

Abstract
Antigen receptor signaling to NF-κB, essential for normal lymphocyte activation, is dysregulated in several types of lymphoma. During normal signaling, the multidomain adapter CARD11 transitions from a closed, inactive state to an open, active scaffold that assembles a multiprotein complex, leading to NF-κB activation. The regulation of CARD11 scaffold function is bypassed by lymphoma-associated oncogenic CARD11 mutations that induce spontaneous signaling. We report an unbiased high-throughput quantitative signaling screen that identifies new CARD11 hyperactive variants and defines a LATCH domain that functions with the CARD to promote CARD11 autoinhibition. Gain-of-function mutations in the LATCH or CARD disrupt inhibitory domain binding, promote Bcl10 association, and induce Bcl10 ubiquitination, NF-κB activation, and human lymphoma cell survival. Our results identify CARD11 mutations with oncogenic potential, provide a mechanistic explanation for their signaling potency, and offer a straightforward method for the discovery of variants that promote the tumorigenesis of NF-κB-dependent lymphomas.
INTRODUCTION
The activation of the NF-κB transcription factor by antigen receptor signaling is critical for lymphocyte activation during the adaptive immune response (1). NF-κB controls a variety of genes that participate in lymphocyte proliferation, survival, and differentiation, and the induction of NF-κB activity by antigen is tightly regulated. Prior to antigen receptor engagement, NF-κB is held inactive in the cytoplasm of cells by the IκB family of inhibitory proteins. Antigen receptor signaling results in the activation of the IκB kinase (IKK) complex, which phosphorylates IκBs, targeting them for ubiquitination and degradation, and allowing NF-κB to accumulate in the nucleus to regulate target genes. The extent and duration of NF-κB activation downstream of antigen recognition at the surface of B and T lymphocytes are controlled by a variety of mechanisms. These include the strength of antigen-receptor interaction, the presence or absence of concomitant costimulatory signaling, and the action of signaling cascade components that control the magnitude of signaling output as it is being induced or that provide negative feedback to terminate signaling (2, 3).
The exquisite regulation of NF-κB activity by the antigen receptor signaling pathway is disrupted in a variety of cancers of the immune system (4). Aberrant NF-κB activity likely contributes to oncogenesis through the unregulated transcriptional induction of proproliferative and antiapoptotic genes that confer a survival advantage to transformed cells. Multiple mechanisms have been described by which lymphoid cancers achieve dysregulated NF-κB activation, including, for example, the overexpression or gain-of-function mutation of proteins that signal upstream of the IKK complex, the deletion or loss of function of inhibitory proteins that downmodulate or terminate signaling, and the overexpression of subunits of NF-κB itself (5).
CARD11 (CARMA1 or BIMP3) is a multidomain scaffold protein that is required for B cell receptor (BCR)- and T cell receptor (TCR)-mediated activation of the IKK complex (6–12). CARD11 contains CARD, coiled-coil, PDZ, SH3, and GUK domains, separated by four intervening regions. As a consequence of BCR or TCR engagement, CARD11 undergoes a conformational transition from a closed, inactive state to an open, active scaffold. This transition is controlled by an inhibitory domain (ID), located between the coiled-coil and PDZ domains, that keeps CARD11 in the closed, latent state through interactions that require the CARD and coiled-coil domains (13–15). Antigen receptor signaling leads to the neutralization of the ID through its phosphorylation at specific serine residues by protein kinase C θ (PKCθ) in T cells, PKCβ in B cells, IKKβ, and at least one additional unidentified kinase (13, 15–18). Subsequent to ID neutralization, CARD11 recruits several positive signaling cofactors, including the adapter Bcl10, the paracaspase MALT1, the TRAF6 E3 ligase, the TAK1 kinase, caspase-8, and IKKγ, into a complex (14). The formation of this complex is thought to elicit the activation of IKK kinase activity through the scaffolding and catalytic activities of each complex component, although the exact mechanistic details of how IKK kinase activity is engaged remain poorly defined. The CARD11-nucleated signaling complex is transient; following IKK activation, the complex disassembles, presumably returning CARD11 to the inactive state.
CARD11-dependent signaling is dysregulated in diffuse large B cell lymphoma (DLBCL), which has been divided into several subtypes based upon gene expression signatures (19). The activated B cell-like (ABC) subtype is characterized by constitutive activation of NF-κB, which is required for the proliferation and survival of ABC-derived cell lines in culture (20). An RNA interference (RNAi) screen for genes required for this aberrant signaling to NF-κB revealed obligate roles for CARD11, Bcl10, and MALT1 (21). In addition, approximately 10% of human ABC DLBCL samples examined by Lenz et al. exhibited gain-of-function mutations in CARD11 that conferred hyperactive signaling ability to the protein, thereby explaining in those cases the origin of the unregulated induction of NF-κB activity (22). Interestingly, most of the hyperactive mutations reported in DLBCL occurred in the coiled-coil domain.
Recently, we characterized two oncogenic CARD11 mutations, F123I and L225LI, found in human DLBCL and provided evidence that these mutations cause hyperactivity by disrupting the normal autoinhibition by the ID that keeps CARD11 inactive prior to receptor engagement, resulting in the spontaneous conversion of CARD11 from the closed, inactive state to the open, active state in the absence of receptor triggering or ID phosphorylation (23). The F123I and L225LI mutations partially disrupted intramolecular binding of the ID and specifically enhanced the ability of CARD11 to associate with Bcl10 but not with several other cofactors examined. Signaling downstream of F123I and L225LI required Bcl10 and the ubiquitination of Bcl10 on K31 and K63 (23), a signaling step required for normal TCR signaling (24).
These findings provided a satisfying explanation for how mutations in the coiled-coil could cause hyperactivity, since the coiled-coil domain is one of the regions targeted by the ID in the inactive state. The results also led to the prediction that any mutation that disrupts the ability of the ID to autoinhibit CARD11 signaling should result in gain-of-function hyperactivity. However, most of the described human DLBCL mutations were found in the coiled-coil domain and not, for example, in the CARD domain, which has also been shown to be required for the intramolecular ID association that keeps CARD11 in the closed inactive state. The apparent concentration of oncogenic mutations in the coiled-coil domain raised the question of how frequently gain-of-function mutations occur outside the coiled-coil domain and, if such mutations occur, whether they confer signaling potencies comparable to those found in human DLBCL and achieve hyperactivity by the same mechanisms. In this report, we address these issues by characterizing novel gain-of-function CARD11 mutants that we identified in a quantitative high-throughput signaling screen designed to detect the dysregulated CARD11-initiated activation of NF-κB.
MATERIALS AND METHODS
Primary and secondary screens for CARD11 gain-of-function mutations.
A library of murine CARD11 variants containing random mutations between amino acid residues 1 and 138 was generated in the context of pc-CARD11 (14) by error-prone PCR under conditions that favored one mutation per clone using a GeneMorph II EZClone domain mutagenesis kit (Stratagene) and primers 5′-GGATCCACTAGTAACGGCCGCC-3′ and 5′-GCAGTTTGATGACCTCGTTCATCAG-3′ according to the manufacturer's protocol. A total of 2,220 individual clones were screened by transiently transfecting 50 ng of each clone, 20 ng of Igκ2-IFN-LUC (where IFN refers to a minimal element containing the TATA box), and 6 ng of CSK-LacZ into HEK293T cells in 24-well plates by the calcium phosphate method and assaying reporter activities as previously described (14). Each clone that activated the Igκ2-IFN-LUC reporter at least 3-fold more than wild-type (WT) CARD11 was considered positive and was sequenced to identify its mutation. The region containing the identified mutation was recloned into the pc-CARD11 backbone and reassayed to confirm hyperactivity.
Determination of specific activities in HEK293T and Jurkat T cells.
Each verified mutant was titrated at subsaturating levels (between 1.5 and 27 ng) into the Igκ2-IFN-LUC reporter assay in HEK293T cells, and the protein concentration was determined by densitometric analysis of Western blots as described previously (23). Relative specific activity was determined by taking the fold Igκ2-IFN-LUC reporter activation elicited by a mutant at the same level of protein expression at which wild-type CARD11 elicited 3-fold activation and dividing by 3. Jurkat T cells in which CARD11 was stably knocked down (KD-CARD11) were transfected with Igκ2-IFN-LUC and CSK-LacZ as previously described (14) using 50 to 90 ng of expression construct for each CARD11 variant to achieve equivalent protein expression levels. The relative activity in T cells was determined by taking the fold Igκ2-IFN-LUC reporter activation elicited by a mutant and dividing by the fold activation achieved by wild-type CARD11 at the comparable level of expression.
