

Abstract
CDT2 targets proteins involved in replication licensing (CDT1), cell cycle control (p21), and chromatin modification (SET8) for destruction by the CUL4-based E3 ligase (CRL4). CRL4CDT2 recruits these substrates through interactions with chromatin-bound PCNA and ubiquitinates them exclusively on chromatin. Rereplication and G2 cell cycle arrest are observed in CDT2-depleted cells. The rereplication phenotype has been attributed to an inability to destroy CDT1, but the molecular target important for G2 cell cycle arrest in CDT2-depleted cells has not been identified. Here we identify CHK1 as a novel CRL4CDT2 substrate and demonstrate that CHK1 activity is required for maintaining G2 arrest in CDT2-depleted cells. We demonstrate that CRL4CDT2 targets the activated form of CHK1 for destruction in the nucleoplasm rather than on chromatin and that this occurs in a PCNA-independent manner. Although both CRL1 and CRL4 ubiquitinate CHK1, we report that they bind CHK1 in distinct cellular compartments. Our study provides insight into how elevated CDT2 expression levels may provide tumors with a proliferative advantage.
INTRODUCTION
The CHK1 protein kinase maintains genome integrity in normal cycling cells and in cells exposed to replication or genotoxic stress (1, 2). Replication stress that occurs during the normal course of DNA replication or following exposure to antimetabolites or certain DNA-damaging agents generates single-stranded DNA (ssDNA). ssDNA is also generated in the course of DNA repair and double-strand break (DSB) end resection. The CHK1 signaling pathway is engaged by checkpoints that detect ssDNA. Replication protein A (RPA) coats ssDNA, thereby recruiting a DNA damage-sensing complex consisting of ATR (ataxia telangiectasia- and RAD3-related protein) and ATRIP (ATR-interacting protein) (3, 4). The ATR/ATRIP module, together with RAD17 and the 9-1-1 complex, activates CHK1 in a claspin-dependent manner on chromatin (5–9). ATR phosphorylates CHK1 on serine 317 (S317) and serine 345 (S345), which in turn activates CHK1 by facilitating autophosphorylation on S296 (10–13). Activated CHK1 is then released from chromatin and phosphorylates downstream effectors to temporarily halt cell cycle progression, stabilize stalled replication forks, and regulate DNA repair (4, 14).
ATR-mediated phosphorylation activates CHK1 and also promotes its ubiquitin-mediated proteolysis by facilitating interactions with two distinct E3 ubiquitin ligases that employ CUL1 and CUL4A (15–17). These cullin proteins function as scaffolds in multisubunit complexes known as cullin-RING ligases (CRLs) (18). CRLs recruit substrates via adaptor proteins specific for each cullin scaffold. CRL1 employs SKP1 (S-phase kinase-associated protein 1), and CRL4 utilizes DDB1 (damaged DNA binding protein 1). Cullin-adaptor complexes often require additional substrate receptors to recruit and ubiquitinate target proteins. Substrate receptors provide E3 ubiquitin ligases with the specificity required to target their diverse repertoire of cellular substrates for ubiquitination. While F-box proteins recruit substrates to CRL1, CRL4 often recruits its substrates via DCAFs (DDB1- and CUL4-associated factors) (19–21). More than a hundred DCAFs and putative DCAF proteins have been identified based on characteristic motifs, including WD40 repeats, WDXR motifs, and DDB boxes (19–23). The DCAF protein CDT2 recognizes substrates containing a specialized PCNA (proliferating cell nuclear antigen) interaction protein motif (PIP box) called a PIP degron (24). Chromatin-bound PCNA mediates the recruitment of PIP degron-containing substrates to CRL4CDT2 (24).
The F-box protein FBX6 facilitates interactions between CHK1 and CRL1 (16), but the substrate receptor mediating interactions between CHK1 and CRL4 has not been identified. Furthermore, it is unclear why two distinct E3 ubiquitin ligases mediate CHK1 degradation. Here we demonstrate that CDT2 targets the activated form of CHK1 to CRL4 using a noncanonical mechanism and that CHK1 stability is regulated in distinct cellular compartments by CRL1FBX6 and CRL4CDT2. We also demonstrate that CHK1 kinase activity is essential for the maintenance of G2 cell cycle arrest in CDT2-depleted cells.
MATERIALS AND METHODS
Cell culture, antibodies, and reagents.
HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 10% bovine growth serum, l-glutamine, and penicillin-streptomycin. HeLa Tet-on cells (Clontech) were grown in DMEM supplemented with 10% Tet system-approved fetal bovine serum (Clontech), l-glutamine, penicillin-streptomycin, and 100 μg/ml Geneticin (Life Technologies). 293T cells were grown in DMEM supplemented with 10% fetal bovine serum and l-glutamine. The following antibodies were purchased: CHK1 (G-4), CUL1 (H-213), CDT2 (B-8), Myc (9E10), PCNA (PC10), SKP1, and FBX6 (7B11) antibodies were purchased from Santa Cruz Biotechnology; actin, Flag (M2), and claspin antibodies were purchased from Sigma; CUL4 and CDT1 antibodies were purchased from Bethyl Laboratories; CUL4A antibody was purchased from Rockland Immunochemicals; V5, CDT2, DDB1, and tubulin antibodies were purchased from Abcam; CHK1 phospho-S296 (pS296) antibody was purchased from Epitomics; CHK1 phospho-S345 (pS345) (133D3) antibody was purchased from Cell Signaling; β-catenin and ORC2 antibodies were purchased from BD Biosciences; and SET8 antibody was purchased from Millipore. The affinity-purified anti-CHK1 antibody was described previously (15). Hydroxyurea (HU), MG132 (Z-Leu-Leu-Leu-al), and cycloheximide (CHX) were purchased from Sigma, doxycycline was purchased from Clontech, and AZD7762 (AZD) was purchased from Axon Medchem. Cells were irradiated by using a UV Stratalinker 2400 instrument (Stratagene).
Plasmid and RNAi transfections.
Plasmids expressing 3×Flag-CHK1 and V5-CUL4A were described previously (15), and a plasmid expressing V5-CUL1 was similarly generated by Van Leung-Pineda. A plasmid encoding Tet-inducible Flag-CHK1 was described previously (16) and was provided by You-Wei Zhang. pEFF-CDT1 was described previously (25) and was provided by Anindya Dutta. pDEST-N-Myc-CDT2 was provided by Wade Harper and Malavika Raman. CHK1 3RE and CHK1 A36F (kinase-inactive) mutants, claspin binding mutants (K54A, R129A, and T153A), a CHK1 phosphorylation site mutant (3SA), CHK1 PIP box mutants (ΔNPIP1, ΔNPIP2, and 2FA), and the CDT2 R246A (RA) mutant were generated by using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). Lipofectamine 2000 transfection reagent (Life Technologies) was used for plasmid transfections. For RNA interference (RNAi) studies, Luciferase GL3 Duplex and SMARTpool small interfering RNAs (siRNAs) specific for CUL1, CUL4, DDB1, CDT2, or PCNA were obtained from Thermo Scientific and transfected with DharmaFECT 1 siRNA transfection reagent (Thermo Scientific). The CDT2 SMARTpool consisted of four siRNAs that were tested individually as well (CDT2-5 [GCGCUUGAAUAGAGGCUUA], CDT2-6 [ACUCCUACGUUCUCUAUUA], CDT2-7 [GAAUUAUACUGCUUAUCGA], and CDT2-8 [GUCAAGACCUGGCCUAGUA]).
Coimmunoprecipitation.
Cells were lysed in mammalian cell lysis buffer (MCLB) (50 mM Tris-HCl [pH 8.0], 0.5% NP-40, 5 mM EDTA, 100 mM NaCl, 2 mM dithiothreitol [DTT]) containing protease (Sigma) and phosphatase (Calbiochem) inhibitor cocktails. Cell lysates were precleared with protein A-agarose (Thermo Scientific) and incubated with anti-Flag M2 affinity gel (Sigma). Immunocomplexes were washed with MCLB, eluted with Flag peptide, and analyzed by Western blotting. Alternatively, endogenous CHK1 was immunoprecipitated from precleared lysates by using protein A-agarose and affinity-purified anti-CHK1 antibody.
Protein phosphatase treatment.
Protein phosphatase treatment of cell lysates was performed according to previously reported protocols (26), with the indicated modifications. Cells were lysed in MCLB lacking EDTA (50 mM Tris-HCl [pH 8.0], 0.5% NP-40, 100 mM NaCl, 2 mM DTT) and supplemented with a protease inhibitor cocktail. Cleared lysates were incubated with MnCl2 and Lambda protein phosphatase (New England BioLabs) at 30°C for 20 min, and reactions were terminated by boiling in SDS-PAGE loading buffer.
Immunofluorescence.
