sedimenting at ∼27S, especially at the lowest concentrations. At the highest concentration (18 μM), two things are occurring: first, we have exceeded the Kd (dissociation constant) for the formation of the 27S species in the absence of nucleotides, and this explains the sudden increase in its formation after 10 μM. Second, the amount of the 27S peak is the same in the absence and presence of nucleotides because we have reached the maximum amount of 27S that can be formed at this concentration. To estimate a value for the dissociation constants of these oligomerizations, each peak was identified by comparing its estimated S value from the peak integrations with S values calculated by hydrodynamic modeling of the structure (see below). Table 1 shows the results of these dissociation constant calculations for the oligomerization steps K2→1, K4→2, K8→4, and K16→8, where the subscripts represent our best estimation as to the minimum oligomerization state described by the peak under investigation. The one peak that is difficult to assess is that assigned to the tetramer, since the signal from both the trimer and the hexamer might overlap the tetramer peak. The values in Table 1 show a degree of consistency both between values determined for the same step in the presence and absence of nucleotides and between those values calculated for what we presume are linear molecules with the same reactive interface (monomer-dimer and dimer-tetramer). There is a larger ∼0.1-fold change in K8→4 than in K4→2, which might indicate an entropic effect caused by ring closure. There seems to be a small decrease in the values of the dissociation constants when either ATP or ADP is added; although the effect is not large, the trend seems to be there. The 27S peak must represent a higher-order oligomer, and based on the electron microscopy results presented below, we tend to identify it as a stack set of rings consisting of two either seven- or eight-member rings or a mixture of both.
Fig 5.

Sedimentation velocity analysis of Rep68*. (A) Sedimentation profiles of Rep68* at five different concentrations obtained using the SEDFIT program. The corresponding values for molecular mass and s20,w are shown in Table 2. (B) Dependence of the weight average sedimentation coefficient on Rep68* loading concentration. ■, weight average sedimentation coefficient for the entire distribution; ▼, weight average for the dimer-monomer equilibrium. (C) Different concentrations of Rep68* (from 1 to 18 μM) were tested in sedimentation analysis assays in the absence or in the presence of nucleotides of ADP and ATP. A final 1 mM concentration of these nucleotides was used in every sedimentation velocity run. (D) Effect of ATP/ADP on the amount of 27S species formed. The area under the 27S peak was integrated at each concentration.
Table 1.
Estimates of dissociation constants for the oligomerization of Rep68* at various concentrations and in the presence of 1 mM ATP or ADPc
| Protein and concna (μM) |
Kd (μM) |
|||
|---|---|---|---|---|
| K2→1 | K4→2 | K8→4 | K16→8 | |
| Apo-Rep68* | ||||
| 1.0 | NDb | ND | ND | ND |
| 2.1 | 1.5 | 1.79 | 0.16 | ND |
| 4.8 | 3.26 | 1.71 | 0.15 | ND |
| 9.3 | 2.35 | 1.98 | 0.17 | 1.6 |
| 18.2 | 1.56 | 2.56 | 0.38 | 0.48 |
| Avg | 2.2 | 2.0 | 0.21 | 1.0 |
| Apo-Rep68* with ATP | ||||
| 0.7 | 4.9 | NDb | ND | ND |
| 1.8 | 2.0 | 0.43 | 0.04 | 0.16 |
| 4.2 | 0.75 | 0.9 | 0.13 | ND |
| 8.6 | 0.87 | 1.26 | 0.09 | 0.31 |
| 17 | 0.96 | 1.1 | 0.06 | 0.07 |
| Avg | 1.9 | 0.9 | 0.08 | 0.18 |
| Apo-Rep68* with ADP | ||||
| 0.9 | 0.9 | 1.1 | 0.02 | ND |
| 2.1 | 1.3 | 0.5 | 0.04 | 0.5 |
| 4.2 | 0.7 | 0.6 | 0.05 | 0.4 |
| 8.8 | 0.9 | ND | ND | 0.7 |
| 18.1 | 1.9 | 1.0 | 0.05 | 0.5 |
| Avg | 1.1 | 0.8 | 0.04 | 0.5 |
Total concentration of the Rep68* monomers in the cell obtained from the integration of the complete c(S) distribution from the interference data.
ND, not determined.