Immunoprecipitations in HEK293T cells.
At 1 day prior to transfection, 5 × 105 HEK293T cells were plated in each well of a six-well plate. A total of 2 μg DNA per well was transfected using the calcium phosphate method. The medium was changed 22 to 26 h posttransfection, and the cells were harvested 42 to 46 h posttransfection. Cells were lysed in 300 or 500 μl of immunoprecipitation lysis buffer (IPLB), and debris was removed as described previously (14). Cell lysate (6 or 20 μl) was saved for Western blot analysis, and 264 or 450 μl was incubated with 1 μg of anti-FLAG antibody (Sigma F7425) for 90 to 120 min at 4°C with rotation. Protein G-Sepharose 4 Fast Flow (GE Healthcare) (7- or 10-μl bed volume) was added, and the mixture was incubated for 90 to 120 min at 4°C with rotation. The resulting immunocomplex was washed with IPLB four times for 5 min at 4°C with rotation, eluted twice, pooled, and resolved for Western blot analysis as described previously (23) using anti-myc (sc-40; Santa Cruz), anti-FLAG (M2; Eastman Kodak IB13026 or Sigma F1804), and anti-Bcl10 (sc-5273; Santa Cruz). The levels of each CARD11 variant in the input and the IP were determined by densitometric analysis of subsaturating exposures of Western blots using ImageJ. To quantitate Bcl10 binding for each variant, the ratio of the quantity of the variant in the IP to that in the input was determined and then normalized to the ratio achieved by the ΔID.
Glutathione-Sepharose pulldowns.
Gain-of-function mutations were introduced into the context of pc-ΔID (14). pEBB-HA-ID-GST (were HA is hemagglutinin and GST is glutathione S-transferase) (14), pEBG (14), and each pc-ΔID variant were transfected separately into HEK293T cells and harvested as described above. Cells from each well of a six-well plate were lysed in 500 μl of IPLB, and debris was removed as described above. Between 0.2% and 5.4% of lysates were first analyzed by Western blotting to quantify the protein expression of each ΔID variant. A titration of ΔID variant-containing extract was mixed with equivalent fractions of extract containing either HA-ID-GST or GST and with extract from pcDNA3-transfected cells to make a total volume of 500 μl. Mixed lysates were precleared twice by incubation with a 10-μl bed volume of protein G-Sepharose for 30 to 50 min at 4°C with rotation. From the precleared mixed lysates, 4.4% was saved as an input for Western blot analysis and the remaining 450 μl was incubated with a 10-μl bed volume of glutathione-Sepharose (GE Healthcare) for 15 to 17 h at 4°C with rotation. Samples were washed and analyzed as described previously (14) using anti-myc (sc-40; Santa Cruz) and anti-GST (sc-459; Santa Cruz). The levels of each CARD11 variant in the input and the pulldown were determined by densitometric analysis of subsaturating exposures of Western blots using ImageJ. To quantitate ID binding for each variant, the ratio of the quantity of the variant in the pulldown to that in the input was determined and then normalized to the ratio achieved by the ΔID at a comparable level of expression.
Bcl10-deficient HEK293T and CARD11-, MALT1-, and TRAF6-deficient Jurkat T cell lines.
The KD-GFP and KD-Bcl10 HEK293T cell lines have been previously described (14). The CARD11 variant and Bcl10 expression were assessed in these lines by Western blot analysis using anti-myc (sc-40; Santa Cruz) and anti-Bcl10 (sc-5273; Santa Cruz). To generate the KD-CARD11 Jurkat T cell line, a self-inactivating lentiviral construct based upon the vector pLKO.1 was constructed to express the sihCARD11-2 short hairpin RNA (shRNA) (sense sequence, 5′-TGGTCAAGAAGCTGACGATTC-3′; loop sequence, 5′-TTCAAGAGA-3′) downstream of the human U6 RNA promoter. Lentiviral particles were packaged as described previously (25). After cell debris was cleared by centrifugation at 2,300 × g for 5 min, 100 μl of viral supernatant was added to 1.5 × 105 Jurkat T cells in a final volume of 300 μl in a 24-well plate. Approximately 24 h postinfection, cells were resuspended in fresh medium containing 0.5 μg/ml puromycin and selected for 1 week. Puromycin-resistant Jurkat T cells were maintained in media containing 0.5 μg/ml puromycin.
To generate the KD-NT, KD-MALT1, and KD-TRAF6 Jurkat T cell lines, pLKO.1-based lentiviruses were constructed to express shNT (Sigma SHC002) (sense sequence, 5′-CAACAAGATGAAGAGCACCAA-3′; loop sequence, 5′-CTCGAG-3′), shM1 (14) (sense sequence, 5′-CCTCACTACCAGTGGTTCAAA-3′; loop sequence, 5′-CTCGAG-3′), or shT6 (26) (sense sequence, 5′-CCACGAAGAGATAATGGAT-3′; loop sequence, 5′-TTCAAGAGA-3′). To package these lentiviruses, HEK293T cells were plated at 5 × 105 cells per well in a 6-well plate and transfected 24 h later using calcium phosphate with 520 ng pMDL-RRE, 182 ng RSV-Rev, 260 ng pCMV-VSVg, and 1,040 ng pLKO.1-based vector. The medium was replaced with 2 ml RPMI medium 22 to 24 h posttransfection. Supernatants containing viral particles were harvested 47 to 48 h posttransfection after cell debris was cleared by centrifugation at 18,000 × g for 5 min. Viral supernatant (1 ml) was added to 6 × 106 Jurkat T cells in a final volume of 12 ml in a 10-cm-diameter plate. Approximately 24 h postinfection, cells were resuspended in fresh RPMI medium containing 0.5 μg/ml puromycin and selected for 10 days. Knockdown was assessed by Western blot analysis using anti-MALT1 (catalog no. 1664-1; Epitomics), anti-TRAF6 (sc-7221; Santa Cruz), and anti-IKKα (sc-7606; Santa Cruz) antibodies.
TAK1 inhibition assays.
(5Z)-7-Oxozeaenol was obtained from Calbiochem (product code 499610). Either dimethyl sulfoxide (DMSO) vehicle or 500 nM (5Z)-7-oxozeaenol was added to each sample every 12 h after transfection for a total of three doses, and samples were harvested 41 h after transfection. Some samples were stimulated with anti-CD3/anti-CD28 cross-linking as described previously (25) for 4 h before harvest.
Stable expression of murine CARD11 variants in Jurkat T cells.
Gain-of-function mutations were introduced in the context of pCLIP3A-myc-CARD11-mCherry (25), a Moloney murine leukemia virus-based vector. To package viruses, HEK293T cells were plated at 5 × 105 cells per well in a 6-well plate and transfected 24 h later using calcium phosphate with 430 ng pCL-SIN-Ampho (27), 430 ng pCMV-VSVg (28), and 1,140 ng pCLIP3A-myc-CARD11-mCherry-based vector. The medium was replaced with 2 ml RPMI complete medium 22 to 24 h posttransfection. Supernatants containing viral particles were harvested 49 to 50 h posttransfection after cell debris was cleared by centrifugation at 200 × g for 5 min. For each sample, 1.8 ml viral supernatant was added to 5.4 × 106 Jurkat T cells in a final volume of 10.8 ml in a 10-cm-diameter plate. Approximately 24 h postinfection, cells were resuspended in fresh medium containing 0.5 μg/ml puromycin and selected for 2 weeks. Puromycin-resistant Jurkat T cell lines were maintained in media containing 0.5 μg/ml puromycin.
NF-κB DNA-binding assays.
Electrophoretic mobility shift assays (EMSA) were done as described previously (11).
Endogenous Bcl10 and IKKγ immunoprecipitations in Jurkat T cell lines.