Cells grown on coverslips were fixed with 4% formaldehyde for 10 min at room temperature and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS). After blocking with 5% normal donkey serum in 0.1% Triton X-100 in PBS, cells were stained with mouse anti-Flag (M2) antibody and rabbit anti-CDT2 antibody followed by donkey anti-mouse IgG (fluorescein isothiocyanate [FITC]) and anti-rabbit IgG (Cy5) secondary antibodies in blocking solution. Coverslips were mounted by using ProLong Gold Antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies). Images were acquired by using an Olympus BX61 microscope, a Hamamatsu c10600 camera, a UPlanFl 40× lens, and Olympus MicroSuite software.
In vitro ubiquitination.
CHK1 ubiquitination in vitro was performed as described previously (15), with the following modifications. Flag-CHK1 was purified from HU-treated 293T cells to promote CHK1 phosphorylation. Immunoprecipitated Flag-CHK1 was sequentially washed with lysis buffer (20 mM Tris-HCl [pH 8.0], 0.5% NP-40, 1 mM EDTA, 150 mM NaCl), LiCl buffer (50 mM Tris-HCl [pH 8.0], 0.5 M LiCl), and ubiquitination buffer (25 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 2 mM NaF) and then eluted with Flag peptide in ubiquitination buffer. CRL4 complexes were purified from HU-treated HeLa cells by using protein A-agarose and an anti-CUL4A antibody (Rockland Immunochemicals). Immunoprecipitated CRL4 complexes were consecutively washed with lysis buffer and ubiquitination buffer. Ubiquitination reactions were carried out with Flag-CHK1 (substrate), CRL4 complex, 0.1 μM E1 (Sigma), 0.4 μM E2 (Sigma), and 10 μg bovine ubiquitin (Sigma) in ubiquitination buffer supplemented with 2 mM ATP and 0.6 mM DTT at 37°C for 75 min, and reactions were terminated by boiling in SDS-PAGE loading buffer.
Cell fractionation.
The fractionation of cellular protein was performed according to previously reported methods, with modifications (14, 27). Cells were harvested by trypsinization, washed in PBS, and resuspended in solution A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT) containing 0.1% Triton X-100 and protease and phosphatase inhibitor cocktails. Supernatants (cytoplasmic fractions) were collected after centrifugation at 1,300 × g for 4 min at 4°C. Nuclear pellets were washed twice with solution A without Triton X-100 and then resuspended in LS buffer (10 mM Tris-HCl [pH 7.4], 0.2 mM MgCl2) containing 1% Triton X-100 and protease and phosphatase inhibitor cocktails. After centrifugation at 1,700 × g for 4 min at 4°C, supernatants (soluble nuclear fractions) were collected, and pellets were washed twice with LS buffer. Chromatin fractions were collected in increasing salt concentrations (0.3, 0.5, and 2.0 M NaCl in LS buffer), which extract proteins that are bound to chromatin weakly, moderately, and strongly, respectively. For coimmunoprecipitations, detergent and salt concentrations of each fraction were matched, and endogenous CHK1 was immunoprecipitated by using affinity-purified anti-CHK1 antibody.
Cell cycle analysis.
Cells were harvested by trypsinization and fixed in 70% ethanol at 4°C. Cells were washed with 1% bovine serum albumin (BSA) in PBS and incubated in 1% BSA in PBS containing 30 μg/ml propidium iodide (Sigma) and 250 μg/ml RNase A for 1 h at room temperature. Flow cytometry was performed by using a FACSCalibur flow cytometer (BD Biosciences) and analyzed by using CellQuest software (BD Biosciences).
Statistical analysis.
The quantitation of Western blots was performed by using ImageJ software (28). Data were analyzed by Student's t test, and statistically significant differences are indicated as P values of <0.05 (∗), P values of <0.01 (∗∗), and P values of <0.005 (∗∗∗). All data are presented as means ± standard errors of the means (SEM).
RESULTS
CRL4 is a major regulator of CHK1 stability.
Individual contributions to CHK1 destruction made by CRL1 and CRL4 were investigated by using HeLa cells. The knockdown of CUL4 but not CUL1 resulted in an increase in CHK1 levels both in normal cycling cells and in cells experiencing replication stress due to hydroxyurea (HU) exposure (Fig. 1A and B). Similar results were obtained when HeLa cells were exposed to UV radiation (Fig. 1C). These results indicate that the function of CRL1 and CRL4 is not entirely redundant and that CUL4 assembles the predominant E3 ligase complex required for CHK1 destruction in HeLa cells.
Fig 1.
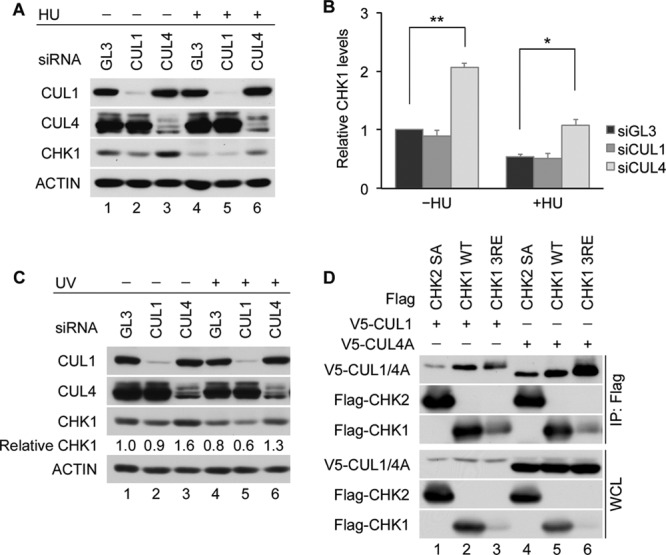
CRL4 is a major regulator of CHK1 stability. (A and B) Contributions made by CRL1 and CRL4 to CHK1 stability. HeLa cells transfected with control siRNA (GL3) or siRNAs specific for either CUL1 or CUL4 were cultured for 48 h and then incubated in the absence or presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. A representative experiment is shown in panel A, and the quantitation of data from 3 independent experiments is shown in panel B. (C) Effects of UV on CHK1 stability. HeLa cells transfected with control siRNA or siRNAs specific for CUL1 or CUL4 were mock irradiated or exposed to 50 J/m2 UV at 48 h posttransfection. Cells were harvested 6 h later and analyzed by Western blotting. Levels of CHK1 in knockdown cells are shown relative to levels in control cells (lane 1). (D) Interactions between CUL4A and CHK1. The Flag-tagged CHK1 WT or the CHK1 3RE or CHK2 SA mutant was coproduced with V5-tagged CUL1 or CUL4A in HeLa cells. Cell lysates were resolved directly by SDS-PAGE or were immunoprecipitated with anti-Flag M2 affinity gel prior to SDS-PAGE. Whole-cell lysates (WCL) and immunoprecipitates (IP: Flag) were then analyzed by Western blotting. CUL1 and CUL4A were detected by using an anti-V5 antibody.
The C terminus of CHK1 contains an autoinhibitory region (AIR) that is relieved during periods of replication stress due to phosphorylation on S317 and S345 (29, 30). The resulting conformational change exposes a degron that facilitates the ubiquitin-mediated proteolysis of CHK1 (16). CHK1 autoinhibition can be constitutively relieved by mutating three highly conserved arginine (R) residues within its C terminus to glutamate (E). This mutant (CHK1 3RE) has a very short half-life in cells (16). As shown in Fig. 1D, the CHK1 3RE mutant was expressed at much lower levels than the CHK1 WT (wild type), but its association with CUL1 and CUL4A (lanes 3 and 6) was greater than that of the CHK1 WT (lanes 2 and 5) (16). A CHK2 mutant encoding a substitution of alanine for serine at position 379 (SA) was used as a negative control (26). This suggests that CRL4-mediated ubiquitination also contributes to the rapid turnover of the CHK1 3RE mutant.
CDT2 is a novel CHK1 regulator.
We identified DDB1 as an adaptor protein that mediates CHK1 ubiquitination by CRL4 (15). However, attempts to reconstitute direct interactions between CHK1 and DDB1 in vitro with purified components were unsuccessful (data not shown). As CRL4 complexes often require additional substrate receptors called DCAFs to recruit substrates to CRL4, we hypothesized that the interaction between CHK1 and CRL4 is mediated by a substrate receptor. CDT2 is a DCAF involved in the S-phase and DNA damage-specific degradation of key cell cycle regulatory proteins such as CDT1, p21, and SET8 (23, 31–41). CHK1 degradation occurs after its phosphorylation and activation by ATR. Given that CHK1 is activated in the S and G2 phases of the cell cycle in normal cycling cells as well as in response to replication stress, we tested whether CDT2 serves as a substrate receptor for CHK1.
