

Abstract
Enterovirus 71 (EV71) is a positive-stranded RNA virus which is capable of inhibiting innate immunity. Among virus-encoded proteins, the 3C protein compromises the type I interferon (IFN-I) response mediated by retinoid acid-inducible gene-I (RIG-I) or Toll-like receptor 3 that activates interferon regulatory 3 (IRF3) and IRF7. In the present study, we report that enterovirus 71 downregulates IRF7 through the 3C protein, which inhibits the function of IRF7. When expressed in mammalian cells, the 3C protein mediates cleavage of IRF7 rather than that of IRF3. This process is insensitive to inhibitors of caspase, proteasome, lysosome, and autophagy. H40D substitution in the 3C active site abolishes its activity, whereas R84Q or V154S substitution in the RNA binding motif has no effect. Furthermore, 3C-mediated cleavage occurs at the Q189-S190 junction within the constitutive activation domain of IRF7, resulting in two cleaved IRF7 fragments that are incapable of activating IFN expression. Ectopic expression of wild-type IRF7 limits EV71 replication. On the other hand, expression of the amino-terminal domain of IRF7 enhances EV71 infection, which correlates with its ability to interact with and inhibit IRF3. These results suggest that control of IRF7 by the 3C protein may represent a viral mechanism to escape cellular responses.
INTRODUCTION
Enterovirus 71 (EV71) is a positive-stranded RNA virus, which encodes a large polyprotein approximately 2,200 amino acids (aa). This precursor is processed into structural (VP1, VP2, VP3, and VP4) and nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) during virus infection (1). The 3C protein expressed by EV71 is essential for viral replication (2, 3). In addition to its activity in viral protein processing (4), the 3C protein is linked to a number of biological processes. It has been reported that EV71 3C acts as an RNA binding protein that interacts the 5′ untranslated region of viral RNA. However, its impact on EV71 infection is not known (2). In neuronal cells, the 3C protein appears to trigger apoptosis, which relies on caspase activation (4). Recent evidence shows that the 3C protein cleaves cellular CstF-64 protein, which subsequently halts host RNA processing and polyadenylation (5). This is postulated to create an advantage for viral replication. Strikingly, the 3C protein inhibits the expression of type I interferon (IFN-I) which mediates antiviral, apoptotic, and immunoregulatory activities (6, 7).
IFN-I production is activated by pathogen-associated molecular patterns (PAMPs) through host pattern recognition receptors (PRRs) (8). In response to viral double-stranded RNA, Toll-like receptor 3 (TLR3) recruits the adaptor TRIF and TANK binding kinase 1 (TBK1), which phosphorylates interferon regulatory factor 3 (IRF3)/IRF7. Alternatively, cytosolic receptors, including retinoic acid-inducible gene-I (RIG-1) and melanoma differentiation-associated gene (MDA5), recruit the adaptor IPS-1 (also known as MAVS, VISA, and CARDIF) and TBK1 to activate IRF3 or IRF7. While IRF3 functions primarily in the priming stage of IFN production, IRF7 has an important role in the amplifying stage, where it provides a positive feedback to the initial response (8, 9). Additionally, upon recognition of single-stranded RNA, TLR7 and TLR8 relay signals to the adaptors MyD88 and TRAF6 (9). As a result, the kinase IRAK1 is activated to phosphorylate the transcription factor IRF7. Once activated, IRF7 translocates to the nucleus and induces IFN-I expression. Therefore, IRF7 represents a converging point of innate immune pathways.
Several studies suggest that MDA-5 and RIG-I play a pivotal role in sensing picornavirus infection (10–12). Moreover, it has been reported that TLR3 detects or limits picornavirus infection (13–15). Therefore, it is not surprising that picornaviruses have evolved mechanisms to escape antiviral immunity. For example, several picornaviruses cleave or interact with these pattern recognition receptors (7, 12, 16, 17). Recently, we reported that EV71 inhibits IFN-I responses mediated by RIG-I and TLR3 (6, 7). Although this involves the 3C protein that negatively regulates RIG-I and TRIF, the precise role of EV71 3C has yet to be defined.
