

Abstract
Maraviroc (MVC) is a CCR5 antagonist that inhibits HIV-1 entry by binding to the coreceptor and inducing structural alterations in the extracellular loops. In this study, we isolated MVC-resistant variants from an HIV-1 primary isolate that arose after 21 weeks of tissue culture passage in the presence of inhibitor. gp120 sequences from passage control and MVC-resistant cultures were cloned into NL4-3 via yeast-based recombination followed by sequencing and drug susceptibility testing. Using 140 clones, three mutations were linked to MVC resistance, but none appeared in the V3 loop as was the case with previous HIV-1 strains resistant to CCR5 antagonists. Rather, resistance was dependent upon a single mutation in the C4 region of gp120. Chimeric clones bearing this N425K mutation replicated at high MVC concentrations and displayed significant shifts in 50% inhibitory concentrations (IC50s), characteristic of resistance to all other antiretroviral drugs but not typical of MVC resistance. Previous reports on MVC resistance describe an ability to use a drug-bound form of the receptor, leading to reduction in maximal drug inhibition. In contrast, our structural models on K425 gp120 suggest that this resistant mutation impacts CD4 interactions and highlights a novel pathway for MVC resistance.
INTRODUCTION
HIV-1 entry into host cells is a complex process characterized by three distinct stages: viral attachment to CD4, coreceptor (CCR5 or CXCR4) binding, and membrane fusion. The viral envelope spike is comprised of trimer of heterodimer subunits of extracellular glycoprotein (gp120) and transmembrane gp41 held together by noncovalent interactions and disulfide bridging (1). During entry, binding of gp120 to CD4 induces conformational changes in the envelope, exposing a coreceptor binding site. Models of these interactions suggest that multiple regions in gp120 are involved with coreceptor binding. The tip of the third variable loop (V3 loop) of gp120 interacts with the second extracellular loop of the coreceptor (ECL2), while the N terminus of the coreceptor interacts with the V3 loop stem, the bridging sheet (between the V1-V2 stem), and the fourth conserved region (C4) of gp120 (2, 3). Coreceptor engagement drives further conformational changes which result in insertion of gp41 fusion peptide (4), formation of the gp41 six-alpha-helix bundle (5), viral and host cell membrane fusion, and release of the viral RNA-containing core into the cell cytoplasm.
As the newest class of antiviral compounds targeting HIV-1 infection, small-molecule entry inhibitors represent a novel generation of drugs targeting a host cell protein rather than an enzymatic process unique to the virus. Although the development of entry inhibitors includes compounds targeting gp41 (T20) as well as gp120 (chemokine derivatives, monoclonal antibodies [MAbs], CD4-IgG2), the observation that naturally occurring polymorphisms in CCR5 can render homozygous individuals resistant to R5-tropic HIV-1 infection (6–8) inspired the development of small-molecule inhibitors of CCR5. CCR5 is the main coreceptor for HIV strains transmitted between individuals and that predominate in early infection. Thus, occluding gp120 engagement of CCR5 was an attractive target for drug development (9). Maraviroc (MVC) became the first, and so far only, FDA-approved small-molecule HIV inhibitor/CCR5 antagonist for use in HIV-infected patients. Other CCR5 antagonists, agonists, and binding antibodies reached various stages of preclinical and clinical development but were eventually abandoned due to off-target complications (10), poor pharmacodynamics and pharmacokinetics (11), and difficulties in screening appropriate patients for treatment due to FDA requirements to counterscreen for CXCR4-using HIV-1 (12, 13).
Maraviroc is an imidazopyridine that binds a hydrophobic transmembrane cavity of CCR5, altering the conformation of the extracellular loops of the receptor and disrupting chemokine binding as well as interactions with the gp120 envelope glycoprotein (14, 15). Vicriviroc (VCV), AD101, TAK-779, and aplaviroc (APL) are additional small-molecule CCR5 inhibitors that bind a transmembrane region similar to that bound by maraviroc and likewise induce altered receptor conformations (15). HIV-1 resistance to such inhibitors is likely to entail unique escape mechanisms given that a host receptor, not a viral enzyme, is the drug target. Potential pathways of resistance to these inhibitors include coreceptor switching to CXCR4-using viruses (16), increased affinity and binding to CD4 and/or CCR5 (17, 18), use of inhibitor-bound conformations of CCR5 (19, 20), and increased kinetics of membrane fusion (21). Although outgrowth of CXCR4-using virus remains a concern for the therapeutic administration of CCR5 antagonists and is why patients are screened for X4-tropic virus prior to starting a maraviroc regimen, de novo mutations altering coreceptor tropism do not appear to be the preferential pathway for resistance (22, 23). Rather, resistant viruses emerging from in vivo and in vitro mutational pathways have been characterized as utilizing an inhibitor-bound conformation of CCR5 for entry (19, 20, 24).
Resistance to MVC and a variety of other small-molecule CCR5 inhibitors has been generated in vitro by passage of inhibitor-sensitive viral isolates in sequential dose escalations of drug (20, 23–25). Resistance is typically characterized as a reduction in the maximal percent inhibition (MPI) indicating usage of an inhibitor-bound conformation of CCR5 for entry. Although resistance is associated with a variety of amino acid changes observed in both gp120 and gp41, changes in the V3 loop have been identified as major contributors to the phenotype of resistance to nearly all CCR5 agonists and antagonists (26). To date, no signature pattern of mutations has been identified as predictive of CCR5 antagonist resistance. Of greater significance, very few specific mutations have been observed more than once in MVC-resistant strains, suggesting that each diverse HIV-1 env gene may provide a different genetic pathway for developing resistance to coreceptor inhibitors.
Here, we report the isolation of MVC-resistant variants from a subtype A HIV-1 primary isolate which developed resistance over 21 weeks of tissue culture passage in the presence of dose escalations of inhibitor. Resistance in this virus appeared to map to a mutation in the C4 region of gp120. Drug sensitivity assays of gp120 chimeric virus clones derived from the resistant virus did not display reductions in MPI but, rather, showed increases in 50% inhibitory concentrations (IC50s) up to 40-fold compared to the parental virus. These pronounced shifts in IC50 as well as the location of the primary resistance mutation in structural modeling predictions suggest a novel MVC resistance mechanism related to altered CD4 binding.
MATERIALS AND METHODS
Cells, viruses, and inhibitors.
U87.CD4.CCR5 cells were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (27). U87.CD4.CCR5 cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 μg/ml penicillin-streptomycin, 300 μg/ml G418, and 1 μg/ml puromycin (Invitrogen, Carlsbad, CA). 293T cells were cultured in DMEM supplemented with 10% FBS and 100 μg/ml penicillin-streptomycin. Peripheral blood mononuclear cells (PBMCs) were obtained from whole blood extracted from an HIV-negative donor by Ficoll-Paque centrifugation. PBMC cultures were stimulated with 1 μg/ml phytohemagglutinin (PHA; Gibco, Carlsbad, CA) for 48 h in RPMI 1640 medium (Gibco) containing 10% FBS and 100 μg/ml penicillin-streptomycin. Cells were maintained thereafter in RPMI 1640 medium with 10% FBS, 100 μg/ml penicillin-streptomycin, and 10,000 U/ml interleukin-2 (IL-2; Gibco).
HIV-1 primary isolates (B6)91US715 and (C8)96USNG58 were obtained from the AIDS Research and Reference Reagent Program, while virus A74 was isolated from a Ugandan patient through the Center for AIDS Research core facility in Kampala, Uganda. Viral swarms were propagated in U87.CD4.CCR5 cells, and titers were determined by the Reed-Muench endpoint titration method (28).
Maraviroc (MVC) and TAK-779 were diluted in phosphate-buffered saline (PBS), while enfuvirtide (T20) was diluted in ethanol.
Resistant virus isolation.
