

Abstract
Foamy viruses are retroviruses whose Pol protein is synthesized without Gag from a spliced mRNA. Unlike orthoretroviruses, reverse transcription occurs during viral assembly, leading to DNA-containing virions. When prototype foamy virus Pol is expressed as an orthoretroviral-like Gag-Pol fusion protein, reverse transcription also occurs late in viral replication, as measured by the timing of reverse transcriptase sensitivity to the inhibitor 3′-azido-3′deoxythymidine (AZT). Thus, timing of reverse transcription is intrinsic to Pol itself.
TEXT
Foamy viruses (FV) are complex retroviruses of the subfamily Spumaretrovirinae. They differ in the mode of Pol expression from orthoretroviruses, such as human immunodeficiency virus (HIV), which synthesize Pol as a Gag-Pol fusion protein. FV Pol expression occurs independently of Gag from a spliced mRNA (1) (Fig. 1A). Gag is not cleaved into separate matrix (MA), capsid (CA), and nucleocapsid (NC) proteins but is partially cleaved once by its own protease (PR) near the C terminus to release a p3 peptide resulting in a 68/71-kDa Gag doublet (Fig. 1B). The Pol precursor is also cleaved only once between reverse transcriptase (RT) and integrase (IN), resulting in IN and a PR-RT fusion protein (Fig. 1C). In orthoretroviruses, RT activity occurs at an early step after infection of new cells. Thus, infections are sensitive to RT inhibitors, such as 3′-azido-3′deoxythymidine (AZT). However, adding an inhibitor late in infection does not affect the ability to produce infectious virions (2). In contrast, FV reverse transcription is a late event in the life cycle, so virions are able to infect cells that have been pretreated with AZT (3). However, the virions have reduced infectivity when produced from AZT-treated cells.
Fig 1.

Diagram of WT Gag, Pol, and Gag-Pol fusion protein. (A) Splice sites (5′ss and 3′ss) for pol mRNA are indicated in the PFV genome; (B) WT Gag with the p3 cleavage site shown by an arrow to produce a 68/71-kDa doublet; (C) WT Pol consists of protease (PR), reverse transcriptase (RT), and integrase (IN) protein domains; (D) in a Gag-Pol fusion mutant, Δ3′ss mp3 G-P, the 3′ splice site in the pol gene, was mutated to eliminate the expression of WT Pol; (E) the Gag-Pol fusion protein was engineered by mutating the p3 cleavage site to abolish PR cleavage. Arrow indicates proteolytic cleavage site.
All three individual enzymes of prototype foamy virus (PFV) Pol (PR, RT, and IN) are required and must be active, and cleavages of both Gag and Pol need to occur by PR for production of infectious particles (4–6). Recently, we and others (7–9) have created a PFV mutant expressing Gag and Pol as a fusion protein. We found that in the absence of wild-type (WT) Pol protein expression, the protease expressed in the Gag-Pol fusion protein did cleave near the C terminus of Gag and in Pol releasing IN, as is seen with WT PR (7). When Gag was coexpressed with the Gag-Pol fusion protein at a molar ratio near that found in orthoretroviruses (20:1), particles had titers similar to that of the WT.
We have now studied the timing of reverse transcription in the PFV Gag-Pol virions to determine whether it resembles that of PFV RT or that of orthoretroviral RT. If the RT expressed in the Gag-Pol fusion protein acts early in infection, it should be sensitive to AZT when cells are pretreated with this inhibitor, similar to what is seen with HIV (2). If the Gag-Pol fusion protein acts like PFV WT Pol, reverse transcription should occur late in viral replication, as in the WT (3). PFV Gag-Pol fusion was created in the context of a full-length proviral clone containing a cytomegalovirus (CMV) immediate-early promoter, pcPFV (10). In the fusion protein mutant, Δ3′ss mp3 G-P, the 3′ splice site (ss) for Pol was mutated to prevent the expression of Pol without Gag (7) (Fig. 1D). The p3 cleavage site near the C terminus of Gag was also mutated to abolish Gag cleavage from the Gag-Pol fusion protein (Fig. 1E).