In this study, we report that EV71 reduces the expression of IRF7 in infected cells. Such effect requires the 3C protein, which mediates IRF7 cleavage. This is independent of capsase, proteasome, lysosome, and autophagy. We provide evidence that H40D substitution in the active site disrupts its activity, whereas R84Q or V154S substitution in the RNA binding motif has no effect. We also demonstrate that IRF7 cleavage depends on its amino acid pair Q189 and S190. Importantly, while wild-type IRF7 limits EV71 replication, cleaved IRF7 has no activity. Therefore, control of IRF7 by 3C may represent an EV71 mechanism to overcome cellular responses.
MATERIALS AND METHODS
Cell lines and viruses.
293T, HeLa, and RD cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA), supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone, Logan, UT), and penicillin/streptomycin at 37°C in a 5% CO2 humidified atmosphere. Human monocytic THP1 cells were cultured in RPMI 1640 media supplemented with 10% FBS. Enterovirus 71 infection was carried out as described previously (7).
Plasmids.
The plasmids pEGFP (where EGFP is enhanced green fluorescent protein), pEGFP-3C, pEGFP-3C variants, Flag-TRIF, Myc-RIG-I, Flag-TBK1, Flag-MDA-5, Flag-IPS-1, and Flag-MyD88 have been described elsewhere (6, 7). The plasmids expressing IRF7, STING, IRF3, TRADD, FADD, RIP1, TRAF2, TRAF3, TRAF6, TAK1, and TANK were purchased from Origene (Rockville, MD). These constructs have the Myc-Flag tag at the C terminus. The IRF7 variants, including the Q151, Q167, Q185, Q188, Q189, and Q215 amino-acid-substitution mutants and deletion mutants, 189N (1 to 189 aa), and 189C (190 to 503 aa), were constructed by site-directed mutagenesis using Pfu DNA polymerase (Strategene, La Jolla, CA). The IRF7 and IRF3 cDNAs also were cloned into the eukaryotic expression vector pGEX 4T-1. All variants were verified by nucleotide sequence analysis.
Antibodies and reagents.
Antibodies against Flag, Myc, IRF3, GFP, and β-actin were purchased from Sigma (St. Louis, MO). Anti-IRF7, anti-TRAF3, and anti-TBK1 antibodies were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibody against GFP was purchased from Roche (Roche, Indianapolis, IN). Mouse anti-enterovirus 71 was purchased from Chemicon (Billerica, MA). Goat anti- mouse or rabbit secondary antibodies were purchased from LI-COR (LI-COR Inc., Lincoln, NE). The general caspase inhibitor Z-VAD-FMK and poly(I·C) were purchased from Sigma (St. Louis, MO). MG132, NH4Cl, and 3-MA were purchased from Sigma (St. Louis, MO). Rupintrivir was purchased from Santa Cruz (Santa Cruz, CA).
Reporter assays.
Luciferase reporter assays were performed as described previously (7). 293T cells were seeded in 24-well plates at a cell density of 3 × 105 cells per well. At 16 h after plating, cells were transfected with a control plasmid or plasmid expressing IRF7, IRF7 variants, and 3C or its variants along with pGL3-IFN-β-luc, ISG56-luc, ISRE-luc, IFN-α4-luc, and pRL-SV40 using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). pRL-SV40 plasmid was used as a control for transfection efficiency. The total amount of DNA was kept constant by adding empty control plasmid. At 24 h after transfection, cells were harvested and cell lysates were used to determine luciferase activities using a Dual-Luciferase reporter system (Promega, Madison, WI) according to the manufacturer's instructions.
Reverse transcription-PCR (RT-PCR).
Cells were mock infected or infected with EV71 or Sendai virus (SeV) individually as described previously (6). At different time points after infection, total RNA was extracted from cells by using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA samples were treated with DNase I (Pierce, Rockford, IL), and reverse transcription was carried out using the Superscript cDNA synthesis kit (Invitrogen) according to the manufacturer's instructions. cDNA samples were subjected to PCR amplification and electrophoresis to detect IFN-β, ISG54, and ISG56 expression. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression was used as a loading control. Primers used were as described previously (6).