The control Jurkat T cell line stably expressing myc–wild-type-CARD11–mCherry was stimulated with 50 ng/ml phorbol myristate acetate (PMA) (Sigma) and 1 μM ionomycin (Iono) (Sigma) for 20 min at 37°C. Cells (108 per sample) were then incubated in an ice water bath for 5 min, spun at 423 × g for 10 min at 4°C, and lysed in a lysis buffer as described previously (29) except that 10% glycerol was added and a different protease inhibitor cocktail (Sigma P8340) was used. Cell lysates were precleared twice with a 20-μl bed volume of protein A-Sepharose (Sigma P9424) or protein G-Sepharose for 1 to 2 h at 4°C with rotation. To analyze Bcl10 ubiquitination, the precleared lysates were denatured and diluted as described previously (24) and then incubated with 4 μg of anti-Bcl10 (sc-5611; Santa Cruz) antibody for 16 h at 4°C with rotation. A 20-μl bed volume of protein A-Sepharose was added to each sample and incubated for 20 to 22 h at 4°C with rotation. To analyze the interaction between IKKγ and K63-linked ubiquitinated Bcl10 [Ubn(K63)-Bcl10], the precleared lysates were incubated with 3 μg of anti-IKKγ (sc-8330; Santa Cruz) antibody for 16 h at 4°C with rotation and then incubated with a 30-μl bed volume of protein G-Sepharose for 4 to 5 h at 4°C with rotation. Samples were washed and analyzed as described previously (14). Western blots were analyzed using anti-Ub-K63 (14-6077-82; eBioscience), anti-Bcl10 (sc-5273; Santa Cruz), anti-myc (sc-40; Santa Cruz), and anti-IKKγ (sc-8032; Santa Cruz).
Stable expression of murine CARD11 variants in OCI-Ly3 cells.
The OCI-Ly3 ABC-DLBCL cell line was a kind gift from R. Eric Davis (M.D. Anderson). Cells were cultured in OCI medium, which contains Iscove's modified Dulbecco medium supplemented with 20% human serum (S40110; Atlanta Biologicals), 50 U/ml each of penicillin and streptomycin, and 55 μM β-mercaptoethanol in humidified 5% CO2 at 37°C. First, OCI-Ly3 cells stably expressing the ecotropic receptor mCAT-1 were generated by infecting cells with the lentivirus pLenti6/Ubc/mSlc7a1 (Addgene Plasmid 17224) (30). To package this lentivirus, HEK293T cells were plated at 5 × 105 cells per well in a 6-well plate and transfected 24 h later using calcium phosphate with 1,040 ng pLenti6/Ubc/mSlc7a1, 520 ng pMDL-RRE (31), 182 ng RSV-Rev (31), and 260 ng pCMV-VSVg (28). The medium was replaced with 2 ml OCI medium 22 to 24 h posttransfection. Supernatants containing viral particles were harvested 47 to 48 h posttransfection after cell debris was cleared by centrifugation at 423 × g for 5 min. An 800-μl volume of viral supernatant was added to 1.2 × 106 OCI-Ly3 cells in a final volume of 4.8 ml in a 6-well plate. Approximately 48 h postinfection, cells were resuspended in fresh OCI medium containing 2 μg/ml blasticidin and selected for 2 weeks.
mCAT-1-expressing OCI-Ly3 cells were then infected with the same panel of CARD11-mCherry-expressing retroviruses as those used to infect Jurkat T cells. pCLIP3A-myc-CARD11-mCherry variants were transfected and packaged as described above. The medium was replaced with 2 ml OCI medium 22 to 24 h posttransfection. Supernatants containing viral particles were harvested 49 h posttransfection after cell debris was cleared by centrifugation at 18,000 × g for 5 min. For each sample, 1 ml viral supernatant was added to 1.5 × 106 mCAT-1-expressing OCI-Ly3 cells in a final volume of 6 ml in a 10-cm-diameter plate. Approximately 25 h postinfection, cells were resuspended in fresh OCI medium containing both 0.5 μg/ml puromycin and 2 μg/ml blasticidin and selected for 2 weeks. Puromycin and blasticidin double-resistant OCI-Ly3 cell lines were maintained in OCI medium containing both 0.5 μg/ml puromycin and 2 μg/ml blasticidin. Relative levels of CARD11 variants were assessed by Western blot analysis using anti-myc (sc-40; Santa Cruz), anti-CARD11 (catalog no. 3189; ProSci), and anti-IKKα (sc-7606; Santa Cruz) antibodies.
OCI-Ly3 proliferation and survival assay.
Retroviral constructs based on the vector pSUPER.retro.neo+GFP (pSRGN; Oligoengine) were constructed to express either sihCARD11-2 (sense sequence, 5′-TGGTCAAGAAGCTGACGATTC-3′; loop sequence, 5′-TTCAAGAGA-3′) or sihNT control (sense sequence, 5′-TTCTCCGAACGTGTCACGT-3′; loop sequence, 5′-TTCAAGAGA-3′) (32) short hairpin RNAs (shRNAs) downstream of the H1 RNA promoter and the enhanced green fluorescent protein (EGFP) gene downstream of the phosphoglycerate kinase promoter. To package retroviruses, HEK293T cells were plated at 5 × 105 cells per well in a 6-well plate and transfected 24 h later using Lipofectamine 2000 (Invitrogen) with 800 ng pCL-Eco (27) and 3,200 ng pSRGN-derived construct. The medium was replaced with 1.25 ml OCI medium 25 h posttransfection. Supernatants containing viral particles were harvested 55 h posttransfection after cell debris was cleared by filtration through a 0.45-μm-pore-size filter. A 500-μl volume of viral supernatant was added to 2 × 105 OCI-Ly3 cells in a final volume of 1 ml in a 12-well plate, followed by the addition of 3 μg/ml Polybrene. At 12 to 18 h postinfection, cells were resuspended in fresh OCI medium. GFP-positive (GFP+) percentages measured by fluorescence-activated cell sorter (FACS) analysis 3 days postinfection ranged from 26.2% to 42.6%. Data were processed as described previously (21), except that the initial GFP+ data were collected 3 days after retroviral transduction.
RESULTS
Identification of novel gain-of-function mutations in CARD11 in a high-throughput signaling screen.
Because of the requirement of the CARD domain for intramolecular ID association, we hypothesized that some CARD domain residues would mediate interactions that maintain CARD11 in the inactive state prior to ID neutralization by antigen receptor signaling. To test this hypothesis, we adapted a screening methodology previously used to identify molecules that signal to NF-κB (11) or that enhance or suppress the signaling activity of CARD11 (25, 33). We generated a library of CARD11 variants in the context of a mammalian expression vector in which residues between M1 and L138 of murine CARD11 were randomly mutated by error-prone PCR. Library clones were screened using a quantitative reporter assay in HEK293T cells using the Igκ2-IFN-LUC reporter in a transient transfection to assess NF-κB activation (Fig. 1A). For normalization of transfection efficiency and extract recovery, each transfection also included CSK-LacZ, which constitutively expresses β-galactosidase. A clone was considered positive if it activated the reporter at least 3-fold more than wild-type CARD11 expressed at a comparable level. Positive clones were sequenced to identify mutations and back-cloned into an otherwise wild-type CARD11 expression vector to confirm that the mutation identified between residues 1 and 138 was responsible for eliciting the enhanced signaling activity. After screening 2,220 clones, we identified 24 gain-of-function mutations in this region of CARD11 (Fig. 1B). Nine of these mutations resided in the CARD domain, and one mutation was located in the coiled-coil domain. Surprisingly, 14 mutations occurred in the 19-residue region located between the CARD and coiled-coil domains (Fig. 1B and C). This region of CARD11 has not been extensively characterized in previous studies. The high density of gain-of-function mutations between residues 112 and 130 defines this region as a functional domain that negatively regulates CARD11 activity. We refer to this region as the LATCH domain.
Fig 1.

A quantitative signaling screen identifies gain-of-function mutations in the CARD and LATCH domains of CARD11. (A) Example of primary screening in which four clones scored as positives. Clones 1, 5, 34, and 36 encoded C49Y, V119E, H129Y, and T117A mutations, respectively. (B) Summary of all mutations identified in the primary sequence at the N terminus of CARD11. CARD domain residues are depicted in black, LATCH domain residues in blue, and the N-terminal end of the coiled-coil domain in gray. (C) Model for CARD11 in the closed inactive state. CARD11 is depicted as a dimer for simplicity, but the oligomerization status has not been determined.
The specific activity of each variant that emerged from the screen was first determined by titrating its expression in the Igκ2-IFN-LUC reporter assay in HEK293T cells. At equivalent levels of protein expression, the collection of variants displayed activities that were between 2.3- and 31-fold greater than that of wild-type CARD11 (see Fig. 2A and B for examples of titrations and Table 1). For comparison, an ID-deleted variant, CARD11 ΔID, was 43.6-fold more active than the wild type under these conditions. The variants displayed similar relative potencies when expressed in a transient-transfection assay in CARD11-deficient Jurkat T cells (Fig. 2C and D and Table 1), indicating hyperactivity in both lymphoid and nonlymphoid cells. The most potent mutations were distributed between the CARD and LATCH domains and included C49Y and F97Y in the CARD domain and G123D, V119E, and G126D in the LATCH domain. Many of the mutations had effects on CARD11 activity that were comparable to or greater than those described previously for the F123I and L225LI mutations found in human DLBCL, which were found to increase specific activity by 12.6- and 28.1-fold in previous studies (23). We note that the numbering scheme used previously (22, 23) to describe F123I and L225LI was based on a human CARD11 sequence that did not account for seven N-terminal residues; F123 and L225 correspond to F130 and L232 in our numbering scheme for murine CARD11 (11).