The turnover of CHK1 was monitored in HeLa cells depleted of CDT2 in both the absence and presence of replication stress (Fig. 2A and B). The ability of replication stress (HU) to induce CHK1 turnover was significantly impaired in CDT2-depleted cells relative to control cells (P value of 0.0065). In the absence of replication stress, the CDT2 knockdown had a more modest effect on CHK1 turnover (P value of 0.088). CHK1 turnover was also impaired in HU-treated HEK293 cells depleted of CDT2 (data not shown). Furthermore, the stabilization of CHK1 in HU-treated HeLa cells was observed when CDT2 was depleted with several distinct siRNAs (Fig. 2C) and when cells were cultured in the presence of 0.5 to 5.0 mM HU (Fig. 2D). Phosphatase experiments were performed in advance of SDS-PAGE and Western blotting in order to dephosphorylate CHK1 prior to determining its relative levels in control and CDT2-depleted cells. This experiment was performed to rule out the possibility that the CHK1 antibody used in our study poorly recognized the phosphorylated forms of CHK1. As shown in Fig. 2E, phosphatase treatment did not alter the conclusion that a CDT2 deficiency impairs CHK1 turnover in HU-treated cells. In addition to HU treatment, the stabilization of CHK1 was also observed in CDT2-depleted cells exposed to UV radiation (Fig. 2F, lanes 3 and 4).
Fig 2.

CDT2 is a novel CHK1 regulator. (A and B) CDT2 is required for CHK1 turnover. HeLa cells transfected with control siRNA (GL3) or pooled siRNAs specific for CDT2 were cultured for 48 h and then incubated in the absence or presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. A representative experiment is shown in panel A, and the quantitation of data from 3 independent experiments is shown in panel B (N.S., not statistically significant by Student's t test). (C) Regulation of CHK1 by CDT2. HeLa cells transfected with control siRNA or individual siRNAs specific for CDT2 were cultured for 48 h and then incubated in the presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. (D) Effects of HU on CHK1 turnover. HeLa cells transfected with control siRNA or siRNA specific for CDT2 were cultured for 48 h and then incubated in the presence of the indicated doses of HU for 6 h. Cell lysates were analyzed by Western blotting. (E) CHK1 levels after dephosphorylation in vitro. HeLa cells transfected with control siRNA or siRNAs specific for CDT2 were cultured for 48 h and then incubated in medium containing 5 mM HU for 6 h. Cell lysates were incubated in the absence or presence of Lambda protein phosphatase (PPase), followed by Western blotting. Levels of CHK1 in cells knocked down for CDT2 are shown relative to levels in control cells (lanes 1 and 3). (F) Effects of UV on CHK1 turnover. HeLa cells transfected with control siRNA or siRNA specific for CDT2 were mock irradiated or exposed to 50 J/m2 UV at 48 h posttransfection. Cells were harvested 6 h later and analyzed by Western blotting. Levels of CHK1 in knockdown cells are shown relative to levels in control cells (lane 1). (G) Regulation of CHK1 stability by CRL1, CRL4, and CDT2. HeLa cells transfected with control siRNA or siRNAs specific for CUL1, CUL4, or CDT2 were cultured for 48 h and then incubated in the presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. Levels of CHK1 in knockdown cells are shown relative to levels in control cells (lane 1).
In the absence of replication stress, the depletion of CDT2 did not increase CHK1 levels as much as did the depletion of CUL4 (Fig. 1B and 2B). However, in the presence of replication stress, CHK1 accumulated to similar levels in CDT2- and CUL4-depleted cells (Fig. 2G). In addition, CHK1 turnover was significantly delayed when CDT2-depleted HeLa cells were exposed to HU (Fig. 3A and B) or UV radiation (Fig. 3C and D). Due to cycloheximide-induced CDT2 turnover (Fig. 4D), it was difficult to compare CHK1 levels in control and CDT2-depleted cells in the presence of cycloheximide. To compare CHK1 destruction in the absence of cycloheximide, we utilized a Tet-on inducible expression system (16) (Fig. 3E). CHK1 expression was induced in control and CDT2-depleted cells by culturing cells in the presence of doxycycline, and levels of ectopic CHK1 were monitored after the removal of doxycycline. As shown in Fig. 3E, the turnover of ectopic CHK1 was observed for control cells (lanes 1 to 3) but not for CDT2-depleted cells (lanes 4 to 6).
Fig 3.

CDT2 regulates CHK1 turnover in response to replication stress. (A and B) HU-induced CHK1 turnover in CDT2-depleted cells. HeLa cells transfected with control siRNA (GL3) or siRNA specific for CDT2 were cultured for 48 h and then incubated in medium containing 5 mM HU in the absence or presence of 10 μM MG132. At the indicated times post-HU treatment, cells were harvested and analyzed by Western blotting. A representative experiment is shown in panel A, and the quantitation of data from 5 independent experiments is shown in panel B. (C and D) UV-induced CHK1 turnover in CDT2-depleted cells. HeLa cells transfected with control siRNA or siRNA specific for CDT2 were exposed to 50 J/m2 UV at 48 h posttransfection. Cells were then cultured in the absence or presence of 10 μM MG132. At the indicated times post-UV exposure, cells were harvested and analyzed by Western blotting. A representative experiment is shown in panel C, and the quantitation of data from 4 independent experiments is shown in panel D. (E) Prolonged half-life of CHK1 in CDT2-depleted cells. HeLa Tet-on cells were transfected with a plasmid encoding Tet-inducible Flag-CHK1 followed by either control siRNA or siRNA specific for CDT2. Cells were cultured in the presence of 0.2 μg/ml doxycycline for 14 h to induce Flag-CHK1 expression. Cells were washed, cultured for 4 h in fresh medium, and then cultured in medium containing 5 mM HU. At the indicated times post-HU treatment, cells were harvested and analyzed by Western blotting. Levels of Flag-CHK1 are shown relative to levels at the 0-h time points (lanes 1 and 4). (F) Effects of CDT1 overexpression on CHK1 stability. HeLa cells expressing Flag-tagged CDT1 for 24 h were incubated in the absence or presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. The arrowhead indicates endogenous CDT1.
Fig 4.

CDT2 serves as the substrate receptor that targets CHK1 to CRL4CDT2. (A) Interactions between CHK1 and CDT2. Flag-tagged WT or a mutant form (3RE) of CHK1 was coproduced with Myc-tagged CDT2 in HeLa cells for 24 h. Cells were then cultured in medium containing 5 mM HU and 10 μM MG132 for 4 h. Cell lysates were resolved directly by SDS-PAGE or were immunoprecipitated with anti-Flag M2 affinity gel prior to SDS-PAGE. Whole-cell lysates (WCL) and immunoprecipitates (IP: Flag) were then analyzed by Western blotting. (B) Subcellular localization of CHK1 and CDT2. Flag-tagged CHK1 was produced in HeLa cells for 48 h. Cells were then cultured in the absence or presence of 5 mM HU or 10 μM MG132 for 4 h. The localization of endogenous CDT2 and ectopic CHK1 was determined by immunofluorescence staining. (C) Interactions between CHK1 and CRL4CDT2. Flag-tagged CHK1 was coproduced with the Myc-tagged CDT2 WT or CDT2 RA mutant in HeLa cells for 24 h. Cells were exposed to 50 J/m2 UV and cultured in the presence of 10 μM MG132 for 2 h. Cell lysates were resolved directly by SDS-PAGE or were immunoprecipitated with anti-Flag M2 affinity gel prior to SDS-PAGE. Whole-cell lysates and immunoprecipitates were then analyzed by Western blotting. (D) Regulation of CDT2 levels in response to replication stress. HeLa cells were cultured in medium containing 50 μg/ml cycloheximide (CHX), 5 mM HU, or both. At the indicated times post-HU/CHX treatment, cells were harvested and analyzed by Western blotting. (E and F) CHK1 ubiquitination in vitro. HeLa cells transfected with control siRNA (GL3) or siRNAs specific for either CDT2 or DDB1 were cultured for 48 h and then incubated in the presence of 10 mM HU for 1 h. Cell lysates were prepared, and a portion was resolved directly by SDS-PAGE followed by Western blotting (E). The remaining lysates were immunoprecipitated with anti-CUL4A antibody. Immunoprecipitated CRL4 complexes were then incubated with purified E1, E2, ubiquitin, and Flag-CHK1 that had been purified from HU-treated cells (F). Long (L) and short (S) exposures are indicated.
The depletion of CDT2 results in CDT1 accumulation, rereplication, and checkpoint activation (23). CDT1 was overproduced in HeLa cells to determine if the CHK1 stabilization observed for CDT2-depleted cells was an indirect effect of the accumulation of CDT1 (Fig. 3F). If this was the case, we would expect to observe the stabilization of CHK1 in CDT1-overproducing cells. However, in three independent experiments, we observed a ∼10% reduction in CHK1 levels in CDT1-overproducing cells relative to control cells in the absence of HU. This is likely due to the generation of phosphorylated CHK1 (the substrate for ubiquitination) in CDT1-overproducing cells (Fig. 3F, lane 2). In the presence of HU, we measured a ∼60% reduction in CHK1 levels in both control and CDT1-overproducing cells. Thus, the effects of the depletion of CDT2 on CHK1 turnover are not explained by the accumulation of CDT1 in CDT2-depleted cells.
