

Abstract
Molecular mechanisms underlying sarcopenia, the age-related loss of skeletal muscle mass and function, remain unclear. To identify molecular changes that correlated best with sarcopenia and might contribute to its pathogenesis, we determined global gene expression profiles in muscles of rats aged 6, 12, 18, 21, 24, and 27 months. These rats exhibit sarcopenia beginning at 21 months. Correlation of the gene expression versus muscle mass or age changes, and functional annotation analysis identified gene signatures of sarcopenia distinct from gene signatures of aging. Specifically, mitochondrial energy metabolism (e.g., tricarboxylic acid cycle and oxidative phosphorylation) pathway genes were the most downregulated and most significantly correlated with sarcopenia. Also, perturbed were genes/pathways associated with neuromuscular junction patency (providing molecular evidence of sarcopenia-related functional denervation and neuromuscular junction remodeling), protein degradation, and inflammation. Proteomic analysis of samples at 6, 18, and 27 months confirmed the depletion of mitochondrial energy metabolism proteins and neuromuscular junction proteins. Together, these findings suggest that therapeutic approaches that simultaneously stimulate mitochondrogenesis and reduce muscle proteolysis and inflammation have potential for treating sarcopenia.
INTRODUCTION
In most mammalian species, including humans, aging is associated with a decline in skeletal muscle mass and function, termed sarcopenia. Sarcopenia leads to a reduced ability to perform activities of daily living and thus causes a loss of independence. Indeed, low skeletal muscle mass or strength is reported to be the most frequent cause of disability in the elderly (1) and to be a predictor of morbidity, loss of independence, frailty, and mortality, independent of other risk factors or disease (2). At the cellular level, sarcopenia is characterized by atrophy of the constituent fibers in the muscle, with a tendency for greater atrophy of fast-twitch type 2 compared to slow-twitch type 1 fibers, an increase in fiber size heterogeneity, and an increase in the composition of noncontractile (adipose and connective) tissues within the muscles (3–6).
The molecular mechanisms underlying sarcopenia have not been elucidated but are thought to be multifactorial, contributed to by a decline in physical activity of idiopathic origin, a decrease in anabolic factors such as growth hormone and androgens, an increase in proinflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) with attendant chronic inflammation and muscle catabolism, and an increase in muscle damage from increased mitochondrial reactive oxygen species produced during energy metabolism in muscles (7–10). This potential multifactorial etiology has confounded efforts to elucidate the primary inciting mechanisms for sarcopenia. Currently, because mechanistic studies in humans are limited by the long human life span and absence of noninvasive methods to assess the molecular changes in human tissues, rodents are often used for mechanistic studies. Studies using both inbred and outbred rats indicate that the age of onset of sarcopenia may vary not only between rat strains but also among individual rats of the same strain with the former attributed to differences in life span and the latter to genomics, the caloric intake, and the incidence of diseases (11). Nonetheless, whenever present, the relative time course, morphological, metabolic, and gene and protein expression features of sarcopenia in the rat (12–14) mimic those of human sarcopenia (5, 15–19). In most if not all of these studies, the morphological, histological, metabolic, transcriptional, and proteomic changes in sarcopenia were not assessed within the same study and/or subjects; hence, the inter-relationships between these parameters remain unclear. Furthermore, most previous studies compared two age groups (usually young, nonsarcopenic versus late middle-age or elderly, sarcopenic subjects); hence, there is dearth of data on the changes in muscle during the onset of sarcopenia. These gaps make it difficult to elucidate and differentiate the causes from the effects of sarcopenia (20). More comprehensive and systematic analyses, involving both assessment of the histological, functional, and molecular changes in muscles from across the life span of the same animal to identify the age of onset of sarcopenia and then further analyses around the identified onset age would facilitate identifying the inciting causes of sarcopenia.
Therefore, in the present study, male Harlan Sprague-Dawley (Harlan SD) rats were profiled for morphological (changes in muscle mass, fiber cross-sectional area, fiber immunohistochemistry), functional (muscle strength), and global muscle gene and protein expression at relatively short intervals across their life span.
MATERIALS AND METHODS
Animals and husbandry.
Male Harlan SD rats (Harlan, Inc.) were obtained at 5, 11, 17, and 20 months of age and studied at 6, 12, 18, 21, 24, or 27 months of age. The rats were fed the 2014S Teklad Global 14% protein rodent maintenance diet, ad libitum. They were randomized into two groups, one to assess evoked muscle contractile properties and the other for postmortem analysis (anatomic pathology and histomorphometric analysis). All animal procedures were approved by the Institutional Animal Care and Use Committee of the Novartis Institutes for Biomedical Research and were in compliance with the Animal Welfare Act Regulations 9 CFR, parts 1, 2 and 3, and U.S. regulations (68).
Evoked EDL muscle strength.
Peak nerve-evoked contractile properties of the extensor digitorum longus (EDL) muscle were determined in a subset of male Harlan SD rats aged 6, 12, 18, and 24 months (n = 4 to 8) using the muscle physiology system from Aurora Scientific, Inc. (Aurora, Ontario, Canada). We measured the peak force of the EDL instead of a larger leg muscle such as the tibialis anterior or gastrocnemius muscle because the peak force generated by the larger leg muscles exceeds the limits (∼1,000 g force) of the available 305C-LR transducer. Briefly, the rats were anesthetized with isoflurane vaporized in oxygen, and the left leg was shaved of fur. Each animal was positioned in dorsal recumbency under a thermocoupled temperature regulated heating lamp to maintain body temperature at ∼37°C as monitored by a rectal temperature probe. The sciatic nerve in the left leg was exposed and attached to a nerve-stimulating electrode via an ∼1-cm incision on the lateral aspect of the thigh. The distal tendon of the EDL muscle was exposed, severed, and attached to a 305C-LR dual-mode muscle lever system (Aurora Scientific). The knee and ankle joints were immobilized with a clamp, and the stimulating electrode was connected to an electrical stimulator (model 604). Sciatic nerve stimulation was performed with square biphasic pulses of 0.2-ms duration and supramaximal strength (100 to 600 mAmp and 20 V). The baseline tension on the muscle was increased gradually during stimulation at 1 to 2 Hz to determine the tension at which peak twitch force was generated (i.e., the optimal length of the muscle). With muscle baseline tension set at optimal (∼15 g), the sciatic nerve was stimulated with 10 sets of stimuli at 2 Hz for 2 s every 12 s, followed by 10 trains of stimuli at 240 Hz for 3 s every 10 s. The force of contraction generated by the muscle in response to electrical stimulation of the sciatic nerve was transmitted via 305C-LR dual-mode muscle lever, acquired using Dynamic Muscle Control data acquisition software and analyzed using Dynamic Muscle Analysis software (Aurora Scientific). The peak force generated during tetanic stimulation at 240 Hz was determined, and the decline in force between the first and tenth tetanic trains was used to calculate the degree of tetanic fatigue. Peak tetanic force was also normalized to muscle weight to determine the specific tetanic force.
Postmortem procedures.
Rats aged 6, 12, 18, 21, 24, or 27 months (n = 10 at each age, except that at 27 months n = 8) were used. Organ weights (liver, kidneys, heart, spleen, brain, and testes) and macroscopic findings were recorded. Microscopic examination was conducted on tissue samples from all organs. Organs were fixed in 10% neutral buffered formalin at necropsy, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin and the Von Kossa special stain for minerals was applied to the sciatic nerve. In addition, specific skeletal muscles (namely, triceps brachii, tibialis cranialis, extensor digitorum longus, soleus, plantaris, gastrocnemius, quadriceps, bulbo-carvenosus, and levator anii) were dissected, weighed, and fixed in 10% buffered formal saline for microscopic examination or snap-frozen in liquid nitrogen and stored at −80°C for subsequent analysis. Microscopic findings were recorded in PATHDATA and graded according to the following scheme: 1 (minimal, very few, very small), 2 (slight, few, small), 3 (moderate, moderate number, moderate size), 4 (marked, many, large), and 5 (severe).
Muscle histomorphometry.
The right plantaris muscle was snap-frozen in 2-methylbutane precooled in liquid nitrogen and stored at −80°C for histomorphometric analysis of fiber type composition and cross-sectional area. Serial cryosections, 8 μm thick, were cut, air dried, and blocked in 10% goat serum for 30 min at room temperature. They were then incubated with antibodies against laminin (Sigma-Aldrich; catalog no. L9393), followed by either myosin heavy chain type 1 (HB283; American Type Culture Collection [ATCC]), type 2A (HB277; ATCC), type 2B (HB283; ATCC), or type 2X/D (6H1; Developmental Studies Hybridoma Bank, University of Iowa) to outline the sarcolemma and differentiate the fiber types, respectively. After washing, the sections were incubated with a mouse-on-mouse horseradish peroxidase polymer (Biocare; catalog no. MM620H) for myosin, followed by a rabbit-on-rodent alkaline phosphatase polymer (Biocare; catalog no. RMR626H) for laminin, followed by incubation first with Vulcan fast red (Biocare; catalog no. FR805CHE) for <10 min to visualize laminin and then with the DAB substrate (Vector; catalog no. SK4100 for MyHC 1 and 2A; Dako; catalog no. K3468; for MyHC 2B). The sections were then rinsed in three changes of phosphate-buffered saline, dehydrated, and mounted with Permount (Fisher; catalog no. SP15-100). Images of the entire muscle section were acquired using Scanscope (Aperio, Vista, CA), and the number and cross-sectional area of each fiber type in serial sections determined. The means and frequency distribution (histogram) of all fibers, typically >10,000, and of the individual types I, IIA, and IIB fibers in the entire section were determined.
To determine whether sarcopenia occurs with increased fibrosis, serial sections were incubated with picrosirius red as described previously to stain collagen (21). The stained sections were digitized and the content of collagen quantified and expressed as intensity of collagen staining per unit area. Muscle collagen content was also estimated by its hydroxyproline content as described previously (22). Serial sections were also stained for alkaline phosphatase activity as described previously (23) to identify and measure viable capillary endothelial cells. The number of capillaries in the section were counted and normalized to fiber cross-sectional to avoid overestimations due to aging-related fiber atrophy (24). To assess potential age-related changes at the neuromuscular junction, serial sections of the plantaris muscle were stained for acetylcholinesterase as described previously (25). The size of the stained area in the section were determined digitally and normalized to fiber cross-sectional area to account for age-related fiber atrophy. Finally, to determine whether fiber oxidative capacity changed with age, serial sections of the plantaris muscle were stained for the mitochondrial oxidative enzyme succinate dehydrogenase (SDH) as described by Dubowitz and Sewry (26). The entire section was digitized to estimate the mean intensity of SDH staining, as well as the proportion of the section occupied by fibers with weak, medium, and strong SDH staining expressed as a percentage of the total number of fibers in the section. All image analysis was automated and performed using a Novartis Institutes for Biomedical Research internal software (ROBIAS ASTORIA, written by Wilfried Frieauff).
Microarray-based transcriptional profiling.
The gastrocnemius muscle was analyzed for age-related changes in RNA expression. The muscle was pulverized under liquid nitrogen and total RNA extracted from a portion using the RNeasy fibrous tissue midi kit according to the manufacturer's protocol (Qiagen). RNA concentration and purity (A260/A280 ratios > 1.9) were assessed using a NanoDrop-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE), and RNA integrity was assured using an Agilent 2100 Bioanalyzer (Santa Clara, CA). To determine global gene expression changes, aliquots of 2 to 4 μg of each RNA sample were subjected to microarray analysis using the GeneChip Rat Genome 230 2.0 Array (Affymetrix, Santa Clara, CA). Statistical analysis of the microarray data was performed within the R statistical environment (27) using dedicated packages from Bioconductor (28). Quality of the array raw data was assessed using the affyPLM package. Of the 57 arrays, 3 were of low quality. The raw intensity files of the remaining 54 arrays were normalized and summarized into probe set level expression index using the PLIER (probe logarithm intensity error) method (29). Affymetrix MAS5 present or absent calls were calculated, and probe sets with “present” calls in <50% of samples within all age groups were removed. The moderated F-test implemented in the limma package (30) was applied to evaluate the significance of individual probe set changes across age groups. Individual moderated t tests were subsequently applied to evaluate the significance of differences between any two relevant age groups. t test values were adjusted using the Benjamini and Hochberg (BH) (31) method to control for false-discovery rate. Expression changes were considered significantly up- or downregulated when the BH adjusted P value was ≤0.01. The area under the curve (AUC) was calculated for all probe sets to summarize the changes over the time course using the log2-fold change of later time points versus 6 months. K-means clustering was performed in SpotFire DXP (TIBCO, Cambridge, MA) on the standardized average expression of the age groups using correlation distances for those probe sets with P < 0.01 and log2 fold change of >±0.5 in at least one of the later age group versus 6 months. Pearson correlations of each probe set to the mean profile of its corresponding cluster were calculated as a measure of membership. The functional enrichment of the clusters was assessed using EASE statistics available through the Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources (32). The enrichment results were visualized using the Cytoscape plug-in enrichment map (67). Ingenuity Pathway Assist (IPA; Ingenuity Systems, Inc.) was used to predict the upstream transcription factors that might be responsible for the changes in expression of the genes in each cluster.
Identification of gene signatures of sarcopenia versus gene signatures of aging.
