

Abstract
Iridoviruses are a family of large double-stranded DNA (dsDNA) viruses that are composed of 5 genera, including the Lymphocystivirus, Ranavirus, Megalocytivirus, Iridovirus, and Chloriridovirus genera. The frog virus 3 (FV3) 75L gene is a nonessential gene that is highly conserved throughout the members of the Ranavirus genus but is not found in other iridoviruses. FV3 75L shows high sequence similarity to a conserved domain found in the C terminus of LITAF, a small cellular protein with unknown function. Here we show that FV3 75L localizes to early endosomes, while LITAF localizes to late endosomes/lysosomes. Interestingly, when FV3 75L and LITAF are cotransfected into cells, LITAF can alter the subcellular localization of FV3 75L to late endosomes/lysosomes, where FV3 75L then colocalizes with LITAF. In addition, we demonstrated that virally produced 75L colocalizes with LITAF. We confirmed a physical interaction between LITAF and FV3 75L but found that this interaction was not mediated by two PPXY motifs in the N terminus of LITAF. Mutation of two PPXY motifs in LITAF did not affect the colocalization of LITAF and FV3 75L but did change the location of the two proteins from late endosomes/lysosomes to early endosomes.
INTRODUCTION
Iridoviruses are large (120 to 200 nm) double-stranded DNA viruses that possess an icosahedral capsid. The Iridoviridae family is composed of 5 genera that infect a wide range of poikilothermic vertebrates (Lymphocystivirus, Ranavirus, and Megalocytivirus) and invertebrate hosts (Iridovirus and Chloriridovirus) (1, 2).
Understanding how iridoviruses cause disease requires an understanding of how these viruses interact with their hosts. Throughout the coevolution of viruses and their hosts, viruses have adapted certain cellular functions to their own advantage. Viruses are particularly adept at manipulating the host cell machinery in order to benefit their own replication and survival. Frog virus 3 (FV3) is the type species of the Ranavirus genus and is commonly used as a model system to study iridoviruses. In addition to the core set of essential iridovirus genes, sequencing of the FV3 genome revealed many genes putatively involved in virus-host interactions (3).
FV3 75L is an 84-amino-acid protein suspected to play a role in virus-host interaction (3). FV3 75L shows striking sequence identity with the C terminus of a cellular protein designated lipopolysaccharide-induced tumor necrosis factor alpha factor (LITAF) (Fig. 1) (3, 4). LITAF is also known as p53-induced gene 7 (PIG7) (5) and small integral membrane protein of the lysosome/late endosome (SIMPLE) (6). LITAF is predicted to encode a 161-amino-acid protein with distinct N and C termini (6). The N terminus of LITAF contains proline-rich binding domains for several cellular proteins, including neural precursor cell expressed, developmentally downregulate 4 (Nedd4) (3, 7), tumor susceptibility gene 101 (TSG101) (8), and Itch (9), which function along a conserved pathway of lysosomal protein degradation. The C terminus of LITAF contains an arrangement of cysteine residues resembling a RING finger domain (4). Interestingly, a stretch of hydrophobic amino acids lies within this predicted RING finger domain. This interrupted RING finger domain is termed the SIMPLE-like domain (SLD) and is found in proteins of many species, including humans, yeasts, and plants (4). The function of the SLD remains unknown.
Fig 1.

FV3 75L shows sequence similarity to the C terminus of cellular LITAF. The sequence alignment compares FV3 75L and LITAF proteins of various species, as well as a homolog in SGIV. The SIMPLE-like domain (SLD) is shown in blue, and the hydrophobic stretch of amino acids within the SLD is highlighted in cyan. Highly conserved cysteine residues are displayed in red, and other conserved amino acid residues are shown in yellow. Amino acids that compose the RING finger motif are shown above the SLD.
In addition to the SLD, FV3 75L contains a tyrosine-based targeting sequence, YXXΦ (YKRL and YKML for LITAF and FV3 75L, respectively), but a dileucine motif which has been characterized for other transmembrane proteins targeted to the endosome/lysosome system (10–12) is present in LITAF only, not FV3 75L. FV3 75L exhibits high levels of sequence identity with the C terminus of cellular LITAF, which encodes the SLD, but FV3 75L lacks the N terminus of LITAF, which contains binding sites for a variety of cellular proteins. FV3 75L therefore appears to represent a truncated version of LITAF. Homologs of FV3 75L are found in other ranaviruses, such as Singapore grouper iridovirus ORF136 (SGIV136), soft-shelled turtle iridovirus ORF37R (STIV 37R), and European catfish virus 94R (ECV 94R) (13–15).
Huang et al. (14) previously characterized the FV3 75L homolog SGIV136. They determined that SGIV136 was expressed from an early viral gene and was localized throughout the cytoplasm and associated with mitochondria in the cell (14). Furthermore, overexpression of SGIV136 resulted in the formation of apoptotic bodies, a change in mitochondrial membrane potential, and activation of caspase-3, suggesting that this product can induce or is involved in apoptosis (14).
Our understanding of the role of FV3 75L is limited by a lack of knowledge regarding the function of LITAF. However, many pieces of evidence suggest a role for LITAF along the conserved pathway of protein degradation. Binding partners of LITAF, such as the E3 ubiquitin ligases Nedd4 and Itch as well as TSG101 (an E2 ubiquitin-conjugating enzyme from the ubiquitin E2 variant family), all function in the ubiquitin pathway. Nedd4 functions to ubiquitinate substrates at the plasma membrane and Golgi apparatus (16), while TSG101 acts at the early endosome to recognize and sort ubiquitinated proteins for future degradation in the lysosome (17, 18). Furthermore, LITAF contains a RING finger domain which is present in many E3 ubiquitin ligases (8, 19, 20). The presence of a transmembrane domain within this RING finger is thought to abolish the function of the RING finger motif; however, this has never been studied (4). Finally, at least 8 mutations of LITAF are associated with Charcot-Marie-Tooth (CMT) disease, a neurological disease in humans. CMT is a hereditary neuropathy characterized by loss of muscle tissue and touch sensation, predominantly in the feet and legs. The mechanism by which mutations in LITAF cause CMT disease is unknown. However, it has been suggested that defects in LITAF function could lead to the inappropriate accumulation of target proteins, including those whose regulated expression is essential to peripheral nerve myelination, suggesting a potential role for LITAF in protein degradation.