The ∼0.1-fold change in K8→4 relative to that for K4→2 might indicate a entropic effect caused by ring closure. Note also that there seems to be a general small decrease in the values of the dissociation constants when either ATP or ADP is added; although the effect is not large, the trend seems to be there.
TEM shows that the 13S population is a mixture of heptameric and octameric rings.
In order to determine the exact nature of the 13S species identified in the sedimentation velocity experiments, we carried out negative-stain TEM. We cross-linked all our samples using DMP to stabilize the protein complexes prior to purification on a Superose 6 gel filtration column. All samples were first analyzed by sedimentation velocity to confirm the formation of the 13S complexes. Figure 6A shows a representative electron microscopic image of a negatively stained Rep68*-13S peak where many ring-shaped particles can be seen. A reference-free 2D alignment without imposing symmetry was carried out, and Fig. 6B and C show a selection of several classes, clearly indicating the presence of seven- and eight-ring structures. The heptameric ring has an external diameter of 140.25 Å and an internal diameter of 63.11 Å; the octameric ring has an external diameter of 144 Å and an internal diameter of 67.65 Å. We also analyzed by TEM the effect of ATP on Rep68*. We observed the same seven- and eight-ring structures seen in apo-Rep68*, and in addition, we observed the formation of large filament-like structures. These higher-order structures may represent a series of stacked rings or a helical arrangement of Rep68 molecules, as has been observed in several other AAA+ proteins and RecA-like proteins (Fig. 6D and E) (35). In addition, we observed the same seven- and eight-ring structures seen in apo-Rep68*.
Fig 6.

Electron microscopic analysis of the Rep68 protein oligomers. (A) Visualization of cross-linked Rep68* particles by electron microscopy at ×50,000 magnification using the negative-stain technique. Image processing of the particles shows the presence of heptameric (B) and octameric (C) rings. Addition of ATP gives a similar mixture of heptamers and octamers (data not show) but also induces the formation of large filament-like structures (D). (E) Dimension details for these structures after the image processing.
Hydrodynamic modeling of Rep68 indicates the presence of multiple oligomers.
Taking advantage of the X-ray structures of the two functional domains of Rep68, we created several oligomeric models of Rep68 and calculated their hydrodynamic properties using the programs HYDRO and UltraScan-SOMO (36, 37). This approach allows us to verify the results from the sedimentation velocity and TEM experiments. First, in order to examine the limits of our Rep68 model, we generated two structures. The first model has the two domains joined by an extended interdomain linker; in the second model, the linker is folded up. The first structure resulted in a calculated sedimentation coefficient of 2.9S s20,w, while the second model predicted a sedimentation coefficient of 3.5S s20,w, which is closer to the experimental value. These results suggest that the interdomain linker is not in an extended conformation but probably folded, bringing the two domains close to each other. Using the second model, we generated models for dimers, trimers, and tetramers. At the same time, we created models for hexameric, heptameric, and octameric ring structures. The theoretical sedimentation velocity coefficients of all models were calculated using the program UltraScan-SOMO, as shown in Table 2. The calculated sedimentation coefficients from these models reproduce the experimental values obtained from the sedimentation velocity experiments remarkably well. Taken together, our modeling results support the notion that Rep68 is present as a mixture of monomer-dimer species at slow equilibrium at low concentrations. Increasing the concentration induces the formation of trimers and tetramers that slowly interconvert to several oligomeric rings, namely, heptamers and octamers (Fig. 7).
Table 2.
Comparison of experimental and calculated sedimentation coefficients (s20,w) for different Rep68 oligomers
| Oligomer | Mass (kDa) |
s20,w (S) |
||
|---|---|---|---|---|
| Sequence | Experimentala | Experimentala | Calculated from modelb | |
| Monomer | 60.8 | 61.4 | 3.7 | 3.4 |
| Dimer | 121.6 | 114 | 5.4 | 5.5 |
| Trimer | 182.4 | 215c | 8.7c | 7 |
| Tetramer | 243.2 | 8.3 | ||
| Hexamer | 364.8 | NDd | ND | 11.1 |
| Heptamer | 425.6 | 412c | 13.3c | 12.5 |
| Octamer | 486.4 | 13.6 | ||
Mass and sedimentation coefficients calculated from the corresponding peak position of the sedimentation profiles and f/f0 values determined with the SEDFIT program.
Sedimentation coefficients calculated from atomic models using the program UltraScan-SOMO.
Calculated from broad peaks that do resolve into two or more peaks with these data.
