

Abstract
Using a simple viral genome enrichment approach, we report the de novo assembly of the Akata and Mutu Epstein-Barr virus (EBV) genomes from a single lane of next-generation sequencing (NGS) reads. The Akata and Mutu viral genomes are type I EBV strains of approximately 171 kb in length. Evidence for genome heterogeneity was found for the Akata but not for the Mutu strain. A comparative analysis of Akata with another four completely sequenced EBV strains, B95-8/Raji, AG876, Mutu, and GD1, demonstrated that the Akata strain is most closely related to the GD1 strain and exhibits the greatest divergence from the type II strain, AG876. A global comparison of latent and lytic gene sequences showed that the four latency genes, EBNA2, EBNA3A, EBNA3B, and EBNA3C, are uniquely defining of type I and type II strain differences. Within type I strains, LMP1, the latency gene, is among the most divergent of all EBV genes, with three insertion or deletion loci in its CTAR2 and CTAR3 signaling domains. Analysis of the BHLF1 and LF3 genes showed that the reading frames identified in the B95-8/Raji genome are not conserved in Akata (or Mutu, for BHLF1), suggesting a primarily non-protein-coding function in EBV's life cycle. The Akata and Mutu viral-genome sequences should be a useful resource for homology-based functional prediction and for molecular studies, such as PCR, RNA-seq, recombineering, and transcriptome studies. As an illustration, we identified novel RNA-editing events in ebv-miR-BART6 antisense transcripts using the Akata and Mutu reference genomes.
INTRODUCTION
The Epstein-Barr virus (EBV) is a human pathogen that causes several malignant diseases, including Burkitt's lymphoma (BL), Hodgkin's disease, and nasopharygneal carcinoma (NPC), as well as nonmalignant diseases such as infectious mononucleosis (1). Two distinct strains of EBV have been identified: the type I strain, which is more prevalent and has a greater cell-immortalizing capacity, and the type II strain.
EBV utilizes two broadly distinct stages in its life cycle: the latency phase, where only a subset of viral genes are expressed, and a viral replication stage (lytic reactivation), where infectious viruses are produced. During latency, the viral genome exists as an episome that is replicated and segregated concordantly with the host genome. During the lytic replication phase, a new set of more than 70 viral genes are expressed and the viral genome is amplified through a rolling circle mechanism (2). In EBV-positive Burkitt's lymphoma cell lines, the virus exists primarily in a latent state. In many of these cell systems, the virus can be efficiently transitioned into the lytic phase through activation of the B-cell receptor signaling pathway, making them ideal for certain lytic cycle-based investigations (3).
Both the Akata and Mutu cell systems are derived from EBV-positive Burkitt's lymphomas (BLs), in which EBV exhibits the type I form of latency. These tissue culture model systems are commonly utilized to study the BL phenotypes as well as the function of latency gene products in viral persistence and maintenance of the tumor phenotype. Their unique ability to exhibit synchronous and robust reactivation following surface immunoglobulin cross-linking also makes them ideal in vitro models to study EBV reactivation and makes them a good source for generating infectious virions. Despite their importance to the EBV field, detailed viral genetic information for these cell model systems is lacking. Investigators needing genomic information for experimental design and/or interpretation typically use the sequence of a surrogate type I strain, B95-8/Raji (4, 5) (GenBank accession no. NC_007605). We were therefore motivated to sequence the Akata and Mutu viral genomes in their entirety to facilitate better experimental design (e.g., the design of PCR primers or genome editing experiments) and analysis (e.g., for transcriptome analysis). Further, these additional genome sequences have allowed us to perform a global evolutionary and comparative analysis of the viral genomes and of EBV genes.
MATERIALS AND METHODS
Cell culture.
The EBV-positive Akata cell line (type I latency) was established from an EBV-positive Burkitt's lymphoma from a Japanese patient and expresses surface IgG and has a t(8:14) chromosome translocation (3). The EBV-positive Mutu (Mutu I) cell line (type I latency) was derived from an EBV-positive Burkitt's lymphoma biopsy specimen from a Kenyan patient and exhibits surface IgM (μK+) expression and a typical t(8:14) chromosome translocation (6). All cells were grown in RPMI 1640 (Thermo Scientific, catalog no. SH30027) plus 10% fetal bovine serum (FBS; Invitrogen-Gibco, catalog no. 16000-069) with 0.5% penicillin and streptomycin (pen/strep; Invitrogen-Gibco, catalog no. 15070). Cells were grown at 37°C in a humidified, 5% CO2 incubator.
Lytic cycle induction and viral genomic DNA extraction.