CDT2 targets CHK1 to CRL4CDT2.
Next, experiments were performed to determine if interactions between CDT2 and CHK1 could be detected. As shown in Fig. 4A, both the CHK1 WT and the CHK1 3RE mutant coprecipitated with CDT2, and these interactions were enhanced when cells were cultured in the presence of HU to induce replication stress (lanes 2 and 4). Despite lower levels of the CHK1 3RE mutant than the CHK1 WT, greater interactions were observed between CDT2 and the CHK1 3RE mutant (Fig. 4A, lanes 3 and 4), consistent with the enhanced interactions between CUL4A and the CHK1 3RE mutant (Fig. 1D). CHK1 and CDT2 exhibited predominant nuclear localization both in the absence and in the presence of replication stress, demonstrating that they are present in the same cellular compartment (Fig. 4B). Next, we examined the ability of CUL4A and DDB1 to coprecipitate with CHK1 in the presence of a CDT2 R246A (RA) mutant, a mutant of CDT2 that is defective for DDB1 binding (23). As shown in Fig. 4C, the ability of DDB1 and CUL4A to coprecipitate with CHK1 was reduced in the presence of the CDT2 RA mutant (lane 3) relative to the CDT2 WT (lane 2). Levels of DDB1 and CUL4A present in CDT2 RA precipitates (Fig. 4C, lane 3) were similar to those present in precipitates from control cells (lane 1) and therefore are likely due to nonspecific binding. These results provide additional evidence that CDT2 serves as a substrate receptor for CHK1.
Interestingly, endogenous CDT2 accumulated under conditions of replication stress (Fig. 2A and D, 3A and E, and 4D). This was not observed when cells were treated with cycloheximide to prevent new protein synthesis (Fig. 4D), and ectopically expressed CDT2 did not accumulate under similar conditions (Fig. 4A). Thus, increases in endogenous CDT2 levels were due to changes at the transcriptional and/or translational level rather than at the level of protein stability. CDT2 accumulation in response to replication stress correlated with enhanced CHK1 turnover (Fig. 3A).
Next, ubiquitination assays were performed to determine if the depletion of CDT2 impaired the ability of the CRL4 complex to ubiquitinate CHK1 in vitro. CHK1 was isolated from HU-treated cells to facilitate CHK1 phosphorylation on S317 and S345 because CHK1 phosphorylation on these residues promotes its binding to CRL4 (15–17). The CRL4 complex was isolated from cells transfected with control siRNA as well as from cells knocked down for CDT2 or DDB1 (Fig. 4E). As shown in Fig. 4F, the CRL4 complex isolated from cells deficient in either CDT2 (lane 5) or DDB1 (lane 6) was less effective at ubiquitinating CHK1 than the CRL4 complex isolated from control cells (lane 4). These results indicate that CDT2 functions as a substrate receptor that targets CHK1 to CRL4CDT2.
Claspin binding is required for CHK1 ubiquitination.
Claspin serves as a mediator that recruits CHK1 to sites of DNA damage, where it is phosphorylated on S317 and S345 by ATR. Phosphorylation relieves CHK1 autoinhibition, leading to the activation of its kinase activity and autophosphorylation on S296 (10, 42). We next asked if the kinase activity of CHK1 or its recruitment to DNA lesions by claspin was required for CHK1 to bind to CRL4CDT2. Several CHK1 mutants were generated for this purpose, including a kinase-inactive CHK1 mutant (A36F) and three claspin binding mutants (K54A, R129A, and T153A) (Fig. 5A).
Fig 5.

Claspin binding is important for CHK1 regulation by CRL4CDT2. (A) Schematic representation of CHK1 indicating the domain structure as well as kinase-inactive (A36F), claspin binding (K54A, R129A, and T153A), and SQ/TQ phosphorylation site (3SA) mutants. (B) Role of kinase activity and claspin binding in mediating interactions between CHK1 and CDT2. Flag-tagged WT or mutant forms of CHK1 were coproduced with Myc-tagged CDT2 for 24 h and then cultured in medium containing 5 mM HU and 10 μM MG132 for 4 h. Cell lysates were resolved directly by SDS-PAGE or were immunoprecipitated with anti-Flag M2 affinity gel prior to SDS-PAGE. Whole-cell lysates (WCL) and immunoprecipitates (IP: Flag) were analyzed by Western blotting. (C and D) Contributions made by kinase activity and claspin binding to CHK1 turnover. HeLa cells expressing the CHK1 WT or the indicated CHK1 mutants were cultured in medium containing 10 μg/ml CHX and 5 mM HU in the absence or presence of 10 μM MG132. At the indicated times post-HU/CHX treatment, cells were harvested and analyzed by Western blotting. A representative experiment is shown in panel C, and the quantitation of data from 4 independent experiments is shown in panel D. (E) Role of ATR-mediated phosphorylation in CHK1 turnover. HeLa cells expressing the CHK1 WT or the CHK1 3SA mutant were cultured in medium containing 10 μg/ml CHX and 5 mM HU in the absence or presence of 10 μM MG132. At the indicated times post-HU/CHX treatment, cells were harvested and analyzed by Western blotting. Levels of Flag-CHK1 are shown relative to levels at the 0-h time points.
WT or mutant forms of CHK1 were coproduced with CDT2 in HeLa cells and examined for their abilities to coprecipitate with one another. As expected, the kinase-inactive mutant (CHK1 A36F) was not phosphorylated on S296, the CHK1 autophosphorylation site (Fig. 5B, lane 3). The CHK1 A36F mutant bound claspin (Fig. 5B, lane 3), its levels of phosphorylation on S345 and binding to CDT2 were higher than those of the CHK1 WT (Fig. 5B, lanes 2 and 3), and it had a shorter half-life than the CHK1 WT (Fig. 5C and D). Thus, CHK1 kinase activity was not required for CHK1 to bind to CRL4CDT2.
As expected, the claspin binding mutants of CHK1 did not interact with claspin (Fig. 5B, lanes 4 to 6). The inability to bind claspin has been shown to reduce the recruitment of CHK1 to ATR (43), and as a result, the claspin binding mutants exhibited less phosphorylation on S345 and S296 than the CHK1 WT (Fig. 5B, lanes 2 and 4 to 6). In addition, the claspin binding mutants exhibited reduced CDT2 binding (Fig. 5B, lanes 2 and 4 to 6) and longer half-lives than the CHK1 WT (Fig. 5C and D). Thus, the recruitment of CHK1 to sites of DNA damage by claspin is an important step in mediating interactions between CHK1 and CRL4CDT2, and this is likely because CHK1 interactions with CRL4CDT2 are facilitated by the ATR-mediated phosphorylation of CHK1. We also included experiments with a CHK1 phosphorylation site mutant (3SA) encoding a substitution of alanine for serine at each of the ATR phosphorylation sites (S317, S345, and S366). As shown in Fig. 5E, the CHK1 3SA mutant was more stable than the CHK1 WT, demonstrating the importance of ATR-mediated phosphorylation in regulating CHK1 turnover.
CRL4CDT2 targets CHK1 for PCNA-independent degradation.
PCNA plays a critical role in recruiting CDT2 substrates to CRL4CDT2 for ubiquitin-mediated proteolysis. In fact, with the exception of SPD1, EPE1, and GCN5, all known targets of CDT2 contain a highly conserved PIP degron that mediates interactions with chromatin-bound PCNA to recruit these substrates to CRL4CDT2 (24, 31, 33, 44–46). In most cases, PCNA knockdown results in substrate accumulation (23, 31–34, 37, 47, 48). CHK1 was reported previously to interact with PCNA through a noncanonical PIP box (49), and therefore, we tested whether the knockdown of PCNA impaired CHK1 proteolysis in HeLa cells. CHK1 levels did not significantly change in cells knocked down for PCNA (Fig. 6A to C), and the rate of CHK1 turnover did not change when PCNA-deficient cells were subjected to replication stress (Fig. 6D and E). This is in contrast to the knockdown of CUL4 (Fig. 1), CDT2 (Fig. 2), or DDB1 (15), where CHK1 accumulation was readily observed. As a positive control for the PCNA knockdown, we monitored the turnover of CDT1, a canonical CDT2 substrate (24). The ubiquitin-mediated proteolysis of CDT1 by CRL4CDT2 is dependent on PCNA binding, whereas its destruction by CRL1SKP2 is PCNA independent (24, 50). Because the turnover of CDT1 in HeLa cells is regulated by both CRL1 and CRL4, we depleted CUL1 so that the effects of the PCNA depletion on the ubiquitin-mediated proteolysis of CDT1 by CRL4CDT2 could be monitored under conditions that minimized compensation by CRL1. The depletion of CUL1 alone or the codepletion of CUL1 and PCNA resulted in CDT1 but not CHK1 stabilization both in the absence and in the presence of replication stress (Fig. 6F and G). A slower-migrating form of CDT1 was observed upon the codepletion of both CUL1 and PCNA (Fig. 6F, lanes 3 and 6). The modification causing this change in electrophoretic mobility is not known but could be due to the stabilization of a phosphorylated or ubiquitinated form of CDT1. Another canonical CDT2 substrate, SET8, was also stabilized by the depletion of PCNA (Fig. 6F, lanes 3 and 6) (31, 33, 35, 38, 41). Taken together, these results indicate that the ability of CRL4CDT2 to target CHK1 for ubiquitin-mediated proteolysis is independent of PCNA binding.