To identify genes whose expression changes best correlate with sarcopenia (i.e., gene signature of sarcopenia) and those that best correlate with aging per se (i.e., gene signature of aging), we determined Pearson correlation coefficients for the distribution of all significantly regulated genes against gastrocnemius muscle mass and age, respectively. Correlations were considered strong and significant at Pearson R ≥ 0.9 or ≤ −0.9 (i.e., absolute R ≥ 0.9) and adjusted P value of <0.0001. A gene was considered to be a signature of sarcopenia compared to aging and vice versa if its correlation coefficients for sarcopenia and aging differed by a factor of 0.04 or more, arbitrarily chosen.
Reverse transcription-PCR validation.
RNA samples from the same gastrocnemius muscles were reverse transcribed to cDNA using a high-capacity cDNA kit (Applied Biosystems, Inc., Foster City, CA). The cDNA was then used for real-time quantitative PCR (qPCR) using an ABI Prism 7900 sequence detection system and gene-specific primers to confirm finding from the microarray study, as well as to determine the profile of selected candidate genes. The transcript levels were normalized to the geometric mean of five housekeeping genes (TBP, Vps26a, Znf830, Tmem1, and Dnttp2) in the same preparation, and the fold change relative to the 6-month-old rats was calculated as 2−ΔΔCT (33). Table S1 in the supplemental material lists the specific probes for the target genes verified by qPCR and the housekeeping genes used for sample normalization.
Proteomics analyses.
Gastrocnemius muscle samples of rats in each of the 6-, 18-, or 27-month-old groups were pooled proportional to muscle weight. The pooled 6-, 18-, and 27-month samples were each homogenized in 8 M urea and 50 mM Tris-HCl (pH 8). Approximately, 500 μg of protein from each sample was reduced, alkylated and digested using 4 μg of Lys-C at 37°C overnight. Peptides from proteolytic digestion were desalted and labeled using reductive dimethylation (34, 35) (see the discussion of methods in the supplemental material). Rat muscle sample for the 6-, 18-, and 27-month-old rats was labeled with light (CH2O-NaBH3CN), medium (CD2O-NaBH3CN), and heavy (CD2O-NaBD3CN) reagents, respectively. The samples were washed with 0.1% formic acid and eluted from the column using 80% ACN and 0.5% acetic acid. Samples were then dried down using a vacuum centrifuge. Peptides (0.75 mg) from light/medium/heavy rat muscle samples were separated by strong cation-exchange chromatography into 20 fractions (see the supplemental material). Each fraction was dried down by vacuum centrifugation and desalted using self-packed C18 STAGE-tips (36). All liquid chromatography-tandem mass spectrometry (LC-MS/MS) data were obtained using an LTQ-Orbitrap Discovery hybrid mass spectrometer (Thermo Fisher, San Jose, CA) (see the supplemental material). Each sample was loaded onto a reverse phase column and separated using 95 min LC gradient of 5 to 27% buffer B at a flow of 0.5 to 1 μl/minute. MS analysis was performed using a top 10 method where the MS1 scan was acquired in the Orbitrap, followed by 10 data depended MS/MS scans on the 10 most intense ions in the LTQ with CID for fragmentation. MS/MS spectra assignments were made with the Sequest algorithm (37) using the entire rat IPI database (version 3.6) (see the supplemental material). Sequest searches were performed using a target-decoy strategy (38) with the rat IPI database in correct orientation (forward database) and the same database but with all sequences in reverse orientation (reverse database). Sequest searching was performed with a precursor ion tolerance of 50 ppm with LysC specificity. For dimethyl labels, a static modification of 28.0313 Da was used on the N-terminus and lysine residues. In addition, differential modifications of 4.02310 and 8.044336 Da were used for medium and heavy proteins, respectively, on the N-terminus and lysine residues. A protein level false-discovery rate of <1% was used as a threshold for protein identifications using the target decoy strategy. Quantification of each protein was determined using the peak heights for light, median, and heavy forms for that protein. The criterion for protein quantification was a signal-to-noise ratio of >5 for at least one of the protein species (light, medium, and heavy). Quantification of protein level was by calculating the median value of the ratios of light to medium and of light to heavy. Using this method of quantification, the contribution of single peptide protein species in the data set was low (<5%), presumably reflecting the high accuracy and sensitivity of the MS methods and the high signal-to-noise cutoff used (see Table S2 in the supplemental material). Additional details of the proteomics methods are provided as supplemental material. Hierarchal clustering was performed on the quantified proteins using Cluster 3.0. Gene ontology analysis was then performed for each cluster using DAVID (32), with each cluster compared to all proteins identified in the experiment. Gene ontology annotations were considered significantly enriched if the BH probability was <0.05.
Mitochondrial DNA content and enzyme and complex activities.
Mitochondrial DNA content and activities of citrate synthase, and complexes I, II, and IV of the electron transport chain were determined in proportionately pooled gastrocnemius muscle samples of rats in each of the 6-, 18-, or 24-month-old groups. For mitochondrial DNA content, total DNA was extracted from 10 to 30 mg of each of the pooled muscle samples using the DNeasy kit (Qiagen, Valencia, CA). To increase the yield, phenol-chloroform extraction was performed instead of the column purification steps. Total DNA was quantified using absorbance at 260 nm; the 260/280 ratios ranged from 1.8 to 2.1. Gene expression assays for cytochrome b (Rn03296746_s1) and ubiquitin C (Rn01789812_g1) (Life Technologies, Carlsbad, CA) were used to quantify mitochondrial and genomic DNA, respectively, by qPCR. PCR efficiencies for all reactions ranged from 90 to 107%. Mitochondrial DNA levels were normalized to genomic DNA levels and fold change relative to the 6-month group was determined using the 2−ΔΔCT method (33). For enzyme activity assays, an equal amount of the pooled 6-, 18-, and 24-month samples was each homogenized in 100 mM potassium phosphate (pH 7.4) containing complete protease inhibitor (Roche Applied Science, Indianapolis, IN). Samples were sonicated at 4°C for 15 s, incubated for 1 min, and sonicated for an additional 15 s using a Sonicator 3000 (Qsonica, Newtown, CT). Homogenates were centrifuged for 20 min at 20,000 × g. The supernatants (S20 fractions) were removed, and the pellets (P20 fractions) resuspended in the homogenization buffer and sonicated for an additional 10 s. Protein concentrations for all fractions (total homogenate, S20, and P20) were determined using the BCA protein assay (Thermo Scientific, Rockford, IL). All samples were stored at −80°C prior to analysis. Citrate synthase activity was measured in the S20 fractions and complexes I, II, and IV activities in the P20 fractions as described previously (39, 40) with the modifications for complexes II and IV (inclusion of dodecyl-β-n-maltoside) described by Birch-Machin and Turnbull (41). For all enzyme activity assays, initial velocities were determined by linear regression analysis of reaction time courses using data corresponding to ≤10% substrate conversion or product formation (initial reaction rates). Rates were normalized to the wet weight of the muscle samples.
Statistical analysis.
Data are presented as mean ± the standard error of the mean. Differences between age groups were evaluated by one-way analysis of variance, followed by Bonferroni's multiple-comparison tests using GraphPad Prism 5. Differences were considered significant at P ≤ 0.05.
RESULTS
Body weight and pathological findings.
The rats used in the present study were in healthy condition for their age. In the last 4 weeks before necropsy, body weight increased in rats 6 to 21 months of age but decreased in 24- and 27-month-old rats (Fig. 1A); mean daily food consumption did not differ between the age groups. Despite clinical evidence of physical frailty, only minor age-related clinical pathological changes were observed in these rats. The main changes noted were an increase in absolute neutrophil count and increased serum globulin concentrations, reflecting an increased incidence of systemic inflammation correlating with age. The clinical pathology and hematological data are summarized in Tables S3 and S4 in the supplemental material. Heart weight increased with age, reaching a plateau at 18 months (Fig. 1B) without detectable macroscopic or microscopic correlates. With the possible exception of the kidney, for which the absolute but not relative (to body or brain weight) weight was increased at 27 months (Fig. 1C), the weights of other organs were comparable between the age groups after excluding one aberrant organ weight increase associated with neoplasia. Macroscopic and microscopic findings in these rats represented a spectrum of changes commonly associated with aging and occurred at the expected incidence and severity for the given age groups. For example, the incidence and severity of chronic progressive nephropathy increased with aging, as did focal (several) and diffuse (rare) pancreatic islet hyperplasia and numerous proliferative processes (e.g., focal C-cell hyperplasia/adenoma of the thyroid and focal hyperplasia/adenoma of the pituitary pars distalis) (data not shown).
Fig 1.

Body, organ, and muscle weights and EDL muscle contractile properties. The percent body weight change over the last 4 weeks before study (A) and the weights of the heart (B), kidneys (C), and tibialis (D), gastrocnemius (E), and quadriceps (F) muscles of rats aged 6, 12, 18, 21, 24, and 27 months. The wet weight of the EDL muscle declined with age (G) in association with approximately twice as much decline in evoked peak tetanic strength (Po) (H); hence, specific strength tended to decline, but not significantly (I). *, **, ***, and **** indicate P < 0.05, 0.01, 0.001, and 0.0001 versus 6 months, respectively. 6M, 6 months; 12M, 12 months, etc.
Skeletal muscle weight.
The weight of individual skeletal muscles of the hind limb declined slowly (11 to 21% decline) between 6 and 18 months of age, but thereafter muscle weights declined rapidly, such that by 27 months some were ∼60% smaller than at 6 months, irrespective of whether or not muscle weight was normalized to end body weight (Fig. 1D to F). In contrast, the weights of the triceps brachii muscle of the fore limb and the nonappendicular bulbocarvenosus levator ani muscle complex declined only ∼19 and 25%, respectively, by 27 months of age (see Fig. S1A and B in the supplemental material).
Evoked EDL muscle strength.
To determine whether changes in muscle mass were associated with changes in strength, the peak force generated by the extensor digitorum longus (EDL) muscle was measured during supramaximal stimulation of the sciatic nerve in another subset of rats aged 6, 12, 18, and 24 months. Relative to 6-month-old rats, the weights of the EDL muscle were 14, 13, and 28% less, while the peak nerve-evoked contractile strengths were 27, 26, and 45% less in the 12-, 18-, and 24-month-old rats, respectively (P < 0.001 only for 24 months, presumably because of the small sample size [n = 4 to 8]) (Fig. 1G and H). Because the decline of evoked peak strength was greater than the decline in EDL muscle weight, there was a tendency for force per unit muscle weight to decline with age, but not significantly (Fig. 1I).
Skeletal muscle histomorphometry.
Histomorphometric analysis of the plantaris muscle of the hind leg indicated that the age-related decline in mass (Fig. 2A) occurred with a decline in mean muscle fiber cross-sectional area (Fig. 2B) and a leftward shift of the fiber cross-sectional area histogram (Fig. 2C). Fiber atrophy was associated with an increase in angularity of fibers as indicated by the age-dependent increase in the mean and the rightward shift of the deformity factor, an index that relates the cross-sectional area to the perimeter of the fiber (Fig. 2D and E). Type 1, 2A, and 2B muscle fibers in the plantaris muscle exhibited an age-dependent decline in size, reflected by the mean fiber cross-sectional area (Fig. 2F and G) and a leftward shift of the fiber cross-sectional area histogram (Fig. 2I to K).
Fig 2.

Plantaris muscle fiber histomorphometry. The age-dependent decline in plantaris muscle mass (A) was associated with a proportional decrease in fiber cross-sectional area (B), a leftward shift of the fiber cross-sectional area histogram (C), and increased deformation of fibers (D and E). Analysis of the individual fiber types indicated no significant differences in the degree (F and G) or patterns (I to K) of atrophy between types 1, 2A, and 2B fiber types. *, **, ***, and **** indicate P < 0.05, 0.01, 0.001, 0.0001 versus 6 months, respectively. 6M, 6 months; 12M, 12 months, etc.
Atrophy of the plantaris muscle was associated with increased fibrosis, indicated by a progressive age-related increase in picrosirius red staining of sections (Fig. 3A) and increased muscle hydroxyproline content (see Fig. S1C in the supplemental material). Alkaline phosphatase staining of serial sections revealed a tendency for the number of capillaries per fiber cross-sectional area to be increased at 24 and 27 months of age (Fig. 3B). The size of the acetylcholinesterase (Ache) stained area relative to fiber cross-sectional area also increased with age—presumably because in muscles from 24- and 27-month-old rats, extrajunctional areas of the sarcolemma were also stain for Ache (Fig. 3C). Cryosections of muscles of 24- and 27-month-old rats were less intensely stained for succinate dehydrogenase (SDH) activity compared to 6- to 21-month-old rat muscles. Categorization of the fibers in each section by intensity of SDH staining further revealed that, compared to 6-month-old rats, the plantaris muscle of 24-and 27-month-old rats had relatively fewer fibers with moderate to strong SDH activity (i.e., types 2A and 1 fibers) and more fibers with low SDH activity (type 2B fibers) (Fig. 3D).
Fig 3.

Representative photomicrographs of 6-month and 27-month plantaris muscles stained for picrosirius red (PSR), alkaline phosphatase (AlkPhos), acetylcholinesterase (Ache), and succinate dehydrogenase (SDH). Analysis (right column) revealed that muscles from 24- and 27-month-old rats have more intense picrosirius staining, a tendency for increased capillary density, increased acetylcholinesterase-stained area per fiber cross-sectional area, and an increase in fibers weakly stained for and a corresponding decrease in fibers strongly stained for SDH activity. *, **, ***, and **** indicate P < 0.05, 0.01, 0.001, and 0.0001 versus 6 months, respectively. 6M, 6 months; 12M, 12 months, etc.
Microarray-based transcriptional profiling.