EBV-positive type I latency Akata and Mutu (Mutu I) cells were grown to approximately 2 × 106 cells per ml, at which time an equal volume of fresh RPMI 1640 (plus 10% FBS and 0.5% pen/strep) was added. The following day, cells were spun down and resuspended in freshly warmed RPMI 1640 (plus 10% FBS and 0.5% pen/strep) with or without 10 μg/ml of anti-IgG (for Akata) or 10 μg/ml anti-IgM (for Mutu). Seventy-two hours postinduction, EBV genomic DNA was extracted using a modified Hirt extrachromosomal DNA extraction protocol (7). Basically, cells were collected, washed once with 1× phosphate-buffered saline (PBS; Invitrogen), and resuspended in Hirt lysis buffer (0.6% SDS, 10 mM EDTA). Cells were incubated in lysis buffer at room temperature for 15 min. Insoluble genomic DNA and cellular debris were then precipitated by adding NaCl (approximately 1.4 M final concentration) and storing at 4°C overnight. The next day, samples were spun for 10 min at 18,000 × g at 4°C. The supernatant was then collected and subjected to phenol-chloroform extractions. Lastly, DNA was ethanol precipitated and suspended in Tris-EDTA (TE) buffer. The efficiency of lytic genome replication/amplification was confirmed by quantitative PCR (qPCR) using primers corresponding to the promoter regions of Zta, Rta, and EBNA1 (data not shown).
Sample preparation and next-generation DNA sequencing.
DNA libraries were prepared using the Illumina Truseq DNA sample preparation protocol (catalog no. FC-121-2002). Paired-end (PE) 100-base sequencing was performed using an Illumina HiSeq instrument. The average fragment size used for sequencing was ∼270 bp (range, 260 to 280 bp).
De novo assembly of EBV genomes.
Raw sequencing data generated from Akata and Mutu DNA preparations were first processed through an in-house deduplication algorithm. In preparation for EBV de novo assembly, the deduplicated sequencing data were precleared of non-EBV sequences. First, reads were aligned using the genome aligner Novoalign (version 2.07.18; Novocraft) with a reference genome containing a human sequence (hg19; Genome Reference Consortium GRCh37) plus abundant additional sequences (including adapters and mitochondrial, ribosomal, and enterobacterial phage phiX174 DNA sequences). Junction-spanning reads were then identified using the junction mapper TopHat (version 1.4.0 [8]). After removing all the mapped and low-quality reads, nonaligned reads were further screened by BLAST (version 2.2.24) using the Human RefSeq (cDNA) database (9) to identify additional human reads that were not identified by Novoalign or TopHat. The remaining reads were then subjected to de novo contig assembly using Velvet (version 1.2.03 [10]), which assembles reads based on the de Bruijin graph algorithm. The generated contigs were then analyzed by BLAST using the NCBI nonredundant nucleotide (NT) database to identify EBV contigs. Results from the BLAST analysis were filtered to eliminate matches with an E value of less than 10e−6. The results were combined and fed into MEGAN4 (MEtaGenome Analyzer [11]) to generate taxonomic data for the de novo assembled contigs. All contigs classified in the human herpesvirus 4 category were collected and subjected to further assembling/scaffolding using SeqMan Pro (DNAStar; Lasergene version 8).
Sequence refinement/validation.
After the initial assembly (see above), the resulting EBV genomes were indexed and raw genomic DNA reads were aligned to the constructed genomes using Novoalign. The aligned reads were visualized using the Integrative Genomics Viewer (IGV) (version 2.1 [12]). Poorly aligned and/or questionable regions (including certain repeat regions) were analyzed by PCR/Sanger sequencing using the primer sets listed in Table S1 in the supplemental material, and the resulting Sanger sequences were inserted into the appropriate regions of the genome scaffold. After the Sanger-solved regions were introduced into the assembled genomes, they were then inspected again by alignment with the raw sequencing reads. Positions showing a 99% or higher difference between the aligned raw reads and the assembled Akata and Mutu viral genomes (including conversions, deletions, and insertions) were corrected or modified accordingly based on the aligned raw reads. For the Mutu EBV genome only, internal repeat 3 and an additional six minor repeat regions remained nonvalidated and were temporarily replaced with the corresponding regions from the B95-8/Raji genome sequence (NCBI GenBank accession no. NC_007605) (these regions are indicated in Table S2 in the supplemental material). Lastly, the finalized Akata and Mutu EBV genomes were further validated by visualization of alignments from both latent and lytic Akata RNA-seq data and from latent Mutu RNA-seq data (13, 14).
Detection of SNV and indels.
Single nucleotide variations (SNVs) and insertions or deletions (indels) found in comparisons of the Mutu, B95-8/Raji, AG876, and GD1 strains and the Akata strain were identified using cross_match/phrap/swat (http://www.phrap.org/phredphrapconsed.html) (15). For ease of interpretation, indels and SNVs identified within the repeat regions were masked.
Comparative analysis of EBV coding genes.
Protein sequences of EBV coding genes from Akata, Mutu, B95-8/Raji (GenBank accession no. NC_007605), GD1 (GenBank accession no. AY961628.3), GD2 (GenBank accession no. HQ020558.1), HKNPC1 (GenBank accession no. JQ009376.1), and AG876 (GenBank accession no. DQ279927.1) strains were aligned with Megalign (DNAStar; Lasergene version 8). The divergence values used for comparisons were taken from the Megalign alignment data, and alignments were visualized directly within Megalign.
RNA-seq analysis of EBV transcriptomes.
Akata and Mutu EBV transcriptome analysis was performed by aligning sequence reads from latent and lytic cytoplasmic and nuclear Akata RNA (reported previously [13]) and from latent Mutu RNA (reported previously [14]) to the assembled Akata or Mutu EBV genome using Novoalign. Aligned reads were visualized using the IGV browser.