ND, not determined.
Fig 7.
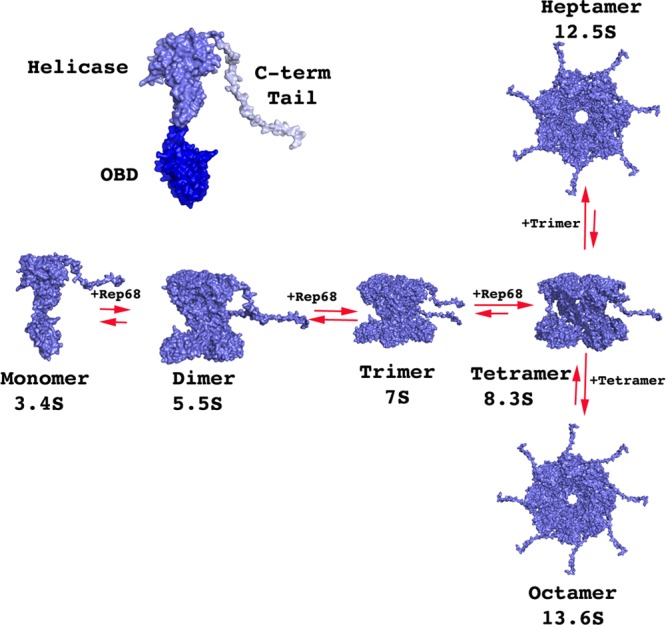
Hydrodynamic modeling of Rep68* oligomerization. A model of full-length Rep68 was generated from the structures of the origin binding domain (dark blue), Rep40 (helicase domain; slate blue), and a flexible C-terminal tail (light blue), as described in Materials and Methods. Models for the different oligomeric rings were generated using the SymmDock server (53). Sedimentation coefficients from each model were calculated using the program UltraScan-SOMO (37).
DISCUSSION
The experiments described here show that in vitro, Rep68 exists as a mixture of multiple species in equilibrium. At low concentrations Rep68 partitions into two populations. The first consist of monomers and dimers interchanging slowly and sedimenting as two clearly defined peaks at most concentrations. The second population is a mixture of heptameric and octameric rings that sediment at ∼13S. The fact that we can detect the individual peaks of the two populations suggests that they interconvert slower than the time scale of our sedimentation velocity experiments. At higher concentrations, the amount of the 13S population increases and we begin to detect the appearance of an intermediate peak at ∼7S. This peak likely represents a mixture of trimers and tetramers. Our data further suggest an oligomerization mechanism where Rep68 heptamers and octamers are assembled through the association of trimers and tetramers from a pool of monomers and dimers (Fig. 7). This process is reversible, as a purified heptamer-octamer mixture dissociates into monomers-dimers upon dilution (data not shown). Similarly, a report by Dignam et al., who used size-exclusion chromatography, showed the concentration-dependent self-association of Rep68 and its tendency to aggregate at moderate salt concentrations, although the nature of the different oligomers was not addressed (38). We have shown here that aggregation of Rep68 is induced by residue Cys151, which has a tendency to form disulfide bridges. In vivo, due to the highly reducing environment found in the nucleus, oxidation of cysteines does not occur. The formation of multiple oligomeric rings displayed by Rep68 is similar to the findings for some AAA+ proteins and other ring helicases. For instance, formation of heptamers has been described for the bacteriophage T7 gene 4 primase/helicase (39), the minichromosome maintenance (MCM) proteins from several species (40–42), the heat shock protein ClpB (43), Pseudomonas aeruginosa Hfq (44), ClpP (45), and Rad52 (46). In many cases, mixtures of heptameric and hexameric rings have been observed to be present at the same time (47). Our data suggest that Rep68 adopts a similar behavior, forming heptameric and octameric rings. The functional role of Rep68 ring structures, however, remains to be addressed, and it is pivotal to investigate it in the context of its interaction with different DNA substrates. In vitro studies of initiator proteins such as DnaA, SV40-LTag, and PV-E1 show that the formation of an initiation complex is a stepwise process that requires the binding of individual protein molecules to several direct repeats. This process is inhibited if the initiator protein is present as a stable ring structure because of the topological constraints of placing a closed ring around DNA. In many cases, this is what has been observed in vitro (13). In addition, formation of rings could provide a more stable structure, increasing protection against degradation. Several groups have postulated that the presence of heptameric rings provides a mechanism for loading the initiator protein onto DNA without the need for an additional loader protein. Thus, upon interaction with DNA, one subunit is lost with the resulting opening of the ring (42, 47). The presence of multiple Rep68 oligomers possibly provides a pool of different functional units that could be used for distinct reactions, such as initiation of DNA replication, recognition of the terminal resolution site (trs) hairpin for nicking, or the binding to the p19 promoter. In this regard, we have shown that Rep68 oligomerization