Fig 6.

CRL4CDT2 targets CHK1 for PCNA-independent degradation. (A and B) Role of PCNA binding in regulating CHK1 stability. HeLa cells transfected with control siRNA (GL3) or siRNA specific for PCNA were cultured for 48 h and then incubated in the absence or presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. A representative experiment is shown in panel A, and the quantitation of data from 3 independent experiments is shown in panel B. (C) PCNA binding is not required for CHK1 turnover in UV-treated cells. HeLa cells transfected with control siRNA or siRNA specific for PCNA were mock irradiated or exposed to 50 J/m2 UV at 48 h posttransfection. Cells were harvested 6 h later and analyzed by Western blotting. Levels of CHK1 in knockdown cells are shown relative to levels in control cells (lane 1). (D and E) HU-induced CHK1 turnover in PCNA-depleted cells. HeLa cells transfected with control siRNA or siRNA specific for PCNA were cultured for 48 h and then incubated in medium containing 5 mM HU in the absence or presence of 10 μM MG132. At the indicated times post-HU treatment, cells were harvested and analyzed by Western blotting. A representative experiment is shown in panel D, and the quantitation of data from 4 independent experiments is shown in panel E. (F and G) PCNA is required for CDT1 and SET8 but not CHK1 turnover. HeLa cells transfected with control siRNA, siRNA specific for CUL1, or siRNAs specific for CUL1 and PCNA were cultured for 48 h and then incubated in the absence or presence of 5 mM HU for 6 h. Cell lysates were analyzed by Western blotting. A representative experiment is shown in panel F, and the quantitation of data from 3 independent experiments is shown in panel G. In panels B, E, and G, differences were not significant between control cells and PCNA, CUL1, or CUL1/PCNA knockdown cells within either treatment condition (Student's t test).
CHK1 PIP boxes are dispensable for CRL4CDT2-mediated degradation.
As mentioned above, CHK1 contains a noncanonical PIP box (residues 371 to 385) within its C terminus (CPIP) (Fig. 7A), and phenylalanine residues at position 380 (F380) and 381 (F381) were shown previously to be necessary for PCNA binding (49). We generated the CHK1 2FA mutant (containing alanine in place of F380 and F381) and monitored its ability to bind to CDT2. If PCNA is required to facilitate the binding of CHK1 to CDT2, we predicted that the 2FA mutant would bind less CDT2 than would the CHK1 WT and that its half-life would be longer. As shown in Fig. 7B, interactions between the CHK1 2FA mutant and CDT2 (lane 3) were actually greater than those observed between the CHK1 WT and CDT2 (lane 2), and the half-life of the CHK1 2FA mutant was shorter than that of the CHK1 WT (Fig. 7C and D). Thus, the C-terminal PIP box of CHK1 was not required for the targeting of CHK1 to CRL4CDT2, and the disruption of key residues within the putative PIP box actually promoted interactions between CHK1 and CDT2. This is likely due to a relief of the C-terminal autoinhibitory region of CHK1 and the exposure of its degron. In support of this, the kinase activities of the CHK1 2FA and 3RE mutants were shown to be greater than that of the CHK1 WT (16, 49), and both mutants exhibited enhanced autophosphorylation on S296 relative to the CHK1 WT (Fig. 7B, lanes 2 to 4), which is indicative of relief from autoinhibition.
Fig 7.

CHK1 PIP boxes are dispensable for CRL4CDT2-mediated destruction. (A) Schematic representation of CHK1 indicating the domain organization. The putative N-terminal PIP degron (NPIP box), C-terminal PIP box (CPIP box), and PIP box mutants (ΔNPIP1, ΔNPIP2, and 2FA) are shown. Conserved residues in the canonical PIP box and PIP degron are shown in black and dark gray, respectively. Additional residues that are highly conserved in CHK1 PIP boxes are shown in dark gray. (B) Role of the CPIP box in mediating CDT2 binding. The indicated Flag-tagged proteins were coproduced with Myc-tagged CDT2 in HeLa cells for 24 h and then cultured in medium containing 5 mM HU and 10 μM MG132 for 4 h. Cell lysates were resolved directly by SDS-PAGE or were incubated with anti-Flag M2 affinity gel prior to SDS-PAGE. Whole-cell lysates (WCL) and immunoprecipitates (IP: Flag) were analyzed by Western blotting. Levels of pS296 were normalized to levels of precipitated Flag-CHK1 and are shown relative to those of the CHK1 WT. (C and D) Role of the CPIP box in regulating CHK1 stability. HeLa cells expressing the CHK1 WT or the CHK1 2FA mutant were cultured in medium containing 10 μg/ml CHX and 5 mM HU in the absence or presence of 10 μM MG132. At the indicated times post-HU/CHX treatment, cells were harvested and analyzed by Western blotting. A representative experiment is shown in panel C, and the quantitation of data from 3 independent experiments is shown in panel D. Long (L) and short (S) exposures are indicated. (E) Role of the CHK1 NPIP box in mediating CDT2 binding. The indicated Flag-tagged proteins were coproduced with Myc-tagged CDT2 in HeLa cells for 24 h and then cultured in medium containing 5 mM HU and 10 μM MG132 for 4 h. Cell lysates were resolved directly by SDS-PAGE or were incubated with anti-Flag M2 affinity gel prior to SDS-PAGE. Whole-cell lysates and immunoprecipitates were analyzed by Western blotting. Levels of pS296 were normalized to levels of precipitated Flag-CHK1 and are shown relative to those of the CHK1 WT. (F and G) Role of the NPIP box in regulating CHK1 stability. HeLa cells expressing the CHK1 3RE or 3RE ΔNPIP2 mutant were cultured in medium containing 10 μg/ml CHX and 5 mM HU. Cells were harvested and analyzed by Western blotting at the indicated times posttreatment. A representative experiment is shown in panel F, and the quantitation of data from 3 independent experiments is shown in panel G.
CHK1 also contains a potential PIP degron (residues 64 to 75) within its N terminus (NPIP) (Fig. 7A). We generated mutants of CHK1, denoted ΔNPIP1 (encoding an alanine substitution for both F70 and Y71) and ΔNPIP2 (encoding an alanine substitution for both R74 and R75), in combination with the 3RE mutations to test the contribution made by the putative N-terminal PIP degron to CDT2 binding and CHK1 proteolysis. The mutation of residues predicted to disrupt the N-terminal PIP box and PCNA binding (ΔNPIP1) enhanced rather than impaired interactions between CHK1 and CDT2 (Fig. 7E, lane 3). The CHK1 ΔNPIP1 mutant was phosphorylated on S345 by ATR, indicating relief from autoinhibition. However, autophosphorylation on S296 was not observed, indicating that this mutant lacked kinase activity. We also tested the importance of the N-terminal PIP degron by mutating two basic residues within this region (ΔNPIP2) in the CHK1 3RE mutant. We predicted that these mutations would impair CDT2 binding and extend the half-life of the CHK1 3RE mutant containing an intact N-terminal PIP degron. This prediction was not met, as CDT2 binding was not impaired when mutations within the N-terminal PIP degron were introduced (Fig. 7E, lane 5), and the half-life of the CHK1 3RE ΔNPIP2 mutant was indistinguishable from that of the CHK1 3RE mutant (Fig. 7F and G).
CRL4CDT2 targets CHK1 for ubiquitination in the nucleoplasm.
Most CDT2 substrates are recruited to CRL4CDT2 by interactions with chromatin-bound PCNA and removed from chromatin by p97 AAA+-ATPase (24, 51). Although the recruitment of CHK1 to sites of DNA damage is a critical step in the targeting of CHK1 for CRL4-mediated destruction, CHK1 did not require PCNA for its association with CDT2. Therefore, we asked where interactions between CHK1 and CRL4CDT2 took place. Cytoplasmic, soluble nuclear, and three different chromatin fractions were isolated from mock- or UV-irradiated HeLa cells. Different chromatin fractions were obtained by sequentially washing chromatin with buffers containing 0.3 M NaCl (fraction 1), 0.5 M NaCl (fraction 2), and 2.0 M NaCl (fraction 3). Each fraction was analyzed for endogenous CHK1 as well as for components of CRL4CDT2 and CRL1FBX6. ORC2 (origin recognition complex subunit 2) was used as a control for chromatin-bound proteins.