Of the 31,099 probe sets on the GeneChip Rat Genome 230 2.0 Array (corresponding to over 30,000 genes and variants), 16,573 (53%) were detected in at least one age group, and 11,097 were statistically differentially expressed among the age groups with BH adjusted F-test P value of <0.01. Furthermore, 3,890 probe sets were progressively and differentially regulated at least 1.4-fold (log2 of 0.5) with a BH-adjusted F-test P value of <0.01 in at least one later time point compared to 6 months.
Discriminating gene signatures of sarcopenia versus gene signature of aging.
To identify specific patterns of sarcopenia-related changes in gene expression, the 3,890 significantly regulated probe sets were subjected to cluster analysis. First, by performing K-means cluster analysis with increasing cluster numbers from 2 to 12 and plotting the cluster score against the number of clusters, we determined that 6 is the optimal number of clusters into which to classify the genes by profile (Fig. 4A). Therefore, the 3,890 significantly regulated probe sets were divided into six clusters by the K-means method. The profiles and number of genes in these six clusters are shown in Fig. 4B and C. Unlike the genes in cluster 1, which declined essentially linearly between 6 and 27 months of age, genes in cluster 2 declined only minimally between 6 and 18 months, followed thereafter by a drastic decline between 21 and 27 months, in concert with the changes in muscle mass. The average profile of genes in cluster 2 correlated with the average age-related changes in muscle mass, with a coefficient of 0.99. Indeed, the changes in gene expression correlated strongly with changes in muscle mass for individual genes in cluster 2 (R ≥ 0.9 for 26% of the 624 genes in cluster 2) compared to R ≤ 0.88 for every gene in cluster 1. The 20 genes in clusters 1 and 2, ranked by correlation with muscle mass, are shown in Table 1. Other genes in cluster 2 that did not make the top 20 list but correlated strongly (R > 0.9) with muscle mass and have putative roles in regulating muscle mass and function include several mitochondrial energy metabolism genes (e.g., Pdhx, NADH dehydrogenase genes, Uqcrc1, Idh3a, etc.), as well as genes not directly associated with mitochondria energy metabolism, including Mylk2, Pde4a, Vegfa, and Mstn.
Fig 4.

Cluster analysis of the 3,890 genes significantly regulated in sarcopenia. The optimal number of clusters, K, was determined to be 6 by the Elbow method after plotting the cluster score against number of clusters (A). (B) K-means clustering of the 3,890 significantly regulated genes into six clusters and expression profiles along the time point in months. (C) The number of genes within each of the six clusters.
Table 1.
Top 20 significantly regulated genes with the strongest correlation with muscle mass in each of clusters 1 and 2
| Symbol | Description | Adjusted P value | AUC | Log2 fold change vs 6 mo |
R vs: |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 mo | 18 mo | 21 mo | 24 mo | 27 mo | Cluster | Mass | Age | ||||
| Cluster 1 | |||||||||||
| Vwa3b | von Willebrand factor A domain containing 3B | 1.5E–15 | −2.7 | −0.2 | −0.4 | −0.7 | −0.9 | −1.2 | 0.97 | 0.88 | −0.97 |
| RGD1309779 | Similar to ENSANGP00000021391 | 6.9E–12 | −1.5 | −0.1 | −0.2 | −0.4 | −0.5 | −0.7 | 0.97 | 0.86 | −0.97 |
| Hras | Harvey rat sarcoma virus oncogene | 6.3E–13 | −1.4 | −0.1 | −0.3 | −0.3 | −0.5 | −0.6 | 0.94 | 0.86 | −0.98 |
| LOC687029 | Similar to differentially expressed in B16F10 1 | 5.5E–19 | −1.6 | −0.1 | −0.3 | −0.4 | −0.5 | −0.6 | 0.97 | 0.85 | −0.99 |
| Erlec1 | Endoplasmic reticulum lectin 1 | 6.0E–15 | −1.3 | −0.1 | −0.2 | −0.3 | −0.4 | −0.5 | 0.96 | 0.85 | −0.98 |
| Mocs2 | Molybdenum cofactor synthesis 2 | 3.0E–12 | −1.9 | −0.2 | −0.4 | −0.5 | −0.7 | −0.8 | 0.96 | 0.83 | −0.99 |
| Sesn1 | Sestrin 1 | 9.3E–12 | −1.7 | −0.1 | −0.2 | −0.5 | −0.6 | −0.8 | 0.97 | 0.83 | −0.07 |
| Gadd45gip1 | Growth arrest and DNA-damage-inducible, gamma interacting protein 1 | 4.8E–13 | −1.4 | −0.1 | −0.3 | −0.3 | −0.5 | −0.6 | 0.96 | 0.83 | −0.98 |
| Hint3 | Histidine triad nucleotide binding protein 3 | 6.5E–13 | −1.3 | −0.1 | −0.2 | −0.3 | −0.5 | −0.5 | 0.95 | 0.83 | −0.98 |
| LOC691921 | Hypothetical protein LOC691921 | 1.1E–11 | −1.6 | −0.2 | −0.3 | −0.4 | −0.5 | −0.7 | 0.96 | 0.82 | −0.96 |
| Mrps15 | Mitochondrial ribosomal protein S15 | 3.0E–14 | −1.5 | −0.2 | −0.3 | −0.4 | −0.5 | −0.6 | 0.97 | 0.82 | −0.98 |
| Aven | Apoptosis, caspase activation inhibitor | 1.2E–14 | −1.4 | −0.2 | −0.3 | −0.3 | −0.5 | −0.5 | 0.95 | 0.82 | −0.98 |
| Zcrb1 | Zinc finger CCHC-type and RNA binding motif 1 | 2.7E–15 | −1.7 | −0.2 | −0.3 | −0.4 | −0.5 | −0.7 | 0.97 | 0.81 | −0.99 |
| Timm44 | Translocase of inner mitochondrial membrane 44 homolog (yeast) | 4.6E–11 | −1.6 | −0.2 | −0.3 | −0.4 | −0.5 | −0.7 | 0.96 | 0.81 | −0.97 |
| Gps2 | G protein pathway suppressor 2 | 2.8E–13 | −1.3 | −0.2 | −0.2 | −0.3 | −0.5 | −0.6 | 0.95 | 0.81 | −0.97 |
| Fnbp4 | Formin binding protein 4 | 1.0E–16 | −2.1 | −0.2 | −0.3 | −0.6 | −0.7 | −0.8 | 0.98 | 0.80 | −0.96 |
| Iqub | IQ motif and ubiquitin domain containing | 1.4E–13 | −1.9 | −0.2 | −0.2 | −0.5 | −0.7 | −0.8 | 0.97 | 0.80 | −0.95 |
| Ggct | Gamma-glutamyl cyclotransferase | 9.9E–15 | −1.9 | −0.3 | −0.3 | −0.4 | −0.7 | −0.7 | 0.94 | 0.80 | −0.95 |
| Hnrnpa2b1 | Heterogeneous nuclear ribonucleoprotein A2/B1 | 9.6E–18 | −1.9 | −0.1 | −0.3 | −0.5 | −0.6 | −0.7 | 0.99 | 0.80 | −0.97 |
| Metap1d | Methionyl aminopeptidase type 1D (mitochondrial) | 6.2E–11 | −1.9 | −0.2 | −0.3 | −0.4 | −0.6 | −0.8 | 0.96 | 0.80 | −0.97 |
| Cluster 2 | |||||||||||
| Pdhx | Pyruvate dehydrogenase complex, component X | 1.7E–17 | −2.1 | −0.1 | −0.2 | −0.3 | −0.9 | −1.3 | 1.00 | 0.94 | −0.86 |
| Rxrg | Retinoid X receptor gamma | 2.2E–19 | −2.5 | −0.1 | −0.2 | −0.4 | −1.1 | −1.6 | 1.00 | 0.94 | −0.86 |
| Ugp2 | UDP-glucose pyrophosphorylase 2 | 1.3E–21 | −1.6 | −0.1 | −0.2 | −0.3 | −0.7 | −0.9 | 1.00 | 0.94 | −0.89 |
| Pgp | Phosphoglycolate phosphatase | 1.3E–16 | −1.8 | 0.0 | −0.1 | −0.3 | −0.8 | −1.3 | 1.00 | 0.94 | −0.83 |
| Tuba8 | Tubulin, alpha 8 | 3.8E–21 | −2.9 | −0.2 | −0.3 | −0.4 | −1.3 | −1.7 | 1.00 | 0.94 | −0.86 |
| Tnks2 | Tankyrase, TRF1-interacting ankyrin-related ADP-ribose polymerase 2 | 2.7E–19 | −2.0 | −0.1 | −0.1 | −0.4 | −0.9 | −1.2 | 1.00 | 0.94 | −0.87 |
| Mylk2 | Myosin light chain kinase 2 | 2.9E–22 | −3.4 | −0.1 | −0.3 | −0.6 | −1.5 | −1.9 | 0.99 | 0.94 | −0.88 |
| Mrpl15 | Mitochondrial ribosomal protein L15 | 5.3E–20 | −1.1 | −0.1 | −0.1 | −0.2 | −0.5 | −0.6 | 0.99 | 0.94 | −0.91 |
| LOC100360936 | rCG22080-like | 7.3E–19 | −1.9 | −0.1 | −0.2 | −0.3 | −0.9 | −1.0 | 0.99 | 0.94 | −0.89 |
| Ppa2 | Pyrophosphatase (inorganic) 2 | 1.6E–18 | −1.8 | −0.1 | −0.2 | −0.4 | −0.7 | −0.9 | 0.99 | 0.94 | −0.92 |
| Lrrc39 | Leucine-rich repeat containing 39 | 1.8E–18 | −2.0 | −0.2 | −0.2 | −0.4 | −0.7 | −1.2 | 0.99 | 0.94 | −0.89 |
| Zfp358 | Zinc finger protein 358 | 4.5E–19 | −1.8 | −0.1 | −0.2 | −0.4 | −0.7 | −0.9 | 0.99 | 0.94 | −0.93 |
| LOC100359995 | rCG63619-like | 1.8E–16 | −1.4 | −0.1 | −0.2 | −0.2 | −0.6 | −0.7 | 0.99 | 0.94 | −0.92 |
| Asb11 | Ankyrin repeat and SOCS box-containing 11 | 1.1E–20 | −3.1 | −0.2 | −0.2 | −0.5 | −1.3 | −1.8 | 1.00 | 0.93 | −0.87 |
| St8sia5 | ST8 α-N-acetylneuraminide α-2,8-sialyltransferase 5 | 3.2E–17 | −3.4 | −0.2 | −0.3 | −0.5 | −1.5 | −2.0 | 1.00 | 0.93 | −0.86 |
| Egln1 | EGL nine homolog 1 (Caenorhabditis elegans) | 1.2E–15 | −1.2 | −0.1 | −0.1 | −0.2 | −0.5 | −0.7 | 1.00 | 0.93 | −0.88 |
| Hfe2 | Hemochromatosis type 2 (juvenile) homolog (human) | 2.5E–19 | −1.7 | 0.0 | −0.1 | −0.3 | −0.7 | −1.1 | 1.00 | 0.93 | −0.87 |
| RGD1564300 | Similar to phosphoseryl-tRNA kinase | 1.5E–18 | −2.2 | −0.1 | −0.2 | −0.3 | −0.9 | −1.3 | 1.00 | 0.93 | −0.87 |
| Pfkfb1 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 1 | 4.1E–19 | −3.5 | −0.1 | −0.2 | −0.5 | −1.6 | −2.4 | 1.00 | 0.93 | −0.84 |
| Pde4a | Phosphodiesterase 4A, cAMP-specific (phosphodiesterase E2 dunce homolog, Drosophila) | 1.0E–21 | −2.3 | 0.0 | −0.1 | −0.3 | −1.1 | −1.6 | 1.00 | 0.93 | −0.93 |
In clusters 3, 4 and 5, there was a more or less moderate increase in gene expression between 6 and 18 or 21 months, followed thereafter by a more drastic increase. Thus, the average expression profiles of genes in these clusters correlated inversely with muscle mass with correlation coefficients of −0.99, −0.97, and −0.97 for clusters 3, 4, and 5, respectively. The gene expression profiles for ∼17% of the 728, ∼5% of the 535, and ∼3% of the 420 genes in clusters 3, 4 and 5, respectively, correlated inversely with muscle mass with correlation coefficients of R ≤ −0.9. That is, the expression profile of genes in cluster 3 correlated the best with changes in muscle mass. The 20 genes with the strongest correlation with muscle mass (R ≤ −0.9) in each of clusters 3, 4, and 5 are shown in Table 2. Other genes in cluster 3 (not captured in Table 2) that inversely correlated strongly with muscle mass (with R ≤ −0.9) and have putative roles in regulating muscle structure and function include Igf2, Chrnd, Chrnb1, Tgfb2, and Fgf7. There were rather few genes in cluster 6; these had a bell-shaped expression profile that did not correlate with muscle mass changes (Fig. 4B). Therefore, cluster 6 was not explored further. Table 3 shows the 20 most differentially up- and downregulated genes ranked by AUC, a method commonly used by previous studies to identify gene signatures of sarcopenia or aging.
Table 2.