Three viruses (A74, B6, and C8) were propagated on U87.CD4.CCR5 cells in a 6-well plate format (1.5 × 105 cells/well) using an initiating inoculum of virus (0.001 infectious unit/cell) in either the presence or absence of MVC. The inhibitor concentration was increased when viral replication was detected using a radiolabeled reverse transcriptase (RT) assay as previously described (29). Equivalent volumes of supernatant (50 to 100 μl) were passaged onto fresh U87.CD4.CCR5 cells either with or without inhibitor each week. Weekly supernatants were collected and frozen.
Multiple-replication-cycle drug susceptibility assays.
U87.CD4.CCR5 cells were plated in a 96-well format (1 × 104 cells/well) for 24 h. Cells were then treated for 2 h with 5-fold dilutions of MVC (100 μM to 2 × 10−6 μM) and then infected at a multiplicity of infection (MOI) of 0.001 infectious unit/cell. All assays were performed in triplicate. After 24 h, cells were washed in 1× PBS to remove free virus, and medium containing the appropriate MVC dilutions was added. Viral replication was measured daily by radiolabeled reverse transcriptase assay using a Packard beta counter to quantify counts per minute. Drug sensitivity curves were generated using nonlinear regression curve fitting features of GraphPad Prism 5 software.
Construction of gp120 chimeric viruses.
gp120 chimeric viruses were cloned into an NL4-3 backbone from passage control and virus passaged in the presence of MVC escalations using the yeast homologous recombination-gap repair technique (30). From this point forward, the viruses and clones are referred to as PC.21.1 or MVC.21.1, where PC stands for “passage control,” MVC stands for “maraviroc selected,” 21 is the number of passages, and 1 is the clone number. The gp120 regions of the PC.1 (starting virus), PC.21, and MVC.21 viruses were PCR amplified using primers F_gp120 (5′-GACAGGTTAATTGATAGACTA-3′) and B_gp120 (5′-CTTCCTGCTGCTCCCAAGAAC-3′). Briefly, these gp120 PCR products were then cotransformed with SacII-linearized pREC_NFL_Δgp120/URA3 into Saccharomyces cerevisiae MYA-906 cells (ATCC). Following homologous recombination, plasmids were extracted from the yeast cells and transformed into electrocompetent Escherichia coli Stbl4 cells (Invitrogen). Individual bacterial colonies were grown, and plasmids were extracted using Qiagen miniprep kits. Chimeric pREC_NFL_gp120 plasmids were cotransfected into 293T cells (3 × 104 cells/well) along with the complementing plasmid pCMV_cplt using Fugene 6 reagent (Promega, Madison, WI) as described previously (30). Heterodiploid virus particles containing one copy of the NFL and cplt HIV-1 RNAs represent approximately half of the virus derived from 293T cell transfections and can be propagated on U87.CD4.CCR5 cells to produce replication-competent virus with a complete virus genome. Titers of virus stocks of each clone were determined as described above and in reference 28.
Oligonucleotide ligation assay (OLA).
The gp120 regions of the bulk passage control (PC.21) and resistant virus (MVC.21) were RT-PCR amplified using envend (5′-CTTTTTGACCACTTGCCACCCAT-3′) for reverse transcription and F_gp120/B_gp120 for subsequent PCR amplification. Oligonucleotides were designed for ligase discrimination reactions to bind upstream or downstream of the mutation sites observed in MVC.21 for envelope codons 117, 396, and 425. For each mutation site, we employed two upstream interrogator oligonucleotides to discriminate between and quantify the sequences RCGA and QCAA at codon 117, sequences LTTA, GGGA, VGTA at codon 396, and sequences NAAT and KAAA at codon 425. A complete description of this ligase discrimination assay has been published previously (31). The oligonucleotides designed specifically for this study are listed in Fig. S1 in the supplemental material.
Site-directed mutagenesis PCR method.
The K425 mutation was introduced into the MVC.21.122 gp120 clone by a nested PCR amplification method. Two PCR fragments were generated from the pREC_NFL_gp120/MVC.21.122 plasmid using primers F_gp120 with B_K425 (5′-CTACTCTTTGCCACATCTTTATAATTTGCTTTATTCTGCATGGAAGA-3′) and F_K425 (5′-TCTTCCATGCAGAATAAAGCAAATTATAAAGATGTGGCAAAGAGTAG-3′) with B_gp120. Underlined nucleotides in B_K425 and F_K425 primer sequences introduce the lysine residue at codon 425. Using products from these PCR amplifications as templates, a nested PCR was performed with primers F_gp120 and B_gp120 to generate the full gp120 region, which was subsequently cloned into pREC_NFL_Δgp120/URA3 by yeast homologous recombination as described above. Individual colonies were sequenced, and a single clone containing the K425 mutation was identified and named pREC_NFL_gp120/MVC.21.122.425K. Replication-competent virus was generated from this plasmid by cotransfection in 293T cells and propagation in U87.CD4.CCR5 cells as described above.
In vitro fitness assays.
The replicative fitness of PC.21 and MVC.21 gp120 chimeric viruses was assessed in head-to-head competition experiments in U87.CD4.CCR5 cells as described in reference 29. Cells were plated in a 48-well format (2 × 105 cells/well) and infected in triplicate with viruses at an MOI of 0.0001 infectious unit/ml. Infected cells were collected 5 days postinfection, and genomic DNA was isolated using a Qiagen kit. The gp120 region was amplified from cellular DNA using F_gp120/B_gp120 as described above. Amplicons were probed for sequence identity at gp120 residues 117 and 425 using OLA as described previously (31, 32). Frequency of wild-type or mutant residue identity versus total signal was used to determine viral fitness. The fitness difference (WD) was calculated as the fitness of the resistant virus divided by the fitness of the sensitive virus.
Structural modeling.
The structural model of the N425K mutant was generated from the crystal structure of gp120 in complex with CD4 and a tyrosine-sulfated antibody, 412d (Protein Data Bank [PDB] ID: 2QAD [3]). The mutation of N425 to K425 was made in the model building program COOT (33), and the local structure around the mutation was regularized using the same program. By a slight torsion of the K425 side chain, two close contacts can be made from the Nε of K425 with residues from CD4, including a cation-π interaction with F43 of CD4.
Nucleotide sequence accession numbers.
The DNA sequences for env (gp120) obtained in the course of this work were deposited in the GenBank/EMBL/DDBJ databases under the following accession numbers: JX993942 to JX993981.
RESULTS
Maraviroc-resistant mutant generated in cell culture.
The concentration of entry inhibitor required to inhibit viral replication by 50% (IC50) can vary up to 1,000-fold for HIV-1 primary isolates that have never been exposed to these drugs (34). In the present study, we assessed “intrinsic” susceptibilities of HIV-1 subtype A, B, and C primary isolates to maraviroc. Multiple-cycle drug susceptibility assays were performed in U87.CD4.CCR5 cells in the presence of 5-fold-decreasing concentrations of MVC starting at 100 μM. The subtype A primary isolate (A74) was less sensitive to MVC inhibition (IC50 = 10 nM) than the subtype B (B6) and C (C8) viruses, which had similar sensitivities (mean IC50 = 2 nM) (Fig. 1A).
Fig 1.

Maraviroc sensitivity of primary HIV-1 isolates. (A) Viruses representing HIV-1 subtypes A, B, and C were used to infect U87.CD4.CCR5 cells in the presence of increasing concentrations of MVC as described in Materials and Methods. Inhibition curves were generated using GraphPad Prism, version 5. The data shown are means of triplicates, with error bars representing standard deviations. (B) Viruses A74, B6, and C8 were passaged in U87.CD4.CCR5 cells weekly in the presence of increasing concentrations of MVC to select for an MVC escape mutant. Cultures were monitored for reverse transcriptase activity as described in Materials and Methods. Cultures for virus C8 were abandoned at week 16 (*) when no RT activity was measured.