Nucleotide sequence accession numbers.
GenBank files containing annotation and genome sequences can be retrieved from GenBank (accession no. KC207813 and KC207814). Sequence data (DNA-seq) used here can be retrieved from the National Center for Biotechnology Information Sequence Read Archive (accession no. SRA060006). Fasta files, bed format annotation files (for visualization of annotation data on a genome browser), and .ann format annotation files (for quantification of EBV RNA-seq data) for the Akata and Mutu genomes are available on request.
RESULTS
One of the challenges of using next-generation sequencing (NGS) to solve a viral genome is the sequencing expense incurred by the presence of the much more abundant cellular genomic DNA inherent in standard DNA preparations. For example, Liu et al. (16) used at least 24 paired-end lanes of Illumina sequence data to obtain 17× coverage of an EBV genome. To overcome this obstacle, we first took advantage of EBV's lytic replication cycle to amplify viral genomic DNA and increase the ratio of viral genome to cellular genome. EBV lytic replication was induced in Akata and Mutu cells by activating B-cell receptor signaling using anti-immunoglobulin antibodies. To further increase the ratios of viral genome to cellular genome, we then used a modified Hirt DNA extraction procedure (7) to enrich for the more soluble viral genomes.
The enriched DNAs were analyzed by paired-end 100-base sequencing on an Illumina HiSeq machine using one lane per sample. Alignment of the sequencing data to the solved/finalized Akata or Mutu EBV genomes (see below) and the hg19 version of the human genome (Genome Reference Consortium GRCh37) showed that 3.41% and 1.58% of the mapped reads corresponded to EBV, with ratios of EBV genome to human genome of 537 and 244, respectively (Fig. 1A). Most importantly, this enrichment approach yielded very high coverage values of 3,278× and 1,450× (Fig. 1A).
Fig 1.
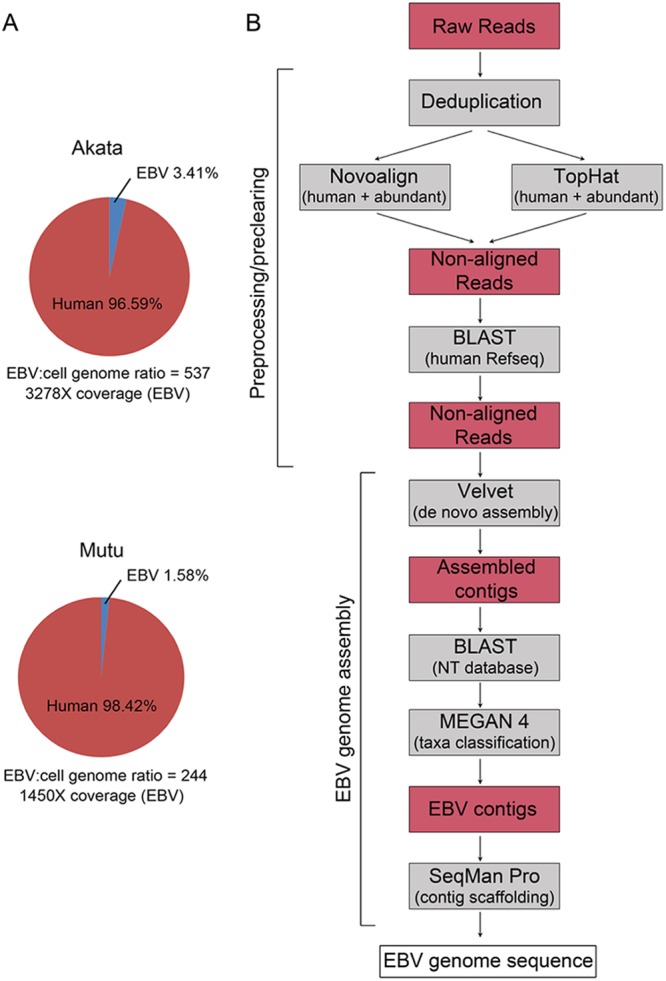
Workflow for Akata and Mutu EBV genome sequencing. (A) Percentages of aligned EBV and human reads in virus genome-enriched DNA fraction. Ratios of EBV genomes to cellular genomes and average fold coverage depth values are shown below pie charts. (B) Data analysis pipeline for de novo EBV genome assembly.
De novo assembly of EBV genomes.