Fig 2.
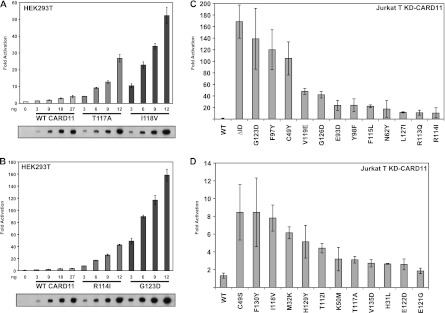
Relative specific activity of CARD11 variants in HEK293T and Jurkat T cells. (A and B) HEK293T cells were transfected with 20 ng of Igκ2-IFN-LUC and 6 ng of CSK-LacZ in the presence of the indicated amounts (in nanograms) of expression vectors for the indicated myc-tagged CARD11 variants. The bottom panel displays Western blots of corresponding lysates probed with anti-myc primary antibody to indicate the relative expression level of each variant. β-Galactosidase activity, driven by CSK-LacZ, was used to normalize luciferase activity and to calculate equivalent amounts of transfected cell lysate for Western analysis. (C and D) Jurkat T cells in which CARD11 was stably knocked down (KD-CARD11) were transfected with 200 ng of CSK-LacZ and 1,500 ng of Igκ2-IFN-LUC in the presence of 50 to 90 ng of expression vectors for the indicated variants. Vector quantities were adjusted to achieve equivalent protein expression levels for all variants, using the relative levels of expression per nanogram in HEK293T cells as a guide.
Table 1.
Properties of hyperactive CARD11 variants
| CARD11 variant | Sp acta | Relative activity in T cellsb | Bcl10 bindingc | ID bindingd |
|---|---|---|---|---|
| WT | 1.0 | 1.0 | − | |
| ΔID | 43.6 | 127.3 | +++++ | +++++ |
| H31L | 2.3 | 2.0 | − | + |
| M32K | 12.9 | 4.6 | − | + |
| C49Y | 31.0 | 79.3 | +++++ | + |
| C49S | 10.7 | 6.4 | + | +++++ |
| K50M | 6.3 | 2.4 | ++ | + |
| N62Y | 8.5 | 13.2 | ++ | + |
| E93D | 11.9 | 17.9 | ++ | + |
| F97Y | 17.5 | 90.7 | +++++ | + |
| Y98F | 12.7 | 18.1 | ++ | +++++ |
| T112I | 10.7 | 3.3 | − | + |
| R113Q | 12.4 | 8.3 | ++ | + |
| R114I | 8.8 | 7.9 | + | + |
| F115L | 15.9 | 16.8 | ++ | ++++ |
| T117A | 3.3 | 2.4 | − | +++ |
| I118V | 8.5 | 5.9 | ++ | + |
| V119E | 9.9 | 36.2 | ++++ | ++ |
| E121G | 3.6 | 1.4 | + | +++ |
| E122D | 6.2 | 2.0 | − | ++ |
| G123D | 20.9 | 104.9 | +++++ | + |
| G126D | 21.0 | 31.8 | ++++ | +++ |
| L127I | 10.8 | 8.9 | +++ | ++ |
| H129Y | 9.3 | 3.9 | ++ | +++++ |
| F130Y | 15.7 | 6.4 | ++ | ++ |
| V135D | 4.4 | 2.1 | ND | ND |
Specific activity (Sp act) in HEK293T cells was determined as described in Materials and Methods.
Relative activity in T cells was determined as fold activation in KD-CARD11 Jurkat T cells as described for Fig. 2C and D and normalized to that observed for the WT.
Bcl10 binding was determined in coimmunoprecipitation studies as described in Materials and Methods and depicted in Fig. 3. −, no enhancement over the WT level; +, 8% to 20% of that observed with ΔID; ++, 21% to 40% of that observed with ΔID; +++, 41% to 60% of that observed with ΔID; ++++, 61% to 80% of that observed with ΔID; +++++, 81% to 100% of that observed with ΔID. ND, not determined.
ID binding was determined in GST pulldown studies as described in Materials and Methods and depicted in Fig. 4. +, 0% to 20% of the level observed with ΔID; ++, 21% to 40% of that observed with ΔID; +++, 41% to 60% of that observed with ΔID; ++++, 61% to 80% of that observed with ΔID; +++++, 81% to 100% of that observed with ΔID.
Gain-of-function mutations selectively confer the ability to associate with Bcl10.
We next determined whether the gain-of-function mutations conferred to CARD11 an enhanced ability to interact with the signaling cofactors that have been previously shown to associate with CARD11 in a signal-dependent and ID-regulated manner (14). We coexpressed each variant with FLAG-tagged Bcl10 in HEK293T cells and assessed the degree to which each mutant coimmunoprecipitated with Bcl10, using wild-type CARD11 and the CARD11 ΔID as negative and positive controls, respectively. Eighteen of the gain-of-function mutants displayed enhanced abilities to associate with Bcl10 in this assay compared to wild-type CARD11 (Fig. 3 and Table 1). The relative ability to associate with Bcl10 roughly correlated with the ability of each variant to activate NF-κB, especially in Jurkat T cells (Table 1). For example, C49Y, F97Y, G123D, G126D, and V119E, the mutations that had the strongest effect on Bcl10 association, were the same mutations that elicited the greatest enhancements of activity in the Igκ2-IFN-LUC reporter assay. Five mutants (H31L, M32K, T112I, T117A, and E122D) did not display any enhanced association with Bcl10, and these were among the weakest gain-of-function mutants for NF-κB activation in Jurkat T cells. These data clearly indicate that most gain-of-function mutations identified enhance Bcl10 association with CARD11. In addition, the results reveal that the LATCH domain plays a critical role in controlling the CARD11-Bcl10 interaction.
Fig 3.

Mutations in the CARD and LATCH domains enhance the ability of CARD11 to associate with Bcl10. (A to F) HEK293T cells were transfected with 70 to 240 ng of expression vectors for the indicated myc-CARD11 variants and 70 to 225 ng of FLAG-Bcl10, as indicated. Anti-FLAG immunoprecipitations (IP) were performed as described in Materials and Methods and analyzed by Western blotting (WB) with the indicated primary antibodies.
We also tested whether the gain-of-function mutants affected the association of CARD11 with TAK1, TRAF6, caspase-8, or IKKγ in similar immunoprecipitation studies. Interestingly, none of the mutations enhanced the interaction with any of these proteins (data not shown), indicating a selective effect on the Bcl10-CARD11 interaction. The F123I and L225LI mutations found in human DLBCL have the same cofactor-selective effect on Bcl10 association (23). Since mutation of 14 of 19 residues in the LATCH domain had no effect in these assays, the results suggest that the LATCH domain does not control the interaction between CARD11 and TAK1, TRAF6, caspase-8, or IKKγ.
MALT1 directly associates with Bcl10 and can be recruited to the F123I and L225LI mutants in a Bcl10-dependent manner (23). We found that none of the gain-of-function mutations identified in the CARD or LATCH domains was able to confer detectable binding to MALT1 in immunoprecipitation studies in the absence of coexpressed Bcl10 (data not shown). We then tested four of the most active variants isolated in our screen to see if their mutations would allow Bcl10-dependent recruitment of MALT1 to CARD11. As shown in Fig. 4, FLAG-tagged MALT1 could associate with the C49Y, F97Y, G123D, and G126D mutants when coexpressed in the presence of Bcl10, indicating that when bound to these mutants, Bcl10 is competent to recruit MALT1 to CARD11. The effect of these mutations in this assay was comparable to that achieved by deletion of the ID.
Fig 4.