Given the differential sensitivities to MVC, these three primary isolates were passaged weekly in the presence of increasing MVC concentrations in U87 human glioma cells expressing CD4 and CCR5 to generate a maraviroc escape mutant (Fig. 1B). As a control, viruses were also passaged in the absence of inhibitor to differentiate changes that occurred as a result of drug pressure versus selection during long-term culture. Reverse transcriptase (RT) activity was monitored in the cell-free supernatant for each weekly passage, and inhibitor concentration increased when RT activity reached 2-fold over background. Virus C8 cultures were abandoned after failure to produce robust RT activity in weeks 15 and 16. Although virus B6 cultures continued to produce RT activity through week 21, viruses collected from this passage remained sensitive to MVC, with IC50s for control and inhibitor-treated cultures of 8 nM and 14 nM, respectively (Table 1). It is important to note that only one other study has selected for MVC resistance in vitro using primary HIV-1 isolates, and in this case, only 2 of 6 viruses challenged with drug developed resistance (20). With other classes of antiretroviral drugs, there are hundreds of published reports on the in vitro selection of drug resistance by multiple research groups. However, in vitro patterns of resistance to CCR5 antagonists with overlapping CCR5 binding sites is limited to just seven published studies, yet each study describes a different mutational pathway to resistance (20, 23–25, 35–37).
Table 1.
Summary of MVC sensitivities in U87.CD4.CCR5 cells and PBMCs
| Virus | U87.CD4.CCR5 cells |
PBMCs |
||
|---|---|---|---|---|
| IC50 (nM) | MPI (%) | IC50 (nM) | MPI (%) | |
| A74 | 10 | 99 | 5 | 100 |
| PC.21 | 1 | 100 | 6 | 98 |
| MVC.21 | 216 | 81 | 300 | 73 |
| B6.PC.21 | 8 | 100 | NDa | ND |
| B6.MVC.21 | 14 | 99 | ND | ND |
ND, not determined.
Drug susceptibility analyses of virus A74 at week 21 (MVC.21) in U87.CD4.CCR5 cells indicated that this virus was able to escape inhibition by MVC at concentrations >1,000-fold higher than the IC50 for the input virus (Fig. 2A). At the highest drug concentration tested (100 μM), MVC.21 was inhibited by 81%, indicating incomplete suppression of virus entry despite saturating levels of inhibitor. In addition to this reduction in the maximal percent inhibition (MPI), a shift in IC50 to 216 nM was also observed for MVC.21, representing a 20-fold level of resistance compared to that of the inoculating virus. In contrast, the passage control virus (PC.21) demonstrated a slight hypersensitivity to MVC, with an IC50 of 1 nM. While the use of cells lacking CXCR4 made a switch in coreceptor usage unlikely, tropism testing performed in U87.CD4.CXCR4 cells confirmed that no dualtropic or X4-tropic viruses arose during the 21 passages (data not shown).
Fig 2.

MVC resistance and cross-resistance to TAK-779 after prolonged culture with MVC inhibitor. (A) Virus A74 derived from passage control week 21 (PC.21) and MVC-treated week 21 (MVC.21) cultures were used to infect U87.CD4.CCR5 cells in the presence of increasing concentrations of MVC as described in Materials and Methods. Inhibition curves were generated in GraphPad Prism software, version 5. (B) The same viruses were also used to infect U87.CD4.CCR5 cells in the presence of maximal inhibitory concentrations (10 μM) of CCR5 antagonists MVC and TAK-779 and fusion inhibitor enfuvirtide (T20). Reverse transcriptase activity was measured 7 days postinfection. Percent infection was calculated relative to the no-drug control for each virus. Data shown are means of triplicates, with error bars representing standard deviations.
The U87 human glioma cell line is stably transfected to express high levels of CD4 and CCR5 which do not accurately reflect levels on natural HIV-1 targets, e.g., CD4+ T cells and macrophages. Therefore, multiple-cycle drug sensitivity assays were performed in peripheral blood mononuclear cells (PBMCs) isolated from an HIV-negative donor with PC.21 and MVC.21. The results confirmed those observed in the U87.CD4.CCR5 cells, with a reduction in MPI of MVC.21 to 73% and an IC50 of 300 nM, compared to 5 nM for PC.21 (Table 1).
MVC.21 cross resistant to another CCR5 antagonist.
MVC.21 showed evidence of cross-resistance to another CCR5 antagonist, TAK-779, as indicated by replication in U87.CD4.CCR5 cells treated with a 99% inhibitory concentration (IC99) (10 μM) that completely blocked PC.21 virus replication (Fig. 2B). TAK-779 (as well as VCV and APL) has been shown to bind a CCR5 transmembrane region similar to that bound by maraviroc and likewise alter coreceptor conformation (15). Cross-resistance to TAK-779 would suggest a mechanism of resistance that permits this virus to overcome inhibition by multiple inhibitors in this class. In contrast, 10 μM enfuvirtide (IC99) mediated similar levels of MVC.21 and PC.21 inhibition in U87.CD4.CCR5 cells. As a fusion inhibitor, enfuvirtide, or T20, inhibits entry by targeting formation of the six-helix gp41 bundle and subsequent membrane fusion (38). Lack of enfuvirtide resistance suggests that MVC resistance in MVC.21 is mediated by a mechanism that precedes membrane fusion and likely relates to receptor interactions.
Multiple mutations throughout gp120 in resistant virus.
Population sequencing of the envelope region of the PC.21 and MVC.21 viruses identified several amino acid substitutions related to MVC selection in the gp120 region but none in gp41. To identify specific mutations and linkage in gp120 related to MVC resistance, the gp120 region of the input (PC.1), passage control (PC.21), and MVC-treated virus from passages 7, 14, and 21 were PCR amplified from viral cDNA and cloned into the pREC_NFL_Δgp120/URA3 construct by yeast homologous recombination-gap repair (Fig. 3A) (30).
Fig 3.

Generation and characterization of gp120 chimeric virus. (A) Viral RNA was isolated from the indicated passage control and MVC-treated culture supernatants, and the gp120 regions were amplified after reverse transcriptase and nested PCR. gp120 regions were recombined into pREC_NFL_Δgp120/URA3 vector using yeast homologous recombination as described in Materials and Methods. Individual bacterial colonies were selected as indicated in the table. (B) Full gp120 regions of clones from PC.1, PC.21, and MVC.21 were sequenced. Nine mutations were identified in MVC.21 clones and are indicated based on HXB2 reference virus numbering. Rows refer to individual gp120 clones, while columns refer to specific gp120 mutations. A black box indicates an average wild-type residue at that gp120 site, whereas a red box indicates that a mutation is present. The asterisks refer to clones harboring only mutations selected primarily in the MVC passage. Numbers of clones harboring mutations at specific gp120 sites are indicated below columns.
Approximately 10 clones from each passage control (PC.1, PC week 7 [PC.7], PC.14, and PC.21) and 25 clones from each MVC-treated passage (MVC week 7 [MVC.7], MVC.14, and MVC.21) were sequenced to identify mutations (>140 clones) to determine the linked amino acids selected under MVC pressure. Nine gp120 amino acid substitutions were more prevalent in the MVC.21 clones than in the PC.1 and PC.21 clones (Fig. 3B). Figure 3B employs a color coding system where a black box at a specific position indicates the predominate average amino acid in the inoculum virus (PC.1), while a red box represents the predominate average sequence in the MVC-treated virus (MVC.21). A total of 23 unique mutational patterns were identified based on this average sequence analyses. Some “mutant” amino acids (red box in Fig. 3B) were observed in PC.1 and PC.21 clones and, likewise, some wild-type amino acids in the MVC.21 clones. Overall, the linkage pattern of these mutations in the MVC.21 population was complex, with only 6 of 23 clones (Fig. 3B) having all nine mutations. Only one of nine clones in the input, or PC.1, virus had wild-type amino acids at all nine positions.