To minimize intrinsic biases associated with reference genome-dependent assembly, we sought to use a de novo assembly approach, which links homologous read sequences into large contigs independently of a reference genome. Through an iterative process of testing different approaches to achieve reference-independent assembly, we found the strategy displayed in Fig. 1B to be particularly effective. Among the most important factors, we noted that EBV genome assembly was more productive (e.g., with significantly longer contigs and overall better coverage) when we minimized the number of nonviral reads prior to assembly. Our pipeline therefore incorporates “preclearing” steps to extract human reads and common artifactual and/or spiked organism reads (Fig. 1B). The remaining reads were then run through the de novo assembler, Velvet, a de Bruijin graphs-based algorithm (10). Using this approach, we found that the de novo assembly step generated contig libraries containing not only EBV contigs but also human contigs from apparently noncleared human reads. The EBV-specific contigs were identified by BLAST analyses performed using the NCBI NT database. EBV contigs were scaffolded using the Lasergene program SeqMan Pro, resulting in a relatively small number of large contigs (seven in the case of Akata) (Fig. 2; see also Fig. S1 and Table S3 in the supplemental material) separated primarily by the major EBV repeats (Fig. 2; see also Fig. S1 in the supplemental material). For the Akata assembly, one or two copies of IR1, IR2, and IR4 and the terminal repeats (TR) were contained within the ends of at least one contig. Each of these repeats was therefore iterated to contain the same number of repeats as represented by the standard B95-8/Raji genome (GenBank accession no. NC_007605). For the Mutu assembly, a total of 8 large scaffolded contigs were generated and the IR1, IR2, IR4, and TR repeat regions were iterated as described above (Fig. 3; see also Fig. S2 and Table S3 in the supplemental material).
Fig 2.

Genome assembly results for Akata EBV. Circos (30) was used to display data and features on the Akata EBV genome. (Track 1 [outermost]) Coding exons for latent genes (red), lytic genes (blue), and RPMS1/A73 (green). (Track 2) Repeat and regulatory elements (black). (Track 3) Signal maps displaying coverage depth at each genomic position (plotted in log scale [0 to 10,000 reads]). (Track 4) Contigs generated by de novo assembly (purple). *, contig containing an artificially expanded (iterated) IR1 repeat region for (better) visualization (the original contig contains approximately 1.5 copies of IR1 repeat units). (Track 5) Repeat regions reconstituted by iterating individual solved repeats (green). (Track 6) Sequences validated or solved by PCR cloning/Sanger sequencing (black). ds-DNA, double-stranded DNA.
Fig 3.

Genome assembly results for Mutu EBV. Circos (30) was used to display data and features on the Mutu EBV genome. (Track 1 [outermost]) Coding exons for latent genes (red), lytic genes (blue), and RPMS1/A73 (green). (Track 2) Repeat and regulatory elements (black). (Track 3) Signal maps displaying coverage depth at each genomic position (plotted in log scale [0 to 10,000 reads]). (Track 4) Contigs generated by de novo assembly (purple). *, contig containing an artificially expanded (iterated) IR1 repeat region for (better) visualization (the original contig contains approximately 1.5 copies of IR1 repeat units). (Track 5) Repeat regions reconstituted by iterating individual solved repeats (green). (Track 6) Sequences validated or solved by PCR cloning/Sanger sequencing (black).
To refine the assembled Akata and Mutu EBV genomes, we aligned the raw sequence data to the constructed Akata and Mutu virus genomes using the aligner Novoalign. The alignments were visualized using the IGV genome browser (12). Poorly aligned and questionable regions were analyzed by PCR cloning and Sanger sequencing (innermost track of Fig. 2 and 3; see also Fig. S1 and S2 in the supplemental material), and these regions of each genome were then corrected using the PCR/Sanger sequencing results. Raw sequence data were then aligned to the corrected genomes, and the alignments were subjected to visual inspection using the IGV. In a final stage, positions showing a 99% or greater difference between the aligned raw reads and the assembled Akata and Mutu viral genome (including conversions, deletions, and insertions) were corrected based on the aligned raw reads. Completion of this final step resulted in the construction of a 171,323-bp Akata EBV genome sequence and a 171,687-bp Mutu EBV genome sequence. The genomes were manually annotated based on homology and positional concordance with the standard B95-8/Raji EBV genome and its annotation. GenBank versions of the annotation were generated for deposition into the GenBank repository (accession no. KC207813 and KC207814), and bed format versions were generated for the display of genome features in genome browsers and are available on request.
Evidence of viral genome heterogeneity in Akata cells.
Relative to traditional Sanger sequencing, an advantage of NGS-based viral genome assembly is great coverage depth arising from the sequencing of multiple different genome fragments. This allows us to assess possible heterogeneity in genome populations by analyzing the nucleotide composition at each position along the genome. After aligning the Akata genomic reads to the assembled Akata EBV genome, we found a total of 20 heterogenous sites within the viral genome (defined by positions with at least 10% of reads showing a variant sequence). Eleven of these sites were located within repeat regions and may represent variations from one repeat to the next instead of intergenome differences. Seven of these sites were found within short homopolymer stretches, which are prone to sequencing errors. Moreover, the quality scores at these positions were typically low for the variant calls but not for the consensus calls. This suggests that the apparent heterogeneity at these positions is due to limitations in homopolymer sequencing rather than truly heterogeneous regions. Lastly, two heterogenous sites, positions 74673 and 134416, are not located in homopolymer stretches and the quality scores for both the variant and the consensus calls at each of these sites are high (Fig. 4). Notably, these variants are predicted to encode different amino acids in the BSRF1 and BVLF1 genes. In contrast to these findings in the Akata genome, no high-confidence variants were observed in the Mutu EBV genome. Together, these data suggest the possibility that low-level genomic evolution occurs in culture.