LITAF has also been proposed to play a role in cellular immune responses. LITAF was first identified as a gene upregulated in the presence of bacterial cell wall components, including lipopolysaccharide (LPS) (6). LPS is a potent stimulator of monocytes and macrophages, causing synthesis of LITAF. These data suggest that the LITAF gene may be a pathogen-associated molecular pattern (PAMP)-induced gene. In addition, LITAF mutations have also been demonstrated to play a pivotal role in inflammation, and high levels of these molecules in fluids and serum have been associated with inflammatory processes such as rheumatoid arthritis, Crohn's disease, multiple sclerosis, and pancreatitis (8, 21, 22).
In this study, we undertook a characterization of the interaction between LITAF and FV3 75L and the role of LITAF in viral infection. In addition, we compared the intracellular localizations of FV3 75L and SGIV136.
MATERIALS AND METHODS
Reagents, cell lines, and antibodies.
Baby green monkey kidney (BGMK), human embryonic kidney 293T (HEK-293T), and fathead minnow (FHM) cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). A fibroblast line derived from the Xenopus adult kidney (A6) was provided by Neils C. Boles (Waterloo, Ontario, Canada). BGMK cells were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone, Ottawa, Ontario, Canada) supplemented with 7% fetal bovine serum (FBS; HyClone), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% CO2. HEK cells were also maintained at 37°C with 5% CO2 in DMEM containing 10% cosmic calf serum (Invitrogen, Burlington, Ontario, Canada), 100 U/ml penicillin, and 100 μg/ml streptomycin. FHM cells were grown in closed flasks and maintained at 30°C without CO2 in minimum essential medium with Hanks' salts (MEM; HyClone) supplemented with 10% FBS and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). A6 cells were maintained at 22°C in L-15 Leibovitz medium (L-15; HyClone) with 10% FBS (HyClone), 100 U/ml penicillin, and 100 μg/ml streptomycin. The following antibodies and probes were used during immunofluorescence assay: rabbit anti-early endosome antigen 1 (anti-EEA1) antibody (1/50 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA), Lysotracker DND-99 (Molecular Probes, Burlington, Ontario, Canada), MitoTrackerRed (Molecular Probes), 9E10 mouse and rabbit myc monoclonal antibody (1/100 dilution; Roche, Indianapolis, IN), monoclonal FLAG (M2) antibody (1/500 dilution; Sigma, Oakville, Ontario, Canada), anti-75L antibody (GenScript, Piscataway, NJ), an affinity-purified antipeptide serum raised against the 75L peptide sequence CMDDKFTTLPCELED, and fluorescein isothiocyanate (FITC)-, Cy3-, and Cy5-conjugated goat anti-mouse or anti-rabbit immunoglobulin G (IgG) (diluted 1/100, 1/200, and 1/100, respectively; Jackson ImmunoResearch Inc., West Grove, PA). Rabbit 9E10 myc monoclonal antibody (1/1,000 dilution; Enzo Life Sciences, Farmingdale, NY), mouse anti-FLAG monoclonal antibody (1/1,000 dilution; Sigma), mouse anti-LITAF antibodies (1/4,000 dilution; BD Biosciences, San Jose, CA), and peroxidase-conjugated AffiniPure F(ab′)2 fragment goat anti-rabbit/mouse IgG (1/10,000 dilution; Jackson ImmunoResearch Inc.) were used during Western blotting.
Isolation of viral DNA.
In order to generate viral DNA for PCR, FHM or A6 cells were grown to 80% confluence and were infected with FV3 at a multiplicity of infection (MOI) of 0.1. The infection was allowed to progress until cytopathic effects (CPE) were seen, at which point cells were harvested and resuspended in phosphate-buffered saline (PBS). Infected cells were freeze-thawed 3 times, and an equal volume of phenol-chloroform was added. The aqueous phase was transferred to a new tube, and 10% (vol/vol) 5 M sodium acetate and 200% (vol/vol) ethanol (96%) were added. The mixture was left for 15 min on ice, followed by centrifugation for 10 min at 10,000 × g. The pellet was air dried and resuspended in PBS.
PCR and expression plasmids.