The frequencies of the wild-type versus mutant residues at these individual mutation sites were determined using the total number of sequenced clones. Three mutations (R117Q, L/G396V, and N425K) were found at high frequency in the MVC.21 clones but at low frequency in either the input or PC.21 clones (Fig. 4A, C, and H, respectively), suggesting their role in MVC resistance. The V3 mutation Q315R was present at the same frequency in the PC.21 clones as in the MVC.21 clones (Fig. 4G), suggesting that this mutation emerged with normal adaptation to U87.CD4.CCR5 tissue culture and not selected under drug pressure. The remaining mutations E33bG, KG138-139ΔΔ, Q290K, and D461E appeared at high frequency in the MVC.21 clones but were also found at frequencies of >25% in the input population (Fig. 4E, F, B, and D, respectively). In regard to linkage, the R117Q, L396V and N425K mutations were found together in 22 of 23 MVC.21 clones but never linked in the PC.1 or PC.21 virus. The N425K mutation was found in all but one of the 23 MVC.21 clones (except MVC.21.122) but was absent from all 17 of the PC.1 or PC.21 clones. The N425K mutation was not found in any of the 22 MVC.7 clones but was present in 8 of 13 MVC.14 clones, suggesting that this mutation arose between passages 7 and 14 (Fig. 5).
Fig 4.

Frequency of gp120 mutations in PC.21- and MVC.21-derived clones. The frequencies of wild-type and mutant amino acids are displayed and represent those individual mutations in the clones derived from PC.1, PC.21, and MVC.21 cultures (A to H). Frequencies were calculated based on number of clones with either wild-type or mutant residues versus the total number of clones sequenced for that population as shown in Fig. 3B. The center panel, containing a schematic of the HIV-1 envelope gene, maps the location of each mutation in the gp120 conserved (C1 to C5) and variable (V1 to V5) regions. Wild-type sequences are shown in black, with mutations shown in red.
Fig 5.

Change of gp120 mutation frequency during passage control and MVC selection. Using the clonal sequences (n = 79) from the inoculum and MVC-treated passages 7, 14, and 21, the frequencies of N425 and K425 were determined.
To confirm the frequency of the putative MVC-resistant mutations in the viral populations, an oligonucleotide ligation assay (OLA) (39, 40) was performed using interrogator oligonucleotides specific to the mutations R117Q, L/G396V, and N425K on gp120 PCR products from cDNA of PC.21 and MVC.21 virus (Table 2). In this study, we could only predict the probability of linkage. Using this quantitative OLA, we found that the Q117 and V396 sequences were detected at frequencies of 0.94 and 0.97 in the MCV.21 virus population but at frequencies of 0.55 and 0.32 in the PC.21 virus population, respectively (Table 2). Based on the very low frequency of K425 in the PC.1 and PC.21 populations (∼7%) (near the limit of detection at 1%), it appears that the N425K mutation either arose as a de novo mutation in the MVC.21 population or was selected for from a very small fraction of the input virus.
Table 2.
Change of gp120 mutation frequency in bulk virus population during passage control and MVC selection
| Viral population | Amino acid frequency |
||||||
|---|---|---|---|---|---|---|---|
| 117 |
396 |
425 |
|||||
| R | Q | L | G | V | N | K | |
| PC.1 | 0.56 | 0.43 | 0.40 | 0.33 | 0.25 | 0.93 | 0.07 |
| PC.21 | 0.45 | 0.55 | 0.41 | 0.27 | 0.32 | 0.93 | 0.07 |
| MVC.21 | 0.06 | 0.94 | 0.02 | 0.01 | 0.97 | 0.08 | 0.92 |
Sensitivity of selected A74 HIV-1 clones to MVC inhibition.
Based on our cloning strategy, we have a diverse array of gp120 clones with different mutational patterns that represent nearly all combinations of single and multiple substitutions associated with MVC resistance (namely, R117Q, Q290K, L/G396V, D461E, E33bG, N425K, and the deletion of K138/G139). This limits the need for site-directed mutagenesis on virus to assess the contribution of each mutation (alone or in combination) to relative MVC resistance. Seven gp120 clones were selected to produce virus from 293T transfections (see Materials and Methods) (Fig. 6A), which were then tested for MVC sensitivity in U87.CD4.CCR5 cells (Fig. 6B to D). Although MVC.21.119 and MVC.21.132 share the same mutation profile for the nine MVC-associated amino acid sites in gp120, they do vary slightly in residues at other gp120 sites and are therefore not identical clones (see Fig. S2 in the supplemental material). The N33c, K187, G335, E337, K344, E351, I377, and A462 mutations found in MVC.21.119 and not in MVC.21.132 were also identified at high frequency in the passage control. Although these sites were not selected for further studies, it is quite possible that these mutations were selected, for example, based on adaptation to tissue culture conditions and, coincidently, also contributed to MVC resistance.
Fig 6.
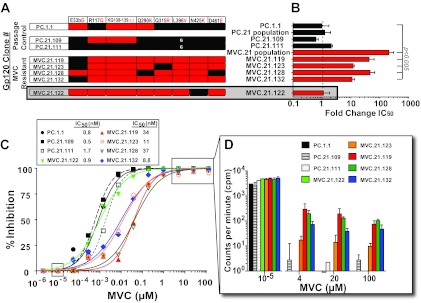
Sensitivity of gp120 chimeric virus to maraviroc inhibition. (A) Individual gp120 clones from PC.21 and MVC.21 were selected based on differential mutation patterns for MVC susceptibility testing in U87.CD4.CCR5 cells. Black indicates a wild-type residue, while red indicates a mutation at that gp120 residue. IC50 fold changes relative to input gp120 clone PC.1.1 are shown in panel B. Data represent means of triplicate wells, with standard deviations. (C) Drug sensitivity curves for chimeric gp120 viruses were performed in U87.CD4.CCR5 cells in triplicate as described in Materials and Methods. The IC50s are shown on the right of panel C, whereas the fold change derived from these IC50s are shown in panel B. Inhibition curves were generated using GraphPad Prism software, version 5. (D) Reverse transcriptase activities, measured as counts per minute for MVC concentrations of 1 × 10−5, 4, 20, and 100 μM, from which the curves in panel C were derived, are shown. Data represent triplicates, with standard deviations shown.
Clones from the input virus (PC.1.1) and week 21 passage control (PC.21.109 and PC.21.111) were sensitive to MVC inhibition, with similar IC50s (Fig. 6B and C). However, clonal viruses derived from MVC.21 varied greatly in IC50s. MVC.21.132 had a moderate (10-fold) shift in IC50, to 8.8 nM, while MVC.21.119 and MVC.21.128 had pronounced (40-fold) shifts in IC50, to 34 and 37 nM, respectively (Fig. 6B and C). Importantly, MVC.21.123 retained the wild-type V3 mutation Q315 yet still demonstrated a moderate (11-fold) IC50 shift and was able to replicate at high concentrations (Fig. 6D), suggesting that the Q315R mutation is not associated with resistance in MVC.21. All four of these clones (119, 123, 128, and 132) had the triad of R117Q-, L396V-, and N425K-linked mutations. Clone MVC.21.122 had an IC50 of 0.9 nM, similar to that observed for the PC.21 clones, despite having the R117Q and L396V mutations.
Despite an apparent “lack” of MPI effect based on the drug susceptibility curves presented in Fig. 6C, viral replication was detected on radiograms (see Fig. S2 in the supplemental material). The counts per minute were quantified by beta counter (see Materials and Methods) for clones 119, 123, 128, and 132 at the highest concentrations of MVC (4, 20, and 100 μM) and at 1 × 10−5 μM MVC, which did not result in significant MVC inhibition (Fig. 6C). At the highest MVC concentrations, the level of RT activity for the MVC-sensitive clone PC.21.109 was just above background (<10 cpm), representing 99.9% inhibition (Fig. 6C and D). With the MVC-resistant clone MVC.21.119 at these MVC concentrations, the level of RT was 100 to 300 cpm or >100-fold above background, representing 97% inhibition. These differences for the MVC-resistant clones were highly significant (P < 0.001) and represented a low but consistent MPI effect.
Relative fitness of MVC-resistant and -sensitive HIV-1 clones.