Virus stocks were prepared by harvesting viruses produced from transfection of 293T cells, using polyethyleneimine (PEI), as previously reported (11). In order to produce infectious Gag-Pol fusion viruses, a Gag expression vector (pGag) was cotransfected with the Δ3′ss mp3 G-P construct at a 20:1 molar ratio, as previously described (7). After 48 h, supernatants were harvested to quantify infectious viruses by using an FV-activated β-galactosidase (β-Gal) expression (FAB) assay (12). FAB cells are BHK-21 cells containing a β-Gal gene under the control of the PFV LTR. After infection with PFV, the viral transactivator, Tas, is produced, and β-Gal activity is detected. Blue cells can be enumerated after staining, and viral titer can be determined. Viral titers of WT PFV ranged from 5 × 105 to 8 × 105 infectious units (IU) per ml. Viral titers for the pGag/Δ3′ss mp3 G-P cotransfection were 3 × 103 to 6 × 103 IU/ml. Both WT and mutant viruses were used at a multiplicity of infection (MOI) of 0.1.
To examine AZT inhibition of viral infection of target cells, FAB cells were treated with 100 μM AZT or left untreated for 4 h prior to infection. In the case of AZT treatment, the WT PFV or pGag/Δ3′ss mp3 G-P virus stocks were suspended in media containing 100 μM AZT and used for infection. This high concentration of AZT was not toxic for viability of FAB cells, as previously observed (3). After 48 h, the cells were fixed and stained, and viral titers were measured using the FAB assay. We found that if WT PFV was used to infect AZT-treated cells, a 5- to 6-fold decrease in viral titer was seen compared to that of untreated cells (Table 1). This modest inhibition of AZT on viral infection confirmed our previous results (3). When using the Gag-Pol mutant viruses to infect cells that were pretreated with AZT, there was a similar reduction in viral titer to that found with WT PFV (Table 1).
Table 1.
Effect of AZT on viral infection of target cells
| Virus | Infectivity (units/ml)a |
Ratio of +AZT to −AZT | |
|---|---|---|---|
| −AZT | +AZT | ||
| WT PFV | 1.3 × 105 | 2.8 × 104 | 0.22 |
| 1.4 × 105 | 2.5 × 104 | 0.18 | |
| 1.3 × 105 | 2.3 × 104 | 0.18 | |
| pGag/Δ3′ss mp3 G-P (20:1) | 9.3 × 102 | 2.1 × 102 | 0.23 |
| 9.0 × 102 | 2.0 × 102 | 0.22 | |
| 1.1 × 103 | 1.3 × 102 | 0.12 | |
Prior to the addition of WT PFV or pGag/Δ3′ss mp3 G-P virus stocks at an MOI of 0.1, cells were pretreated for 4 h with 100 μM AZT or left untreated. Forty-eight hours postinfection, the cells were fixed and stained, and viral titers were measured using the FAB assay as described in reference 12.
To examine the inhibitory effect of AZT on virus production, FAB cells were treated with 100 μM AZT or left untreated for 4 h prior to infection. Forty-eight hours postinfection, supernatants were harvested, and viral titer was measured by the FAB assay. WT virus produced from AZT-treated cells showed a 40- to 60-fold decrease in infectivity (Table 2). When the mutant fusion viruses were produced from AZT-treated cells, there was a 17- to 125-fold decrease in infectivity (Table 2). Unlike WT virus stocks, in the case of Gag-Pol fusion virus stocks, we had to cotransfect 293T cells with the Gag-Pol proviral construct and pGag plasmid. This led to concern that there might not be enough Gag during infection of the FAB cells with the pGag/Δ3′ss mp3 G-P virus stocks described above. Therefore, we did a transfection experiment in the presence or absence of AZT, in order to see the effect of AZT on viral infectivity, as another laboratory previously reported (13). 293T cells were transfected with either pcPFV or cotransfected with pGag and Δ3′ss mp3 G-P proviral DNAs at a 20:1 molar ratio in the presence or absence of 100 μM AZT. After 48 h of transfection, cells and supernatants were collected and cell lysates and viral pellets were prepared as described in reference 7 and run on 9% SDS-polyacrylamide gels. Expression and assembly of Gag and Pol proteins into virions were analyzed by Western blotting. The amounts of Gag and Pol protein expressed under AZT treatment were equivalent to those in the absence of AZT (Fig. 2A and B, cell). Viral release measured by the amount of Gag proteins in supernatants was greatly reduced in pGag/Δ3′ss mp3 G-P cotransfection compared to that of WT viruses (Fig. 2A, virus). This reduction was caused by a requirement for Env for PFV particle release (14). Less Env was expressed in the cotransfection with Gag than in the transfection with WT proviral DNA alone. However, there was no difference in viral protein expression and assembly in the AZT-treated and untreated cells. The results indicated that 100 μM AZT was not toxic for 293T cells. Viral titers were measured by the FAB assay (Table 3). Similar to WT virus, when the mutant fusion viruses were produced from AZT-treated cells, there was a greater than 50-fold decrease in infectivity, demonstrating results obtained from the transfection assay consistent with those from the infection assay.