As shown in Fig. 8A, ORC2 was found in chromatin fractions 1 and 2. CHK1 was present in cytoplasmic, nuclear, and chromatin fractions, and its phosphorylated form was enriched in cytoplasmic and nuclear fractions, as previously reported (14). DDB1 was distributed in cytoplasmic, nuclear, and chromatin fractions, as previously reported (52). Neddylated and nonneddylated forms of CUL4A were present in all three fractions. Interestingly, in the chromatin fractions, the neddylated (active) form of CUL4A partitioned predominantly in fractions 2 and 3 (Fig. 8A, lanes 13 to 20), indicating that it was tightly bound to chromatin. CDT2, the substrate receptor for CRL4, was found predominantly in the nuclear fraction and chromatin fractions 1 and 2 (Fig. 8A, lanes 5 to 16), whereas FBX6, the substrate receptor for CRL1, was found predominantly in the cytoplasmic fraction (lanes 1 to 4), as previously reported (16). Levels of CDT2 in chromatin fractions increased after UV treatment, demonstrating that while a fraction of CDT2 was redistributed from the nuclear pool to the chromatin-bound pool, a major fraction of CDT2 remained in the nucleoplasm. In contrast, SKP1 and FBX6 were enriched in cytoplasmic fractions. Thus, CRL4CDT2 components were enriched in the nucleoplasm, while CRL1FBX6 components were enriched in the cytoplasm. These results indicate that these E3 ubiquitin ligases function in distinct cellular compartments.
Fig 8.

CRL4CDT2 interacts with CHK1 in the nucleoplasm. (A) Spatial regulation of CHK1 ubiquitination by CRL1 and CRL4. HeLa cells were exposed to 20 J/m2 UV and then harvested at the indicated time points post-UV exposure. Subcellular fractionation was performed, and individual fractions were analyzed by Western blotting. The arrowheads indicate the neddylated form of cullins. (B) Subcellular location of CHK1 interactions with CDT2. HeLa cells were exposed to 50 J/m2 UV. Cytoplasmic (C), soluble nuclear (N), and chromatin (CH) fractions were isolated 2 h later and either resolved directly by SDS-PAGE (input) or incubated with protein A-agarose either alone (IP: Control) or with affinity-purified anti-CHK1 antibody (IP: CHK1) prior to SDS-PAGE. (C) The checkpoint activation/termination cycle. In response to replication stress, CHK1 is recruited to sites of DNA damage in a claspin-dependent manner, where it is phosphorylated by ATR. Phosphorylated CHK1 is then released from the chromatin to phosphorylate downstream effectors. Two E3 ubiquitin ligases target CHK1 for destruction. CRL4CDT2 ubiquitinates CHK1 in the nucleoplasm, whereas CRL1FBX6 ubiquitinates CHK1 in the cytoplasm. This terminates checkpoint signaling through the ATR/CHK1 pathway.
To examine where interactions between CHK1 and CRL4CDT2 take place, endogenous CHK1 was immunoprecipitated from cytoplasmic fractions, soluble nuclear fractions, and chromatin fractions 1 and 2. As shown in Fig. 8B, CHK1 and CDT2 complexes were enriched in nuclear fractions (lane 8), which are also where the phosphorylated form of CHK1 resided (Fig. 8A, lanes 6 to 8). These results indicate that CHK1 ubiquitination by CRL4CDT2 occurs mainly in the nucleoplasm, in contrast to interactions between CHK1 and the CUL1 substrate receptor FBX6, which occur in the cytoplasm (16). Thus, CRL1FBX6 and CRL4CDT2 regulate CHK1 stability in distinct cellular compartments (Fig. 8C).
CHK1 is required for G2 arrest in CDT2-depleted cells.
CDT2 depletion results in rereplication and G2 cell cycle arrest in cultured cells (23, 36, 40). The stabilization of CDT1 by CDT2 depletion contributes to the ability of cells to undergo rereplication (23). We investigated the role of CHK1 in regulating G2 cell cycle arrest in CDT2-depleted cells. Since the active form of CHK1 strongly associates with CRL4CDT2, replication stress-induced CHK1 activation is accompanied by a decrease in the total CHK1 levels (Fig. 1B, 2B, and 6B). However, as shown in Fig. 9A, the S296 phosphorylated (active) form of CHK1 accumulated in CDT2-depleted cells without a decrease in total CHK1 levels (Fig. 9A, lane 3, and B), and this correlated with a reduction in the number of G1- and S-phase cells coincident with an increase in the percentage of G2/M-phase cells (Fig. 9C and D). When CDT2-depeleted cells were treated with AZD7762 (AZD), a selective CHK1 inhibitor (53), cells bypassed the G2 arrest and moved into the G1 phase (Fig. 9C and D). The loss of S296 phosphorylation confirmed the inhibition of CHK1 by AZD7762 (Fig. 9A, lanes 2 and 4). These results indicate that CHK1 kinase activity is required for maintaining G2 cell cycle arrest in CDT2-depleted cells.
Fig 9.
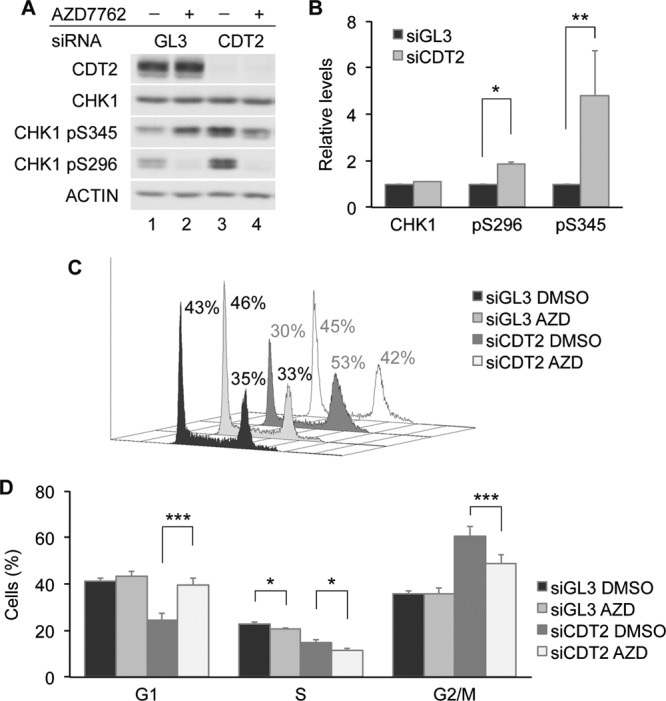
CHK1 maintains G2 arrest in CDT2-depleted cells. (A and B) Stabilization of the active form of CHK1 in CDT2-depleted cells. HeLa cells transfected with control siRNA (GL3) or siRNA specific for CDT2 were cultured for 48 h and then incubated in medium containing dimethyl sulfoxide (DMSO) or 30 nM AZD7762 (AZD) for 2 h. Cells were harvested and analyzed by Western blotting. A representative experiment is shown in panel A, and the quantitation of data from 3 independent experiments is shown in panel B. (C and D) G2 checkpoint regulation by CHK1 and CDT2. HeLa cells transfected with control siRNA or siRNA specific for CDT2 were cultured for 48 h and then incubated in medium containing dimethyl sulfoxide or 30 nM AZD7762 for 4 h. Cells were analyzed by flow cytometry. A representative experiment is shown in panel C, and the quantitation of data from 3 independent experiments is shown in panel D.
DISCUSSION
In this study, we identified CDT2 as a novel CHK1 regulator. CDT2 functions as a substrate receptor that targets CHK1 to CRL4CDT2 for ubiquitin-mediated proteolysis. CHK1 turnover was observed in cells exposed to replication stress, and CDT2 depletion abrogated replication stress-induced CHK1 turnover in HeLa cells. This indicates that CDT2 is the predominant substrate receptor for regulating replication stress-induced CHK1 proteolysis in these cells. The predominant role played by CRL4CDT2, as opposed to CRL1FBX6, in regulating CHK1 destruction in HeLa cells is likely due to the low levels of FBX6 in these cells (16).