Top 20 significantly regulated genes with the strongest correlation with muscle mass in each of clusters 3, 4, and 5
| Symbol | Description | Adjusted P value | AUC | Log2 fold change vs 6 mo |
R vs: |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 mo | 18 mo | 21 mo | 24 mo | 27 mo | Cluster | Mass | age | ||||
| Cluster 3 | |||||||||||
| Sirpa | Signal-regulatory protein alpha | 5.6E–21 | 4.1 | 0.3 | 0.5 | 0.7 | 1.6 | 2.2 | 1.00 | −0.95 | 0.91 |
| Tspan13 | Tetraspanin 13 | 1.2E–19 | 2.3 | 0.1 | 0.3 | 0.4 | 1.0 | 1.2 | 1.00 | −0.94 | 0.90 |
| Map3k7 | Mitogen-activated protein kinase kinase kinase 7 | 8.6E–21 | 2.3 | 0.1 | 0.3 | 0.4 | 0.9 | 1.3 | 1.00 | −0.94 | 0.90 |
| Hmgn2 | High-mobility group nucleosomal binding domain 2 | 2.3E–19 | 1.9 | 0.1 | 0.2 | 0.3 | 0.8 | 1.1 | 1.00 | −0.94 | 0.87 |
| Rb1 | Retinoblastoma 1 | 2.7E–19 | 1.8 | 0.1 | 0.3 | 0.3 | 0.7 | 1.0 | 0.99 | −0.94 | 0.92 |
| Ccni | Cyclin I | 6.8E–20 | 1.5 | 0.2 | 0.1 | 0.2 | 0.6 | 0.9 | 0.99 | −0.94 | 0.84 |
| Krt18 | Keratin 18 | 2.2E–22 | 6.4 | 0.0 | 0.7 | 1.1 | 2.7 | 3.7 | 1.00 | −0.93 | 0.89 |
| Mgmt | O-6-Methylguanine-DNA methyltransferase | 5.0E–23 | 4.9 | 0.3 | 0.5 | 0.9 | 2.1 | 2.4 | 0.99 | −0.93 | 0.91 |
| Tmem38b | Transmembrane protein 38B | 6.0E–24 | 3.4 | 0.0 | 0.3 | 0.7 | 1.5 | 1.7 | 0.98 | −0.93 | 0.91 |
| Fst | Follistatin | 1.4E–17 | 3.3 | 0.2 | 0.3 | 0.5 | 1.5 | 1.9 | 1.00 | −0.93 | 0.88 |
| Cd151 | CD151 molecule (Raph blood group) | 1.7E–21 | 2.4 | 0.2 | 0.2 | 0.5 | 1.0 | 1.2 | 0.99 | −0.93 | 0.92 |
| Cd63 | Cd63 molecule | 2.9E–22 | 2.2 | 0.1 | 0.2 | 0.5 | 0.9 | 1.1 | 0.99 | −0.93 | 0.91 |
| Iffo1 | Intermediate filament family orphan 1 | 4.0E–15 | 2.0 | 0.1 | 0.2 | 0.3 | 0.8 | 1.2 | 1.00 | −0.93 | 0.88 |
| Slain2 | SLAIN motif family, member 2 | 6.6E–22 | 1.9 | 0.0 | 0.2 | 0.3 | 0.8 | 1.1 | 1.00 | −0.93 | 0.88 |
| Adcy2 | Adenylate cyclase 2 (brain) | 2.1E–17 | 1.7 | 0.1 | 0.3 | 0.2 | 0.8 | 1.0 | 0.99 | −0.93 | 0.86 |
| Ppp1r14b | Protein phosphatase 1, regulatory (inhibitor) subunit 14B | 3.3E–20 | 1.6 | 0.1 | 0.1 | 0.3 | 0.6 | 0.9 | 0.99 | −0.93 | 0.88 |
| Prkar1a | Protein kinase, cAMP-dependent, regulatory, type I, alpha | 8.8E–20 | 1.4 | 0.0 | 0.0 | 0.3 | 0.7 | 0.9 | 0.98 | −0.93 | 0.85 |
| Sec62 | SEC62 homolog (S. cerevisiae) | 4.0E–18 | 1.3 | 0.1 | 0.2 | 0.3 | 0.5 | 0.6 | 0.99 | −0.93 | 0.92 |
| Nono | Non-POU domain containing, octamer-binding | 2.9E–18 | 1.3 | 0.1 | 0.1 | 0.3 | 0.5 | 0.7 | 0.99 | −0.93 | 0.90 |
| Dclk1 | Doublecortin-like kinase 1 | 6.7E–22 | 7.2 | 0.4 | 1.0 | 1.4 | 2.9 | 3.4 | 0.99 | −0.92 | 0.94 |
| Cluster 4 | |||||||||||
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A | 6.0E–24 | 10.7 | 0.6 | 1.6 | 2.5 | 4.0 | 4.6 | 0.98 | −0.92 | 0.96 |
| B2 m | β2-Microglobulin | 6.0E–24 | 3.9 | 0.3 | 0.5 | 0.9 | 1.5 | 1.7 | 0.99 | −0.92 | 0.95 |
| Ftl | Ferritin, light polypeptide | 7.7E–23 | 3.3 | 0.3 | 0.4 | 0.7 | 1.3 | 1.6 | 0.99 | −0.92 | 0.93 |
| Slc40a1 | Solute carrier family 39 (iron-regulated transporter), member 1 | 3.7E–16 | 3.0 | 0.4 | 0.4 | 0.7 | 1.1 | 1.2 | 0.99 | −0.92 | 0.95 |
| Nedd4 | Neural precursor cell expressed, developmentally downregulated 4 | 8.3E–20 | 2.8 | 0.3 | 0.4 | 0.7 | 1.0 | 1.3 | 0.99 | −0.92 | 0.92 |
| Arpp19 | cAMP-regulated phosphoprotein 19 | 2.0E–20 | 2.4 | 0.1 | 0.2 | 0.6 | 1.0 | 1.3 | 0.99 | −0.92 | 0.92 |
| Ppia | Peptidylprolyl isomerase A (cyclophilin A) | 5.5E–20 | 1.8 | 0.2 | 0.2 | 0.4 | 0.7 | 0.9 | 1.00 | −0.92 | 0.93 |
| Snx1 | Sorting nexin 1 | 4.2E–19 | 1.4 | 0.1 | 0.1 | 0.3 | 0.6 | 0.7 | 0.99 | −0.92 | 0.93 |
| Hnrnpa3 | Heterogeneous nuclear ribonucleoprotein A3 | 6.4E–17 | 1.3 | 0.1 | 0.1 | 0.3 | 0.5 | 0.6 | 1.00 | −0.92 | 0.92 |
| Ninj1 | Ninjurin 1 | 1.1E–19 | 3.1 | 0.2 | 0.4 | 0.7 | 1.2 | 1.6 | 0.99 | −0.91 | 0.93 |
| Hspb8 | Heat shock protein B8 | 9.4E–23 | 3.0 | 0.2 | 0.3 | 0.7 | 1.1 | 1.5 | 0.99 | −0.91 | 0.94 |
| Rpl3 | Ribosomal protein L3 | 3.8E–20 | 2.7 | 0.3 | 0.3 | 0.6 | 1.0 | 1.2 | 1.00 | −0.91 | 0.92 |
| Tmem30a | Transmembrane protein 30A | 2.5E–15 | 1.2 | 0.1 | 0.1 | 0.3 | 0.5 | 0.6 | 1.00 | −0.91 | 0.93 |
| Prrt4 | Proline-rich transmembrane protein 4 | 2.8E–21 | 7.4 | 0.5 | 1.0 | 1.7 | 2.8 | 3.4 | 0.99 | −0.90 | 0.95 |
| Ostalpha | Organic solute transporter alpha | 3.6E–16 | 5.1 | 0.4 | 0.7 | 1.1 | 1.9 | 2.4 | 0.99 | −0.90 | 0.95 |
| Phlda3 | Pleckstrin homology-like domain, family A, member 3 | 2.4E–20 | 4.3 | 0.2 | 0.5 | 0.9 | 1.7 | 2.0 | 0.99 | −0.90 | 0.94 |
| Tp63 | Tumor protein p63 | 8.3E–20 | 3.4 | 0.1 | 0.4 | 0.8 | 1.4 | 1.6 | 0.98 | −0.90 | 0.95 |
| Asgr2 | Asialoglycoprotein receptor 2 | 7.3E–16 | 2.8 | 0.3 | 0.4 | 0.6 | 1.1 | 1.3 | 0.99 | −0.90 | 0.94 |
| Tubb5 | Tubulin, beta 5 | 4.1E–21 | 2.6 | 0.2 | 0.3 | 0.6 | 1.0 | 1.2 | 0.99 | −0.90 | 0.94 |
| App | Amyloid beta (A4) precursor protein | 4.2E–21 | 2.5 | 0.3 | 0.2 | 0.6 | 1.0 | 1.2 | 0.99 | −0.90 | 0.90 |
| Cluster 5 | |||||||||||
| Stxbp1 | Syntaxin binding protein 1 | 3.4E–19 | 2.8 | 0.3 | 0.5 | 0.5 | 1.0 | 1.3 | 0.99 | −0.93 | 0.95 |
| Il6st | Interleukin-6 signal transducer | 4.9E–18 | 2.4 | 0.3 | 0.4 | 0.5 | 0.8 | 1.1 | 0.99 | −0.93 | 0.96 |
| Creld1 | Cysteine-rich with EGF-like domains 1 | 5.8E–16 | 2.1 | 0.2 | 0.4 | 0.4 | 0.8 | 1.0 | 0.99 | −0.91 | 0.93 |
| Tmem111 | Transmembrane protein 111 | 2.3E–17 | 1.7 | 0.2 | 0.3 | 0.4 | 0.7 | 0.8 | 0.98 | −0.91 | 0.95 |
| Csnk1d | Casein kinase 1, delta | 1.6E–16 | 1.5 | 0.2 | 0.2 | 0.2 | 0.5 | 0.7 | 0.98 | −0.91 | 0.91 |
| Ids | Iduronate 2-sulfatase | 2.3E–15 | 1.1 | 0.1 | 0.2 | 0.2 | 0.4 | 0.5 | 0.99 | −0.91 | 0.95 |
| Chrna1 | Cholinergic receptor, nicotinic, alpha 1 (muscle) | 9.4E–23 | 11.6 | 0.4 | 1.9 | 2.6 | 4.4 | 5.1 | 0.97 | −0.90 | 0.96 |
| Hdac4 | Histone deacetylase 4 | 1.1E–20 | 4.4 | 0.3 | 0.7 | 0.9 | 1.7 | 1.9 | 0.99 | −0.90 | 0.96 |
| Gsta3 | Glutathione S-transferase A3 | 9.7E–19 | 4.0 | 0.4 | 0.7 | 0.8 | 1.5 | 1.6 | 0.99 | −0.90 | 0.96 |
| Rab27a | RAB27A, member RAS oncogene family | 1.7E–13 | 2.5 | 0.3 | 0.4 | 0.5 | 0.9 | 1.2 | 0.99 | −0.90 | 0.94 |
| Ddr1 | Discoidin domain receptor tyrosine kinase 1 | 2.1E–15 | 1.9 | 0.1 | 0.3 | 0.4 | 0.7 | 0.9 | 0.99 | −0.90 | 0.95 |
| Prim1 | DNA primase, p49 subunit | 3.1E–15 | 1.5 | 0.0 | 0.3 | 0.3 | 0.6 | 0.7 | 0.97 | −0.90 | 0.95 |
| Runx1 | Runt-related transcription factor 1 | 5.7E–23 | 8.1 | 0.4 | 1.3 | 2.0 | 2.9 | 3.4 | 0.97 | −0.89 | 0.98 |
| Ltbp1 | Latent transforming growth factor beta binding protein 1 | 1.2E–17 | 4.7 | 0.5 | 0.9 | 0.9 | 1.5 | 2.1 | 0.99 | −0.89 | 0.96 |
| Plxnb2 | Plexin B2 | 1.7E–17 | 2.2 | 0.3 | 0.4 | 0.5 | 0.8 | 1.0 | 0.99 | −0.89 | 0.95 |
| Grina | Glutamate receptor, ionotropic, N-methyl d-aspartate-associated protein 1 (glutamate binding) | 1.8E–16 | 1.8 | 0.2 | 0.3 | 0.3 | 0.6 | 0.8 | 0.99 | −0.89 | 0.94 |
| Lrp4 | Low-density lipoprotein receptor-related protein 4 | 3.7E–13 | 1.4 | 0.1 | 0.3 | 0.2 | 0.5 | 0.8 | 0.96 | −0.89 | 0.92 |
| Sln | Sarcolipin | 1.2E–21 | 9.8 | 0.4 | 1.6 | 2.1 | 3.8 | 4.1 | 0.98 | −0.88 | 0.96 |
| Musk | Muscle, skeletal, receptor tyrosine kinase | 1.8E–17 | 3.9 | 0.1 | 0.6 | 0.8 | 1.5 | 1.7 | 0.98 | −0.88 | 0.96 |
| Abca1 | ATP-binding cassette, subfamily A (ABC1), member 1 | 1.6E–15 | 3.0 | 0.3 | 0.6 | 0.4 | 1.2 | 1.3 | 0.96 | −0.88 | 0.89 |
Table 3.