Fig 4.

Akata EBV genomes are heterogenous. Screen shots of IGV-displayed aligned reads are shown at the two high-confidence variant locations in the Akata EBV genome. Reads are represented by horizontal gray bars, and variant bases are indicated within the read bars. The genomic position and heterogenous percentage of each variant are indicated above the read bars. The constructed Akata reference genome sequence is displayed at the bottom of the alignments.
Comparative analysis of EBV genomes.
To investigate EBV genome variations between viral isolates, single nucleotide variations (SNVs), deletions, and insertions in the B95-8/Raji (type I), AG876 (type II), Mutu (type I), and GD1 (type I) genomes relative to the Akata (type I) genome were identified using Cross_Match (15) (Fig. 5; see also Fig. S3 in the supplemental material). The previously published GD2 (16) and HKNPC1 (17) genome sequences were not included in our analysis due to the presence of ambiguous residues (Ns). In our comparisons, repeat regions were masked to avoid errors resulting from variations between individual repeats. A total of 1,009, 2,014, 1,185, and 504 variations were detected between the B95-8/Raji, AG876, Mutu, and GD1 genomes and the Akata genome. This analysis showed that the Akata genome is most closely related to the GD1 genome which was isolated from the saliva of a nasopharyngeal carcinoma patient (18). In contrast, the Akata genome has the greatest divergence from the type II strain, AG876, as expected. The high-density variations between the AG876 sequence and the Akata sequence in the areas encoding the latency proteins EBNA2, EBNA3A, EBNA3B, and EBNA3C are notable (Fig. 5; see also Fig. S3 in the supplemental material). This is in line with previous studies that reported significant type I to type II strain divergence in these genes (19, 20).
Fig 5.
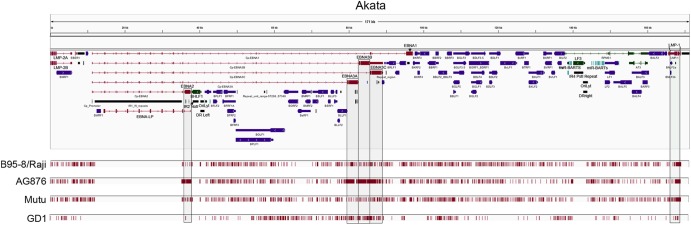
Comparative analysis of type I and type II viral genomes with the Akata EBV genome. Variations (indels and SNVs) between each viral genome and the Akata EBV genome were identified using cross_match (15). The position of each variation is displayed below the Akata EBV gene annotations and is indicated by vertical red bars. Regions with high variation densities (at EBNA2, EBNA3A, EBNA3B, EBNA3C, and LMP1) are shaded for better visualization.
Although divergence of EBNA2, EBNA3A, EBNA3B, and EBNA3C between type I and type II strains has been previously reported (19, 20), to our knowledge, a comprehensive analysis of type I and type II variations for all EBV coding genes has not been carried out. To assess divergence globally, we determined the amino acid differences in coding genes between five type I strains and the B95-8/Raji genome (intra-type I divergence; Fig. 6A, left panel, and Fig. 6B) and between six type I strains and the AG876 type II strain (type I-type II divergence; Fig. 6A, right panel, and Fig. 6C). To visualize these data in greater detail, see File S1 in the supplemental material. Strikingly, the lytic genes show considerable conservation between strains compared to that seen with the EBNA2, EBNA3A, EBNA3B, and EBNA3C latency gene products, and they are generally well conserved within the type I class (Fig. 6 [see also File S1 in the supplemental material]; note the scale differences between panel A and panels B and C). Among the latency genes, EBNA2, EBNA3A, EBNA3B, and EBNA3C are uniquely and significantly less conserved between type I and type II strains (Fig. 6A, right panel) but nevertheless show strong intrastrain conservation (Fig. 6A, left panel).
Fig 6.

Phylogenetic analysis of EBV coding genes at the amino acid level. Divergence data were obtained using Megalign (DNAStar). Intratype I divergence values represent the divergence between the indicated strain genome and the B95-8/Raji genome (NC007605.1) (4, 5). Type I-type II divergence values represent the divergence between the indicated strain and the type II strain, AG876 (DQ279927.1) (19). (A) Divergence at latency gene loci; (B) intratype I divergence of lytic genes; (C) interstrain divergence of lytic genes.
Variations in the major EBV oncogene, LMP1.
Unlike the EBNA2, EBNA3A, EBNA3B, and EBNA3C differences, the interstrain differences for LMP1 are not greater than the intrastrain differences. Nevertheless, LMP1 has the highest divergence among the type I strains of all the genes analyzed here except EBNA1 (which shows divergence primarily due to IR3 repeat variations) (Fig. 6A). In addition to amino acid differences in all five functional domains (Fig. 7A), LMP1 has three regions of insertions/deletions within the CTAR2 and CTAR3 signaling domains, which presumably influence one or more of LMP1's signaling functions.
Fig 7.