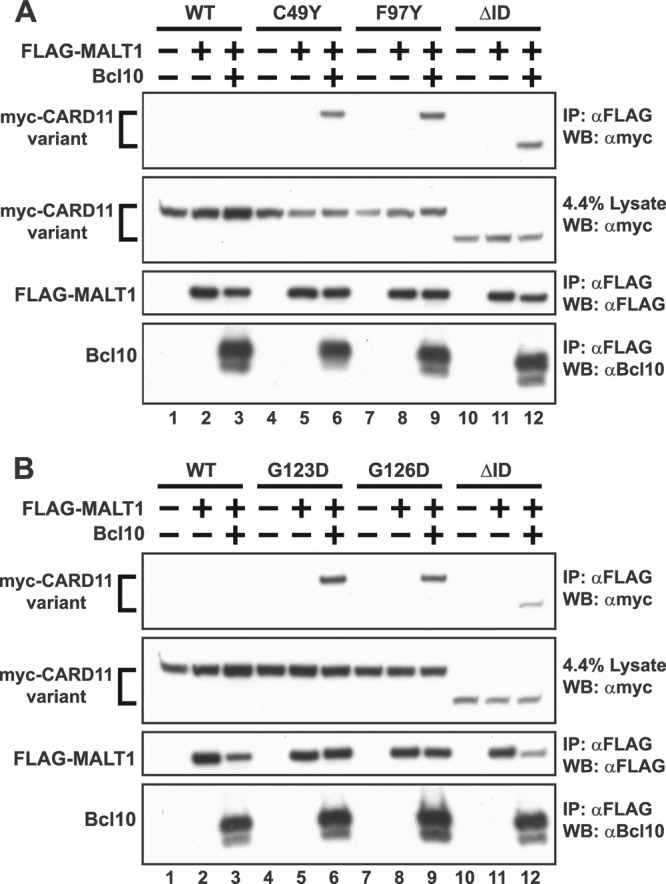
Gain-of-function mutations enhance Bcl10-mediated association of MALT1 with CARD11. (A and B) HEK293T cells were transfected with 400 to 700 ng of expression vectors for the indicated myc-CARD11 variants, 500 to 800 ng of FLAG-MALT1, and 300 to 500 ng of untagged Bcl10, as indicated. Anti-FLAG immunoprecipitations were performed as described in Materials and Methods and analyzed by Western blotting with the indicated primary antibodies.
Gain-of function mutations perturb ID binding.
We have previously shown that the ID can associate in trans with the CARD11 ΔID construct in a manner that depends upon the CARD and coiled-coil domains (14). The F123I and L225LI oncogenic mutations found in human DLBCL reduce this association (23). To test the hypothesis that the gain-of-function mutations we isolated also disrupt ID binding, we introduced each mutation in the context of the CARD11 ΔID construct and tested for their association with an ID-GST fusion protein under subsaturating conditions after expression in HEK293T cells. All 24 mutations reduced ID binding in trans in this assay (Fig. 5 and Table 1) to various degrees. The strongest effects on ID binding were observed for 12 mutants, 7 in the CARD domain (H31L, M32K, C49Y, K50M, N62Y, E93D, and F97Y) and 5 in the LATCH domain (T112I, R113Q, R114I, I118V, and G123D), all of which displayed 0% to 20% of the ID binding observed with the unmutated CARD11 ΔID. These data are consistent with the prediction that gain-of-function mutations would impact the ability of the ID to participate in intramolecular inhibitory interactions. In addition, the results reveal a previously unappreciated role for the LATCH domain in promoting ID binding.
Fig 5.

Mutations in the CARD and LATCH domains disrupt ID binding in trans. (A to D) HEK293T cells were transfected with 800 to 2,000 ng of expression vectors for the indicated CARD11 ΔID variants or with 2,000 ng or 1,500 ng of expression vectors for ID-GST or GST, respectively. Lysates were mixed as indicated and incubated, and glutathione-Sepharose pulldowns were performed as described in Materials and Methods and analyzed by Western blotting with the indicated primary antibodies.
Hyperactive mutants require Bcl10, MALT1, TRAF6, and TAK1 for signaling to NF-κB.
Bcl10 is a required signaling cofactor for CARD11 in normal antigen receptor signaling (34) as well as for the F123I and L225LI oncogenic variants that signal independently of receptor triggering (23). To confirm that the novel hyperactive mutants we identified also require Bcl10 for signaling to NF-κB, we tested the effect of knocking down Bcl10 on their activities in the Igκ2-IFN-LUC reporter assay in HEK293T cells. As expected, the activity of all mutants, regardless of their potency, was severely diminished as a result of Bcl10 deficiency (Fig. 6 and data not shown).
Fig 6.

Hyperactive CARD11 variants require Bcl10 for NF-κB activation. (A to D) HEK293T cells stably expressing a shRNA that targets either Bcl10 (KD-Bcl10) or GFP (KD-GFP) as a control were transfected with 20 ng of Igκ2-IFN-LUC, 6 ng of CSK-LacZ, and 8 to 100 ng of expression vectors for the indicated myc-tagged CARD11 variants. Western blots of corresponding lysates probed with anti-myc or anti-Bcl10 primary antibody to indicate the relative expression levels of each variant and Bcl10 in each sample are displayed at the bottom of each panel. β-Galactosidase activity, driven by CSK-LacZ, was used to normalize luciferase activity and to calculate equivalent amounts of transfected cell lysate for Western blot analysis.
For several of the most active variants, we also tested the dependence of their signaling activity on MALT1, TRAF6, and TAK1. We compared the activities of the G123D, F97Y, C49Y, G126D, V119E, and Y98F mutants in Jurkat T cell lines that stably expressed a control short hairpin RNA (KD-NT) or one that targeted MALT1 (KD-MALT1) or TRAF6 (KD-TRAF6). As shown in Fig. 7, the 75% and >75% reductions in MALT1 and TRAF6 levels, respectively, reduced the apparent activity of all of the mutants tested, as assessed in the Igκ2-IFN-LUC reporter assay (Fig. 7A to C). To test the requirement for TAK1, we treated Jurkat T cells with (5Z)-7-oxozeaenol, a specific inhibitor of TAK1 (35, 36), after transient expression of these variants in the reporter assay. As shown in Fig. 7D, the signaling activity of all variants tested was inhibited by (5Z)-7-oxozeaenol to a level similar to that observed after ΔID expression or treatment with anti-CD3/anti-CD28 antibodies.
Fig 7.

Hyperactive variants require MALT1, TRAF6, and TAK1 for NF-κB activation. (A and B) Jurkat T cells stably expressing hairpins that target MALT1 (KD-MALT1) or TRAF6 (KD-TRAF6), or a control hairpin (KD-NT), were transfected with 200 ng of CSK-LacZ and 1,500 ng of Igκ2-IFN-LUC in the presence of the indicated amounts of expression vectors for the indicated CARD11 variants and assayed as described in Materials and Methods. (C) Lysates from KD-MALT1 and KD-TRAF6 Jurkat T cell lines were assayed by Western blot analysis with the indicated primary antibodies. (D) Jurkat T cells were transfected with 200 ng of CSK-LacZ and 1,500 ng of Igκ2-IFN-LUC in the presence of 100 ng of expression vectors for the indicated CARD11 variants. Either DMSO vehicle (−) or 500 nM (5Z)-7-oxozeaenol (5Z-7-o) (+) was added to each sample as indicated every 12 h after transfection for a total of 3 doses. The indicated samples were stimulated with 1 μg/ml anti-CD3/anti-CD28 cross-linking in the absence or presence of (5Z)-7-oxozeaenol for 4 h prior to harvest.
Gain-of-function variants induce K63-linked ubiquitination of Bcl10 and the association of Ubn(K63)-Bcl10 with IKKγ.
The K63-linked ubiquitination of the CARD domain of Bcl10 on K31 and K63 is a required step downstream of CARD11 action in the TCR signaling pathway to NF-κB (24). Furthermore, we demonstrated that the F123I and L225LI oncogenic variants fail to activate NF-κB in the presence of a Bcl10 mutant containing arginine substitutions at K31 and K63 (23), suggesting that F123I and L225LI also depend upon Bcl10 ubiquitination for their dysregulated signaling. To test whether gain-of-function mutations in the CARD and LATCH domains spontaneously induce the K63-linked ubiquitination of Bcl10, we used retrovirus-mediated infection to establish a panel of Jurkat T cell lines that stably expressed 12 variants with a range of specific activities. Five of these variants (M32K, C49Y, N62Y, E93D, and Y98F) harbored mutations in the CARD domain, while 7 (T112I, E121G, E122D, G123D, G126D, L127I, and F130Y) contained LATCH domain mutations. To confirm that each variant spontaneously activated NF-κB after stable expression, we transfected each line with the Igκ2-IFN-LUC reporter and compared the fold induction to that observed with a control line expressing wild-type CARD11 in the absence and presence of PMA-Iono treatment, which mimics signaling downstream of the TCR. As shown in Fig. 8A, stable expression of wild-type CARD11 did not induce appreciable reporter activation, while PMA-Iono treatment of this line resulted in 27-fold reporter activation. As expected, many of the gain-of-function variants spontaneously activated the reporter at levels of expression comparable to the expression level seen with the wild-type control (Fig. 8A). The C49Y and G123D variants were the most potent, achieving levels of activation similar to that observed with PMA-Iono treatment of the control line. The activity of the stably expressed gain-of-function variants also led, as expected, to the spontaneous induction of nuclear NF-κB DNA binding activity, as revealed by EMSA analysis of nuclear extracts using a consensus κB site DNA probe (Fig. 8B). The extent of induction of NF-κB DNA-binding activity closely correlated with the activities exhibited in the reporter assay.