FV3 75L, SGIV ORF136, and LITAF were amplified using a 50-μl PCR mixture containing 1× PCR buffer (Invitrogen), 3.0 mM MgCl2 (Invitrogen), 0.2 mM (each) forward and reverse primers, and 2.5 U Taq DNA polymerase (5 U/μl; Invitrogen). Specifically, FV3 75L was amplified and a myc tag was added using 10 ng of FV3 DNA and the following primers: 5′-AAGCTTATTAAAGATGGACGACAAG-3′ (FV3 75L-forward) and 5′-CTCGAGCTACAGATCTTCTTCAGAAATAAGTTTTTGTTCTAAAATTTTGTACACAAACAC-3′ (FV3 75L-reverse). SGIV ORF136 was synthesized by Genescript (Piscataway, NJ) as the template DNA and amplified with the following primers: 5′-ATGGAGCAGAAACTGATTAGT-3′ (SGIV136-forward) and 5′TTAGACAATCTTATAGACGGC-3′ (SGIV136-reverse). LITAF was amplified and a FLAG tag added using mouse LITAF cDNA (MGC-6569; ATCC) as the template DNA and the following primers: 5′-AAGCTTATGGATTACAAGGATGACGACGATAAGTCGGTTCCAGGACCTTACC-3′ (LITAF-forward) and 5′-CTCGAGCTAAAAGCGTTGTAGGTG-3′ (LITAF-reverse). The following cycling conditions were used: 94°C for 30 s, 52°C for 30 s, and 72°C for 90 s for 30 cycles. The resulting PCR product was initially cloned into pGEM-T Easy (Promega, Madison, WI), followed by cloning into the XhoI and HindIII sites of pcDNA3.1 (Invitrogen). A green fluorescent protein (GFP) plasmid expressing LITAF was also generated through a PCR using FLAG-LITAF as the template DNA, the forward primer 5′-GAGACTCGAGAATGTCGGTTCCAGGACC-3′, and the reverse primer 5′-GAGAAAGCTTCTACAAACGCTTGTAGGTG-3′ and was then cloned into the plasmid pEGFP-C2 (Clontech, Mountain View, CA) through the XhoI and HindIII restriction sites. The GFP LITAF plasmid was used as a template to create a LITAF Y23,61A double mutant plasmid. Site-directed mutagenesis was completed using a QuikChange Lightning Multi site-directed mutagenesis kit (Stratagene, La Jolla, CA) per the manufacturer's directions. The forward primer 5′-AAGCTTATGGAACAAAAAGTTATTTCTGAAGAAGATCTGTCGGTTCCAGGACCTTACC-3′ and the reverse primer 5′-CTCGAGCTAAAAGCGTTGTAGGTG-3′ were used.
Transfections.
BGMK, HEK, or A6 cells were grown to 70% confluence and transfected using a polyethylenimine (PEI) reagent at a concentration of 0.2 mM. DNA (5 μg) was resuspended in 400 μl of serum-free DMEM. PEI was added at a 4:1 ratio of PEI to DNA and left to incubate at room temperature for 15 min. Transfection complexes were then added to cells, along with fresh DMEM.
Immunofluorescence assay.
At 24 h posttransfection, cells were fixed using a 3.7% paraformaldehyde solution in PBS and permeabilized using a 0.1% Triton X-100 solution in PBS. Cells were blocked for 2 h at room temperature in blocking buffer (5% [wt/vol] bovine serum albumin [BSA], 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% [vol/vol] NP-40), followed by several 5-min washes in wash buffer (1% [wt/vol] BSA, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% [vol/vol] NP-40). Primary antibody diluted in wash buffer was incubated on cells for 1 h at room temperature. The primary antibody was removed following several 5-min washes in wash buffer, and secondary antibody was diluted in wash buffer and applied to cells. The secondary antibody was left on cells for 1 h at room temperature in darkness before removal with several 5-min washes in wash buffer. Cells were mounted using Vectashield (Vector Laboratories, Burlington, Ontario, Canada), and fluorescence was detected using a Leica DM SP2 confocal microscope (Leica, Wetzlar, Germany). Images were assembled using Adobe Photoshop CS4 (Adobe, San Jose, CA).
FV3 lysates.
FHM cells were grown to 90% confluence and then infected with FV3 at an MOI of 20. Mock-infected cells and cells infected for 2, 4, and 6 h were harvested and collected by centrifugation after 5 min at 10,000 × g. The cell pellets containing cell lysate were resuspended in Laemmli buffer and boiled for 5 min, dithiothreitol (DTT) was added to a final concentration of 1%, and proteins were resolved by Western blotting.
Western blotting.
Lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using SDS running buffer (125 mM Tris, 1.25 M glycine, 0.5% SDS), followed by transfer to a polyvinylidene difluoride (PVDF) membrane by use of a semidry transfer apparatus (Fisher Biotech, Pittsburgh, PA). The membrane was blocked for a minimum of 24 h at 4°C in TBST buffer (140 mM NaCl, 24 mM Tris [pH 7.4], 0.2% Tween 20, 3 mM KCl) containing 5% nonfat milk powder. Following several washes in TBST, the membrane was incubated in primary antibody for 1 h at room temperature. The membrane was washed several times with TBST and then incubated in secondary antibody for 1 h at room temperature. Following several more washes in TBST, proteins were detected using a chemiluminescence reagent (Chemiluminescence Reagent Plus; Perkin-Elmer, Boston, MA). Images were viewed using a Genius2 Bio imaging system (Syngene, Frederick, MD).
Pulldown assay.
HEK-293T cells were transfected using a calcium-phosphate technique (22). At 24 h posttransfection, cells were washed in PBS and resuspended in buffer A (20 mM HEPES, pH 7.4, 150 mM NaCl) and 1 mM phenylmethylsulfonyl fluoride (PMSF) (MP Biomedicals, Solon, OH). Cells were lysed via sonication, and Triton X-100 was added to a final concentration of 1%. Lysed cells were incubated for 20 min at 4°C, followed by centrifugation (in a microcentrifuge) at 15,000 rpm for 10 min at 4°C. Extracts were incubated with 20 μg of glutathione S-transferase (GST) fused to the WW domain of Itch (23) for 16 h at 4°C, along with glutathione Sepharose 4B (Bioworld, Dublin, OH), and then washed extensively with buffer A containing 1% Triton X-100 solution, and proteins were resolved by Western blotting.
RESULTS
Upregulation of LITAF upon viral infection.
LITAF was first identified as a gene upregulated in the presence of LPS, a major component of the outer membranes of Gram-negative bacteria (6). LPS is an endotoxin that elicits a strong response from the host immune system, and LITAF was therefore identified as a PAMP-induced gene (6). We were therefore interested in examining whether LITAF was also upregulated following a viral infection. FHM cells were infected with FV3 at an MOI of 20, and at various times postinfection LITAF was detected using anti-LITAF antibodies in a Western blot (Fig. 2). In mock-infected cells, the level of LITAF remained low (Fig. 2). However, the level of LITAF increased substantially at 2 h postinfection and remained elevated at 4 and 6 h postinfection (Fig. 2). These results suggest that LITAF is rapidly induced following an FV3 infection.