Immunoprecipitation.
At 24 h after transfection, cells were lysed with a radioimmunoprecipitation assay (RIPA) (25 mM Tris-HCl buffer [pH 7.4] containing 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate) containing a protease inhibitor cocktail (Roche, Indianapolis, IN) as described previously (7). Cell lyates were incubated with anti-GFP antibody (Roche, Indianapolis, IN) or anti-Myc antibody (Sigma, St. Louis, MO) at 4°C overnight on a rotator in the presence of protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoprecipitates were subjected to electrophoresis and Western blot analysis (6, 7).
Western blot analysis.
Cells were pelleted by centrifugation and lysed in buffer containing 150 mM NaCl, 25 mM Tris (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, and 1 mM EDTA with a protease inhibitor cocktail (Roche, Indianapolis, IN). Aliquots of cell lysates were electrophoresed on 12% SDS-PAGE gels and transferred to a nitrocellulose membrane (Pall, Port Washington, NY). The membranes were blocked with 5% nonfat dry milk and then proteins on the membrane were incubated with indicated primary antibodies at 4°C overnight. This was followed by incubation with corresponding IRD Fluor 800-labeled IgG or IRD Fluor 680-labeled IgG secondary antibody (LI-COR Inc., Lincoln, NE) for 1 h at room temperature. After washing, the membranes were scanned with the Odyssey infrared imaging system (LI-COR, Lincoln, NE) at a wavelength of 700 to 800 nm and the molecular sizes of the developed proteins were determined by a comparison with prestained protein markers (Fermentas, MD).
In vitro cleavage assays.
Recombinant EV71 3C protease was produced by prokaryotic expression as described previously (18). To express recombinant IRF7 and IRF3, pGEX4T-1 expressing IRF7 or IRF3 was transformed into competent Escherichia coli BL21(DE3) cells, and protein expression was induced by treatment with 200 μM IPTG (isopropyl-β-d-thiogalactopyranoside) at 16°C overnight. Glutathione S-transferase (GST)-fused IRF7 or IRF3 protein was purified with a glutathione-agarose column. To examine IRF7 cleavage in vitro, aliquots of recombinant 3C and lysates of cells expressing IRF7, GST-IRF7, and GST-IRF3 were incubated at 37°C in 50 mM Tris-HCl buffer (pH 7.0) containing 200 mM NaCl. At 12 h after incubation, samples were subjected to Western blot analysis.
RESULTS
Expression of EV71 3C induces cleavage of IRF7 in mammalian cells.
We previously reported that EV71 3C mediates cleavage of TRIF, a key adaptor of the TLR3 pathway (6). To explore whether EV71 3C has an additional target(s), we examined a panel of components in the innate immune pathways (data not shown). Cells were transfected with GFP-3C along with plasmids expressing the host factors individually. At 24 h after transfection, cell lysates were processed to assess 3C-mediated cleavage by Western blotting. We found that GFP-3C induced cleavage of IRF7, TRAF3, and TAK1, in addition to TRIF. As such, the effect on TRAF3 and TAK1 was modest (data not shown); we therefore focused on IRF7. Figure 1A illustrates that when coexpressed, GFP-3C did not affect the expression of IRF3 (lanes 1 and 2). Under this condition, GFP-3C reduced IRF7 expression compared to the GFP control (lanes 3 and 4). This paralleled with the appearance of a smaller protein band (45 kDa) (lane 4). Further analysis showed that EV71 3C acted in a dose-dependent manner (Fig. 1B). As expected, GFP-3C induced the cleavage of TRIF and in transfected cells (Fig. 1C).
Fig 1.