is regulated by DNA structure and found that single-stranded DNA and helicase substrates support the formation of double-octameric Rep68 (19). Moreover, we determined that Rep68 is capable of unwinding a helicase substrate when purified as a double-octamer Rep68-helicase substrate complex. Other reports have found that Rep68 forms hexamers, when bound to dsDNA (22). Another level of complexity that could regulate the oligomerization properties of Rep proteins in vivo involves posttranslational modifications. Studies have determined that Rep proteins are highly modified, with residues being phosphorylated, ubiquitinated, and sumoylated. The last two modifications are used primarily to target the Rep proteins for proteosomal degradation (48–50). In contrast, phosphorylation inhibits several enzymatic properties of Rep proteins, such as DNA binding and helicase activity, and may regulate the assembly-disassembly of Rep proteins to DNA substrates (51, 52). The final answer to the functional oligomeric state of Rep68 will be to determine the structure of Rep68 bound to different DNA substrates, such as the AAV ori, AAV S1, and p19 promoter and to characterize the effect of phosphorylation on the formation of these complexes.
ACKNOWLEDGMENTS
We thank Rahul Jaiswal, Faik Musayeb, and Soumya G. Remesh for helpful discussions in the preparation of the manuscript. We thank Sergio B. Kaufman and Jeremias Incicco for the ATPase assay protocol.
This work was supported by NIH grant RO1-GM092854 (to C.R.E.) and United Kingdom Medical Research Council grant 1001764 (to R.M.L.).
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Hickman AB, Dyda F. 2005. Binding and unwinding: SF3 viral helicases. Curr. Opin. Struct. Biol. 15:77–85 [DOI] [PubMed] [Google Scholar]
- 2. Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76:23–50 [DOI] [PubMed] [Google Scholar]
- 3. Erzberger JP, Berger JM. 2006. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35:93–114 [DOI] [PubMed] [Google Scholar]
- 4. Ogura T, Wilkinson AJ. 2001. AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6:575–597 [DOI] [PubMed] [Google Scholar]
- 5. James JA, Escalante CR, Yoon-Robarts M, Edwards TA, Linden RM, Aggarwal AK. 2003. Crystal structure of the SF3 helicase from adeno-associated virus type 2. Structure 11:1025–1035 [DOI] [PubMed] [Google Scholar]
- 6. Owens RA, Weitzman MD, Kyostio SR, Carter BJ. 1993. Identification of a DNA-binding domain in the amino terminus of adeno-associated virus Rep proteins. J. Virol. 67:997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoon-Robarts M, Blouin AG, Bleker S, Kleinschmidt JA, Aggarwal AK, Escalante CR, Linden RM. 2004. Residues within the B′ motif are critical for DNA binding by the superfamily 3 helicase Rep40 of adeno-associated virus type 2. J. Biol. Chem. 279:50472–50481 [DOI] [PubMed] [Google Scholar]
- 8. Chiorini JA, Wiener SM, Owens RA, Kyostio SR, Kotin RM, Safer B. 1994. Sequence requirements for stable binding and function of Rep68 on the adeno-associated virus type 2 inverted terminal repeats. J. Virol. 68:7448–7457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiorini JA, Yang L, Safer B, Kotin RM. 1995. Determination of adeno-associated virus Rep68 and Rep78 binding sites by random sequence oligonucleotide selection. J. Virol. 69:7334–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Im DS, Muzyczka N. 1990. The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with DNA helicase activity. Cell 61:447–457 [DOI] [PubMed] [Google Scholar]
- 11. Kyostio SR, Wonderling RS, Owens RA. 1995. Negative regulation of the adeno-associated virus (AAV) P5 promoter involves both the P5 rep binding site and the consensus ATP-binding motif of the AAV Rep68 protein. J. Virol. 69:6787–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iyer LM, Koonin EV, Leipe DD, Aravind L. 2005. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 33:3875–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dean FB, Borowiec JA, Eki T, Hurwitz J. 1992. The simian virus 40 T antigen double hexamer assembles around the DNA at the replication origin. J. Biol. Chem. 267:14129–14137 [PubMed] [Google Scholar]