Fig 2.
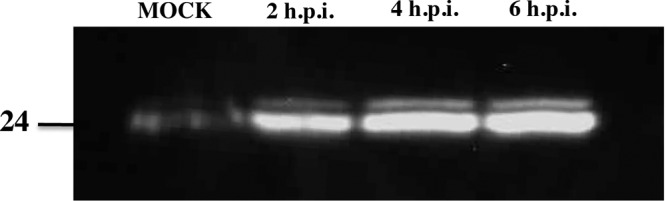
LITAF is induced following FV3 infection. FHM cells were harvested 0, 2, 4, and 6 h following an FV3 infection. LITAF (24 kDa) was detected using an anti-LITAF antibody in a Western blot.
FV3 75L localizes to early endosomes.
Cellular localization often helps to reveal a potential function for a specific protein. Therefore, to determine the subcellular localization of FV3 75L, we transiently transfected a myc-tagged FV3 75L construct into BGMK cells. FV3 75L exhibited a punctate staining pattern that was found to overlap that of the early endosome marker EEA1 (Fig. 3A). The FV3 75L pattern did not overlap those of other cellular markers, including LysoTracker, which stains late endosomes/lysosomes (Fig. 3A). Similar results were seen in human HEK-293T cells (data not shown). These data suggest that FV3 75L localizes to the early endosome.
Fig 3.

FV3 75L and LITAF localize to different cellular compartments. BGMK cells were transiently transfected with myc-tagged FV3 75L (A) or FLAG-tagged LITAF (B). LysoTracker was applied to live cells at 24 h posttransfection, and cells were then fixed and permeabilized. Cells then underwent indirect immunofluorescence assay, and FV3 75L was detected using anti-myc antibodies (red) and LITAF was visualized using anti-FLAG antibodies (green). Late endosomes/lysosomes (LysoTracker) and early endosomes (EEA1) are shown in blue. Nuclei were visualized using differential interference contrast (DIC) microscopy. All images were taken using a laser scanning confocal microscope.
LITAF alters the subcellular localization of FV3 75L.
LITAF was previously found to localize to the Golgi apparatus, plasma membrane, and late endosome/lysosome, and its localization is thought to be cell type specific (6, 8, 24, 25). As previously demonstrated, a transfected FLAG-tagged LITAF construct localized with LysoTracker but not EEA1 (Fig. 3B). Early endosomes mature into late endosomes, and both types of endosomes are part of a dynamic system involving ubiquitin-mediated lysosomal protein degradation. The possibility therefore exists that FV3 75L and LITAF exist at least transiently within the same compartment. In order to determine whether any subcellular overlap exists between FV3 75L and LITAF, myc-tagged FV3 75L and FLAG-tagged LITAF were cotransfected into BGMK cells, and indirect immunofluorescence assay was completed along with LysoTracker and EEA1 staining. Instead of individual localization patterns, we found that FV3 75L and LITAF staining overlapped within the cell and that this overlap colocalized with LysoTracker and not EEA1 (Fig. 4A). In addition, we observed the colocalization of 75L and LITAF in Xenopus laevis A6 cells (data not shown). These data suggest a potential interaction between 75L and LITAF that results in altered subcellular localization of FV3 75L from early to late endosomes in the presence of LITAF.
Fig 4.
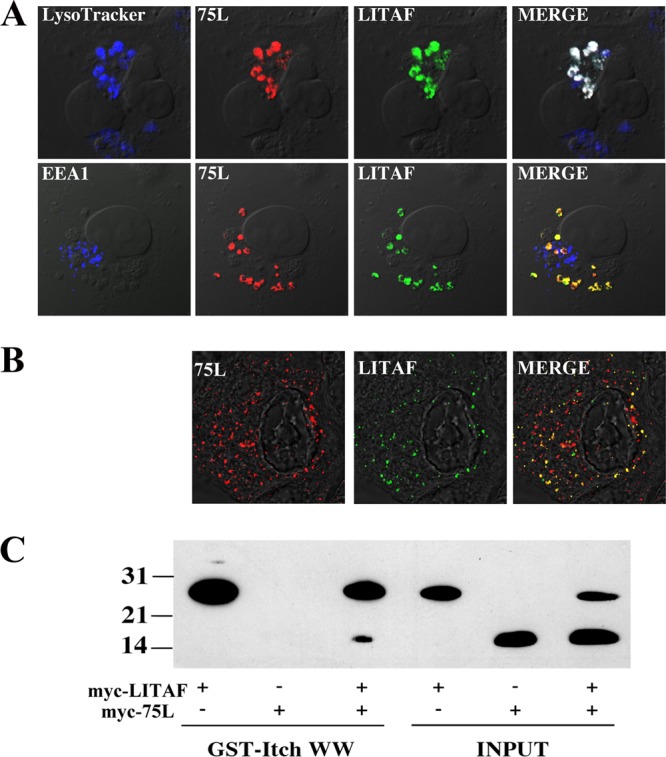
Alteration of cellular LITAF localization and its interaction with FV3 75L. (A) BGMK cells were transiently cotransfected with myc-tagged LITAF and myc-tagged 75L. At 24 h posttransfection, cells were incubated with LysoTracker and processed for indirect immunofluorescence assay. 75L was detected using anti-myc antibodies (red), LITAF was detected with anti-FLAG antibodies (green), and early (EEA1) and late endosomes/lysosomes (LysoTracker) are shown in blue. (B) A6 cells were transfected with myc-tagged LITAF, and at 10 h posttransfection, the cells were infected with FV3. At 16 h posttransfection, the cells were fixed, LITAF was detected using anti-myc antibodies (green), and FV3-produced 75L was detected using anti-FV3 75L antibodies (red). DIC microscopy was used to visualize cell nuclei. (C) HEK-293T cells were transfected with myc-tagged FV3 75L, myc-tagged LITAF, or myc-tagged FV3 75L and myc-tagged LITAF together. Transfected lysates were incubated with a GST fusion protein (GST fused to the Itch WW domain) precoupled to glutathione Sepharose. Cell lysate and bound protein were detected following application of myc and FLAG antibodies during Western blotting. An aliquot of transfected lysates without the GST fusion protein or glutathione Sepharose was probed with myc and FLAG antibodies during Western blotting to verify the presence of the target protein. Molecular weights are shown to the left of the blot.