The R117Q, Q290K, L/G396V, and D461E mutations have a higher frequency (5 to 30%) in the HIV-1 subtype A population in the human epidemic, whereas the lysine at 425 was found in less than 2% of HIV-1 subtype A envelope sequences (Table 3) (Los Alamos HIV Sequence Database [http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html]). The prevalence of N425K was even lower in other HIV-1 subtypes (B, C, and D), with an occurrence of <0.2% (Table 3). These findings suggest that R117Q, Q290K, L/G396V, D461E, and E33bG have a lower genetic barrier for mutation and less impact on replicative fitness than the N425K mutation. However, we have previously shown that env mutations conferring resistance to entry inhibitors may not always confer a replicative fitness cost. Their low frequency in the HIV-1 population (i.e., low population fitness) may be attributable to other factors such as cellular tropism and sensitivity to neutralizing antibodies or cell-mediated cytotoxic T cell killing. We performed pairwise competitions between three MVC-sensitive clones (PC.1.1, PC.21.109, and MVC.21.122) and three MVC-resistant clones (MVC.21.119, MVC.21.128, and MVC.21.132) in U87.CD4.CCR5 cells (Fig. 7). It is important to note that even though MVC.21.122 was isolated from the MVC selection, this clone remained MVC sensitive (Fig. 6B to D). Interestingly, all of the MVC-resistant clones were actually more fit than MVC sensitive clones PC.1.1 and PC.21.109. The clone (MVC.21.132) with the lowest level of MVC resistance (10-fold) was also the least fit of the MVC-resistant clones, losing the competition against PC.21.109. In contrast, the MVC-sensitive clone MVC.21.122 (lacking N425K) could compete against the MVC-resistant clones, suggesting that the mutations (R117Q, Q290K, L/G396V, D461E, and E33bG) may actually confer fitness increases and compensate for the loss of fitness conferred by N425K.
Table 3.
Summary of Los Alamos HIV Database gp120 sequence identity at positions 117, 396, and 425
| HIV lineage | Total no. of sequences | Amino acid frequency (%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 117 |
396 |
425 |
||||||||||
| R | Q | K | L | G | V | H | K | N | R | Other | ||
| Group M | 61,096 | 0.2 | 1.7 | 97.1 | 0.01 | 0.5 | 99.2 | 0.3 | 0.1 | 97.1 | 2.0 | 0.5 |
| Subtype A | 1,065 | 0.2 | 6.3 | 86.7 | 0.1 | 0.1 | 99.8 | 7.8 | 2.0 | 77.0 | 12.3 | 0.9 |
| Subtype B | 42,585 | 0.26 | 1.0 | 98.1 | 0.1 | 4.0 | 86.1 | 0.2 | 0.01 | 99.1 | 0.3 | 0.3 |
| Subtype C | 11,451 | 0.3 | 0.45 | 97.6 | 0.0 | 1.0 | 77.5 | 0.1 | 0.1 | 98.5 | 0.9 | 0.4 |
| Subtype D | 1,450 | 0.2 | 5.8 | 91.8 | 0.0 | 0.1 | 99.9 | 0.6 | 0.2 | 98.3 | 0.2 | 0.6 |
Fig 7.

Replicative fitness of HIV-1 gp120 chimeric virus clones derived from MVC selection and passage control experiments. The infectious titers of three MVC-resistant clones (MVC.21.119, MVC.21.128, and MVC.21.132) and three MVC-sensitive clones (MVC.21.122, PC.21.109, and PC.1.1) were measured using a standard 50% tissue culture infective dose assay (45). A pairwise competition was performed, using competition between the MVC-resistant and -sensitive gp120 clones in U87.CD4.CCR5 cells using equal MOIs of the viruses (0.0001 infectious unit/ml). Infected cells were harvested at peak viremia (day 5), and the relative frequency of each virus in the competition was measured using OLA to distinguish and quantify the amount of one virus versus the other (as described in reference 21). The fitness difference (or ratio of the relative fitness values) was calculated as described, and the results are presented. Shaded bars indicate that the resistant clone won the competition, whereas the absence of shading indicates that the resistant clone lost the competition.
K425 is the primary resistance mutation.
MVC.21.122 was the only clone from the MVC-resistant population that lacked the N425K mutation in the C4 region of gp120 and the only clone not to exhibit an MVC resistance phenotype (Fig. 6B and C). This observation indicated that the K425 mutation was likely essential for resistance in MVC.21. Site-directed mutagenesis was performed to introduce the K425 mutation into the MVC.21.122 clone and to confirm the role of this mutation in MVC resistance. Using the drug inhibition assays described above, we observed a dramatic decrease in susceptibility of MVC.21.122.425K to maraviroc, i.e., a 36-fold shift in IC50 from 0.9 nM to 33 nM (Fig. 8A), as well as replication of MVC.21.122.K425 at 100 μM MVC (Fig. 8B).
Fig 8.

Effect of the N425K mutation on MVC resistance. (A) Drug sensitivity assays were performed for clone MVC.21.122 and clone MVC.21.122.425K, in which the K425 mutation was introduced by site-directed mutagenesis as described in Materials and Methods. U87.CD4.CCR5 cells were infected in triplicate, with standard deviations shown. Inhibition curves were generated using GraphPad Prism software, version 5. (B) Reverse transcriptase activities, measured as counts per minute for MVC concentrations of 1 × 10−5, 4, 20, and 100 μM, from which the curves in panel A were derived, are shown. Data represent triplicate experiments, and means and standard deviations are shown. Reverse transcriptase activity was detected at 100 μM MVC for MVC.21.122.425K.
Shift in IC50 versus the MPI effect denotes MVC resistance.
As described above, resistance to CCR5 antagonist is generally related to continued virus replication at even the highest achievable drug concentrations in culture. For example, the CC1/85 MVC-resistant virus containing the T316 and V323 V3 loop mutations is capable of replicating at a 25% level in the presence of 1 μM MVC compared to the absence of drug; however, no shift in IC50 is apparent (20). When changes in IC50s have been associated with CCR5 resistance, they have been in the context of reduced maximal inhibition, which can be modulated by cell receptor density (41). With our MVC-resistant virus, there is a mixed resistant phenotype. By simply plotting the percent relative inhibition versus drug concentration, the shift in IC50 is quite clear and similar to the increases in IC50 characteristic of resistance to all other antiretroviral drugs aside from CCR5 antagonists. In addition to this drug resistance phenotype, the MVC-resistant clones bearing the K425 mutation showed low but significant levels of virus replication at even the highest drug concentrations (Fig. 6D; see also Fig. S2 in the supplemental material). However, this MPI effect was much less pronounced than with other MVC-resistant viruses, as previously reported (37). Despite MPI values of >95% for MVC.21 clones 119, 123, 128, and 132, virus replication was detectable even with 100 μM MVC, whereas no replication was observed with the MVC-sensitive clones 1, 109, and 122 (Fig. 6D; see also Fig. S2).
Model of K425 in gp120 structure suggests a role in CD4 binding affinity.
The N425 residue is located in the β20 sheet of the gp120 bridging sheet, as illustrated in Fig. 9 using the crystal structure of HIV-1 gp120Yu-2 complexed with CD4 and a tyrosine-sulfated 412d antibody (3). These four anti-parallel beta sheets of gp120 comprise the complete CD4 and partial coreceptor binding sites. This 425 position as either an asparagine or lysine is found distal from the gp120 region thought to interact with the N terminus of CCR5. However, N425 is specifically located within a cavity known to interact with phenylalanine 43, found within the D1 domain of CD4. N425 gp120 and F43 CD4 are not predicted to form direct interactions. However, when K425 is modeled into the structure using the COOT modeling program, the side chain of K425 is predicted to form a hydrogen bond with the oxygen of S42 CD4 as well as generate a new cation-π interaction between the aromatic ring of F43 CD4 and the Nε side chain of K425. Since F43 of CD4 is known to be a critical residue for gp120 binding (42–44), our model would suggest that enhanced interactions with CD4 may play a role in the resistance mechanism of A74.MVC.21 virus.
Fig 9.

Modeling of K425 in gp120 HIV-1YU-2 virus structure suggests a role in CD4 binding affinity. The structure of gp120 HIV-1YU-2 complexed with CD4 and 412 antibody (not shown) was used to model the N425-to-K mutation. The N425 residue lines a cavity in gp120 into which F43 of CD4 projects (inset). Mutation of N425 to K is predicted to form new interactions with F43 (PDB: 2QAD [3]).