Variation of LMP1 protein sequences. (A) Alignment of LMP1 protein sequences derived from 6 EBV genomes was generated using Megalign (DNAStar). Variant positions are highlighted in yellow. Functional domains are indicated above the consensus sequence. C-term, C terminus. (B) Phylogenetic tree of LMP1 sequences from the indicated 6 EBV genomes.
BHLF1 and LF3 are noncoding in Akata and Mutu EBV genomes.
The BHLF1 gene in the B95-8/Raji genome has a predicted 660-amino-acid open reading frame (ORF) (4, 5). Despite this observation, whether the BHLF1 transcript is translated into a functional protein is questionable (4, 5), and Rennekamp and Lieberman (21) have shown that the BHLF1 transcript plays a noncoding function through its association with the lytic replication complex. Analysis of the corresponding BHLF1 region in the Akata genome revealed a stop codon just downstream from the corresponding methionine start codon (Fig. 8A). Further, there is an additional upstream start codon which similarly has an in-frame stop codon soon after. Analysis of the Mutu BHLF1 gene showed only an upstream translation initiation codon with a stop codon soon after (Fig. 8A). Together, these findings support the idea that only noncoding functions of BHLF1 are conserved across these viral genome species.
Fig 8.

The BHLF1 and LF3 genes are noncoding in the Akata and Mutu EBV genomes. Predicted BHLF1 ORFs (A) and LF3 ORFs (B) in the Akata and Mutu EBV genomes are indicated above and below the predicted B95-8/Raji BHLF1 and LF3 ORFs. Start codons (blue) and stop codons (red) are displayed for all three frames in zoomed regions of the Akata and Mutu BHLF1 gene (A) and LF3 gene (B).
The BHLF1 cognate gene, LF3, similarly has a predicted reading frame in the B95-8/Raji genome (4, 5). Nevertheless, in strains Akata and Mutu, there are several start codons with nearby downstream stop codons located at the 5′ end of the transcript (Fig. 8B) which likely prevent downstream initiation. Further, if the downstream initiation codon in the Akata genome is in fact functional, there is a frameshift relative to B95-8/Raji reading frame after the first 23 amino acids (aa) (Fig. 8B). Together, these data show that in strain Akata, the ORF is not conserved, and the findings of upstream start-stop codons in the Akata and the Mutu EBV genomes suggest that, like BHLF1, LF3 may primarily serve a noncoding function in the virus life cycle.
Detection of RNA-editing events in Akata and Mutu viral transcriptomes.
RNA editing modifies select transcripts posttranscriptionally, resulting in transcript variants with alternative biological functions (22). Using Akata and Mutu EBV genome sequences as references, we assessed RNA-editing events in Akata and Mutu viral transcriptomes. Moderate to high levels of RNA-editing events in transcripts derived from the Akata and Mutu ebv-miR-BART6 region were observed (Fig. 9). A-to-I/G editing of the primary ebv-miR-BART6 transcripts (at position 139637 of the Akata genome and position 139956 of the Mutu genome) was observed, which is consistent with previous studies (23). Surprisingly, high levels of A-to-I/G editing were also detected in apparent antisense transcripts spanning the ebv-miR-BART6 region (shown as T to C in the genome browser window). The orientation of this transcript and the associated RNA-editing event were further verified using strand-specific sequencing from induced Akata cells (unpublished data).
Fig 9.

Detection of RNA-editing events in ebv-miR-BART-6 regions in Akata and Mutu EBV transcriptomes. A-to-I (G) RNA-editing evidence for sense and antisense transcripts spanning the ebv-miR-BART6-3p region is shown. T-to-C changes displayed as genome browser data represent antisense A-to-I (G) changes (validated by strand-specific sequence data [unpublished]).
DISCUSSION
Here we report the sequencing of EBV genomes from two widely used Burkitt's lymphoma cell systems. Before settling on the lytic replication-based enrichment method that we utilized for this work, we initially tested the use of secreted viruses as a source for viral-genome sequencing. However, we found it cumbersome to obtain enough DNA for sequencing without significant scaling up. We reasoned that progression through the lytic cycle to the point of viral secretion is inefficient relative to the level of intracellular viral genomic replication that occurs following induction of the lytic cycle. Indeed, we are able to capture high levels of viral genomes using intracellular genomic material. Despite the fact that our method leads to the sequencing of unwanted cellular genomic material, the level of EBV coverage is still robust.
The amplification strategy used here achieved a robust level of coverage. We envision that viral mutants could easily be validated by sequencing their entire genomes using only a fraction of this level of coverage. This is especially true for validation sequencing, because reference genome-based alignment, which requires much lower coverage values (versus the de novo strategy used here), would be sufficient. Assuming a coverage requirement of 50× for genome sequence validation, multiplexing of up to 25× would be sufficient to accurately verify genome integrity, thereby reducing the sequencing cost to a nominal level.