Virally produced 75L colocalizes with LITAF.
To determine if virally expressed 75L colocalizes with LITAF in amphibian cell lines, natural host cells for the virus, we transfected A6 cells with myc-tagged LITAF, and 10 h after transfection, we infected cells with FV3 for 6 h. All of the virally produced 75L colocalized with transfected LITAF (Fig. 4B). These data confirm that transfected 75L colocalizes in a manner similar to that of 75L produced during an FV3 infection.
FV3 75L and LITAF physically interact.
The ability of LITAF to alter the subcellular localization of FV3 75L suggests that these two proteins may directly interact. In order to investigate a potential interaction, a pulldown assay was performed. We have previously demonstrated that Itch, a cellular ubiquitin ligase, is able to interact with LITAF through its WW domain (24). WW domains are hydrophobic domains that are found in many cellular proteins and that facilitate protein-protein interactions. Specifically, the Itch WW domain can interact with two proline-rich (PPXY [where X is any amino acid]) motifs found in the N terminus of LITAF (24). A GST fusion protein that included GST fused to the WW domain of Itch (23) was incubated alongside a cellular lysate expressing myc-tagged LITAF, myc-tagged FV3 75L, or myc-tagged FV3 75L and myc-tagged LITAF. As predicted, the GST fusion protein was able to precipitate LITAF through the interaction mediated by the WW domains of Itch and the PPXY motifs of LITAF (Fig. 4C). The GST fusion protein was unable to precipitate FV3 75L alone, suggesting that no motif present on FV3 75L is able to physically interact with WW domains (Fig. 4C). However, when the cell lysate included both myc-tagged FV3 75L and myc-tagged LITAF, both FV3 75L and LITAF were detected by Western blotting (Fig. 4C). The fact that the GST-WW fusion protein cannot precipitate FV3 75L alone suggests that FV3 75L is precipitated through a direct interaction with LITAF.
Mutation of two PPXY domains in LITAF induces a change in its cellular localization.
LITAF has two proline-rich motifs (PPXY) in the N terminus. PPXY motifs mediate a wide range of protein-protein interactions, including the interactions between LITAF, Nedd4, Itch, and the putative tumor suppressor WWOX (7–9, 25). We were interested in determining whether the PPXY motifs in LITAF were responsible for the cellular relocalization of FV3 75L. Using site-directed mutagenesis, the tyrosine (Y) was mutated to an alanine (A) in both PPXY motifs. This was previously demonstrated to abolish all ability of this motif to interact with other proteins (26). The LITAF double mutant (LITAF Y23,61A) was transiently cotransfected into cells along with myc-tagged FV3 75L. Very high levels of overlap between LITAF Y23,61A and FV3 75L were evident; however, LITAF Y23,61A and FV3 75L were found to colocalize with the early endosome marker EEA1 and not with LysoTracker (Fig. 5). This suggests that mutation of LITAF's PPXY motifs abolishes the ability of LITAF to change the cellular localization of FV3 75L, and instead results in a change in cellular localization of LITAF from late to early endosomes. The fact that both proteins still strongly colocalize within the cell suggests, however, that the PPXY motifs are not responsible for the interaction between LITAF and FV3 75L.
Fig 5.
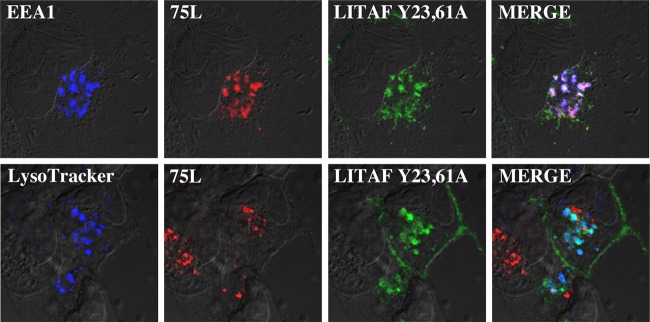
Mutations to PPXY domains of LITAF alter subcellular localization. BGMK cells were transiently transfected with LITAF Y23,61A. Site-directed mutagenesis was used to specifically mutate the tyrosine in the two PPXY motifs in LITAF to alanine. At 24 h posttransfection, live cells were stained with LysoTracker and/or processed for indirect immunofluorescence assay. FV3 75L was detected using anti-myc antibodies (red), LITAF was detected with anti-FLAG antibodies (green), and early (EEA1) and late endosomes/lysosomes (LysoTracker) are shown in blue. DIC microscopy was used to visualize cell nuclei.
SGIV136 localization is cell type specific.