DISCUSSION
HIV-1 enters a host cell by engaging two receptors, CD4 and one of two seven-transmembrane G-coupled protein receptors, CCR5 or CXCR4. With a few exceptions, nearly all new HIV-1 infections are established with a CCR5-using virus, while the CXCR4-using virus emerges only in late disease in approximately 50% of infected individuals. The clear predominance of R5 HIV-1 during asymptomatic disease led to the rapid preclinical development of several CCR5 agonists and antagonists. The CCR5 agonists, such as AOP-RANTES or PSC-RANTES, displayed higher per-mole potency than the small-molecule CCR5 antagonists but also resulted in receptor downregulation, some aberrant signal transduction, and increased potential for inflammatory responses (45–50). Of the various CCR5 antagonists, only three reached advanced stages of clinical trials and only maraviroc was approved first for salvage-based treatment and then for first-line treatment in combination with nucleoside analogs (51, 52). For the most part, virologic failures with MVC-based treatment regimens have been poorly characterized and generally complicated by the preexistence of multidrug-resistant genotypes in these heavily treated patients (16, 53, 54). When MVC is used as a salvage-based treatment regimen, resistance commonly relates to the rapid selection of preexisting X4-using HIV-1 in the intrapatient HIV-1 population (16). These CXCR4-using clones can be detected, even at low frequencies, by phenotypic assays (55). Consequently, MVC resistance via altered CCR5 binding or increased receptor affinity is rarely observed in MVC treatment failures (19, 20). However, in the absence of X4-using virus prior to treatment, altered CCR5 usage may be a more common pathway for MVC resistance than a de novo emergence of X4-using virus, i.e., mutations resulting in higher genetic or fitness barriers.
In addition to inefficient screening for X4-using viruses, there are several problems in measuring MVC resistance during treatment. First, nearly 30% of the MVC treatment failures (without resistance to the other drugs in the regimen) did not show characteristic resistance to MVC (53, 56). Second, current phenotypic assays using single-cycle entry may be skewed for the detection of the MPI effect (16, 20). Third, a different evolutionary pathway in env was associated with every MVC-resistant virus that retains CCR5 binding (19, 20, 37). Fourth, it is commonly assumed that these MVC-resistant viruses can utilize drug-bound forms of the receptor based on incomplete inhibition even at the highest MVC concentrations (20). However, there are other studies to suggest that increased kinetics of host cells could lead to resistance to CCR5 agonists and antagonists (25, 39, 45, 57). Two independent studies have shown that mutations in the gp41 domain may enhance virus-host membrane fusion, the last step in host cell entry (39, 58). In these cases, rapid formation of the six-alpha-helix bundle may help the transition from the rate-limiting step of Env engagement with CCR5, with or without the inhibitor. Additionally, improved CD4 binding affinity has been associated with resistance to CCR5 antagonists in laboratory-adapted viruses (17, 59). In these studies, deletion of portions of the V3 loop region resulted in virus with enhanced CD4 binding and the ability to utilize CCR5 antagonist-bound CCR5. Collectively, these observations, caveats, and exceptions with HIV-1 resistance to CCR5 antagonist suggest that divergent evolution within drug-sensitive env genes does not necessarily result in convergence to a specific resistance mechanism.
Our in vitro selection experiments for MVC resistance started with three primary HIV-1 isolates, but only a subtype A virus (A74) developed MVC resistance over a 6-month period. This MVC-resistant virus retains CCR5 binding and does not switch coreceptor usage. Approximately 6 million individuals are infected worldwide with HIV subtype A as a pure clade or in a recombinant form (60). Unlike in previous studies (19, 20, 37), a V3 loop mutation is not associated with resistance and our MVC-resistant virus displays only a weak MPI effect. Instead, resistance is described as a >20-fold shift in the concentrations required to inhibit 50% of the resistant versus wild-type virus. This increase in IC50s is typical for HIV-1 resistance to nearly all antiretroviral drugs but is rarely observed with HIV-1 resistant to CCR5 antagonists. As in previous studies, our MVC-resistant A74 virus had mutations scattered throughout the gp120 coding region that appeared to be selected due to drug pressure. However, most of these mutations had minimal effect on the resistance phenotype but may stabilize the N425K mutation found in the C4 domain. This observation was clearly demonstrated with the MVC-sensitive MVC.21.122 clone, which harbored the R117Q, Q290K, L396V, and D461E mutations but lacked the N425K mutation. When the N425K mutation was introduced into the MVC.21.122 clone, this virus became highly resistant to MVC (>36-fold increase in IC50s). It is important to note that the level of MVC resistance and the MPI effect of the virus population appear greater than the sum of MVC resistance and the MPI effects for each of the selected clones. This is partly related to disproportional testing of clones with unique mutational patterns rather than repeat testing of the dominant clones with the same mutational pattern. In addition, there is no conclusive evidence that the level of “drug resistance” in the HIV-1 population always reflects the cumulative drug resistance of the individual clones. A complex interplay and competition between clones within the virus population may affect the outcome of drug susceptibility assays.
As described above, most diverse HIV-1 isolates appear to follow different evolutionary pathways to MVC resistance, but all have been linked to emergence of a V3 loop mutation. These V3 loop mutations are often found at high frequency in the untreated HIV-1 populations, suggesting higher entropy, low cost on replicative fitness, and a lower genetic barrier to resistance. We have previously shown that env mutations conferring resistance to entry inhibitors were often associated with increased replicative fitness, i.e., directly related to enhanced host cell entry efficiency (39, 45, 57). This is in sharp contrast to decreased replicative fitness observed with acquired resistance to nearly all other antiretroviral drugs. However, drug resistance mutations conferring lower fitness costs are often found at higher frequencies in the untreated HIV-1 population or within the intrapatient population. For example, there is a low genetic and fitness barrier to nevirapine (NVP) resistance via the K103N mutation (61, 62), which reflects (i) the slow reversion of this mutation following cessation of NVP treatment (63–65) and (ii) increasing circulation and transmission of the K103N virus within the human population where NVP-based treatment regimens are most prevalent (66). In this study, we have shown that the collection of E33bG, R117Q, Q290K, L/G396V, N425K, and D461E selected under MVC pressure resulted in virus of higher replicative fitness than the wild-type passage control virus. With the exception of the N425K mutation, these mutations are found in relatively high proportions of HIV-1 among the subtype A population and within the group M HIV-1 population in general. Thus, these findings suggest a low genetic barrier for these mutations and limited fitness cost. We did not observe a wild-type virus containing all of these natural polymorphisms. Thus, the emergence of all mutations may follow a complex fitness landscape which, in this case, is directed by MVC selective pressure and emergence of resistance. In contrast to the relatively high frequency of these polymorphisms, the N425K mutation is found in <1% of all HIV-1 sequences in the Los Alamos Database (n = 61,096) and has never been associated with resistance to any entry inhibitor. Interestingly, the unique MVC-sensitive clone which emerged under MVC selective pressure, i.e., MVC.21.122, had a higher replicative fitness than the highly MVC-resistant viruses MVC.21.119 and MVC.21.128. All three of the viruses contained the R117Q mutation, deletion of residues 138 and 139, and the L/G396V mutation, but the more fit MVC.21.122 lacked the primary drug resistance mutation, N425K. These findings suggest that N425K mutation may confer a fitness cost which is compensated by the linked R117Q and L/G396V mutations.