Since the Akata and Mutu cell lines are commonly used for many EBV-related studies, we think that their genome sequences, along with their associated annotation, will be a useful resource for EBV researchers. This should be of value for the design of PCR oligonucleotides and should be useful for RNA-seq experiments using these systems, since alignments that are more accurate and robust would be achieved. Further, it should provide better clarity for assessing RNA-seq-based transcript variations. For example, using RNA-seq data from reactivated Akata cells (13), we find evidence of A-to-I (G) RNA editing in a lytic antisense transcript spanning the ebv-miR-BART6-3p region of the genome (Fig. 9; note that, on the sense strand shown in the genome browser view, this is depicted as a T-to-C change). These editing events were also observed using RNA-seq data from latent Mutu cells (14), and the orientation of this transcript and the associated RNA-editing event were verified using strand-specific sequencing data from induced Akata cells (unpublished). Nevertheless, in addition to the high abundance of RNA editing of the antisense transcripts, we also detected A-to-I (G) editing at the primary transcripts (pri-microRNA [pri-miRNA] and/or pre-miRNA) of ebv-miR-BART6 (position 139637 of the Akata genome and position 139956 of the Mutu genome) consistent with previous studies in other cell lines (23). At this point, we do not know the functions of these newly identified antisense transcripts. One possibility, however, is that they may act like microRNA sponges/inhibitors to sequester a subset of BART miRNAs generated from this region during reactivation. There is an extensive level of RNA editing observed on both the antisense and the microRNA strands from the ebv-miR-BART6-3p region (which is even more evident in the strand-specific sequencing data) (not shown). The resulting structure and thermodynamics of an interaction between these RNAs are likely to differ substantially from the homologous interactions between the antisense strand and microRNAs at surrounding BART microRNA loci. Whether this can sufficiently destabilize the interaction and allow ebv-miR-BART6-3p to escape suppression during reactivation, or whether it facilitates unique secondary structures that facilitate alternative processing of ebv-miR-BART6-3p, is an open question that may warrant further investigation.
In the Akata genome, two significant heterogenous sites which are predicted to encode different amino acids in the BSRF1 and BVLF1 genes were identified (Fig. 4). Both BSRF1 and BVLF1 function as lytic genes, and BSRF1 encodes a viral tegument protein. Although the exact function of BVLF1 is still unknown, it is believed that BVLF1 may function as the cytomegalovirus (CMV)-encoded UL79 protein which plays an important role for the accumulation of late viral transcripts. Such mutations may cause phenotype variations (such as differences in infectious virion production), and these results demonstrate possible genomic evolution in culture systems.
Among sequenced type I strains, sequence relatedness better correlated with geography than with tissue of origin, with the Akata genome (BL from a Japanese patient) being most similar to that of the GD1 virus (saliva from a Chinese NPC patient) and most distant from that of strain Mutu (BL from a Kenyan patient). A global analysis of EBV coding sequences of type I and type II viral strains highlighted the uniquely significant level of divergence at the EBNA2 loci, with lower but also significant divergence at the EBNA3A, EBNA3B, and EBNA3C loci. Notably, this high-level sequence variation is localized to two clusters (an EBNA2 cluster and an EBNA3 cluster), with approximately 40 kb of relatively conserved sequences separating the two (Fig. 5; see also Fig. S3 in the supplemental material). Intrastrain comparisons show good conservation at these loci, indicating that they are not mutagenic “hot spots” (Fig. 5 and 6; see also Fig. S3 in the supplemental material). The physical separation of these loci raises the possibility that their codivergence may be more related to a functional relationship rather than a spatial arrangement. EBNA2, EBNA3A, and EBNA3C are known to function through the cellular transcription factor RBP-J kappa (24, 25). Further, while EBNA2 transactivates Cp, the latency promoters, EBNA3A and EBNA3C, have been shown to be negative regulators of Cp (24). Since the transactivation activity of the type II EBNA2 is significantly weaker than that of the type I EBNA2, it is possible that the negative regulatory functions of the EBNA3 proteins in type II strains were attenuated to compensate for a weaker EBNA2. Conversely, if the type I strain was evolutionarily derived from the type II strain, it is possible that there was a selective increase in the negative impact of EBNA3 on EBNA2 functions evolved to prevent supraphysiological activity of EBNA2.
LMP1 sequence variation has been extensively studied in the context of its clinical significance in disease phenotype and progression (26–29). By assessing divergence at all known EBV genes, we find that within the type I strain, LMP1 does in fact show the greatest divergence (except for EBNA1, which appears divergent due to differences in the IR3 repeat sequences) (Fig. 6). Nevertheless, for the genomes analyzed here, the principal divergence lies between two evolutionarily distinct clusters (Fig. 7), with the Asian isolates, GD2, HKNPC1, and Akata, forming one relatively compact cluster and the African/American isolates, AG876, B95-8, and Mutu, forming another. This suggests that geography may be a key driver of LMP1 sequence differences (Fig. 7). The localization of the most substantial differences (single amino acid differences and deletions/insertions) to the carboxyl-terminal CTAR2 and CTAR3 signaling domains supports the idea that LMP1 function may differ between these two LMP1 subtypes, possibly contributing to region-specific EBV-associated disease propensities.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01CA124311, R01CA130752, and R01CA138268 to E.K.F. and P20GM103518 to Prescott Deininger.
Footnotes
Published ahead of print 14 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02517-12.