FV3 75L localizes to early endosomes. In contrast, the FV3 75L homolog SGIV136 localizes (at least partially) to the mitochondria (13). SGIV136 and FV3 75L both contain the highly conserved SLD, suggesting similar functions between the two proteins, and it would be expected that they would have the same subcellular localization. Since FV3 75L localizes to the early endosomes and SGIV136 localizes to the mitochondria (13), we wanted to test whether the localization of SGIV136 is cell type specific. BGMK cells were transfected with myc-SGIV136, and indirect immunofluorescence assay demonstrated that SGIV136 localized with the mitochondrial marker MitoTracker and not with the late endosome/lysosome marker, in agreement with the work of Huang et al. (14) (Fig. 6A). In a fibroblast line derived from the Xenopus adult kidney (A6), SGIV136 colocalized with the late endosomal/lysosomal marker CD63 (Fig. 6A). These data suggest that SGIV136 localization is cell type specific, confirming that SGIV136 is localized to the late endosome/lysosome. Interestingly, FV3 75L did not show different subcellular localizations: FV3 75L localized to the early endosome in both BGMK cells (Fig. 3A) and A6 cells (Fig. 6B).
Fig 6.
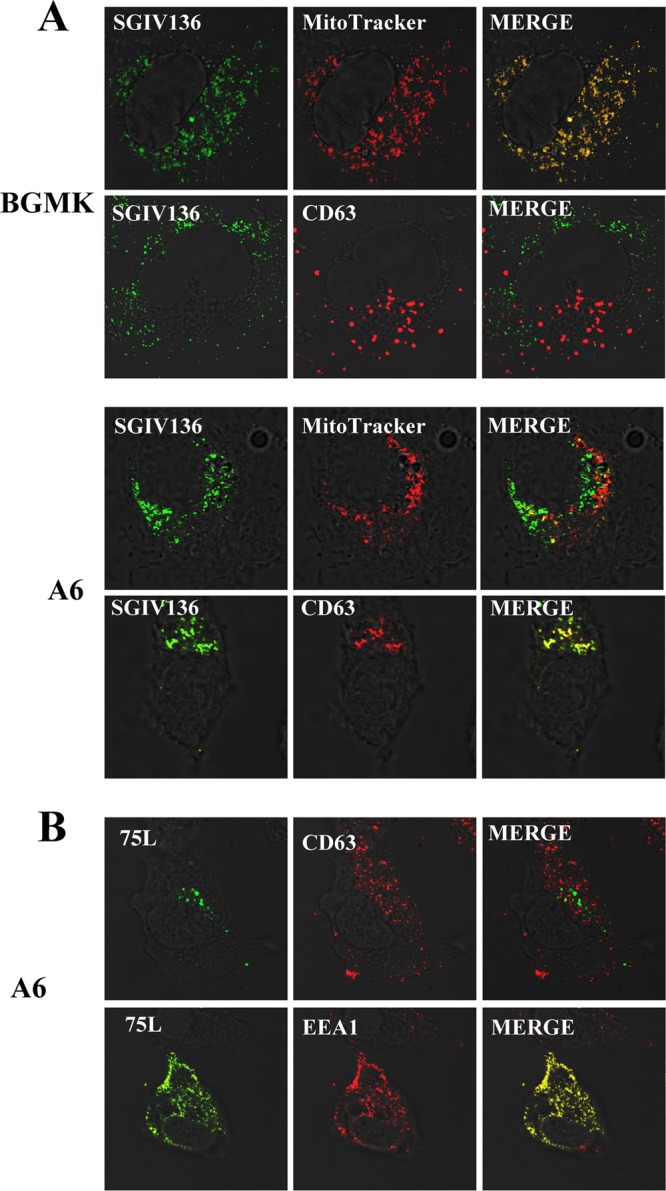
SGIV136 cellular localization is cell type specific. BGMK and/or A6 cells were transfected with myc-SGIV136 (A) or myc-FV3 75L (B). At 24 h posttransfection, the cells were incubated with MitoTracker (red). Cells were fixed and stained for SGIV136 or FV3 75L (anti-myc antibodies; green), lysosomes (anti-CD63; red), and mitochondria (MitoTracker; red). DIC microscopy was used to visualize cells, and the images were captured using a laser scanning microscope.
DISCUSSION
In this study, we demonstrated a novel interaction between the viral protein FV3 75L and the cellular homolog LITAF. Both proteins were found to localize independently to different compartments, but when they were cotransfected into cells, LITAF was able to alter the subcellular localization of FV3 75L. We demonstrated that this change in cellular localization is mediated through direct binding between FV3 75L and LITAF. Furthermore, the presence or absence of the two PPXY motifs in the N terminus of LITAF determines whether LITAF and FV3 75L localize to early or late endosomes. LITAF and FV3 75L are both found within organelles that play essential roles in ubiquitin-mediated lysosomal protein degradation. The dynamic nature of this pathway suggests that LITAF and FV3 75L are at least transiently present in the same cellular compartment.
Mutation of both PPXY motifs in LITAF changed the subcellular localization of LITAF and FV3 75L but did not alter the colocalization between the two proteins. This suggests that the interaction between LITAF and FV3 75L is not mediated through the PPXY motifs in the N terminus of LITAF but instead may be mediated through the C terminus (SLD) of LITAF. No known protein-protein interaction motifs are present in the SLD of LITAF, which therefore suggests that the highly conserved SLD may function in mediating protein-protein interactions. In order to determine whether the SLD mediates an interaction between LITAF and FV3 75L, mutant constructs lacking sections of or the entire SLD can be tested along with FV3 75L to determine which domains are responsible for the interaction. Changes in the ability of the mutant LITAF and FV3 75L constructs to interact can also be confirmed using a GST pulldown assay or immunoprecipitation followed by Western blotting.