Fig 8.

Hyperactive CARD11 variants induce K63-linked ubiquitination of Bcl10 and the association of Ubn(K63)-Bcl10 with IKKγ. (A) Jurkat T cell lines stably expressing the indicated CARD11 variants fused to mCherry were generated by retrovirus infection, transiently transfected with 200 ng of CSK-LacZ and 1,500 ng of Igκ2-IFN-LUC, and assayed as described in Materials and Methods. The line expressing wild-type CARD11 was treated with 50 ng/ml PMA and 1 uM ionomycin (P/I) for 4 h as indicated. (B) Electrophoretic mobility shift assays were performed using 3 μg of nuclear extracts from the stably expressing Jurkat T cell lines described for panel A and a 32P-labeled DNA fragment containing a consensus κB site. The line expressing wild-type CARD11 was treated with 50 ng/ml PMA and 1 μM ionomycin for 30 min as indicated. (C) Immunoprecipitations were performed using denatured lysates [IP(den)] from the stably expressing Jurkat T cell lines described for panel A using anti-Bcl10 antibodies and analyzed by Western blotting with antiubiquitin (K63 linkage specific) [αUb(K63)], anti-Bcl10, or anti-myc primary antibodies as indicated. The line expressing wild-type CARD11 was treated with 50 ng/ml PMA and 1 μM ionomocyin for 20 min as indicated. (D) Immunoprecipitations were performed using lysates from the stably expressing Jurkat T cell lines described for panel A using anti-IKKγ antibodies and analyzed by Western blotting with anti-Bcl10 or anti-IKKγ primary antibodies as indicated. The line expressing wild-type CARD11 was treated with 50 ng/ml PMA and 1 μM ionomocyin for 20 min as indicated. The relative expression level of CARD11 variants in this experiment is also indicated in the anti-myc Western blot in panel C, since the same cell lines were used in panels C and D. (E) Quantitation of the levels of Bcl10-Ub detected in each sample in panel C, divided by the levels of unconjugated Bcl10, normalized to the ratio observed in the WT sample. (F) Quantitation of the levels of Bcl10-Ub detected in the IP with IKKγ in panel D, divided by the levels of unconjugated Bcl10, normalized to the ratio observed in the WT sample.
To probe for K63-linked ubiquitination of Bcl10, Bcl10 was immunoprecipitated from denatured cell lysates from these lines (24) and assessed for the presence of conjugated ubiquitin using K63-linkage-specific antibodies. K63-linked ubiquitinated Bcl10 [Ubn(K63)-Bcl10] was detected in the presence of 10 of the variants but not in the presence of the 2 weakest variants (Fig. 8C and E). The levels of Ubn(K63)-Bcl10 correlated remarkably well with the specific activities displayed in the NF-κB reporter and DNA-binding assays. In addition, the levels of unconjugated Bcl10 inversely correlated with the activity of each variant (Fig. 8C).
Since Ubn(K63)-Bcl10 has been shown to associate with IKKγ during TCR signaling (24), we immunoprecipitated IKKγ from lysates from these stable lines and probed for the presence of associated Ubn(K63)-Bcl10. Indeed, we observed that Ubn(K63)-Bcl10 associated with IKKγ to an extent that correlated with the degree of NF-κB activation in the reporter and DNA-binding assays (Fig. 8D and F). These data indicate that several of the gain-of-function variants induce K63-linked ubiquitination of Bcl10 and the association of Ubn(K63)-Bcl10 with IKKγ, even in the absence of antigen receptor engagement or signaling upstream of CARD11. In addition, they strongly suggest that residues in both the CARD (C49, N62, E93, and Y98) and LATCH (T112, E122, G123, G126, L127, and F130) domains function to prevent spontaneous Bcl10 ubiquitination and the association of Ubn(K63)-Bcl10 with IKKγ, two critical CARD11-dependent steps in IKK complex activation by antigen receptor signaling.
Gain-of-function mutations in the CARD and LATCH domains can promote the growth of a human DLBCL line in culture.
The OCI-Ly3 cell line, which is representative of the ABC subtype of DLBCL, harbors the L244P oncogenic variant of CARD11 and relies on CARD11-dependent signaling to NF-κB for survival and proliferation in culture (22). We adapted an assay originally developed by Lenz et al. (22) to assess the potential for hyperactive CARD11 variants to substitute for the L244P variant in this OCI-Ly3 line. We stably introduced 10 murine CARD11 variants into the OCI-Ly3 line by retroviral transduction and then superinfected the stable lines with a retrovirus that coexpressed GFP with an shRNA that could target the endogenous human L244P CARD11 mRNA by RNA interference (shC11). As previously demonstrated (22), the parental OCI-Ly3 line was highly sensitive to CARD11 L244P knockdown, as indicated by the 56% loss of GFP+ cells over 18 days in culture (Fig. 9A). The presence of the hairpin-resistant murine L244P oncogenic mutant served as a positive control for the assay and rescued a significant fraction of the cells, resulting in increased survival of GFP+ cells over the same time course (Fig. 9A). Among the nine gain-of-function mutants tested, the G123D, G126D, C49Y, and L127I mutants showed the best abilities to rescue the OCI-Ly3 cells (Fig. 9A). The E93D, Y98F, and F130Y mutants also displayed significant rescue ability, while the T112I and N62Y mutants conferred only a slight cell survival advantage compared to the OCI-Ly3 parental control (Fig. 9B). An assessment of the expression levels of the different hyperactive variants (Fig. 9C) indicated that the level of the G123D variant was slightly higher than that of the other variants, while those of L244P, N62Y, and T112I variants were slightly lower. Analysis of GFP+ cells from four lines revealed that expression of the shC11 hairpin resulted in a 50% to 60% reduction in endogenous CARD11 expression under the conditions of this experiment (Fig. 9D). Overall, there was a good correlation between the specific activities of the variants for rescue in this assay and their abilities to activate NF-κB when stably expressed in Jurkat T cells (Fig. 8A), with the exception of the L127I mutant, which had greater relative OCI-Ly3 rescue activity than expected. The results indicate that gain-of-function mutations in both the CARD (C49Y, E93D, Y98F) and the LATCH (G123D, G126D, L127I, F130Y) domains can provide sufficient activity to promote the survival of human DLBCL-derived cells that depend on NF-κB activation for proliferation in culture.
Fig 9.

CARD11 variants containing gain-of-function mutations in the CARD and LATCH domains can promote the survival of OCI-Ly3 human DLBCL-derived cells. (A and B) OCI-Ly3 cells stably expressing the indicated CARD11 variants were generated by retrovirus infection as described in Materials and Methods. Each line was superinfected with a retrovirus that coexpresses GFP and a shRNA that targets the endogenous human CARD11 mRNA (shC11). The percentages of GFP+ cells were normalized to the percentage observed on day 3 after infection. The survival curves for the parental OCI-Ly3 line and the line rescued with the L244P positive control are depicted in each panel for ease of comparison. (C) Lysates from the OCI-Ly3 lines stably expressing the indicated CARD11 variants were analyzed by Western blotting using anti-myc and anti-IKKα antibodies as indicated to reveal the relative expression level of each variant. The leftmost lane represents the parental mCAT-1-expressing OCI-Ly3 line. (D) OCI-Ly3 cells stably expressing the indicated myc-CARD11-mCherry variants were infected with a retrovirus coexpressing GFP with either a hairpin targeting endogenous human CARD11 (shC11) or a control hairpin (shNT). Seven days after infection, GFP+ cells were isolated by FACS sorting and lysates were analyzed by Western blotting with anti-CARD11 or anti-IKKα antibodies.