In the absence of replication stress, the CUL4 depletion (Fig. 1A and B) had a more robust effect on CHK1 levels than did the CDT2 depletion (Fig. 2A and B). In the absence of HU, there was a 2-fold increase in CHK1 levels in CUL4A/B-depleted cells relative to those in control cells, and there was still a 50% reduction in CUL4A/B-depleted cells after HU treatment. This could be interpreted to mean that CUL4 is not responsible for CHK1 destruction in the presence of replication stress. However, interactions between CUL4A and CHK1 were well characterized in two previous studies (15, 17). Zhang et al. previously identified CUL4A (in addition to CUL1) as an E3 ligase that targets CHK1 for ubiquitin-mediated proteolysis (17). They also presented evidence that CUL4B does not regulate CHK1, but this may vary depending on the cell line studied. We subsequently identified DDB1 in a proteomic screen looking for CHK1-interacting proteins and demonstrated that CUL4A, together with DDB1, regulates CHK1 turnover (15). Here we demonstrated that in the presence of HU, cells knocked down for either CDT2 or CUL4 accumulate 2-fold more CHK1 than do control cells (Fig. 1A and B and 2A and B). In addition, CUL4A coprecipitates with CHK1 (Fig. 1D), a CUL4 deficiency extends the half-life of CHK1 (15), and CUL4A immunoprecipitates ubiquitinate CHK1 in vitro (Fig. 4F). Taken together, these data provide strong evidence that CRL4CDT2 targets CHK1 for ubiquitination under conditions of replication stress. What, then, accounts for the relatively higher level of accumulation of CHK1 in CUL4-depleted cells (Fig. 1A and B) than in CDT2-depleted cells (Fig. 2A and B) in the absence of replication stress and the 50% reduction in CHK1 levels in HU-treated CUL4-depleted cells? One explanation for this might be that the CUL4 depletion indirectly stimulates CHK1 transcription/translation in the absence but not in the presence of HU treatment. This would account for the higher level of accumulation of CHK1 in CUL4-depleted cells than in CDT2-depleted cells in the absence of replication stress. In addition, if this hypothesis is correct, the reduction in CHK1 levels observed for HU-treated CUL4-depleted cells would be due to reduced transcription/translation as opposed to ubiquitin-mediated proteolysis.
Most known substrates of CDT2 contain a specialized PIP box, and interactions with chromatin-bound PCNA mediate their recruitment to CRL4CDT2 (24). A canonical PIP degron consists of a PCNA interaction protein motif (PIP box), a TD motif within the PIP box, and a basic amino acid (K/R) in the +4 position (B+4) relative to the PIP box. Havens et al. reported previously that CRL4CDT2 interacts not only with the substrate via B+4 but also with an acidic residue on PCNA (D122) (24, 50). In the absence of a basic residue in the +4 position, basic residues upstream of the PIP degron can contribute to the CRL4CDT2-mediated destruction of the substrate. In addition, a TD motif with the PIP box contributes to the high-affinity binding of the substrate and PCNA. It was proposed that CRL4CDT2 is recruited to the PIP degron-PCNA complex rather than to either protein independently.
The C-terminal PIP box of CHK1 encodes aspartic acid (D385) in the B+4 position and does not contain basic residues upstream of the PIP box. Furthermore, the CPIP box lacks a TD motif. The putative N-terminal PIP box of CHK1 contains arginines (R74 and R75) in the B+3 and B+4 positions as well as basic residues upstream of the PIP box but lacks a TD motif. We found that the depletion of PCNA did not increase CHK1 stability and that CHK1 recruitment to CRL4CDT2 did not require either of its PIP boxes. Interactions between CHK1 and PCNA were shown previously to decrease during periods of replication stress (49), yet we observed enhanced interactions between CHK1 and CDT2 during periods of replication stress. This provides further evidence that PCNA binding does not mediate interactions between CHK1 and CRL4CDT2. In fact, enhanced interactions between CHK1 and CDT2 were observed when conserved residues within the PIP boxes of CHK1 were mutated. Thus, CHK1 joins a noncanonical class of CDT2 substrates, including GCN5, that do not require PCNA binding in order to be targeted to CRL4CDT2 (44–46).
Mutations present in the CHK1 2FA and CHK1 3RE mutants neighbor the autoinhibitory region (AIR) of CHK1. Previous studies reported that structural changes within this region relieve CHK1 autoinhibition and that this in turn activates the kinase activity of CHK1 while at the same time exposing its degron (16). Indeed, the CHK1 2FA and CHK1 3RE mutants exhibited elevated kinase activities (16, 49), enhanced autophosphorylation on S296, and enhanced turnover relative to the CHK1 WT. However, the enhanced turnover did not require CHK1 kinase activity or phosphorylation on S296, as the kinase-inactive mutant of CHK1 also exhibited enhanced CDT2 binding and enhanced turnover compared with the CHK1 WT. This is likely because the kinase-inactive mutant is highly phosphorylated on S345, which is sufficient to relieve the AIR of CHK1 (29, 30). Thus, the enhanced binding to CDT2 observed with these CHK1 mutants is likely due to a conformation change in CHK1 that relieves CHK1 autoinhibition and exposes the CDT2 binding domain.
The accumulation of CDT2 was observed in HU-treated cells. CDT2 accumulation in response to prolonged replication stress was not observed when cells were treated with cycloheximide to block new protein synthesis. This indicates that increases in CDT2 levels that accompany replication stress are due to enhanced transcription and/or translation. Increases in CDT2 levels in HU-treated cells correlated with an enhanced turnover of CHK1. This finding indicates that changes in substrate receptor levels also contribute to the enhanced turnover of certain substrates during checkpoint activation. In the fission yeast Schizosaccharomyces pombe, CDT2 levels also increase in response to HU treatment (46).
Substrates of CRL4CDT2 are also often targeted to CRL1. For example, the CUL1 substrate receptor SKP2 recruits CDT1, p21, and SET8 to CRL1 for ubiquitination (54–56), and these substrates are also regulated by CRL4CDT2. CDT1 has distinct binding sites for each E3 ligase (57, 58). While CRL1SKP2 regulates CDT1 destruction in the S and G2 phases, CRL4CDT2 regulates CDT1 destruction during DNA replication and in G1-phase cells with DNA damage (57). CDT1 is ubiquitinated on chromatin by CRL4CDT2 in a PCNA-dependent manner, whereas it is ubiquitinated by the CRL1SKP2 complex in the nucleoplasm (24, 31, 33, 59).
We demonstrated that CRL1FBX6 and CRL4CDT2 target CHK1 in distinct subcellular locations, and we identified CRL4CDT2 as the major E3 ligase targeting CHK1 for destruction during periods of replication stress in HeLa cells. The replication stress-induced phosphorylation of CHK1 occurs on chromatin, and the phosphorylated/activated from of CHK1 is then released from chromatin (4, 14), where it then phosphorylates effectors before being degraded by CRL4CDT2. Thus, the regulation of substrate ubiquitination by distinct E3 ligases is determined by substrate receptors and the subcellular compartmentalization of the substrate and ligase and can be impacted by the phase of the cell cycle and whether checkpoints have been activated or not.
The essential function of CHK1 in cell survival and early embryonic development has been demonstrated by the constitutive and tissue-specific deletion of CHK1 in mice (11, 60–63). However, a partial loss of CHK1 by hemizygous deletion was reported previously to promote tumorigenesis and tumor progression in the mouse mammary gland and skin (61, 64). Elevated expression levels of CDT2 have been reported for several human cancers, including hepatocellular carcinoma, breast and gastric cancers, and Ewing sarcoma (65–68). The identification of CHK1 as a CRL4CDT2 substrate may explain, in part, how CDT2 overexpression provides cancer cells with a proliferative advantage. By reducing CHK1 levels, CDT2 overexpression is expected to weaken checkpoint barriers and enable cancer cells to progress through the cell cycle in the presence of DNA damage.
ACKNOWLEDGMENTS
We thank Kendall Blumer, Peter Burgers, Zhongsheng You, and Anurag Agarwal for scientific discussions; Johannes Walter and Courtney Havens for communicating the presence of a putative PIP box within the N terminus of CHK1 and for scientific discussions; You-Wei Zhang for providing the Tet-inducible Flag-CHK1 construct; Anindya Dutta for providing the pEFF-CDT1 construct; Wade Harper and Malavika Raman for providing the pDEST-N-Myc-CDT2 construct; and Diana Owyoung, Emily Powell, and W. Timothy Schaiff for manuscript editing.
J.H. was supported in part by the Cancer Biology Pathway program administered through the Siteman Cancer Center at Washington University. The Siteman Cancer Center is supported, in part, by NCI Cancer Center support grant P30 CA91842. This study was supported by NIH grant GM047017 to H.P.-W., who is a research professor of the American Cancer Society.