Top 20 most up- or downregulated annotated genes, sorted by AUC
| Symbol | Description | Adjusted P value | AUC | Log2 fold change vs 6 mo |
R vs: |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| 12 mo | 18 mo | 21 mo | 24 mo | 27 mo | Mass | Age | ||||
| Upregulated genes | ||||||||||
| Chrna1 | Cholinergic receptor, nicotinic, alpha 1 (muscle) | 9.43E–23 | 11.55 | 0.37 | 1.87 | 2.56 | 4.41 | 5.06 | −0.90 | 0.96 |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A | 5.99E–24 | 10.70 | 0.55 | 1.61 | 2.47 | 4.04 | 4.62 | −0.92 | 0.96 |
| Sln | Sarcolipin | 1.16E–21 | 9.75 | 0.44 | 1.63 | 2.08 | 3.80 | 4.05 | −0.88 | 0.96 |
| Klk1l | Kallikrein 1-like peptidase | 3.21E–20 | 9.61 | 0.43 | 1.48 | 1.80 | 4.04 | 4.15 | −0.90 | 0.93 |
| Ankrd1 | Ankyrin repeat domain 1 | 1.06E–19 | 9.24 | 0.29 | 1.04 | 2.10 | 3.87 | 4.17 | −0.87 | 0.93 |
| Gadd45a | Growth arrest and DNA-damage-inducible, alpha | 4.86E–21 | 9.00 | 0.39 | 1.46 | 2.28 | 3.22 | 3.68 | −0.86 | 0.98 |
| Emb | Embigin homolog (mouse) | 1.10E–22 | 8.39 | 0.04 | 0.89 | 1.55 | 3.80 | 4.26 | −0.90 | 0.90 |
| Runx1 | Runt-related transcription factor 1 | 5.66E–23 | 8.13 | 0.40 | 1.33 | 1.99 | 2.89 | 3.43 | −0.89 | 0.98 |
| Prrt4 | Proline-rich transmembrane protein 4 | 2.82E–21 | 7.36 | 0.49 | 1.00 | 1.65 | 2.75 | 3.42 | −0.90 | 0.95 |
| Ppp1r14c | Protein phosphatase 1, regulatory (inhibitor) subunit 14c | 9.43E–23 | 7.20 | −0.05 | 1.02 | 1.57 | 2.95 | 3.34 | −0.89 | 0.94 |
| Dclk1 | Doublecortin-like kinase 1 | 6.70E–22 | 7.15 | 0.37 | 1.01 | 1.39 | 2.86 | 3.42 | −0.92 | 0.94 |
| Mt1a | Metallothionein 1a | 5.93E–18 | 6.98 | 0.10 | 0.90 | 1.24 | 3.07 | 3.43 | −0.87 | 0.91 |
| Chrnd | Cholinergic receptor, nicotinic, delta | 2.56E–22 | 6.93 | 0.19 | 1.06 | 1.45 | 2.76 | 3.14 | −0.92 | 0.94 |
| Myog | Myogenin | 1.21E–21 | 6.71 | 0.34 | 1.06 | 1.69 | 2.40 | 2.78 | −0.88 | 0.98 |
| Gpnmb | Glycoprotein (transmembrane) nmb | 3.46E–21 | 6.65 | 0.45 | 1.08 | 1.68 | 2.40 | 2.53 | −0.84 | 0.98 |
| Krt18 | Keratin 18 | 2.18E–22 | 6.39 | 0.03 | 0.71 | 1.14 | 2.69 | 3.67 | −0.93 | 0.89 |
| RT1-Bb | RT1 class II, locus Bb | 2.48E–01 | 6.36 | −0.04 | 1.36 | 2.32 | 2.30 | 0.83 | −0.19 | 0.73 |
| Myl4 | Myosin, light chain 4 | 1.08E–08 | 6.09 | 0.13 | 1.20 | 1.20 | 2.37 | 2.52 | −0.71 | 0.95 |
| Ampd3 | AMP deaminase 3 | 5.17E–21 | 5.98 | 0.16 | 0.95 | 1.53 | 2.17 | 2.51 | −0.85 | 0.97 |
| Cst7 | Cystatin F (leukocystatin) | 5.61E–16 | 5.97 | 0.51 | 1.13 | 1.22 | 2.26 | 2.22 | −0.83 | 0.97 |
| Downregulated genes | ||||||||||
| Tfrc | Transferrin receptor | 2.28E–19 | −6.28 | −0.84 | −1.22 | −1.07 | −2.16 | −2.84 | 0.89 | −0.93 |
| Nr4a3 | Nuclear receptor subfamily 4, group A, member 3 | 3.14E–03 | −6.09 | −0.76 | −1.38 | −1.51 | −1.97 | −1.70 | 0.41 | −0.96 |
| Col1a1 | Collagen, type I, alpha 1 | 4.39E–09 | −4.84 | −0.81 | −1.40 | −1.47 | −0.98 | −1.16 | 0.33 | −0.76 |
| Slc41a3 | Solute carrier family 41, member 3 | 1.17E–16 | −4.02 | −0.29 | −0.54 | −0.54 | −1.77 | −2.04 | 0.92 | −0.88 |
| Cish | Cytokine inducible SH2-containing protein | 3.71E–02 | −4.01 | −0.84 | −1.12 | −0.68 | −1.02 | −1.53 | 0.36 | −0.85 |
| Cdh22 | Cadherin 22 | 8.79E–17 | −3.94 | −0.47 | −0.81 | −1.08 | −1.14 | −1.34 | 0.75 | −0.99 |
| Palld | Palladin, cytoskeletal associated protein | 4.89E–11 | −3.87 | −0.37 | −0.72 | −0.92 | −1.18 | −1.73 | 0.79 | −0.98 |
| RGD1306119 | Similar to transcriptional regulating protein 132 | 4.56E–16 | −3.84 | −0.30 | −0.57 | −0.74 | −1.39 | −2.01 | 0.88 | −0.92 |
| Chac1 | ChaC, cation transport regulator homolog 1 (Escherichia coli) | 5.21E–06 | −3.83 | −0.83 | −0.69 | −0.41 | −1.47 | −1.70 | 0.68 | −0.82 |
| Tnmd | Tenomodulin | 8.18E–05 | −3.71 | −0.47 | −1.14 | −0.93 | −0.92 | −0.98 | 0.41 | −0.86 |
| Zmynd17 | Zinc finger, MYND-type containing 17 | 1.08E–20 | −3.61 | 0.00 | −0.16 | −0.61 | −1.79 | −2.11 | 0.92 | −0.86 |
| Eef2k | Eukaryotic elongation factor-2 kinase | 1.45E–12 | −3.55 | −0.26 | −0.68 | −0.92 | −1.12 | −1.38 | 0.75 | −0.99 |
| Neb | Nebulin | 6.28E–15 | −3.52 | −0.46 | −0.64 | −1.14 | −0.95 | −1.12 | 0.63 | −0.95 |
| Pfkfb1 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 1 | 4.09E–19 | −3.48 | −0.05 | −0.20 | −0.45 | −1.61 | −2.40 | 0.93 | −0.84 |
| Chad | Chondroadherin | 4.31E–02 | −3.41 | −0.25 | −0.97 | −0.98 | −0.85 | −1.00 | 0.28 | −0.91 |
| Mylk2 | Myosin light chain kinase 2 | 2.94E–22 | −3.37 | −0.06 | −0.32 | −0.56 | −1.52 | −1.89 | 0.94 | −0.88 |
| Col1a2 | Collagen, type I, alpha 2 | 4.02E–07 | −3.36 | −0.57 | −1.07 | −1.00 | −0.61 | −0.81 | 0.26 | −0.70 |
| St8sia5 | ST8 α-N-acetyl-neuraminide α-2,8-sialyltransferase 5 | 3.22E–17 | −3.35 | −0.19 | −0.31 | −0.48 | −1.47 | −1.99 | 0.93 | −0.86 |
| Sfxn5 | Sideroflexin 5 | 1.00E–19 | −3.31 | −0.18 | −0.45 | −0.66 | −1.30 | −1.62 | 0.90 | −0.93 |
| Gbas | Glioblastoma amplified sequence | 1.73E–17 | −3.29 | −0.36 | −0.45 | −0.76 | −1.06 | −1.68 | 0.85 | −0.93 |
Pathways and biological processes perturbed in sarcopenia.
To identify the pathways and biological processes dysregulated in sarcopenia, the genes in each of clusters 2 to 5 were subjected to enrichment cluster analysis using the online DAVID tool and visualized using the Cytoscape plug-in enrichment map.
(i) Energy metabolism pathways are depressed in sarcopenia.
Functional annotation analysis using DAVID showed that cluster 2 was comprised predominantly of genes involved in mitochondrial energy metabolism, including oxidative phosphorylation (OXPHOS), glucose metabolism, the tricarboxylic acid (TCA) cycle, and fatty acid metabolism, as well as genes associated with muscle structural components (Fig. 5A). To further verify depression of the OXPHOS pathway, the regulation of genes in its five complexes was examined. Although the fold decline of the individual genes versus the 6-month values was small (range, −0.12 to −1.24 [by AUC]), 87% of the 71 OXPHOS genes identified in the five complexes of the pathway were downregulated (see Fig. S2 in the supplemental material). It is noteworthy that the few upregulated (e.g., Atp6v isoforms and Cox7a2l) are genes not normally expressed in healthy muscles from young adult rats. The expression profile of some of the energy metabolism related genes was further verified by qPCR (Fig. 5B). In addition to the depression of genes involved with mitochondrial biogenesis and energy metabolism, sarcopenia was associated with downregulation of genes that regulate mitochondrial fission and fusion, including Mfn1, Mfn2, Opa1, Fis1, and Drp1_Dmn1 (Fig. 5C). Together, these results indicate that sarcopenia is characterized by a depressed mitochondrial energy metabolism and dynamics.
Fig 5.

Map of enriched gene sets for genes in cluster 2 based on DAVID output. Each node represents a gene set. Clusters of functionally related gene sets were manually framed and summarized into key findings in the text frame with the same color. (A) Functional annotation analysis of the genes in cluster 2 indicates that pathways and processes involved in energy metabolism are depressed in sarcopenia. (B) Quantitative PCR analyses verified downregulation of some genes in cluster 2 and other genes involved in energy metabolism (B) and mitochondrial dynamics (C). *, **, ***, and **** indicate P < 0.05, 0.01, 0.001, and 0.0001 versus 6 months, respectively. 6M, 6 months; 12M, 12 months, etc.
(ii) Pathways and processes upregulated with sarcopenia.
Functional enrichment analysis indicated that genes in clusters 3, 4, and 5, which are inversely correlated with muscle mass, fall into several pathways and biological processes. In cluster 3, the most enriched pathways were those involved with the cell cycle, ribosome, translation, protein synthesis, immune response, and cell death/apoptosis (Fig. 6A). The expression profiles of some of the genes in this cluster (Fst, Chrnd, Igf2, Casp3, and Bad) were verified by qPCR (Fig. 6B). Cluster 4 was comprised of pathways and processes involved with immune response, cytoskeleton, extracellular matrix, and response to various stimuli (see Fig. S3A in the supplemental material), while cluster 5 comprised of pathways and processes involved with antigen processing and presentation, transcription, phosphate metabolism, cytoskeleton, muscle development, neuromuscular junction, apoptosis/cell death, and the regulation of cellular biosynthetic processes, among others (see Fig. S3B in the supplemental material). Thus, genes in clusters 3, 4, and 5 contributed jointly to some pathways including cytoskeletal development and immune response (see Fig. S3C in the supplemental material). The expression profiles of some of the genes in these clusters were subsequently verified by qPCR: for Myog (in cluster 4) and Hdac4, Chrna1, Musk, and Fbxo30 (in cluster 5) (see Fig. S3D in the supplemental material). That several neuromuscular junction genes were enriched—including Chrna1, Chrnb, Chrnd, Chrne, Chrng, Musk, Myog, the utrophin gene (Fig. 6B; also see Fig. S3D in the supplemental material), and MyoD (data not shown)—is indicative of neuromuscular junction functional denervation in sarcopenia. Table S5 in the supplemental material summarizes the microarray data for other genes associated with the neuromuscular junction.
Fig 6.
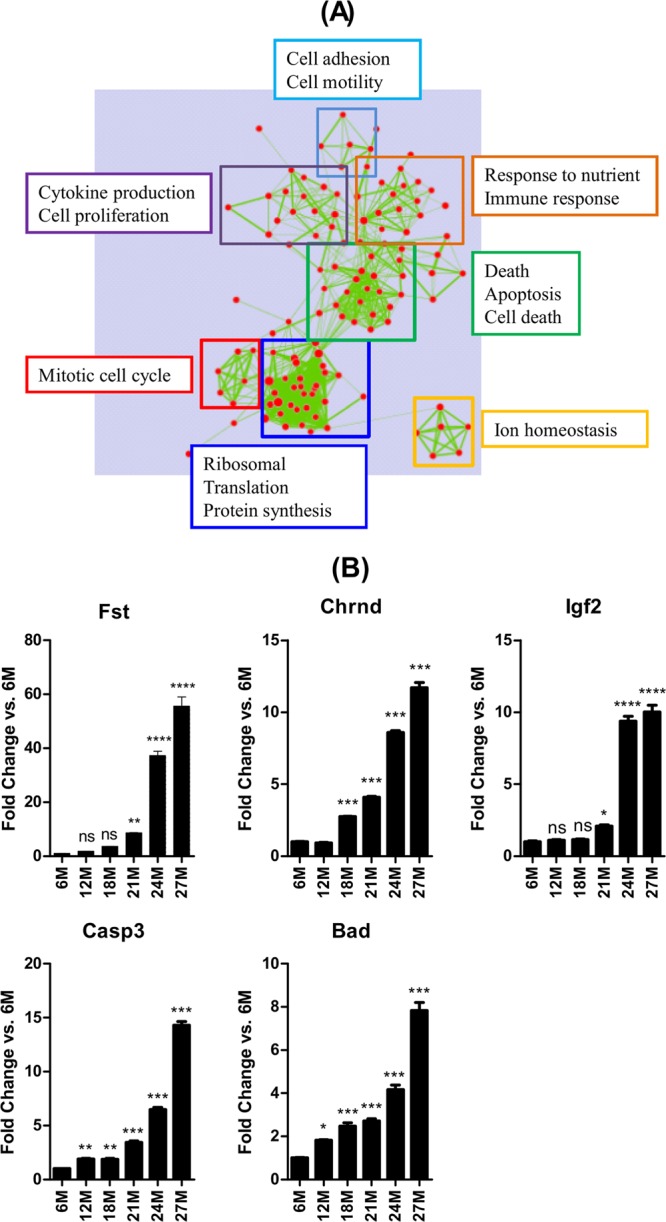
(A) Functional annotation analysis of genes in cluster 3 indicates that pathways and processes involved in regulating the cell cycle, protein metabolism, immune response, and cell death/apoptosis are enriched in sarcopenia. (B) Quantitative PCR analyses verified upregulation of some of the genes in cluster 3 and candidate genes involved in apoptosis. *, **, ***, and **** indicate P < 0.05, 0.01, 0.001, and 0.0001 versus 6 months, respectively. 6M, 6 months; 12M, 12 months, etc.