It is important to stress that resistance to entry inhibitors based on Env mutations is less predictable in terms of replicative fitness costs. Nearly all mutations for resistance to the RT inhibitors, protease inhibitors, and integrase inhibitors confer a fitness cost (67). In contrast, higher replicative fitness is often related to resistance to CCR5 antagonists and agonists even when the drug is absent (39, 45, 57). This increased replicative fitness of the virus resistant to the CCR5 agonist or antagonist is commonly related to enhanced CCR5 binding affinity (with or without bound inhibitor) and faster CCR5 binding kinetics, which is the rate-limiting step in the host cell entry process. We are now attempting to introduce the N425K mutation into the env gene of other subtypes, such as subtype B, but have failed to obtain an infectious virus. It is quite likely that the compensatory mutations, E33bG, R117Q, Q290K, L/G396V, and D461E, are required to stabilize N425K. However, these compensatory mutations may be subtype A specific, and another set of compensatory mutations may be required. As part of more detailed studies on the N425K mutation and its interaction with CD4, we are currently exploring the direct impact of this mutation on replicative fitness, entry efficiency, CD4 affinity, and other parameters.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant AI49170. All virus work was performed in the biosafety level 2 and 3 facilities of the Case Western Reserve/University Hospitals Center for AIDS Research (AI36219).
Footnotes
Published ahead of print 7 November 2012
Supplemental material for this article can be found at http://dx.doi.org/10.1128/JVI.01863-12.
REFERENCES
- 1. Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Da LT, Wu YD. 2011. Theoretical studies on the interactions and interferences of HIV-1 glycoprotein gp120 and its coreceptor CCR5. J. Chem. Inf. Model. 51:359–369 [DOI] [PubMed] [Google Scholar]
- 3. Huang CC, Lam SN, Acharya P, Tang M, Xiang SH, Hussan SS, Stanfield RL, Robinson J, Sodroski J, Wilson IA, Wyatt R, Bewley CA, Kwong PD. 2007. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317:1930–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gallaher WR. 1987. Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell 50:327–328 [DOI] [PubMed] [Google Scholar]
- 5. Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430 [DOI] [PubMed] [Google Scholar]
- 6. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86:367–377 [DOI] [PubMed] [Google Scholar]
- 7. Paxton WA, Kang S, Koup RA. 1998. The HIV type 1 coreceptor CCR5 and its role in viral transmission and disease progression. AIDS Res. Hum. Retroviruses 14(Suppl 1):S89–S92 [PubMed] [Google Scholar]
- 8. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. 1996. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382:722–725 [DOI] [PubMed] [Google Scholar]
- 9. Berger EA, Murphy PM, Farber JM. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17:657–700 [DOI] [PubMed] [Google Scholar]
- 10. Nichols WG, Steel HM, Bonny T, Adkison K, Curtis L, Millard J, Kabeya K, Clumeck N. 2008. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob. Agents Chemother. 52:858–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Esté JA. 2002. Sch-351125 and Sch-350634. Schering-Plough. Curr. Opin. Investig. Drugs 3:379–383 [PubMed] [Google Scholar]
- 12. Abbate I, Rozera G, Tommasi C, Bruselles A, Bartolini B, Chillemi G, Nicastri E, Narciso P, Ippolito G, Capobianchi MR. 2011. Analysis of coreceptor usage of circulating viral and proviral HIV genome quasispecies by ultra-deep pyrosequencing in patients who are candidates for CCR5 antagonist treatment. Clin. Microbiol. Infect. 17:725–731 [DOI] [PubMed] [Google Scholar]
- 13. Poveda E, Alcami J, Paredes R, Cordoba J, Gutierrez F, Llibre JM, Delgado R, Pulido F, Iribarren JA, Garcia DM, Hernandez QJ, Moreno S, Garcia F. 2010. Genotypic determination of HIV tropism—clinical and methodological recommendations to guide the therapeutic use of CCR5 antagonists. AIDS Rev. 12:135–148 [PubMed] [Google Scholar]
- 14. Dragic T, Trkola A, Thompson DA, Cormier EG, Kajumo FA, Maxwell E, Lin SW, Ying W, Smith SO, Sakmar TP, Moore JP. 2000. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. U. S. A. 97:5639–5644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M. 2008. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 73:789–800 [DOI] [PubMed] [Google Scholar]
- 16. Westby M, Lewis M, Whitcomb J, Youle M, Pozniak AL, James IT, Jenkins TM, Perros M, van der Ryst E. 2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 80:4909–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agrawal-Gamse C, Lee FH, Haggarty B, Jordan AP, Yi Y, Lee B, Collman RG, Hoxie JA, Doms RW, Laakso MM. 2009. Adaptive mutations in a human immunodeficiency virus type 1 envelope protein with a truncated V3 loop restore function by improving interactions with CD4. J. Virol. 83:11005–11015 doi:10.1128/JVI.01238-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pfaff JM, Wilen CB, Harrison JE, Demarest JF, Lee B, Doms RW, Tilton JC. 2010. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. J. Virol. 84:6505–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tilton JC, Wilen CB, Didigu CA, Sinha R, Harrison JE, Agrawal-Gamse C, Henning EA, Bushman FD, Martin JN, Deeks SG, Doms RW. 2010. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 84:10863–10876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81:2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reeves JD, Gallo SA, Ahmad N, Miamidian JL, Harvey PE, Sharron M, Pohlmann S, Sfakianos JN, Derdeyn CA, Blumenthal R, Hunter E, Doms RW. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 99:16249–16254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pastore C, Ramos A, Mosier DE. 2004. Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J. Virol. 78:7565–7574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trkola A, Kuhmann SE, Strizki JM, Maxwell E, Ketas T, Morgan T, Pugach P, Xu S, Wojcik L, Tagat J, Palani A, Shapiro S, Clader JW, McCombie S, Reyes GR, Baroudy BM, Moore JP. 2002. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. U. S. A. 99:395–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pugach P, Marozsan AJ, Ketas TJ, Landes EL, Moore JP, Kuhmann SE. 2007. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology 361:212–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marozsan AJ, Kuhmann SE, Morgan T, Herrera C, Rivera-Troche E, Xu S, Baroudy BM, Strizki J, Moore JP. 2005. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D). Virology 338:182–199 [DOI] [PubMed] [Google Scholar]
- 26. Lobritz MA, Ratcliff AN, Arts EJ. 2010. HIV-1 entry, inhibitors, and resistance. Viruses 2:1069–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Björndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman DR, Fenyo EM. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71:7478–7487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marozsan AJ, Fraundorf E, Abraha A, Baird H, Moore D, Troyer R, Nankja I, Arts EJ. 2004. Relationships between infectious titer, capsid protein levels, and reverse transcriptase activities of diverse human immunodeficiency virus type 1 isolates. J. Virol. 78:11130–11141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ball SC, Abraha A, Collins KR, Marozsan AJ, Baird H, Quinones-Mateu ME, Penn-Nicholson A, Murray M, Richard N, Lobritz M, Zimmerman PA, Kawamura T, Blauvelt A, Arts EJ. 2003. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J. Virol. 77:1021–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dudley DM, Gao Y, Nelson KN, Henry KR, Nankya I, Gibson RM, Arts EJ. 2009. A novel yeast-based recombination method to clone and propagate diverse HIV-1 isolates. Biotechniques 46:458–467 [DOI] [PubMed] [Google Scholar]