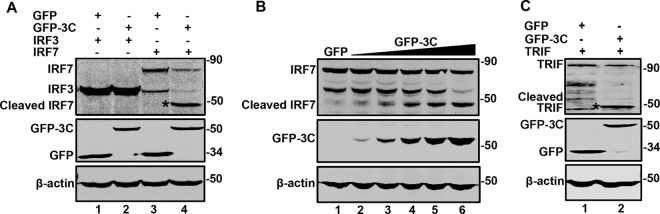
The 3C protease of EV71 cleaves IRF7 and TRIF. (A) 293T cells were transfected with plasmids encoding Myc-Flag-IRF3 (lanes 1 and 2) and Myc-Flag-IRF7 (lanes 3 and 4) or alone with GFP (lanes 1 and 3) or GFP-3C (lanes 2 and 4). At 24 h after transfection, cells were lysed and then cell lysates were analyzed by Western blotting with antibodies specific for Flag and GFP. β-Actin was used as a protein loading control. (B) 293T cells were transfected with Myc-Flag-IRF7 along with GFP (lanes 1) or increasing amounts of GFP-3C (lanes 2 to 6). The cells were then processed for Western blot analysis as described for panel A. (C) 293T cells were transfected with plasmids encoding Flag-TRIF (lanes 1 and 2) or alone with GFP (lanes 1) or GFP-3C (lanes 2). The cells were processed for Western blot analysis as described for panel A. Data shown are representative of three separate experiments.
Next, we assessed the impact of EV71 3C on endogenous IRF7. When ectopically expressed in 293T cells (Fig. 2A), GFP-3C reduced the level of IRF7 in a dose-dependent fashion (lanes 2 to 4). Due to a low level of IRF7 expression, the cleaved product was not detectable. However, ectopic expression of GFP-3C had no effect on endogenous of IRF3, TBK1, and β-actin. A similar phenotype was seen in HeLa cells, although the kinetics of IRF7 reduction was delayed (lanes 2 to 5, Fig. 2B). These experimental results suggest that IRF7 is a target of EV71 3C in mammalian cells.
Fig 2.
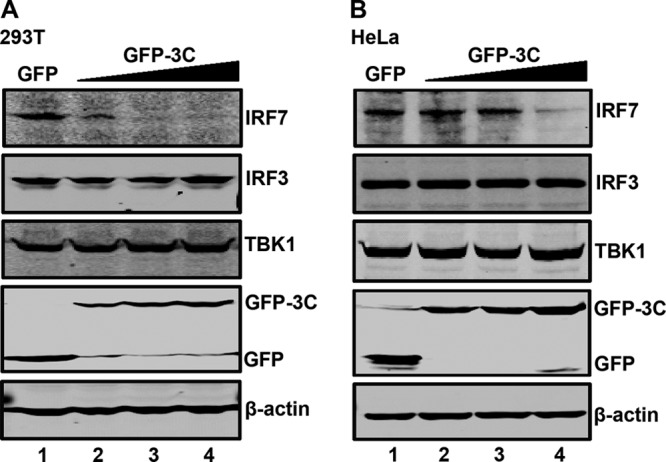
The protease 3C mediates endogenous IRF7 cleavage. 293T (A) and HeLa (B) cells were transfected with the control GFP or increasing amounts of GFP-3C. At 24 h after transfection, cell lysates were subjected to Western blot analysis with antibodies against IRF7, IRF3, TBK1, GFP, and β-actin.
The steady-state level of IRF7 is reduced in cells infected with EV71.
Based on the above analysis, we reasoned that EV71 may reduce IRF7 expression in infected cells. To test this notion, RD cells were mock infected or infected with EV71. At different time points postinfection, cells were processed for Western blot analysis. As shown in Fig. 3A, RD cells constitutively expressed IRF7, IRF3, TBK1, and β-actin (lane 1). EV71 infection had a different effect on these host proteins (lanes 2 to 5). Notably, the level of IRF7 was decreased as the virus infection progressed. This was most evident at 24 h after infection (lane 5). However, the level of IRF3 and TBK1 was marginally reduced in infected cells. Western blot analysis verified the expression of 3C, VP0, and VP2 (lanes 3 to 5). We also examined the impact of EV71 3C in 293T cells. As shown in Fig. 3B, EV71 infection initially stimulated IRF7 expression, but as the viral infection continued, the level of IRF7 was similarly decreased (lanes 1 to 5). This was paralleled with 3C expression. Little change was seen on the expression of IRF3, TBK1, and β-actin. Consistently, EV71 failed to induce IFN-β and RANTES mRNA expression in infected cells (Fig. 3C). In THP1 cells, the IRF7 protein also was decreased after EV71 infection while its mRNA was not (Fig. 3D and E). We conclude that EV71 reduces the expression of IRF7 in infected cells, which coincides with 3C expression.