Since denervation would be expected to alter muscle fiber type composition and fiber type switching has been reported in sarcopenia, we determined the expression of transcripts for the different isoforms of myosin heavy chain (MyHC) by qPCR. There was a progressive decrease in expression of transcripts for Myh7 (MyHC 1) and Myh2 (MyHC 2a) and an increase in Myh1 (MyHC 2x/d) and Myh4 (MyHC 2b), as well as an increased expression of Myh3 (the embryonic) and Myh8 (the perinatal) isoforms of MyHC (see Fig. S4A in the supplemental material). We also determined by qPCR that transcripts for connective tissue growth factor (Ctgf) was upregulated consistent with enrichment of the extracellular matrix processes and that the E3 ligase, MuRF1 (which is usually associated with muscle atrophy but does not have probe sets on the microarray chip), was also upregulated in sarcopenia; the other atrophy-associated E3 ligase, MAFbx (also known as Atrogin1) (42, 43), was not significantly upregulated (see Fig. S4B in the supplemental material).
(iii) Transcription factors upstream of the regulated genes.
To predict the transcription factors upstream of the significantly regulated genes, IPA (Ingenuity Systems, Inc.) was used to determine the relationship between a transcription factor and its target genes, based on previously published data in the IPA knowledge database. The transcription factors most associated with the regulated genes in cluster 2 are shown in Table 4. This cluster of regulated genes predicted a set of transcription factors that may have been perturbed, thus inducing this regulatory change. The predicted transcription factors included the following: (i) FoxO1, which is required for muscle atrophy (44); (ii) NRF1 and TFAM, which activate key metabolic genes required for mitochondrial DNA transcription and replication; (iii) KLF15, which contributes to fatty acid metabolism in muscles; (iv) histone deacetylase 5 (HDAC5), which represses PPARGC1A (PGC-1α) expression via inhibiting MEF2; and (v) PPARGC1A, a transcriptional coactivator of several genes involved in mitochondrial energy metabolism (45).
Table 4.
Transcription factors in promoter regions of regulated genes in cluster 2 (sorted by P value)
| Upstream regulator | No. of targets | P value of overlap | Target molecules in data set |
|---|---|---|---|
| PPARGC1A | 23 | 6.91E–11 | ACADM, ALAS1, ATP5O, C12orf5, CKMT2, CYCS, DLAT, ESRRA, FABP3, IDH3A, LPIN1, MDH2 (includes EG:17448), NDUFB5, NDUFS1, NDUFV2, OXCT1, PDK2, PPARGC1A, PPARGC1B, PRDX5, PRKAA2, SLC2A4, SOD2 |
| MED30 | 7 | 6.91E–07 | ESRRA, NDUFS2, NDUFS7, PPARGC1A, SDHC, SDHD, SOD2 |
| HNF4A | 92 | 4.07E–06 | ACN9 (includes EG:362323), ACO2 (includes EG:11429) ACOT13, ACSL1, ADCK3, AGTPBP1, AKAP13, AMACR, ATF4, BNIP1, BPHL, C12orf52, C21orf33, C3orf26, CD55, CDKN1B, CHCHD3 (includes EG:296966), CLYBL, CMC2 (includes EG:100363376) COQ3, COX14 (includes EG:100428926) CPED1, PT2, CRB3, PF3 EMC9, FASTKD2, FOXRED1, G0S2, GFM1, GOT1, GPLD1, GPT, GRB14, GRHPR, HOOK3, HPN, IL-15 (includes EG:16168), CNJ11, KLHDC3, MCCC1, MCEE, MDH1, MDH2 (includes EG:17448) MGST3, MLXIPL, MRM1, MRPL11 (includes EG:293666), MRPL15 (includes EG:27395), MRPS11, MRPS14, MRPS28 (includes EG:28957), MUT, NDUFA3, NDUFA5, DUFB5, NDUFS1, NDUFS3, NR0B2, NR1D1, OPA3, PCMT1, PDK2, PHKB, PIP5K1A, PKM, PPARGC1A, PPP1R3C, PRDX5, PRKCE, QRSL1, RABGEF1, RASGRP3, RORA, RTN4IP1, SAMM50, SDR39U1, SLC37A4, SLC38A4, SUCLA2, SUCLG1, TCF7L2, TFB2M, TIMM17A, TIMM21, TTC19, TXNL1, UBE2B, USP15, VAMP1, VDAC, XRCC5 |
| TFAM | 6 | 1.74E–05 | ACADM, ACADS, AUH (includes EG:11992), CPT2, FABP3, PGK1 |
| HTT | 41 | 2.20E–05 | ACADM, AMACR, ATL2, ATP5G1, ATP5O, ATPAF1, CAMK2A, Cd24a, CITED2, CYCS, ESRRA, FKBP4, GJC1, GPI, IL-15 (includes EG:16168), Ldhb, MDH2 (includes EG:17448), MED1 (includes EG:19014), NAA15, NDUFA12, NDUFA3, NDUFA5, NDUFB8, NDUFS3, NDUFS7, NR4A1, NREP, PFKM, PPARGC1A, PPARGC1B, PPP1R1A (includes EG:5502), PRKAA2, RXRG, SATB1, SCN4B, SOD2, ST3GAL2, ST8SIA5, TFRC, TUBA4A, VAMP1 |
| ARNT | 11 | 2.80E–05 | ATP5G1, ATP5O, CITED2, GPI, NDUFS2, NDUFS3, PGK1, RGS5, TFRC, TUBA4A, VEGFA |
| KLF15 | 7 | 1.49E–04 | ACADM, ACSS1, CPT2, FABP3, MLYCD, PPARGC1A, SLC2A4 |
| FOXO1 | 15 | 1.34E–03 | ALAS1, CDKN1B, CITED2, CKMT2, CYCS, GPD1, GPD2, GPI, GRHPR, MYL3, PPARGC1A, PPARGC1B, SLC2A4, SOD2, WNT16 |
| SREBF1 | 13 | 1.72E–03 | ABCD2, ACADS, ACSL1, ACSS2, ETHE1, FAM189B, LPIN1, MLYCD, NR0B2, SC5DL, SUCLG1, UQCRFS1, VEGFA |
| NRF1 | 6 | 1.74E–03 | ALAS1, COX6C, CYCS, SDHD, TFB2M, VDAC1 |
| HNF1A | 24 | 1.76E–03 | CD55, CDKN1B, CLYBL, CMC2 (includes EG:100363376), COQ7, FBP2, G0S2, GOT1, GRHPR, GRTP1, MCCC1, ME3, MLXIPL, MRPL15 (includes EG:27395), NDUFS2, NR0B2, NR1D1, PDHX, PGK1, PPP1R1A (includes EG:5502), SLC37A4, SLC38A4, TFRC, VLDLR |
| HDAC5 | 5 | 3.42E–03 | ACADM, ACSL1, CPT2, PPARGC1A, PYGM |
| STAT4 | 14 | 4.25E–03 | ACSS1, AKAP8L, ATF4, CDKN1B, GRTP1, LPIN1, MLLT3, PGP, RAMP1, RORA, RRAGD, VDAC1, VEGFA, VLDLR |
Proteomic changes in sarcopenia were consistent with transcriptome changes.
About 1,563 proteins (1,509 of which are annotated) were identified at least once as differentially expressed in the gastrocnemius muscle between 6- versus 18- or 27-month-old rats. Cluster analysis of the 1,509 regulated annotated proteins identified four clusters based on pattern of protein expression changes at 18 and 27 months compared to 6 months (Fig. 7A). Proteins in clusters 1 and 2 were depressed, whereas proteins in clusters 3 and 4 were enriched at 27 compared to 6 months. Gene ontology analysis indicated that the depleted proteins belong in pathways associated with energy metabolism (oxidative phosphorylation, TCA cycle, and gluconeogenesis/glycolysis) and Parkinson's, Huntington's, and Alzheimer's diseases, whereas the upregulated proteins belong in pathways associated with ribosome, translation, proteasome, focal adhesion, and complement and coagulation cascades (Table 5). Consistent with the microarray findings, there was depletion of many of the identified mitochondrial proteins (Fig. 7B). All skeletal muscle isoforms of myosin heavy chain detected (Myh1 and Myh4 were not detected), including the perinatal (Myh8) and embryonic (Myh3) isoforms, were depleted, whereas the nonmuscle (Myh9) and smooth muscle (Myh11) isoforms were enriched (Fig. 7C). Most of the proteins associated with the ribosome and translation were enriched at 27 compared to 6 months (Fig. 7D). The altered expression of some of these proteins was further confirmed by Western blotting (Fig. 7E). Western blotting of the same samples also indicated depletion of peroxisome proliferator coactivator alpha (PGC1α), a key regulator of mitochondrial biogenesis. Furthermore, the expression of Mfn1, Mfn2, and Fis1, which play critical roles in regulating mitochondrial fusion and fission, was also depressed, as was the expression of Chrna1, a subunit of the acetylcholine receptor. On the other hand, there was enrichment of phosphorylated S6 in the absence of overt changes in phosphorylation of AKT (Fig. 7F).
Fig 7.

Gene ontology analysis of differentially regulated proteins in gastrocnemius muscles from 6-, 18-, and 27-month-old rats. (A) Cluster analysis identified four profiles. (B to D) Proteins in clusters 1 and 2, comprising many mitochondrial (B) and muscle structural proteins (C), were mostly repressed, whereas proteins in clusters 3 and 4, comprising ribosomal and translation proteins, were enriched (D) in muscles from 27-month-old rats relative to 6-month-old rats. (E and F) The expression of some of these (E) and other proteins (F) was verified by Western blotting. M, months.
Table 5.
Pathways identified by proteomic analysis to be significantly repressed or enriched in sarcopenia (with an FDR of <0.05)a
| Category | Term | Count | % | P | BH value | FDR |
|---|---|---|---|---|---|---|
| Depleted | ||||||
| KEGG_PATHWAY | Parkinson's disease | 34 | 12.3 | 1.2E–20 | 1.3E–18 | 1.4E–17 |
| KEGG_PATHWAY | Huntington's disease | 37 | 13.4 | 2.6E–19 | 1.4E–17 | 2.9E–16 |
| KEGG_PATHWAY | Oxidative phosphorylation | 32 | 11.6 | 4.8E–19 | 1.7E–17 | 5.3E–16 |
| KEGG_PATHWAY | Alzheimer's disease | 36 | 13.0 | 7.6E–18 | 2.0E–16 | 8.5E–15 |
| KEGG_PATHWAY | Citrate cycle (TCA cycle) | 16 | 5.8 | 1.5E–15 | 3.0E–14 | 1.6E–12 |
| KEGG_PATHWAY | Glycolysis/gluconeogenesis | 23 | 8.3 | 3.8E–15 | 6.5E–14 | 4.2E–12 |
| KEGG_PATHWAY | Cardiac muscle contraction | 21 | 7.6 | 1.1E–13 | 1.7E–12 | 1.3E–10 |
| KEGG_PATHWAY | Pyruvate metabolism | 13 | 4.7 | 1.1E–09 | 1.5E–08 | 1.3E–06 |
| KEGG_PATHWAY | Valine, leucine, and isoleucine degradation | 13 | 4.7 | 9.4E–09 | 1.1E–07 | 1.0E–05 |
| KEGG_PATHWAY | Propanoate metabolism | 11 | 4.0 | 3.5E–08 | 3.6E–07 | 3.9E–05 |
| KEGG_PATHWAY | Butanoate metabolism | 9 | 3.3 | 5.6E–06 | 5.3E–05 | 6.2E–03 |
| KEGG_PATHWAY | Hypertrophic cardiomyopathy | 13 | 4.7 | 9.6E–06 | 8.3E–05 | 1.1E–02 |
| KEGG_PATHWAY | Dilated cardiomyopathy | 13 | 4.7 | 2.0E–05 | 1.6E–04 | 2.2E–02 |
| KEGG_PATHWAY | Fatty acid metabolism | 9 | 3.3 | 3.7E–05 | 2.8E–04 | 4.2E–02 |
| Enriched | ||||||
| KEGG_PATHWAY | Ribosome | 37 | 3.7 | 5.4E–19 | 8.5E–17 | 6.5E–16 |
| KEGG_PATHWAY | Proteasome | 24 | 2.4 | 3.4E–13 | 2.7E–11 | 4.1E–10 |
| KEGG_PATHWAY | Focal adhesion | 35 | 3.5 | 6.5E–06 | 3.4E–04 | 7.9E–03 |
| KEGG_PATHWAY | Complement and coagulation cascades | 18 | 1.8 | 2.0E–05 | 7.8E–04 | 2.4E–02 |
FDR, false-discovery rate.