- 14. Sedman J, Stenlund A. 1998. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J. Virol. 72:6893–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wessel R, Schweizer J, Stahl H. 1992. Simian virus 40 T-antigen DNA helicase is a hexamer which forms a binary complex during bidirectional unwinding from the viral origin of DNA replication. J. Virol. 66:804–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berns KI. 1990. Parvovirus replication. Microbiol. Rev. 54:316–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou X, Zolotukhin I, Im DS, Muzyczka N. 1999. Biochemical characterization of adeno-associated virus rep68 DNA helicase and ATPase activities. J. Virol. 73:1580–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brister JR, Muzyczka N. 2000. Mechanism of Rep-mediated adeno-associated virus origin nicking. J. Virol. 74:7762–7771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mansilla-Soto J, Yoon-Robarts M, Rice WJ, Arya S, Escalante CR, Linden RM. 2009. DNA structure modulates the oligomerization properties of the AAV initiator protein Rep68. PLoS Pathog. 5:e1000513 doi:10.1371/journal.ppat.1000513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maggin JE, James JA, Chappie JS, Dyda F, Hickman AB. 2012. The amino acid linker between the endonuclease and helicase domains of adeno-associated virus type 5 Rep plays a critical role in DNA-dependent oligomerization. J. Virol. 86:3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarate-Perez F, Bardelli M, Burgner JW, II, Villamil-Jarauta M, Das K, Kekilli D, Mansilla-Soto J, Linden RM, Escalante CR. 2012. The interdomain linker of AAV-2 Rep68 is an integral part of its oligomerization domain: role of a conserved SF3 helicase residue in oligomerization. PLoS Pathog. 8:e1002764 doi:10.1371/journal.ppat.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith RH, Spano AJ, Kotin RM. 1997. The Rep78 gene product of adeno-associated virus (AAV) self-associates to form a hexameric complex in the presence of AAV ori sequences. J. Virol. 71:4461–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Packman LC, Perham RN. 1982. Quaternary structure of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus studied by a new reversible cross-linking procedure with bis(imidoesters). Biochemistry 21:5171–5175 [DOI] [PubMed] [Google Scholar]
- 24. Schuck P. 2003. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320:104–124 [DOI] [PubMed] [Google Scholar]
- 25. Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, Schuck P. 2004. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326:234–256 [DOI] [PubMed] [Google Scholar]
- 26. Bjornson KP, Amaratunga M, Moore KJ, Lohman TM. 1994. Single-turnover kinetics of helicase-catalyzed DNA unwinding monitored continuously by fluorescence energy transfer. Biochemistry 33:14306–14316 [DOI] [PubMed] [Google Scholar]
- 27. Baginski ES, Foa PP, Zak B. 1967. Microdetermination of inorganic phosphate, phospholipids, and total phosphate in biologic materials. Clin. Chem. 13:326–332 [PubMed] [Google Scholar]
- 28. Zeltner N, Kohlbrenner E, Clement N, Weber T, Linden RM. 2010. Near-perfect infectivity of wild-type AAV as benchmark for infectivity of recombinant AAV vectors. Gene Ther. 17:872–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29:291–325 [DOI] [PubMed] [Google Scholar]
- 30. Kim DE, Chivian D, Baker D. 2004. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32:W526–W531 doi:10.1093/nar/gkh468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brookes E, Demeler B, Rosano C, Rocco M. 2010. The implementation of SOMO (SOlution MOdeller) in the UltraScan analytical ultracentrifugation data analysis suite: enhanced capabilities allow the reliable hydrodynamic modeling of virtually any kind of biomacromolecule. Eur. Biophys. J. 39:423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hickman AB, Ronning DR, Kotin RM, Dyda F. 2002. Structural unity among viral origin binding proteins: crystal structure of the nuclease domain of adeno-associated virus Rep. Mol. Cell 10:327–337 [DOI] [PubMed] [Google Scholar]