Table 2.
Effect of AZT on viral infectivity produced from infection of FAB cells
| Virus | Infectivity (units/ml)a |
Ratio of +AZT to −AZT | |
|---|---|---|---|
| −AZT | +AZT | ||
| WT PFV | 3.9 × 104 | 6.7 × 102 | 0.017 |
| 3.2 × 104 | 7.8 × 102 | 0.024 | |
| 3.3 × 104 | 7.2 × 102 | 0.022 | |
| pGag/Δ3′ss mp3 G-P (20:1) | 1.2 × 103 | 10 | 0.008 |
| 2.5 × 102 | 15 | 0.06 | |
| 1.5 × 102 | <10b | <0.067 | |
Cells were pretreated with 100 μM AZT or left untreated for 4 h prior to infection with each virus stock at an MOI of 0.1. Forty-eight hours postinfection, supernatants were collected, and then viral titers were measured by the FAB assay as described in reference 12.
No blue cells were observed in a 10−1 dilution of viruses for the FAB assay.
Fig 2.
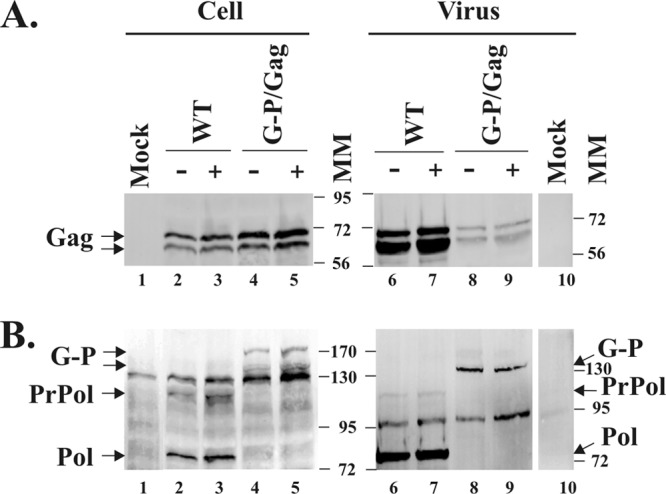
Analyses of intracellular and extracellular viral proteins by Western blotting. 293T cells were treated with 100 μM AZT or left untreated during transfection with WT PFV or cotransfection of Δ3′ss mp3 G-P with pGag at a molar ratio of 1:20. Forty-eight hours after transfection, cells and supernatants were collected. Cell lysates and viral pellets were resuspended in 1× SDS sample buffer and run on 9% SDS-polyacrylamide gels. The proteins were transferred to a membrane, and the membrane was probed with anti-Gag antibody (A) and anti-Pol antibody (B). The cellular protein GAPDH was used as a loading control (data not shown). Negative and positive signs indicate cells left untreated or treated with AZT during transfection, respectively. G-P, both Gag-PR-RT-IN and Gag-PR-RT; PrPol, precursor Pol (PR-RT-IN); Pol, cleaved Pol (PR-RT). Molecular mass markers (MM) are shown in kilodaltons.
Table 3.
Effect of AZT on viral infectivity produced from 293T cell transfection
| Virus | Infectivity (units/ml)a |
Ratio of +AZT to −AZT | |
|---|---|---|---|
| −AZT | +AZT | ||
| WT PFV | 9.6 × 104 | 5 × 102 | 0.005 |
| 2.2 × 105 | 4 × 103 | 0.018 | |
| 2.1 × 105 | 2 × 103 | 0.009 | |
| pGag/Δ3′ss mp3 G-P (20:1) | 4.9 × 102 | <10b | <0.02 |
| 1.2 × 103 | <10 | <0.008 | |
| 1.8 × 103 | <10 | <0.006 | |
293T cells were treated with 100 μM AZT or left untreated during transfection with WT PFV or cotransfection of Δ3′ss mp3 G-P with pGag at a molar ratio of 1:20. Forty-eight hours posttransfection, supernatants were collected, and then viral titers were measured by the FAB assay as described in reference 12.