Fig 3.

IRF7 is cleaved in EV71-infected cells. RD (A) and 293T (B) cells were mock infected or infected with EV71 at a multiplicity of infection (MOI) of 2. At the indicated time points, cell lysates were analyzed by Western blotting with antibodies for IRF7, IRF3, TBK1, EV71, EV71 3C, and β-actin. (C) 293T cells were mock infected or infected with EV71 (MOI = 2). At different time points after infection, total RNA extracted from cells were subjected to RT-PCR analysis for IFN-β, ISG54, ISG56, RANTES, and GAPDH mRNA. As a control, Sendai virus was used to infect cells. (D) THP1 cells were mock infected or infected with EV71 at an MOI of 10. At the indicated time points, cell lysates were analyzed by Western blotting with antibodies against IRF7, IRF3, TBK1, EV71, and β-actin. (E) THP1 cells were treated as described for panel D. Total RNAs extracted from cells were subjected to RT-PCR analysis for IRF7 and GAPDH mRNA.
The 3C protein inhibits IRF7-mediated promoter activation.
IRF7 serves as a transcription factor to activate IFN-I responses (8, 9). Since EV71 3C induced IRF7 cleavage, we evaluated whether it affected the activity of IRF7. As such, we carried out luciferase reporter assays. As shown in Fig. 4A, ectopically expressed IRF7 stimulated IFN-β promoter activation in 293T cells. Coexpression of GFP-3C inhibited IFN-β promoter activation in a dose-dependent manner. As expected, the control GFP had no effect. Consistently, GFP-3C antagonized ISRE, ISG56, and IFN-α4 promoter activation by IRF7 (Fig. 4B, C, and D). Taken in combination, these results suggest that upon expression, the 3C protein of EV71 inhibits IRF7-mediated promoter activation.
Fig 4.
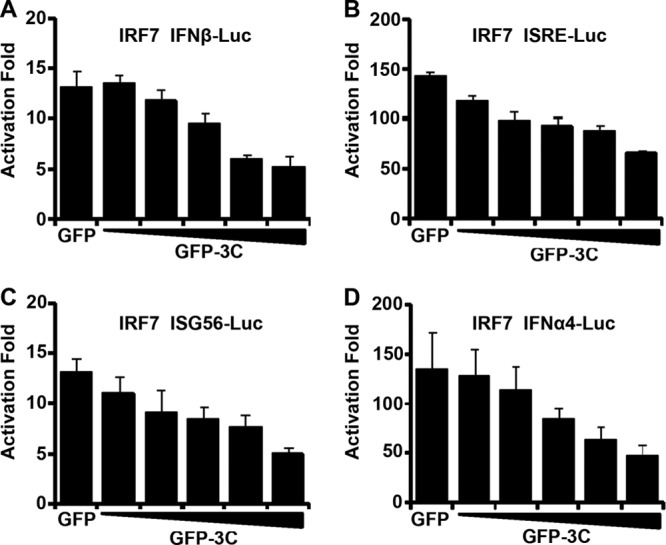
EV71 3C inhibits promoter activation by IRF7. 293T cells were transfected with IRF7 and GFP-3C along with IFN-β-Luc (A), ISRE-Luc (B), ISG56-Luc (C), and IFN-α4-Luc (D). A plasmid expressing GFP or pRL-SV40 was used as a control. At 24 h after transfection, cell lysates were assayed for luciferase activities. Data are representative of three independent experiments with triplicate samples.
The protease activity of 3C is required for cleavage and inhibition of IRF7.