REFERENCES
- 1. Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Takada K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27–32 [DOI] [PubMed] [Google Scholar]
- 4. Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211 [DOI] [PubMed] [Google Scholar]
- 5. Parker BD, Bankier A, Satchwell S, Barrell B, Farrell PJ. 1990. Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179:339–346 [DOI] [PubMed] [Google Scholar]
- 6. Gregory CD, Rowe M, Rickinson AB. 1990. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. J. Gen. Virol. 71(Pt 7):1481–1495 [DOI] [PubMed] [Google Scholar]
- 7. Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365–369 [DOI] [PubMed] [Google Scholar]
- 8. Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pruitt KD, Tatusova T, Brown GR, Maglott DR. 2012. NCBI reference sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40:D130–D135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thorvaldsdottir H, Robinson JT, Mesirov JP. 19 April 2012. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. [Epub ahead of print.] doi:10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Concha M, Wang X, Cao S, Baddoo M, Fewell C, Lin Z, Hulme W, Hedges D, McBride J, Flemington EK. 2012. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. J. Virol. 86:1458–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin Z, Xu G, Deng N, Taylor C, Zhu D, Flemington EK. 2010. Quantitative and qualitative RNA-Seq-based evaluation of Epstein-Barr virus transcription in type I latency Burkitt's lymphoma cells. J. Virol. 84:13053–13058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gordon D, Abajian C, Green P. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195–202 [DOI] [PubMed] [Google Scholar]
- 16. Liu P, Fang X, Feng Z, Guo YM, Peng RJ, Liu T, Huang Z, Feng Y, Sun X, Xiong Z, Guo X, Pang SS, Wang B, Lv X, Feng FT, Li DJ, Chen LZ, Feng QS, Huang WL, Zeng MS, Bei JX, Zhang Y, Zeng YX. 2011. Direct sequencing and characterization of a clinical isolate of Epstein-Barr virus from nasopharyngeal carcinoma tissue by using next-generation sequencing technology. J. Virol. 85:11291–11299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kwok H, Tong AH, Lin CH, Lok S, Farrell PJ, Kwong DL, Chiang AK. 2012. Genomic sequencing and comparative analysis of Epstein-Barr virus genome isolated from primary nasopharyngeal carcinoma biopsy. PLoS One 7:e36939 doi:10.1371/journal.pone.0036939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeng MS, Li DJ, Liu QL, Song LB, Li MZ, Zhang RH, Yu XJ, Wang HM, Ernberg I, Zeng YX. 2005. Genomic sequence analysis of Epstein-Barr virus strain GD1 from a nasopharyngeal carcinoma patient. J. Virol. 79:15323–15330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dolan A, Addison C, Gatherer D, Davison AJ, McGeoch DJ. 2006. The genome of Epstein-Barr virus type 2 strain AG876. Virology 350:164–170 [DOI] [PubMed] [Google Scholar]
- 20. Sample J, Young L, Martin B, Chatman T, Kieff E, Rickinson A. 1990. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 64:4084–4092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rennekamp AJ, Lieberman PM. 2011. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA-DNA hybrid molecule at OriLyt. J. Virol. 85:2837–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G. 2011. Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis 32:1569–1577 [DOI] [PubMed] [Google Scholar]
- 23. Iizasa H, Wulff BE, Alla NR, Maragkakis M, Megraw M, Hatzigeorgiou A, Iwakiri D, Takada K, Wiedmer A, Showe L, Lieberman P, Nishikura K. 2010. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their Dicer targeting and consequently affects viral latency. J. Biol. Chem. 285:33358–33370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waltzer L, Perricaudet M, Sergeant A, Manet E. 1996. Epstein-Barr virus EBNA3A and EBNA3C proteins both repress RBP-J kappa-EBNA2-activated transcription by inhibiting the binding of RBP-J kappa to DNA. J. Virol. 70:5909–5915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zimber-Strobl U, Strobl LJ, Meitinger C, Hinrichs R, Sakai T, Furukawa T, Honjo T, Bornkamm GW. 1994. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 13:4973–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kingma DW, Weiss WB, Jaffe ES, Kumar S, Frekko K, Raffeld M. 1996. Epstein-Barr virus latent membrane protein-1 oncogene deletions: correlations with malignancy in Epstein-Barr virus-associated lymphoproliferative disorders and malignant lymphomas. Blood 88:242–251 [PubMed] [Google Scholar]
- 27. Miller WE, Edwards RH, Walling DM, Raab-Traub N. 1994. Sequence variation in the Epstein-Barr virus latent membrane protein 1. J. Gen. Virol. 75(Pt 10):2729–2740 [DOI] [PubMed] [Google Scholar]
- 28. Saechan V, Settheetham-Ishida W, Kimura R, Tiwawech D, Mitarnun W, Ishida T. 2010. Epstein-Barr virus strains defined by the latent membrane protein 1 sequence characterize Thai ethnic groups. J. Gen. Virol. 91:2054–2061 [DOI] [PubMed] [Google Scholar]
- 29. Sample J, Kieff EF, Kieff ED. 1994. Epstein-Barr virus types 1 and 2 have nearly identical LMP-1 transforming genes. J. Gen. Virol. 75(Pt 10):2741–2746 [DOI] [PubMed] [Google Scholar]
- 30. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19:1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.