No blue cells were observed in a 10−1 dilution of viruses for the FAB assay.
We can conclude that when PFV Pol is expressed as a Gag-Pol fusion, RT is active at the same stage of the infection cycle as in WT PFV. Reverse transcription is a late event in the viral life cycle during viral assembly and release. We found a modest reduction in viral titer (about 5-fold) following infection under AZT treatment for both WT and the Gag-Pol fusion mutant viruses. It suggests the possibility that there is some requirement for early DNA synthesis as observed for orthoretroviruses. Previously, other laboratories (15, 16) have reported that an early reverse transcription is indispensable for FV infectivity at the low MOI of 0.1 that we used, when the amount of incoming viral DNA was not sufficient for productive infection. Taken together, the timing of reverse transcription is not influenced by the presence or absence of Gag sequences in the Pol protein. It suggests that the information in the Pol protein itself determines the timing of reverse transcription.
ACKNOWLEDGMENTS
This research was partially funded by NIH grant R01 CA 18282 to M.L.L.
We thank Xiaoxing Wang and Karen Craig for critical reading of the manuscript.
Footnotes
Published ahead of print 7 November 2012
REFERENCES
- 1. Yu SF, Baldwin DN, Gwynn SR, Yendapalli S, Linial ML. 1996. Human foamy virus replication—a pathway distinct from that of retroviruses and hepadnaviruses. Science 271:1579–1582 [DOI] [PubMed] [Google Scholar]
- 2. Arts EJ, Mak J, Kleiman L, Wainberg MA. 1994. DNA found in human immunodeficiency virus type 1 particles may not be required for infectivity. J. Gen. Virol. 75:1605–1613 [DOI] [PubMed] [Google Scholar]
- 3. Yu SF, Sullivan MD, Linial ML. 1999. Evidence that the human foamy virus genome is DNA. J. Virol. 73:1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enssle J, Fischer N, Moebes A, Mauer B, Smola U, Rethwilm A. 1997. Carboxy-terminal cleavage of the human foamy virus gag precursor molecule is an essential step in the viral life cycle. J. Virol. 71:7312–7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roy J, Linial ML. 2007. Role of the foamy virus Pol cleavage site in viral replication. J. Virol. 81:4956–4962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zemba M, Wilk T, Rütten T, Wagner A, Flügel RM, Löchelt M. 1998. The carboxy-terminal p3(Gag) domain of the human foamy virus gag precursor is required for efficient virus infectivity. Virology 247:7–13 [DOI] [PubMed] [Google Scholar]
- 7. Lee EG, Sinicrope A, Jackson DL, Yu SF, Linial ML. 2012. Foamy virus Pol protein expressed as a Gag-Pol fusion retains enzymatic activities, allowing for infectious virus production. J. Virol. 86:5992–6001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spannaus R, Hartl M, Wohrl B, Rethwilm A, Bodem J. 2012. The prototype foamy virus protease is active independently of the integrase domain. Retrovirology 9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swiersy A, Wiek C, Reh J, Zentgraf H, Lindemann D. 2011. Orthoretroviral-like prototype foamy virus Gag-Pol expression is compatible with viral replication. Retrovirology 8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stenbak CR, Linial ML. 2004. Role of the C terminus of foamy virus Gag in RNA packaging and Pol expression. J. Virol. 78:9423–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Durocher Y, Perret S, Kamen A. 2002. High-level and high-throughput recombinant protein production by transient-transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu SF, Linial ML. 1993. Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J. Virol. 67:6618–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moebes A, Enssle J, Bieniasz PD, Heinkelein M, Lindemann D, Bock D, McClure MO, Rethwilm A. 1997. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 71:7305–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baldwin DN, Linial ML. 1998. The roles of Pol and Env in the assembly pathway of human foamy virus. J. Virol. 72:3658–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delelis O, Saïb A, Sonigo P. 2003. Biphasic DNA synthesis in Spumaviruses. J. Virol. 77:8141–8146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zamborlini A, Renault Nm., Saïb A, Delelis O. 2010. Early reverse transcription is essential for productive foamy virus infection. PLoS One 5:e11023 doi:10.1371/journal.pone.0011023 [DOI] [PMC free article] [PubMed] [Google Scholar]