Due to the fact that FV3 75L appears to be a truncated version of LITAF, with FV3 75L having sequence similarity to the C terminus of LITAF (3), it is possible that FV3 75L acts in a dominant-negative manner to antagonize the function of LITAF. While the N terminus of LITAF appears to be involved in mediating protein-protein interactions through several proline-rich binding motifs, the function of the C terminus in LITAF and FV3 75L remains unknown (4). The ability of FV3 75L and LITAF to interact and the fact that this interaction does not appear to require the two PPXY protein-protein interaction motifs in the N terminus of LITAF suggest that the C terminus may be able to mediate homotypic binding. It is possible that LITAF typically requires substrate binding at both the N terminus (through proline-rich motifs) and the C terminus (through the SLD) in order to elicit some sort of response. FV3 75L may act to outcompete other substrates or bind preferentially to LITAF through the SLD, thereby reducing or eliminating the typical response of LITAF.
It is interesting that SGIV136 localizes to either the mitochondria or early endosomes, depending on the cell type, since the C terminus of LITAF contains a YXXΦ motif (where X is any amino acid and Φ is any bulky hydrophobic amino acid). This motif is known to interact with adaptor proteins involved in sorting of membrane proteins as well as targeting proteins to the lysosome or trans-Golgi network (27, 28). Lee et al. (29) identified a C-rich domain in LITAF that is responsible for its transmembrane association. The absence of an endoplasmic reticulum (ER)-targeting signal sequence and the fact that the transmembrane domain is localized close to the C terminus of LITAF suggest that LITAF undergoes posttranslational insertion as a C-tail-anchored membrane protein (30, 31). Based on this, it is possible to speculate that the different protein compositions present in different cell lines could play a role in the differences in cellular localization of SGIV136 but not FV3 75L. It is possible that the N terminus of SGIV136 interacts with other proteins in A6 cells that target it for lysosomal localization.
The FV3 75L gene was identified as a late viral gene (32) by use of an oligonucleotide microarray, in contrast to the case for the SGIV136 gene, which was found to be an immediate-early gene. We previously found that FV3 75L is expressed as early as 3 h postinfection (33), and upregulation of LITAF occurs as early as 2 h postinfection, suggesting that the FV3 75L gene may be an early gene. Growing evidence suggests that apoptosis plays a critical role in viral pathogenesis and that viruses are able to carry genes that trigger apoptosis. Considering that the mitochondria are the central control point of apoptosis, Huang et al. (14) suggested that mitochondria may be involved in LITAF-induced apoptosis. Viruses that induce apoptosis represent an important step in the spread of progeny to neighboring cells while also evading the host immune and inflammatory responses (3), leading Huang et al. (14) to suggest that apoptosis induced by SGIV136 may contribute to virus transmission during SGIV replication. Future experiments will focus on confirming the temporal classes of FV3 75L and SGIV 136, as well as exploring whether FV3 75L plays a role in the induction or inhibition of apoptosis during a viral infection in FHM and BGMK cells. If FV3 encodes a protein to antagonize the function of LITAF, then we would speculate that the FV3 75L gene product should be an early transcript. Future studies need to determine whether LITAF has antiviral functions. A reduction in cellular LITAF levels through the use of RNA interference or antisense morpholinos could be used to compare FV3 titers in cell lines containing and lacking LITAF. If LITAF does have antiviral activity, then one would expect higher FV3 titers following a reduction in cellular LITAF.
ACKNOWLEDGMENTS
We thank Dylan Drummond for providing feedback on the manuscript.
This work was supported by discovery grants (Natural Science and Engineering Research Council [NSERC] of Canada) to C.R.B. H.E.E. is the recipient of an NSERC postgraduate scholarship.
Footnotes
Published ahead of print 24 October 2012
REFERENCES
- 1. Williams T. 1996. The iridoviruses. Adv. Virus Res. 46:345–412 [DOI] [PubMed] [Google Scholar]
- 2. Williams T, Chinchar G, Darai G, Hyatt A, Kalmakoff J, Seligg V. 2000. Family Iridoviridae, p 167–182 In van Regenmortel MHV. (ed), Virus taxonomy: 7th report of the International Committee on Taxonomy of Viruses Academic Press, New York, NY [Google Scholar]
- 3. Tan WG, Barkman TJ, Chinchar G, Essani K. 2004. Comparative genomic analyses of frog virus 3, type species of the genus Ranavirus (family Iridoviridae). Virology 323:70–84 [DOI] [PubMed] [Google Scholar]
- 4. Myokai F, Takashiba S, Lebo R, Amar S. 1999. A novel lipopolysaccharide-induced transcription factor regulating tumor necrosis factor alpha gene expression: molecular cloning, sequencing, characterization, and chromosomal assignment. Proc. Natl. Acad. Sci. U. S. A. 96:4518–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. 1997. A model for p53-induced apoptosis. Nature 389:300–305 [DOI] [PubMed] [Google Scholar]
- 6. Moriwaki Y, Begum NA, Kobayashi M, Matsumoto M, Toyoshima K, Seya T. 2001. Mycobacterium bovis bacillus Calmette-Guerin and its cell wall complex induce a novel lysosomal membrane protein, SIMPLE, that bridges the missing link between lipopolysaccharide and p53-inducible gene, LITAF(PIG7), and estrogen-inducible gene, EET-1. J. Biol. Chem. 276:23065–23076 [DOI] [PubMed] [Google Scholar]