Footnotes
Published ahead of print 29 October 2012
REFERENCES
- 1. Sorensen CS, Syluasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou B-B, Bartek J, Lukas J. 2003. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3: 247–258 [DOI] [PubMed] [Google Scholar]
- 2. Zhao H, Watkins JL, Piwnica-Worms H. 2002. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. U. S. A. 99: 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smits VA, Warmerdam DO, Martin Y, Freire R. 2010. Mechanisms of ATR-mediated checkpoint signalling. Front. Biosci. 15: 840–853 [DOI] [PubMed] [Google Scholar]
- 4. Stracker TH, Usui T, Petrini JH. 2009. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst.) 8: 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cortez D, Guntuku S, Qin J, Elledge SJ. 2001. ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- 6. Kumagai A, Kim SM, Dunphy WG. 2004. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem. 279: 49599–49608 [DOI] [PubMed] [Google Scholar]
- 7. Sorensen CS, Syljuasen RG, Lukas J, Bartek J. 2004. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle 3: 941–945 [PubMed] [Google Scholar]
- 8. Yang XH, Zou L. 2006. Recruitment of ATR-ATRIP, Rad17, and 9-1-1 complexes to DNA damage. Methods Enzymol. 409: 118–131 [DOI] [PubMed] [Google Scholar]
- 9. Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
- 10. Clarke CA, Clarke PR. 2005. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem. J. 388: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 12. Walworth NC, Bernards R. 1996. rad-dependent responses of the chk1-encoded protein kinase at the DNA damage checkpoint. Science 271: 353–356 [DOI] [PubMed] [Google Scholar]
- 13. Zhao H, Piwnica-Worms H. 2001. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smits VA, Reaper PM, Jackson SP. 2006. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 16: 150–159 [DOI] [PubMed] [Google Scholar]
- 15. Leung-Pineda V, Huh J, Piwnica-Worms H. 2009. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 69: 2630–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T. 2009. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol. Cell 35: 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. 2005. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 19: 607–618 [DOI] [PubMed] [Google Scholar]
- 18. Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6: 9–20 [DOI] [PubMed] [Google Scholar]
- 19. Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. 2006. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443: 590–593 [DOI] [PubMed] [Google Scholar]
- 20. He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. 2006. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 20: 2949–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. 2006. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 8: 1277–1283 [DOI] [PubMed] [Google Scholar]
- 22. Fukumoto Y, Dohmae N, Hanaoka F. 2008. Schizosaccharomyces pombe Ddb1 recruits substrate-specific adaptor proteins through a novel protein motif, the DDB-box. Mol. Cell. Biol. 28: 6746–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin J, Arias EE, Chen J, Harper JW, Walter JC. 2006. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23: 709–721 [DOI] [PubMed] [Google Scholar]
- 24. Havens CG, Walter JC. 2009. Docking of a specialized PIP box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 35: 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teer JK, Dutta A. 2008. Human Cdt1 lacking the evolutionarily conserved region that interacts with MCM2-7 is capable of inducing re-replication. J. Biol. Chem. 283: 6817–6825 [DOI] [PubMed] [Google Scholar]
- 26. Lovly CM, Yan L, Ryan CE, Takada S, Piwnica-Worms H. 2008. Regulation of Chk2 ubiquitination and signaling through autophosphorylation of serine 379. Mol. Cell Biol. 28: 5874–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang QE, Zhu Q, Wani G, Chen J, Wani AA. 2004. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis 25: 1033–1043 [DOI] [PubMed] [Google Scholar]
- 28. Abramoff MD, Magelhaes PJ, Ram SJ. 2004. Image processing with Image J. Biophotonics Int. 11: 36–42 [Google Scholar]
- 29. Chen P, Luo C, Deng Y, Ryan K, Register J, Margosiak S, Tempczyk-Russell A, Nguyen B, Myers P, Lundgren K, Kan CC, O'Connor PM. 2000. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell 100: 681–692 [DOI] [PubMed] [Google Scholar]
- 30. Chen Y, Caldwell JM, Pereira E, Baker RW, Sanchez Y. 2009. ATRMec1 phosphorylation-independent activation of Chk1 in vivo. J. Biol. Chem. 284: 182–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. 2010. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40: 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. 2008. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22: 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. 2010. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40: 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Higa LA, Banks D, Wu M, Kobayashi R, Sun H, Zhang H. 2006. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 5: 1675–1680 [DOI] [PubMed] [Google Scholar]
- 35. Jorgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MS, Kousholt AN, Syljuasen RG, Trelle MB, Jensen ON, Helin K, Sorensen CS. 2011. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J. Cell Biol. 192: 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim Y, Starostina NG, Kipreos ET. 2008. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 22: 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. 2008. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 283: 29045–29052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. 2010. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 40: 364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ralph E, Boye E, Kearsey SE. 2006. DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep. 7: 1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, Hopkins N, Lees JA. 2006. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 20: 3117–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 12: 1086–1093 [DOI] [PubMed] [Google Scholar]
- 42. Ma CX, Janetka JW, Piwnica-Worms H. 2011. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 17: 88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jeong SY, Kumagai A, Lee J, Dunphy WG. 2003. Phosphorylated claspin interacts with a phosphate-binding site in the kinase domain of Chk1 during ATR-mediated activation. J. Biol. Chem. 278: 46782–46788 [DOI] [PubMed] [Google Scholar]
- 44. Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. 2011. The Cul4-Ddb1(Cdt)(2) ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144: 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Y, Jaramillo-Lambert A, Hao J, Yang Y, Zhu W. 2011. The stability of histone acetyltransferase general control non-derepressible (Gcn) 5 is regulated by Cullin4-RING E3 ubiquitin ligase. J. Biol. Chem. 286: 41344–41352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu C, Poitelea M, Watson A, Yoshida SH, Shimoda C, Holmberg C, Nielsen O, Carr AM. 2005. Transactivation of Schizosaccharomyces pombe cdt2+ stimulates a Pcu4-Ddb1-CSN ubiquitin ligase. EMBO J. 24: 3940–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arias EE, Walter JC. 2006. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8: 84–90 [DOI] [PubMed] [Google Scholar]
- 48. Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A. 2006. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281: 6246–6252 [DOI] [PubMed] [Google Scholar]
- 49. Scorah J, Dong MQ, Yates JR, III, Scott M, Gillespie D, McGowan CH. 2008. A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J. Biol. Chem. 283: 17250–17259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Havens CG, Shobnam N, Guarino E, Centore RC, Zou L, Kearsey SE, Walter JC. 2012. Direct role for proliferating cell nuclear antigen (PCNA) in substrate recognition by the E3 ubiquitin ligase CRL4-Cdt2. J. Biol. Chem. 287: 11410–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raman M, Havens CG, Walter JC, Harper JW. 2011. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell 44: 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS. 2002. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 30: 2588–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM. 2008. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 7: 2955–2966 [DOI] [PubMed] [Google Scholar]
- 54. Li X, Zhao Q, Liao R, Sun P, Wu X. 2003. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J. Biol. Chem. 278: 30854–30858 [DOI] [PubMed] [Google Scholar]
- 55. Yin Y, Yu VC, Zhu G, Chang DC. 2008. SET8 plays a role in controlling G1/S transition by blocking lysine acetylation in histone through binding to H4 N-terminal tail. Cell Cycle 7: 1423–1432 [DOI] [PubMed] [Google Scholar]
- 56. Yu ZK, Gervais JL, Zhang H. 1998. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl. Acad. Sci. U. S. A. 95: 11324–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, Lygerou Z, Nishimoto T. 2006. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 25: 1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takeda DY, Parvin JD, Dutta A. 2005. Degradation of Cdt1 during S phase is Skp2-independent and is required for efficient progression of mammalian cells through S phase. J. Biol. Chem. 280: 23416–23423 [DOI] [PubMed] [Google Scholar]
- 59. Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschlager M. 2009. Skp2 inversely correlates with p27 and tuberin in transformed cells. Amino Acids 37: 257–262 [DOI] [PubMed] [Google Scholar]
- 60. Greenow KR, Clarke AR, Jones RH. 2009. Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene 28: 1443–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lam MH, Liu Q, Elledge SJ, Rosen JM. 2004. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6: 45–59 [DOI] [PubMed] [Google Scholar]
- 62. Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M, Nakayama K. 2000. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 14: 1439–1447 [PMC free article] [PubMed] [Google Scholar]
- 63. Zaugg K, Su YW, Reilly PT, Moolani Y, Cheung CC, Hakem R, Hirao A, Liu Q, Elledge SJ, Mak TW. 2007. Cross-talk between Chk1 and Chk2 in double-mutant thymocytes. Proc. Natl. Acad. Sci. U. S. A. 104: 3805–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tho LM, Libertini S, Rampling R, Sansom O, Gillespie DA. 2012. Chk1 is essential for chemical carcinogen-induced mouse skin tumorigenesis. Oncogene 31: 1366–1375 [DOI] [PubMed] [Google Scholar]
- 65. Li J, Ng EK, Ng YP, Wong CY, Yu J, Jin H, Cheng VY, Go MY, Cheung PK, Ebert MP, Tong J, To KF, Chan FK, Sung JJ, Ip NY, Leung WK. 2009. Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br. J. Cancer 101: 691–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mackintosh C, Ordonez JL, Garcia-Dominguez DJ, Sevillano V, Llombart-Bosch A, Szuhai K, Scotlandi K, Alberghini M, Sciot R, Sinnaeve F, Hogendoorn PC, Picci P, Knuutila S, Dirksen U, Debiec-Rychter M, Schaefer KL, de Alava E. 2012. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31: 1287–1298 [DOI] [PubMed] [Google Scholar]
- 67. Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC. 2006. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle 5: 2676–2687 [DOI] [PubMed] [Google Scholar]
- 68. Ueki T, Nishidate T, Park JH, Lin ML, Shimo A, Hirata K, Nakamura Y, Katagiri T. 2008. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene 27: 5672–5683 [DOI] [PubMed] [Google Scholar]