Mitochondrial DNA content and enzyme and complex activities.
Muscle mitochondrial DNA content was comparable at 6 and 18 months but significantly reduced at 24 months (Fig. 8A). In addition to the decline in muscle mass with age (Fig. 8B), the protein yield per mg of muscle also declined with age (Fig. 8C and D). Consequently, the activities of citrate synthase in the soluble (S20) fraction and of complexes I, II, and IV in the pellet (P20) fraction of the gastrocnemius muscle homogenates declined progressively with age when normalized to muscle weight (Fig. 8E to H) but not when normalized to mg of protein (data not shown).
Fig 8.

Mitochondrial DNA content and activity. (A to D) Mitochondrial DNA content (A), muscle weight (B), and protein yield in the soluble (S20 [C]) and pellet (P20 [D]) fractions of proportionately pooled gastrocnemius muscle samples from the 6-, 18-, and 24-month-old age groups. (E to H) The activities of citrate synthase, complex I, complex II, and complex IV of the electron transport chain were determined and normalized to the muscle weight. The pooled samples for the 6-, 18-, and 24-month-old groups were analyzed three times, and the mean plus the standard error of the mean plotted. *, **, ***, and **** indicate P < 0.05, 0.01, 0.001, and 0.0001 versus 6 months, respectively. M, months.
DISCUSSION
While the age-related decline in skeletal muscle mass and function, known as sarcopenia, has been clinically described, it was unclear by what molecular processes it occurred, and whether these mechanisms were distinct from those previously observed during other settings of skeletal muscle loss, such as acute skeletal muscle atrophy caused either by denervation or immobilization, or cachectic cytokines in settings of cancer cachexia, or perturbations such as upregulation of cortisol observed in renal cachexia or burns (8); thus, it was not clear whether the loss of muscle that occurred coincident with old age even deserved a unique name. To determine gene perturbations, proteomic changes, and functional and physical changes that occur at the onset and during sarcopenia, a rat model was used.
The male Harlan SD rat exhibited many features of human sarcopenia, including the relative age of onset, rate of progression, and clinical manifestations. With an average life span of 30 months for these rats (46), compared to 75 years for humans, muscle mass peaked by the second quintile of life (∼10 months in rats compared to 30 years in humans), remained fairly stable in the middle quintile (13 to 18 months in rats versus 31 to 45 years in humans), and then declined drastically thereafter (after 18 months in rats compared to 50 years in humans). Thus, phenotypically, these ad libitum fed male Harlan SD rats mimicked the sarcopenia phenotype of the relatively sedentary and ad libitum fed average “Western” human.
At the cellular level, the rat faithfully mimicked the preferential type 2 fiber atrophy and the absence of significant fiber type switching seen in human sarcopenia (5, 6, 47, 48). The findings of increased extrajunctional staining for acetylcholinesterase, together with the decreased size of the acetylcholinesterase-stained neuromuscular junction, indicate that sarcopenia in this rat model is also associated with perturbations at the neuromuscular junction, as seen in human sarcopenia. Another feature of human sarcopenia is increased fatigability due, in part, to muscle fatigue but also to cardiac, respiratory, and other system failures (49). In the present rat study, there was an increase in the proportion of fibers with weak SDH staining and a decrease in the proportion of fibers with moderate or strong SDH staining with age consistent with a decrease in mitochondrial oxidative enzyme activity.
There are several reports on the global gene expression changes associated with sarcopenia in rodents, rhesus monkeys, and humans (50–54). However, these studies typically compared young, nonsarcopenic subjects versus a single time point of old, sarcopenic subjects and therefore did not study enough time points to be able to correlate gene changes to the onset of morphological and functional changes in sarcopenia, making it difficult to ascertain which molecular changes are potentially causally related versus merely sequelae of sarcopenia (20). Given that the age-related clinical and morphological presentation of sarcopenia in the male Harlan SD rat mimicked the human condition, we determined the global gene expression profile in muscles from these rats at 6, 12, 18, 21, 24, and 27 months of age (spanning almost their entire life span and including the negative inflection point in muscle mass at 21 months) and correlated the gene profile to the changes in muscle mass. This unbiased analysis revealed first that, as with the changes in muscle mass and strength, the most drastic changes in gene expression occurred at 21 months of age, suggesting that the perturbations that trigger sarcopenia occur prior to or at about 21 months of age in these rats. Second, the analysis identified clusters of genes whose expression profiles correlated strongly and directly (cluster 2; R ≥ 0.9) or inversely (cluster 3; R ≤ −0.9) with changes in skeletal muscle mass as distinct from genes or clusters whose expression profiles correlated better (clusters 1 and 5) or equally well, albeit reciprocally (cluster 4), with age compared to muscle mass. The genes in clusters 2 and 3, whose expression profiles correlate better with muscle mass than with age, were considered gene signatures of sarcopenia, whereas the genes in clusters 1, 4, and 5 whose expression profiles correlated better with age than with muscle mass were considered gene signatures of aging.
Functional annotation analysis indicated that genes whose expression changes most directly correlated with sarcopenia (e.g., Pdhx, Rxrg, Ugp2, Pgp, Tuba8, Tnks2, Mylk2, Lrrc39, Asb11, St8sia5, Egln1, Hfe2, RGD1564300, and Pfkfb1 in cluster 2) belong to pathways associated with mitochondrial energy metabolism (e.g., oxidative phosphorylation, TCA cycle, fatty acid metabolism, and glycolysis/gluconeogenesis) and skeletal muscle structure. The timing and almost absolute correlation between the decline in mitochondrial gene expression and the onset of sarcopenia provide strong evidence that mitochondriogenesis in particular may be driving sarcopenia. The relevance of the mitochondrial decline in sarcopenia is highlighted by reports that manipulating this pathway through overexpression of PGC1α in skeletal muscles significantly improved muscle mass, function, and metabolism in mice. In MCK-PGC1α mice (in which PGC1α is expressed in all muscles at a level comparable to endogenous PGC-1α in type I muscles), muscle mass, function (treadmill performance), and mitochondrial COX enzyme activity were significantly greater at 22 months of age than in age-matched wild-type mice. Moreover, these improvements in muscle morphology and function were associated with reduced markers of inflammation, muscle protein degradation, and neuromuscular junction disintegration, as well as systemic metabolic benefits and extended life span (55, 56). MCK–PGC-1α mice were also partially protected from denervation-induced atrophy, in part, through the inhibition of expression of FoxO3, which in turn inhibits the expression of atrogenes (MAFBx and MuRF1) and muscle proteolysis (57). Similarly, the upregulation of inflammation markers in sarcopenia observed in the present study is consistent with previous reports in animals and humans associating elevated blood inflammatory markers (e.g., α2-macroglobulin, C-reactive proteins, and cytokines) with age-related decreases in muscle mass, physical performance, and even mortality (58). Indeed, some studies in rodents have reported that anti-inflammatory agents protect against sarcopenia, in part, through increased phosphorylation of FoxO3 (59). These reports lend credence to the relevance in sarcopenia of the pathways identified in the present study. Taken together, these would indicate that the loss of mitochondria and increased inflammation are present upon initiation of sarcopenia and that restoring mitochondriogenesis, which itself reduces inflammation, is sufficient to rescue the sarcopenia phenotype and extend life.
Genes in the sarcopenia cluster 2 were downregulated coincident with the onset and progression of sarcopenia and thus represent the molecular signatures for the decline in muscle fiber oxidative capacity demonstrated histochemically in the present study. On the other hand, genes with expression changes most inversely correlated with sarcopenia (e.g., Hmgn2, Ccni, Krt18, Fst, Iffo1, Slain2, Adcy2, Ppp1r14b, and Prkar1a in cluster 3) were associated with the cell cycle, ribosome, translation, immune response, cytokine production, proteolysis-ubiquitin, apoptosis, and survival. Thus, at the level of biological pathways and processes, underexpression of genes for mitochondrial energy metabolism and muscle structural proteins and overexpression of genes for cell cycle, ribosome, translation, immune response, cytokine production, proteolysis, and apoptosis represent signatures of sarcopenia. It is noteworthy, however, that the specific genes in the signatures of sarcopenia identified here by correlating changes in gene expression against changes in muscle mass differ from, albeit with some overlap with, sarcopenia gene signatures identified based on the magnitude of gene expression changes in previous studies (50–54). For example, in a meta-analysis of previous data, the transferrin receptor (Tfrc) and collagen type 1 alpha 1 (Col1A1) genes were identified to be the most and third most downregulated genes, respectively, and considered part of a common signature of muscle aging (60). However, despite ranking similarly in the present study, the changes in expression of Tfrc correlated better with age than with muscle mass, whereas changes in expression of Col1A1 correlated poorly with age and worst still with muscle mass (Table 3). Similarly, although upregulation of genes for inflammation, immune response and complement activation (e.g., C1qa), Hdac4, S100a4, and Il6r have been identified as part of the gene signature for muscle aging, changes in expression of these genes were better correlated with age than with muscle mass in the present study. However, consistent with previous studies, the present study found that downregulation of genes encoding some mitochondrial proteins (e.g., Pdh) and upregulation of Hmgn2 and Fst are members of the gene signature of sarcopenia (50–53, 60). It is noteworthy that there were several regulated genes whose expression profiles correlated strongly with age but not with muscle mass, whereas the converse (i.e., genes with expression profiles that correlated strongly with muscle mass but not with age) was rare. These observations probably reflect the fact that sarcopenia is age dependent, whereas age is not sarcopenia dependent.
Further confirmation for many of the above microarray data was provided by proteomics analysis of muscle samples from rats at 6, 18, and 27 months of age which independently identified sarcopenia-related depletion of proteins associated with mitochondrial energy metabolism (the oxidative phosphorylation and TCA pathways) and depletion of myofibrillar proteins, as well as the overexpression of proteins associated with the ribosome and protein translation. These findings are consistent with those of Piec et al. (14), obtained also in rats (but with only a single group in the sarcopenic window—at 30 months—and therefore without an ability to correlate findings to the onset and progression of the phenotype), that sarcopenia was associated with selective downregulation of proteins involved with myofibrillar filament proteins, mitochondrial Krebs cycle and oxidative phosphorylation, as well as with selective upregulation of several proteins that regulate RNA and protein metabolism suggestive of increased protein synthesis. Indeed, we showed that the decline in mitochondrial gene and protein expression in sarcopenia was associated with reduced mitochondrial content with functional significance—reductions in citrate synthase and complexes I, II, and IV enzyme activities per unit of muscle mass but not per unit of protein. These findings indicate that the decline in mitochondrial enzyme activity in sarcopenia was probably due to a decline in mitochondrial content rather than to reduced efficiency of existing mitochondria. Also depressed in sarcopenia was expression of the genes, and their protein products, that regulate mitochondrial fission and fusion, suggesting that mitochondrial dynamics is also perturbed in sarcopenia.
The present observation of upregulation of genes involved with protein synthesis in sarcopenia, although paradoxical and inconsistent with some previous reports (see, for example, reference 60), agrees with reports that muscle protein synthesis, RNA content, and S6K1 phosphorylation all increased at 24 and 27 months compared to 6 months despite progressing sarcopenia in male Harlan SD rats (61). The coexistence of increased signaling for protein synthesis, protein degradation, and muscle atrophy has also been reported in intensive care unit patients with sepsis and was explained as reflecting failure of the increase in anabolic signal (i) to compensate for the increase in protein degradation or (ii) to result in synthesis of the appropriate proteins (62, 63). The present findings that all myosin heavy-chain proteins were depleted in sarcopenia despite increased expression of transcripts for Myh1, Myh4, Myh3, and Myh8 are consistent with a failed compensatory mechanism and a resultant loss of skeletal muscle sarcomeres, which is sufficient to explain the decline in muscle strength observed during sarcopenia. Similarly, the findings of increased cell cycle genes might have derived from increased numbers of fibroblasts (hence, the increased fibrosis observed in sacropenic muscles), vascular cells, and proliferation-competent inflammatory cells (hence, the increase in cytokine genes), as opposed to skeletal muscle fibers.
As to how the gene expression changes in sarcopenia compare to those in other forms of muscle atrophy (e.g., disuse- or denervation-atrophy or cancer cachexia), such comparison is complicated by differences in the muscle, atrophy-inducing stimuli, duration of atrophy, microarray platform, and animal species between studies. Nonetheless, a meta-analysis of data from seven previous studies indicates that some of the gene changes are unique to sarcopenia, whereas others are common signatures. Common to sarcopenia, denervation, and systemic wasting (cachexia) is depression of mitochondrial energy metabolism pathways and variable upregulation of protein degradation pathways (64), whereas upregulation of neuromuscular junction genes is common to both sarcopenia and denervation but not cachexia, and overexpression of inflammation genes is common to both sarcopenia and cachexia but not denervation (65). Thus, the uniqueness of sarcopenia might be in the multiplicity of pathways perturbed (mitochondrial energy metabolism, neuromuscular junction, proteolysis, immune response, and inflammation) and the insidious nature which allows for initiation of compensatory albeit futile mechanisms (e.g., depression of Mstn and increase in Fst and IGF-1 genes).