- 34. Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78:1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mueller-Cajar O, Stotz M, Wendler P, Hartl FU, Bracher A, Hayer-Hartl M. 2011. Structure and function of the AAA+ protein CbbX, a red-type Rubisco activase. Nature 479:194–199 [DOI] [PubMed] [Google Scholar]
- 36. Garcia de la Torre J, Navarro S, Lopez Martinez MC, Diaz FG, Lopez Cascales JJ. 1994. HYDRO: a computer program for the prediction of hydrodynamic properties of macromolecules. Biophys. J. 67:530–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rai N, Nollmann M, Spotorno B, Tassara G, Byron O, Rocco M. 2005. SOMO (SOlution MOdeler) differences between X-ray- and NMR-derived bead models suggest a role for side chain flexibility in protein hydrodynamics. Structure 13:723–734 [DOI] [PubMed] [Google Scholar]
- 38. Dignam SS, Correia JJ, Nada SE, Trempe JP, Dignam JD. 2007. Activation of the ATPase activity of adeno-associated virus Rep68 and Rep78. Biochemistry 46:6364–6374 [DOI] [PubMed] [Google Scholar]
- 39. Crampton DJ, Ohi M, Qimron U, Walz T, Richardson CC. 2006. Oligomeric states of bacteriophage T7 gene 4 primase/helicase. J. Mol. Biol. 360:667–677 [DOI] [PubMed] [Google Scholar]
- 40. Costa A, Pape T, van Heel M, Brick P, Patwardhan A, Onesti S. 2006. Structural basis of the Methanothermobacter thermautotrophicus MCM helicase activity. Nucleic Acids Res. 34:5829–5838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pape T, Meka H, Chen S, Vicentini G, van Heel M, Onesti S. 2003. Hexameric ring structure of the full-length archaeal MCM protein complex. EMBO Rep. 4:1079–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu X, VanLoock MS, Poplawski A, Kelman Z, Xiang T, Tye BK, Egelman EH. 2002. The Methanobacterium thermoautotrophicum MCM protein can form heptameric rings. EMBO Rep. 3:792–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akoev V, Gogol EP, Barnett ME, Zolkiewski M. 2004. Nucleotide-induced switch in oligomerization of the AAA+ ATPase ClpB. Protein Sci. 13:567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nikulin A, Stolboushkina E, Perederina A, Vassilieva I, Blaesi U, Moll I, Kachalova G, Yokoyama S, Vassylyev D, Garber M, Nikonov S. 2005. Structure of Pseudomonas aeruginosa Hfq protein. Acta Crystallogr. D Biol. Crystallogr. 61:141–146 [DOI] [PubMed] [Google Scholar]
- 45. Thompson MW, Miller J, Maurizi MR, Kempner E. 1998. Importance of heptameric ring integrity for activity of Escherichia coli ClpP. Eur. J. Biochem. 258:923–928 [DOI] [PubMed] [Google Scholar]
- 46. Stasiak AZ, Larquet E, Stasiak A, Muller S, Engel A, Van Dyck E, West SC, Egelman EH. 2000. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 10:337–340 [DOI] [PubMed] [Google Scholar]
- 47. Crampton DJ, Mukherjee S, Richardson CC. 2006. DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell 21:165–174 [DOI] [PubMed] [Google Scholar]
- 48. Farris KD, Fasina O, Sukhu L, Li L, Pintel DJ. 2010. Adeno-associated virus small rep proteins are modified with at least two types of polyubiquitination. J. Virol. 84:1206–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sukhu L, Pintel D. 2011. The large Rep protein of adeno-associated virus type 2 is polyubiquitinated. J. Gen. Virol. 92:2792–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weger S, Hammer E, Heilbronn R. 2004. SUMO-1 modification regulates the protein stability of the large regulatory protein Rep78 of adeno associated virus type 2 (AAV-2). Virology 330:284–294 [DOI] [PubMed] [Google Scholar]
- 51. Collaco R, Prasad KM, Trempe JP. 1997. Phosphorylation of the adeno-associated virus replication proteins. Virology 232:332–336 [DOI] [PubMed] [Google Scholar]
- 52. Narasimhan D, Collaco R, Kalman-Maltese V, Trempe JP. 2002. Hyper-phosphorylation of the adeno-associated virus Rep78 protein inhibits terminal repeat binding and helicase activity. Biochim. Biophys. Acta 1576:298–305 [DOI] [PubMed] [Google Scholar]
- 53. Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. 2005. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33:W363–W367 doi:10.1093/nar/gki481 [DOI] [PMC free article] [PubMed] [Google Scholar]