- 31. Tebit DM, Lobritz M, Lalonde M, Immonen T, Singh K, Sarafianos S, Herchenroder O, Krausslich HG, Arts EJ. 2010. Divergent evolution in reverse transcriptase (RT) of HIV-1 group O and M lineages: impact on structure, fitness, and sensitivity to nonnucleoside RT inhibitors. J. Virol. 84:9817–9830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lalonde MS, Troyer RM, Syed AR, Bulime S, Demers K, Bajunirwe F, Arts EJ. 2007. Sensitive oligonucleotide ligation assay for low-level detection of nevirapine resistance mutations in human immunodeficiency virus type 1 quasispecies. J. Clin. Microbiol. 45:2604–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Labrosse B, Labernardiere JL, Dam E, Trouplin V, Skrabal K, Clavel F, Mammano F. 2003. Baseline susceptibility of primary human immunodeficiency virus type 1 to entry inhibitors. J. Virol. 77:1610–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baba M, Miyake H, Wang X, Okamoto M, Takashima K. 2007. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob. Agents Chemother. 51:707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ogert RA, Wojcik L, Buontempo C, Ba L, Buontempo P, Ralston R, Strizki J, Howe JA. 2008. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology 373:387–399 [DOI] [PubMed] [Google Scholar]
- 37. Yuan Y, Maeda Y, Terasawa H, Monde K, Harada S, Yusa K. 2011. A combination of polymorphic mutations in V3 loop of HIV-1 gp120 can confer noncompetitive resistance to maraviroc. Virology 413:293–299 [DOI] [PubMed] [Google Scholar]
- 38. Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML. 1995. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. J. Virol. 69:3771–3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dudley DM, Wentzel JL, Lalonde MS, Veazey RS, Arts EJ. 2009. Selection of a simian-human immunodeficiency virus strain resistant to a vaginal microbicide in macaques. J. Virol. 83:5067–5076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lalonde MS, Arts EJ. 2010. DNA suspension arrays: silencing discrete artifacts for high-sensitivity applications. PLoS One 5:e15476 doi:10.1371/journal.pone.0015476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pugach P, Ray N, Klasse PJ, Ketas TJ, Michael E, Doms RW, Lee B, Moore JP. 2009. Inefficient entry of vicriviroc-resistant HIV-1 via the inhibitor-CCR5 complex at low cell surface CCR5 densities. Virology 387:296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ashkenazi A, Presta LG, Marsters SA, Camerato TR, Rosenthal KA, Fendly BM, Capon DJ. 1990. Mapping the CD4 binding site for human immunodeficiency virus by alanine-scanning mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 87:7150–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moebius U, Clayton LK, Abraham S, Harrison SC, Reinherz EL. 1992. The human immunodeficiency virus gp120 binding site on CD4: delineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resolution CD4 atomic structure. J. Exp. Med. 176:507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lobritz MA, Marozsan AJ, Troyer RM, Arts EJ. 2007. Natural variation in the V3 crown of human immunodeficiency virus type 1 affects replicative fitness and entry inhibitor sensitivity. J. Virol. 81:8258–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marozsan AJ, Torre VS, Johnson M, Ball SC, Cross JV, Templeton DJ, Quinones-Mateu ME, Offord RE, Arts EJ. 2001. Mechanisms involved in stimulation of human immunodeficiency virus type 1 replication by aminooxypentane RANTES. J. Virol. 75:8624–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pastore C, Picchio GR, Galimi F, Fish R, Hartley O, Offord RE, Mosier DE. 2003. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother. 47:509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodríguez-Frade JM, Vila-Coro AJ, Martin A, Nieto M, Sanchez-Madrid F, Proudfoot AE, Wells TN, Martinez A, Mellado M. 1999. Similarities and differences in RANTES- and (AOP)-RANTES-triggered signals: implications for chemotaxis. J. Cell Biol. 144:755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, Buser R, Wells TN, Proudfoot AE. 1997. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 276:276–279 [DOI] [PubMed] [Google Scholar]
- 50. Torre VS, Marozsan AJ, Albright JL, Collins KR, Hartley O, Offord RE, Quinones-Mateu ME, Arts EJ. 2000. Variable sensitivity of CCR5-tropic human immunodeficiency virus type 1 isolates to inhibition by RANTES analogs. J. Virol. 74:4868–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sierra-Madero J, Di PG, Wood R, Saag M, Frank I, Craig C, Burnside R, McCracken J, Pontani D, Goodrich J, Heera J, Mayer H. 2010. Efficacy and safety of maraviroc versus efavirenz, both with zidovudine/lamivudine: 96-week results from the MERIT study. HIV Clin. Trials 11:125–132 [DOI] [PubMed] [Google Scholar]
- 53. Hardy WD, Gulick RM, Mayer H, Fätkenheuer G, Nelson M, Heera J, Rajicic N, Goodrich J. 2010. Two-year safety and virologic efficacy of maraviroc in treatment-experienced patients with CCR5-tropic HIV-1 infection: 96-week combined analysis of MOTIVATE 1 and 2. J. Acquir. Immune. Defic. Syndr. 55:558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Swenson LC, Mo T, Dong WW, Zhong X, Woods CK, Thielen A, Jensen MA, Knapp DJ, Chapman D, Portsmouth S, Lewis M, James I, Heera J, Valdez H, Harrigan PR. 2011. Deep V3 sequencing for HIV type 1 tropism in treatment-naive patients: a reanalysis of the MERIT trial of maraviroc. Clin. Infect. Dis. 53:732–742 [DOI] [PubMed] [Google Scholar]
- 55. Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, Chappey C, Kiss LD, Paxinos EE, Petropoulos CJ. 2007. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob. Agents Chemother. 51:566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fätkenheuer G, Nelson M, Lazzarin A, Konourina I, Hoepelman AI, Lampiris H, Hirschel B, Tebas P, Raffi F, Trottier B, Bellos N, Saag M, Cooper DA, Westby M, Tawadrous M, Sullivan JF, Ridgway C, Dunne MW, Felstead S, Mayer H, van der Ryst E. 2008. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 359:1442–1455 [DOI] [PubMed] [Google Scholar]
- 57. Anastassopoulou CG, Marozsan AJ, Matet A, Snyder AD, Arts EJ, Kuhmann SE, Moore JP. 2007. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss. PLoS Pathog. 3:e79 doi:10.1371/journal.ppat.0030079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Anastassopoulou CG, Ketas TJ, Depetris RS, Thomas AM, Klasse PJ, Moore JP. 2011. Resistance of a human immunodeficiency virus type 1 isolate to a small molecule CCR5 inhibitor can involve sequence changes in both gp120 and gp41. Virology 413:47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Laakso MM, Lee FH, Haggarty B, Agrawal C, Nolan KM, Biscone M, Romano J, Jordan AP, Leslie GJ, Meissner EG, Su L, Hoxie JA, Doms RW. 2007. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 3:e117 doi:10.1371/journal.ppat.0030117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tebit DM, Arts EJ. 2011. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 11:45–56 [DOI] [PubMed] [Google Scholar]
- 61. Armstrong KL, Lee TH, Essex M. 2011. Replicative fitness costs of nonnucleoside reverse transcriptase inhibitor drug resistance mutations on HIV subtype C. Antimicrob. Agents Chemother. 55:2146–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Iglesias-Ussel MD, Casado C, Yuste E, Olivares I, Lopez-Galindez C. 2002. In vitro analysis of human immunodeficiency virus type 1 resistance to nevirapine and fitness determination of resistant variants. J. Gen. Virol. 83:93–101 [DOI] [PubMed] [Google Scholar]
- 63. Eshleman SH, Guay LA, Wang J, Mwatha A, Brown ER, Musoke P, Mmiro F, Jackson JB. 2005. Distinct patterns of emergence and fading of K103N and Y181C in women with subtype A vs. D after single-dose nevirapine: HIVNET 012. J. Acquir. Immune. Defic. Syndr. 40:24–29 [DOI] [PubMed] [Google Scholar]
- 64. Gianotti N, Galli L, Boeri E, Maillard M, Serra G, Ratti D, Gallotta G, Vacchini D, Tremolada Y, Lazzarin A, Clementi M, Castagna A. 2005. In vivo dynamics of the K103N mutation following the withdrawal of non-nucleoside reverse transcriptase inhibitors in human immunodeficiency virus-infected patients. New Microbiol. 28:319–326 [PubMed] [Google Scholar]
- 65. Palmer S, Boltz V, Maldarelli F, Kearney M, Halvas EK, Rock D, Falloon J, Davey RT, Jr, Dewar RL, Metcalf JA, Mellors JW, Coffin JM. 2006. Selection and persistence of non-nucleoside reverse transcriptase inhibitor-resistant HIV-1 in patients starting and stopping non-nucleoside therapy. AIDS 20:701–710 [DOI] [PubMed] [Google Scholar]
- 66. Turner D, Wainberg MA. 2006. HIV transmission and primary drug resistance. AIDS Rev. 8:17–23 [PubMed] [Google Scholar]
- 67. Weber J, Henry KR, Arts EJ, Quinones-Mateu ME. 2007. Viral fitness: relation to drug resistance mutations and mechanisms involved: nucleoside reverse transcriptase inhibitor mutations. Curr. Opin. HIV AIDS 2:81–87 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.