DISCUSSION
The central role that CARD11 serves in relaying signaling from the antigen receptor to NF-κB depends upon its signal-induced transition from a closed inactive state to an open active scaffold that can recruit signaling cofactors to activate the IKK complex. Previous studies established that this transition is regulated by the ID through intramolecular interactions involving the CARD and coiled-coil domains (14) and that two DLBCL-associated oncogenic mutations achieve hyperactive signaling by disrupting ID-mediated autoinhibition (23). These findings led us to hypothesize that, in addition to those described in the coiled-coil domain, hyperactive mutations should occur in other domains of CARD11 involved in the ID-mediated repression. Our data confirm that this hypothesis is correct. The screen for gain-of-function CARD11 variants yielded several mutations in the CARD domain that disrupt ID binding, as we predicted. Unexpectedly, the screen also identified a strikingly high density of mutations in the LATCH domain, a short 19-residue region between the CARD and coiled-coil domains that clearly plays a requisite role in autoinhibition by the ID.
Mutations in both the CARD and LATCH domains resulted in the disruption of ID binding, the selective enhanced association with Bcl10, the spontaneous ubiquitination of Bcl10, and the subsequent activation of the IKK complex and NF-κB (Fig. 10). These mutants cause hyperactivity by the same mechanisms ascribed to the F123I and L225LI mutants that were identified in human DLBCL.
Fig 10.

Model depicting how gain-of-function mutations in the CARD, LATCH, and coiled-coil domains spontaneously induce NF-κB activation.
The chief conclusion that emerges from our data is that the intramolecular regulation of CARD11 that allows for its signal-dependent activation by antigen receptor signaling also results in a vulnerability to mutation that can be exploited by lymphoid cancers to bypass normal controls and induce NF-κB and its proproliferative and antiapoptotic target genes.
All of the gain-of-function mutations in the CARD and LATCH domains reduce ID binding to the CARD11 ΔID variant in trans. The effects on ID binding likely account for the enhanced ability of 18 of the mutants to associate with Bcl10. It is clear, however, that there is not an absolute quantitative correlation between the positive effect on Bcl10 binding and the degree to which a mutation disrupted ID binding for all mutants. This is probably due to the fact that the ID and Bcl10 bind to partially overlapping determinants in the CARD–LATCH–coiled-coil region such that some mutations that deleteriously affect ID binding may also partially interfere with Bcl10 association. The net effect of such mutations would still be expected to increase Bcl10 association compared to what would occur with the closed, inactive wild-type CARD11.
It is intriguing that, among the gain-of-function mutations that emerged in the screen, 18 of 23 increased Bcl10 binding and none appreciably increased the association of other signaling cofactors whose recruitment to CARD11 is also controlled by the ID. This suggests that selectively enhancing Bcl10 association with CARD11 is sufficient to initiate signaling, presumably by promoting the K63-linked ubiquitination of Bcl10. It is important to point out, however, that the binding of other signaling cofactors that we did not examine, or that remain unknown, could also be affected by some or all of the mutations and may be important in determining signaling output. This possibility is suggested by differences in the specific activity of some mutants in the NF-κB reporter assay that otherwise display very similar abilities to bind Bcl10 and by the five mutants that did not enhance Bcl10 binding in the coimmunoprecipitation assay.
The enhanced association of Bcl10 with CARD11 that is induced by most of the mutations appears to promote the same K63-linked ubiquitination of Bcl10 that transiently occurs during normal antigen receptor signaling and that licenses Bcl10 for its action on the IKK complex through its association with IKKγ. For the 12 mutants analyzed after stable expression in Jurkat T cells, there was a good correlation between their enhancements in Bcl10 binding and the levels of Ubn(K63)-Bcl10 that were spontaneously produced in these cells. The ability of CARD11 mutations to elicit Bcl10 ubiquitination suggests that, during normal antigen receptor signaling, there are no CARD11-independent signaling events upstream of CARD11 activation that are required to induce this modification of Bcl10. The hyperactive CARD11 mutants must be sufficient to directly or indirectly promote the association between Bcl10 and the as-yet-unidentified E3 ligase that mediates conjugation. The levels of Ubn(K63)-Bcl10 produced as a result of hyperactive variant expression correlated well with the observed binding of endogenous Ubn(K63)-Bcl10 to IKKγ and with the relative degree of NF-κB activation. These data emphasize the importance of K63-linked polyubiquitinated Bcl10 as a signaling intermediate in this pathway that can dictate the extent of signal output. During normal antigen receptor signaling, polyubiquitinated Bcl10 is produced only transiently (24) and is removed by degradative mechanisms that serve to terminate signaling (37–39). In cells stably expressing the most active hyperactive variants, we readily detected a steady-state level of Ubn(K63)-Bcl10, indicating that its production must override these normal negative-feedback mechanisms. It is clear that, in order to fully understand how both normal and dysregulated CARD11 signaling occurs, further studies are necessary to identify the E3 ligase that ubiquitinates Bcl10 through K63 linkages, explain how the association with CARD11 promotes this modification, and elucidate exactly how Ubn(K63)-Bcl10 activates IKK kinase activity.
The six highly active variants tested also required MALT1, TRAF6, and TAK1 for signaling to NF-κB. These factors have been shown by others to be required for IKK kinase induction downstream of antigen receptor engagement. MALT1 is required for K63-linked ubiquitination of Bcl10 (24) and has also been implicated in K63-linked ubiquitination of IKKγ (40). TRAF6 also contributes to IKKγ K63-linked ubiquitination (26) but does not appear to be required for the induction of K63-linked ubiquitination of Bcl10 (data available upon request). TAK1 has been shown to mediate IKK kinase subunit phosphorylation during signaling (36). Our data indicate that the activities of these factors must function in concert with Ubn(K63)-Bcl10 to effect maximal NF-κB activation downstream of hyperactive CARD11 variants.
Most of the hyperactive variants that we tested had enough enhanced activity to promote the proliferation and survival of the ABC DLBCL-derived OCI-Ly3 line in culture. There was a good correlation between the ability to activate NF-κB after stable expression in Jurkat T cells and the ability to protect OCI-Ly3 cells from apoptosis in culture. However, the L127I mutant had more potential to enhance the survival of OCI-Ly3 cells than we would have expected on the basis of its relative activity after stable expression in Jurkat T cells. This observation may suggest that cell-type-specific aspects of the cellular environment in lymphoma can influence the potency of hyperactive CARD11 variants, such as the absence or presence of proteins that modify CARD11 signaling to NF-κB and whose functional interaction with CARD11 could be affected by some mutations and not others. The T112I and N62Y mutants did not provide much protection for OCI-Ly3 cells, but their enhanced abilities to activate NF-κB may be sufficient to contribute to the growth and survival of lymphoid cancers in other settings that have sustained other genetic alterations that could cooperate with weaker CARD11 mutants.
While this work was in progress, several studies reported the presence of CARD11 mutations in the CARD and LATCH domains in human biopsy specimens of DLBCL. Six of these are identical to mutations that we isolated in our screen, including C49Y (41), C49S (42), E93D (42), F115L (42, 43), G123D (44), and G126D (45). Four other reported mutations, including R114I (42), F115I (43), T117P (42), and F130V (45), occurred at the same position as those we found in our screen but mutated to a different residue. Although these studies did not demonstrate a functional effect of the mutations in the context of the cancers in which they were sequenced, our data provide a mechanistic characterization that can explain how the substitutions could lead to dysregulated NF-κB activity and promote proliferation and survival.
Our quantitative high-throughput screen could be easily expanded to detect other gain-of-function mutations in other domains of CARD11, including the coiled-coil domain. A database of such mutations would provide functional insights that would complement the catalog of cancer biopsy-associated CARD11 mutations that is likely to grow as more samples are surveyed. The screening methodology is modular, making it straightforward to substitute the protein that undergoes mutation and screening, and therefore is easily adaptable to the analysis of other proteins, such as MyD88, CD79A, and CD79B, that incur gain-of-function mutations in NF-κB-dependent cancers (4). With the substitution of the NF-κB reporter with that of another transcription factor, the screen could readily be applied to molecules that influence the activity of other gene regulators that promote tumorigenesis in other cell types.
ACKNOWLEDGMENTS
We thank R. Jattani for assistance in generating the KD-CARD11 Jurkat T cell line, H. Hu and G. Semenza for pSRGN, R. Davis for the OCI-Ly3 line, R. Jattani, D. Mackie, J. Tritapoe, and Z. Wang for critical reading of the manuscript, and M. Meffert and S. Desiderio for helpful discussions and advice.
This work was supported by RO1AI078980 and PO1AI072677 from the NIH, RSG-06-172-01-LIB from the American Cancer Society, and funds from the Johns Hopkins University Institute for Cell Engineering. J.L.P. is a recipient of a Leukemia and Lymphoma Society Scholar Award.