Taken together, the depletion of genes and proteins associated with mitochondrial energy metabolism, the overexpression of genes for the ribosome, transcription, protein synthesis, protein breakdown, immune response, and inflammation suggest that therapeutic interventions for sarcopenia should ideally involve simultaneously increasing mitochondrogenesis, decreasing muscle proteolysis and inflammation, and increasing the patency of the neuromuscular junction. One potential approach to achieve these multitude of effects is through modulation of PGC-1α, which in addition to regulating mitochondrial content has been reported to be a positive modulator of metabolism, the neuromuscular junction, apoptosis, and even inflammatory responses (for a review, see reference 66).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Muscle Diseases Group at the Novartis Institutes for Biomedical Research (NIBR) for their enthusiastic support, along with the rest of the NIBR community, and in particular Mark Fishman, Brian Richardson, and Andrew Mackenzie. We also thank Nicole Hartmann of the Novartis Genomics Factory for performing the microarray hybridization experiment and Wilfried Frieauff for the internal software tool (ROBIAS ASTORIA).
Footnotes
Published ahead of print 29 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01036-12.
REFERENCES
- 1. Janssen I, Baumgartner RN, Ross R, Rosenberg IH, Roubenoff R. 2004. Skeletal muscle cutpoints associated with elevated physical disability risk in older men and women. Am. J. Epidemiol. 159:413–421 [DOI] [PubMed] [Google Scholar]
- 2. Metter EJ, Talbot LA, Schrager M, Conwit R. 2002. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J. Gerontol. A Biol. Sci. Med. Sci. 57:B359–B365 [DOI] [PubMed] [Google Scholar]
- 3. Balagopal P, Rooyackers OE, Adey DB, Ades PA, Nair KS. 1997. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy- chain and sarcoplasmic protein in humans. Am. J. Physiol. 273:E790–E800 [DOI] [PubMed] [Google Scholar]
- 4. Gray C, MacGillivray TJ, Eeley C, Stephens NA, Beggs I, Fearon KC, Greig CA. 2011. Magnetic resonance imaging with K-means clustering objectively measures whole muscle volume compartments in sarcopenia/cancer cachexia. Clin. Nutr. 30:106–111 [DOI] [PubMed] [Google Scholar]
- 5. Lexell J. 1995. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 50(Spec No):11–16 [DOI] [PubMed] [Google Scholar]
- 6. Nikolic M, Malnar-Dragojevic D, Bobinac D, Bajek S, Jerkovic R, Soic-Vranic T. 2001. Age-related skeletal muscle atrophy in humans: an immunohistochemical and morphometric study. Coll. Antropol. 25:545–553 [PubMed] [Google Scholar]
- 7. Aagaard P, Suetta C, Caserotti P, Magnusson SP, Kjaer M. 2010. Role of the nervous system in sarcopenia and muscle atrophy with aging: strength training as a countermeasure. Scand. J. Med. Sci. Sports 20:49–64 [DOI] [PubMed] [Google Scholar]
- 8. Glass D, Roubenoff R. 2010. Recent advances in the biology and therapy of muscle wasting. Ann. N. Y. Acad. Sci. 1211:25–36 [DOI] [PubMed] [Google Scholar]
- 9. Lee CE, McArdle A, Griffiths RD. 2007. The role of hormones, cytokines and heat shock proteins during age-related muscle loss. Clin. Nutr. 26:524–534 [DOI] [PubMed] [Google Scholar]
- 10. Lenk K, Schuler G, Adams V. 2010. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J. Cachex Sarcopenia Muscle 1:9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rice KM, Linderman JK, Kinnard RS, Blough ER. 2005. The Fischer 344/NNiaHSd X Brown Norway/BiNia is a better model of sarcopenia than the Fischer 344/NNiaHSd: a comparative analysis of muscle mass and contractile properties in aging male rat models. Biogerontology 6:335–343 [DOI] [PubMed] [Google Scholar]
- 12. Hagen JL, Krause DJ, Baker DJ, Fu MH, Tarnopolsky MA, Hepple RT. 2004. Skeletal muscle aging in F344BN F1-hybrid rats. I. Mitochondrial dysfunction contributes to the age-associated reduction in VO2max. J. Gerontol. A Biol. Sci. Med. Sci. 59:1099–1110 [DOI] [PubMed] [Google Scholar]
- 13. Hepple RT, Ross KD, Rempfer AB. 2004. Fiber atrophy and hypertrophy in skeletal muscles of late middle-aged Fischer 344 X Brown Norway F1-hybrid rats. J. Gerontol. A Biol. Sci. Med. Sci. 59:108–117 [DOI] [PubMed] [Google Scholar]
- 14. Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D. 2005. Differential proteome analysis of aging in rat skeletal muscle. FASEB J. 19:1143–1145 [DOI] [PubMed] [Google Scholar]
- 15. Arts IM, Pillen S, Overeem S, Schelhaas HJ, Zwarts MJ. 2007. Rise and fall of skeletal muscle size over the entire life span. J. Am. Geriatr. Soc. 55:1150–1152 [DOI] [PubMed] [Google Scholar]
- 16. Frontera WR, Reid KF, Phillips EM, Krivickas LS, Hughes VA, Roubenoff R, Fielding RA. 2008. Muscle fiber size and function in elderly humans: a longitudinal study. J. Appl. Physiol. 105:637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lexell J, Taylor CC. 1991. Variability in muscle fibre areas in whole human quadriceps muscle: effects of increasing age. J. Anat. 174:239–249 [PMC free article] [PubMed] [Google Scholar]
- 18. Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J, Tarnopolsky MA. 2010. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One 5:e10778 doi:10.1371/journal.pone.0010778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan LJ, Liu SL, Lei SF, Papasian CJ, Deng HW. 2012. Molecular genetic studies of gene identification for sarcopenia. Hum. Genet. 131:1–31 [DOI] [PubMed] [Google Scholar]
- 20. Murphy CT. 2006. Using whole-genome transcriptional analyses to identify molecular mechanisms of aging. Drug Discov. Today Dis. Mechanisms 3:41–46 [Google Scholar]
- 21. Dolber PC, Spach MS. 1987. Picrosirius red staining of cardiac muscle following phosphomolybdic acid treatment. Stain Technol. 62:23–26 [DOI] [PubMed] [Google Scholar]
- 22. Reddy GK, Enwemeka CS. 1996. A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 29:225–229 [DOI] [PubMed] [Google Scholar]
- 23. Josza L, Lehto MU, Jarvinen M, Kvist M, Reffy A, Kannus P. 1993. A comparative study of methods for demonstration and quantification of capillaries in skeletal muscle. Acta Histochem. 94:89–96 [DOI] [PubMed] [Google Scholar]
- 24. Inoue N, Kondo T, Kobayashi K, Aoki M, Numaguchi Y, Shibuya M, Murohara T. 2007. Therapeutic angiogenesis using novel vascular endothelial growth factor-E/human placental growth factor chimera genes. Arterioscler. Thromb. Vasc. Biol. 27:99–105 [DOI] [PubMed] [Google Scholar]
- 25. Karnovsky MJ, Roots L. 1964. A “direct-coloring” thiocholine method for cholinesterases. J. Histochem. Cytochem. 12:219–221 [DOI] [PubMed] [Google Scholar]
- 26. Dubowitz V, Sewry CA. 2007. Muscle biopsy: a practical approach, 3rd ed. Saunders Elsevier, London, England [Google Scholar]
- 27. Team RDC. 2004. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 28. Ihaka R, Gentleman RR. 1996. A language for data analysis and graphics. J. Comput. Graphical Statist. 5:299–314 [Google Scholar]
- 29. Affymetrix 2005. Guide to probe logarithmic intensity error (PLIER) estimation. Affymetrix, Santa Clara, CA: http://media.affymetrix.com/support/technical/technotes/plier_technote.pdf [Google Scholar]
- 30. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 57:289–300 [Google Scholar]
- 32. Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 33. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 34. Boersema PJ, Aye TT, van Veen TA, Heck AJ, Mohammed S. 2008. Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8:4624–4632 [DOI] [PubMed] [Google Scholar]
- 35. Hsu JL, Huang SY, Chow NH, Chen SH. 2003. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75:6843–6852 [DOI] [PubMed] [Google Scholar]
- 36. Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670 [DOI] [PubMed] [Google Scholar]
- 37. McCormack AL, Yates JR., III 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989 [DOI] [PubMed] [Google Scholar]
- 38. Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4:207–214 [DOI] [PubMed] [Google Scholar]
- 39. Janssen AJM, Sengers TFRCA, Smeitink JAM, van den Heuvel LP, Wintjes LTM, Stoltenborg-Hogenkamp BJM, Rodenburga RJT. 2007. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 53:729–734 [DOI] [PubMed] [Google Scholar]
- 40. Trounce IA, Kim YL, Jun AS, Wallace DC. 1996. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 264:484–509 [DOI] [PubMed] [Google Scholar]
- 41. Birch-Machin MA, Turnbull DM. 2001. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65:97–117 [DOI] [PubMed] [Google Scholar]
- 42. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. 2001. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294:1704–1708 [DOI] [PubMed] [Google Scholar]
- 43. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. 2001. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U. S. A. 98:14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. 2004. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 14:395–403 [DOI] [PubMed] [Google Scholar]
- 45. Hock MB, Kralli A. 2009. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 71:177–203 [DOI] [PubMed] [Google Scholar]
- 46. Masoro EJ. 1980. Mortality and growth characteristics of rat strains commonly used in aging research. Exp. Aging Res. 6:219–233 [DOI] [PubMed] [Google Scholar]
- 47. Korhonen MT, Cristea A, Alen M, Hakkinen K, Sipila S, Mero A, Viitasalo JT, Larsson L, Suominen H. 2006. Aging, muscle fiber type, and contractile function in sprint-trained athletes. J. Appl. Physiol. 101:906–917 [DOI] [PubMed] [Google Scholar]
- 48. Lexell J, Sjostrom M, Nordlund AS, Taylor CC. 1992. Growth and development of human muscle: a quantitative morphological study of whole vastus lateralis from childhood to adult age. Muscle Nerve 15:404–409 [DOI] [PubMed] [Google Scholar]
- 49. Theou O, Jones GR, Overend TJ, Kloseck M, Vandervoort AA. 2008. An exploration of the association between frailty and muscle fatigue. Appl. Physiol. Nutr. Metab. 33:651–665 [DOI] [PubMed] [Google Scholar]
- 50. Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, Kandarian SC. 2005. Identification of a molecular signature of sarcopenia. Physiol. Genomics 21:253–263 [DOI] [PubMed] [Google Scholar]
- 51. Kayo T, Allison DB, Weindruch R, Prolla TA. 2001. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. U. S. A. 98:5093–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee CK, Klopp RG, Weindruch R, Prolla TA. 1999. Gene expression profile of aging and its retardation by caloric restriction. Science 285:1390–1393 [DOI] [PubMed] [Google Scholar]
- 53. Welle S, Brooks AI, Delehanty JM, Needler N, Thornton CA. 2003. Gene expression profile of aging in human muscle. Physiol. Genomics 14:149–159 [DOI] [PubMed] [Google Scholar]
- 54. Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK. 2006. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2:e115 doi:10.1371/journal.pgen.0020115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Handschin C, Spiegelman BM. 2008. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454:463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. 2009. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. U. S. A. 106:20405–20410 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57. Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. 2006. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U. S. A. 103:16260–16265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roubenoff R. 2003. Catabolism of aging: is it an inflammatory process? Curr. Opin. Clin. Nutr. Metab. Care 6:295–299 [DOI] [PubMed] [Google Scholar]
- 59. Rieu I, Magne H, Savary-Auzeloux I, Averous J, Bos C, Peyron MA, Combaret L, Dardevet D. 2009. Reduction of low grade inflammation restores blunting of postprandial muscle anabolism and limits sarcopenia in old rats. J. Physiol. 587:5483–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. de Magalhaes JP, Curado J, Church GM. 2009. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 25:875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kimball SR, O'Malley JP, Anthony JC, Crozier SJ, Jefferson LS. 2004. Assessment of biomarkers of protein anabolism in skeletal muscle during the life span of the rat: sarcopenia despite elevated protein synthesis. Am. J. Physiol. Endocrinol. Metab. 287:E772–780 [DOI] [PubMed] [Google Scholar]
- 62. Fredriksson K, Tjader I, Keller P, Petrovic N, Ahlman B, Scheele C, Wernerman J, Timmons JA, Rooyackers O. 2008. Dysregulation of mitochondrial dynamics and the muscle transcriptome in ICU patients suffering from sepsis induced multiple organ failure. PLoS One 3:e3686 doi:10.1371/journal.pone.0003686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jespersen JG, Nedergaard A, Reitelseder S, Mikkelsen UR, Dideriksen KJ, Agergaard J, Kreiner F, Pott FC, Schjerling P, Kjaer M. 2011. Activated protein synthesis and suppressed protein breakdown signaling in skeletal muscle of critically ill patients. PLoS One 6:e18090 doi:10.1371/journal.pone.0018090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Calura E, Cagnin S, Raffaello A, Laveder P, Lanfranchi G, Romualdi C. 2008. Meta-analysis of expression signatures of muscle atrophy: gene interaction networks in early and late stages. BMC Genomics 9:630 doi:10.1186/1471-2164-9-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. 2007. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 21:140–155 [DOI] [PubMed] [Google Scholar]
- 66. Wenz T. 2011. Mitochondria and PGC-1α in aging and age-associated diseases. J. Aging Res. 2011:810619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Merico D, Isserlin R, Stueker O, Emili A, Bader GD. 2010. Enrichment Map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5:e13984 doi:10.1371/journal.pone.0013984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.