- 7. Ingham RJ, Gish G, Pawson T. 2004. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23:1972–1984 [DOI] [PubMed] [Google Scholar]
- 8. Shirk AJ, Anderson SK, Hashemi SH, Chance PF, Bennett CL. 2005. SIMPLE interacts with NEDD4 and TSG101: evidence for a role in lysosomal sorting and implications for Charcot-Marie-Tooth disease J. Neurosci. Res. 82:43–50 [DOI] [PubMed] [Google Scholar]
- 9. Eaton HE, Desrochers G, Drory SB, Metcalf J, Angers A, Brunetti CR. 2011. SIMPLE/LITAF expression induces the translocation of the ubiquitin ligase Itch towards the lysosomal compartments. PLoS One 6:e16873 doi:10.1371/journal.pone.0016873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Letourneur F, Klausner RD. 1992. A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell 69:1143–1157 [DOI] [PubMed] [Google Scholar]
- 11. Sandoval IV, Arredondo JJ, Alcalde J, Gonzalez Noriega A, Vandekerckhove J, Jimenez MA, Rico M. 1994. The residues Leu(Ile)475-Ile(Leu, Val, Ala)476, contained in the extended carboxyl cytoplasmic tail, are critical for targeting of the resident lysosomal membrane protein LIMP II to lysosomes. J. Biol. Chem. 269:6622–6631 [PubMed] [Google Scholar]
- 12. Tikkanen R, Obermuller S, Denzer K, Pungitore R, Geuze HJ, von Figura K, Honing S. 2000. The dileucine motif within the tail of MPR46 is required for sorting of the receptor in endosomes. Traffic 1:631–640 [DOI] [PubMed] [Google Scholar]
- 13. Hong YH, Lillehoj HS, Lee SH, Park DW, Lillehoj EP. 2006. Molecular cloning and characterization of chicken lipopolysaccharide-induced TNF-alpha factor (LITAF). Dev. Comp. Immunol. 30:919–929 [DOI] [PubMed] [Google Scholar]
- 14. Huang X, Huang Y, Gong J, Yan Y, Qin Q. 2008. Identification and characterization of a putative lipopolysaccharide-induced TNF-alpha factor (LITAF) homolog from Singapore grouper iridovirus. Biochem. Biophys. Res. Commun. 373:140–145 [DOI] [PubMed] [Google Scholar]
- 15. Mavian C, López-Bueno A, Fernández Somalo MP, Alcamí A, Alejo A. 2012. Complete genome sequence of the European sheatfish virus. J. Virol. 86:6365–6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang Y, Huang X, Liu H, Gong J, Ouyang Z, Cui H, Cao J, Zhao Y, Wang X, Jiang Y, Qin Q. 2009. Complete sequence determination of a novel reptile iridovirus isolated from soft-shelled turtle and evolutionary analysis of Iridoviridae. BMC Genomics 10:224–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dupre S, Volland C, Haguenauer-Tsapis R. 2001. Membrane transport: ubiquitylation in endosomal sorting. Curr. Biol. 11:R932–R934 [DOI] [PubMed] [Google Scholar]
- 18. Liu G, Cao G, Xue J, Wan J, Wan Y, Jiang Q, Yao J. 2012. Tumor necrosis factor-alpha (TNF-α)-mediated in vitro human retinal pigment epithelial (RPE) cell migration mainly requires Akt/mTOR complex 1 (mTORC1) but not mTOR complex 2 (mTORC2) signaling. Eur. J. Cell Biol. 91:728–737 [DOI] [PubMed] [Google Scholar]
- 19. Jackson PK, Eldridge AG, Freed E, Furstenthal L, Hsu JY, Kaiser BK, Reimann JD. 2000. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 10:429–439 [DOI] [PubMed] [Google Scholar]
- 20. Liu YC. 2004. Ubiquitin ligases and the immune response. Annu. Rev. Immunol. 22:81–127 [DOI] [PubMed] [Google Scholar]
- 21. Brennan FM, Feldmann M. 1996. Cytokines in autoimmunity. Curr. Opin. Immunol. 8:872–877 [DOI] [PubMed] [Google Scholar]
- 22. Kingston RE, Chen CA, Rose JK. 2003. Calcium phosphate transfection. Curr. Protoc. Mol. Biol. 63:9.1.1–9.1.11 [DOI] [PubMed] [Google Scholar]
- 23. Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, Angers A. 2006. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J. Biol. Chem. 281:38738–38747 [DOI] [PubMed] [Google Scholar]
- 24. Eaton HE, Desrochers G, Metcalf J, Ferreira Lacerda A, Angers A, Brunetti CR. 2012. Accumulation of endogenous LITAF in aggresomes. PLoS One 7:e30003 doi:10.1371/journal.pone.0030003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ludes-Meyers JH, Kil H, Bednarek AK, Drake J, Bedford MT, Aldaz CM. 2004. WWOX binds the specific proline-rich ligand PPXY: identification of candidate interacting proteins. Oncogene 23:5049–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen HI, Sudol M. 1995. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. U. S. A. 92:7819–7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonifacino JS, Dell'Angelica EC. 1999. Molecular bases for the recognition of tyrosine-based sorting signals. J. Cell Biol. 145:923–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simmen T, Schmidt A, Hunziker W, Beermann F. 1999. The tyrosinase tail mediates sorting to the lysosomal compartment in MDCK cells via a di-leucine and a tyrosine-based signal. J. Cell Sci. 112:45–53 [DOI] [PubMed] [Google Scholar]
- 29. Lee SM, Olzmann JA, Chin L, Li L. 2011. Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J. Cell Sci. 124:3319–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borgese N, Fasana E. 2011. Targeting pathways of C-tail-anchored proteins. Biochim. Biophys. Acta 1808:937–946 [DOI] [PubMed] [Google Scholar]
- 31. Borgese N, Brambillasca S, Colombo S. 2007. How tails guide tail-anchored proteins to their destination. Curr. Opin. Cell Biol. 19:368–375 [DOI] [PubMed] [Google Scholar]
- 32. Majji S, Thodima V, Sample R, Whitley D, Deng Y, Mao J, Chinchar VG. 2009. Transcriptome analysis of frog virus 3, the type species of the genus Ranavirus, family Iridoviridae. Virology 391:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eaton H, Metcalf J, Brunetti CR. 2008. Expression of frog virus 3 genes is impaired in mammalian cell lines. Virol. J. 5:83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]