Footnotes
Published ahead of print 12 November 2012
REFERENCES
- 1. Schulze-Luehrmann J, Ghosh S. 2006. Antigen-receptor signaling to nuclear factor kappa B. Immunity 25: 701–715 [DOI] [PubMed] [Google Scholar]
- 2. Jiang C, Lin X. 2012. Regulation of NF-kappaB by the CARD proteins. Immunol. Rev. 246: 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaileh M, Sen R. 2012. NF-kappaB function in B lymphocytes. Immunol. Rev. 246: 254–271 [DOI] [PubMed] [Google Scholar]
- 4. Lim KH, Yang Y, Staudt LM. 2012. Pathogenetic importance and therapeutic implications of NF-kappaB in lymphoid malignancies. Immunol. Rev. 246: 359–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Staudt LM. 2010. Oncogenic activation of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2: a000109 doi:10.1101/cshperspect.a000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O'Brien W, Thome M, Littman DR. 2003. Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Curr. Biol. 13: 1252–1258 [DOI] [PubMed] [Google Scholar]
- 7. Gaide O, Favier B, Legler DF, Bonnet D, Brissoni B, Valitutti S, Bron C, Tschopp J, Thome M. 2002. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat. Immunol. 3: 836–843 [DOI] [PubMed] [Google Scholar]
- 8. Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, Nghiem M, Griffiths EK, Krawczyk C, Bauer B, D'Acquisto F, Ghosh S, Yeh WC, Baier G, Rottapel R, Penninger JM. 2003. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity 18: 763–775 [DOI] [PubMed] [Google Scholar]
- 9. Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, Cook MC, Kucharska EM, Hara H, Penninger JM, Domashenz H, Hong NA, Glynne RJ, Nelms KA, Goodnow CC. 2003. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity 18: 751–762 [DOI] [PubMed] [Google Scholar]
- 10. Newton K, Dixit VM. 2003. Mice lacking the CARD of CARMA1 exhibit defective B lymphocyte development and impaired proliferation of their B and T lymphocytes. Curr. Biol. 13: 1247–1251 [DOI] [PubMed] [Google Scholar]
- 11. Pomerantz JL, Denny EM, Baltimore D. 2002. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J. 21: 5184–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang D, You Y, Case SM, McAllister-Lucas LM, Wang L, DiStefano PS, Nunez G, Bertin J, Lin X. 2002. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat. Immunol. 3: 830–835 [DOI] [PubMed] [Google Scholar]
- 13. Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Lin X. 2005. Phosphorylation of CARMA1 plays a critical role in T cell receptor-mediated NF-kappaB activation. Immunity 23: 575–585 [DOI] [PubMed] [Google Scholar]
- 14. McCully RR, Pomerantz JL. 2008. The protein kinase C-responsive inhibitory domain of CARD11 functions in NF-kappaB activation to regulate the association of multiple signaling cofactors that differentially depend on Bcl10 and MALT1 for association. Mol. Cell. Biol. 28: 5668–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ. 2005. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23: 561–574 [DOI] [PubMed] [Google Scholar]
- 16. Shinohara H, Maeda S, Watarai H, Kurosaki T. 2007. IkappaB kinase beta-induced phosphorylation of CARMA1 contributes to CARMA1 Bcl10 MALT1 complex formation in B cells. J. Exp. Med. 204: 3285–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thome M, Charton JE, Pelzer C, Hailfinger C. 2010. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb. Perspect. Biol. 2: a003004 doi:10.1101/cshperspect.a003004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, Ferch U, Mak TW, Ruland J, Heissmeyer V, Krappmann D. 2006. Essential role for IkappaB kinase beta in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol. Cell 23: 13–23 [DOI] [PubMed] [Google Scholar]
- 19. Rosenwald A, Staudt LM. 2003. Gene expression profiling of diffuse large B-cell lymphoma. Leuk. Lymphoma 44(Suppl 3): S41–S47 [DOI] [PubMed] [Google Scholar]
- 20. Davis RE, Brown KD, Siebenlist U, Staudt LM. 2001. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 194: 1861–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. 2006. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441: 106–110 [DOI] [PubMed] [Google Scholar]
- 22. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM. 2008. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319: 1676–1679 [DOI] [PubMed] [Google Scholar]
- 23. Lamason RL, McCully RR, Lew SM, Pomerantz JL. 2010. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry 49: 8240–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu CJ, Ashwell JD. 2008. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. U. S. A. 105: 3023–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lamason RL, Kupfer A, Pomerantz JL. 2010. The dynamic distribution of CARD11 at the immunological synapse is regulated by the inhibitor kinesin GAKIN. Mol. Cell 40: 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. 2004. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14: 289–301 [DOI] [PubMed] [Google Scholar]
- 27. Naviaux RK, Costanzi E, Haas M, Verma IM. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70: 5701–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naldini L, Blomer U, Gage FH, Trono D, Verma IM. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U. S. A. 93: 11382–11388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. 2006. NEMO is a sensor of Lys 63-linked polyubiquitination and functions in NF-kappaB activation. Nat. Cell Biol. 8: 398–406 [DOI] [PubMed] [Google Scholar]
- 30. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872 [DOI] [PubMed] [Google Scholar]
- 31. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72: 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luo W, Wang Y, Reiser G. 2007. p24A, a type I transmembrane protein, controls ARF1-dependent resensitization of protease-activated receptor-2 by influence on receptor trafficking. J. Biol. Chem. 282: 30246–30255 [DOI] [PubMed] [Google Scholar]
- 33. Lamason RL, Lew SM, Pomerantz JL. 2010. Transcriptional target-based expression cloning of immunoregulatory molecules. Immunol. Res. 47: 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A, Ohashi PS, Mak TW. 2001. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell 104: 33–42 [DOI] [PubMed] [Google Scholar]
- 35. Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. 2003. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 278: 18485–18490 [DOI] [PubMed] [Google Scholar]
- 36. Shambharkar PB, Blonska M, Pappu BP, Li H, You Y, Sakurai H, Darnay BG, Hara H, Penninger J, Lin X. 2007. Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. EMBO J. 26: 1794–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lobry C, Lopez T, Israel A, Weil R. 2007. Negative feedback loop in T cell activation through IkappaB kinase-induced phosphorylation and degradation of Bcl10. Proc. Natl. Acad. Sci. U. S. A. 104: 908–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scharschmidt E, Wegener E, Heissmeyer V, Rao A, Krappmann D. 2004. Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappa B signaling. Mol. Cell. Biol. 24: 3860–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zeng H, Di L, Fu G, Chen Y, Gao X, Xu L, Lin X, Wen R. 2007. Phosphorylation of Bcl10 negatively regulates T-cell receptor-mediated NF-kappaB activation. Mol. Cell. Biol. 27: 5235–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. 2004. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature 427: 167–171 [DOI] [PubMed] [Google Scholar]
- 41. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, Pasqualucci L. 2009. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459: 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, Saksena G, Stransky N, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Hernandez-Lemus E, Schwarz-Cruz y Celis A, Imaz-Rosshandler I, Ojesina AI, Jung J, Pedamallu CS, Lander ES, Habermann TM, Cerhan JR, Shipp MA, Getz G, Golub TR. 2012. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. U. S. A. 109: 3879–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Montesinos-Rongen M, Schmitz R, Brunn A, Gesk S, Richter J, Hong K, Wiestler OD, Siebert R, Kuppers R, Deckert M. 2010. Mutations of CARD11 but not TNFAIP3 may activate the NF-kappaB pathway in primary CNS lymphoma. Acta Neuropathol. 120: 529–535 [DOI] [PubMed] [Google Scholar]
- 44. Bu R, Bavi P, Abubaker J, Jehan Z, Al-Haqawi W, Ajarim D, Al-Dayel F, Uddin S, Al-Kuraya KS. 2012. Role of NF-kappaB regulators TNFAIP3 and CARD11 in Middle Eastern Diffuse Large B cell lymphoma. Leuk. Lymphoma 53: 1971–1977 [DOI] [PubMed] [Google Scholar]
- 45. Dong G, Chanudet E, Zeng N, Appert A, Chen YW, Au WY, Hamoudi RA, Watkins AJ, Ye H, Liu H, Gao Z, Chuang SS, Srivastava G, Du MQ. 2011. A20, ABIN-1/2, and CARD11 mutations and their prognostic value in gastrointestinal diffuse large B-cell lymphoma. Clin. Cancer Res. 17: 1440–1451 [DOI] [PubMed] [Google Scholar]