To investigate the underlying events of IRF7 cleavage, we examined protein degradation pathways using specific inhibitors. As illustrated in Fig. 5A, GFP-3C mediated IRF7 cleavage, resulting in a 45-kDa protein band (lanes 2 and 3). This was blocked in the presence of rupintrivir, which is an inhibitor EV71 3C protease (lanes 5 and 6). However, the caspase inhibitor Z-VAD-FMK (Fig. 5B), the lysosome inhibitor NH4Cl (Fig. 5C), the proteasome inhibitor MG132 (Fig. 5D), or the autophagy inhibitor 3-MA (Fig. 5E) virtually had no effect on 3C-mediated IRF7 cleavage. These results suggest that the 3C protease activity is involved in IRF7 cleavage.
Fig 5.

The effect of selected inhibitors on IRF7 cleavage. 293T cells were transfected with Myc-Flag-IRF7 along with GFP or GFP-3C. At 24 h after transfection, cells were incubated with the protease inhibitor rupintrivir (2 μM) (A) (31), caspase inhibitor Z-VAD (20 μM) (B) (6, 32), lysosome inhibitor NH4Cl (10 mM) (C) (33), proteasome inhibitor MG132 (0.1 μM) (D) (33), and autophagy inhibitor 3-MA (500 μM) (E) (34) for 24 h. Cell lysates were then processed for Western blot analysis.
To further address this issue, we carried out a mutational analysis. H40D substitution in the active site of EV71 3C disrupts the protease activity, whereas R84Q or V154S substitution abolishes its RNA binding activity (2). Accordingly, we compared 3C variants in relation to IRF7. As shown in Fig. 6A, when expressed in 293T cells, wild-type 3C induced cleavage of IRF7 compared to the GFP control (lanes 1 and 2). In stark contrast, H40D failed to mediate IRF7 cleavage (lane 3). R84Q and V154S behaved like wild-type 3C (lanes 4 and 5). These variants expressed at comparable levels as measured by Western blotting (lanes 2 to 5, bottom panel). Thus, the protease activity of EV71 3C seems essential for IRF7 cleavage.
Fig 6.

(A) The effect of 3C variants on IRF7 cleavage. 293T cells were transfected with Myc-Flag-IRF7 (lanes 1 to 5), GFP, or GFP-3C variants as indicated. Cell lysates were subjected to Western blot analysis with antibodies as indicated using the LI-COR Odyssey dual-color system (LI-COR, Lincoln, NE). Antibodies recognizing Myc-Flag-IRF7 (Myc, C-terminal of IRF7, 800 nm, green; IRF7, 1 to 150 amino acids, 700 nm, red) were used. The merged images of the two channels were shown below (yellow). 3C or its variants were detected by using GFP antibody. β-Actin was included as a loading control. The effect of 3C variants on the IFN-β (B), ISRE (C), ISG56 (D), and IFN-α4 (E) promoter activation. 293T cells were transfected with plasmids encoding IRF7 and pIFN-β-Luc, pISRE-Luc, pISG56-Luc, or pIFN-α4-Luc, along with GFP or GFP-3C variants. pRL-SV40 was included as an internal control. At 24 h after transfection, cells were harvested to determine luciferase activities.
We next evaluated the effects of 3C variants on IRF7 activity in luciferase reporter assays. As illustrated in Fig. 6B, wild-type 3C, R84Q, or V154S inhibited the IFN-β promoter activation mediated by IRF7. However, H40D could not inhibit the IFN-β promoter activation. This paralleled with IRF7 cleavage. Similar phenotypes were observed for ISRE, ISG56, or IFN-α4 promoter activation (Fig. 6C, D, and E). These experimental results suggest that the protease activity of 3C is necessary to cleave and inhibit IRF7.
The Q189-S190 pair on IRF7 is necessary for 3C-mediated cleavage.
As IRF7 cleavage produced 45-kDa (C-terminal) and 25-kDa (N-terminal) products, we inferred that a cleavage site(s) may exist between amino acids 150 and 220 (Fig. 7A). This region bears several glutamines which resemble a signature Q-G sequence of proteolytic sites for enterovirus 3C. To define the putative cleavage site, we generated a series of IRF7 substitution mutants (Fig. 7B). These IRF7 mutants were expressed along with GFP or GFP-3C in 293T cells. Cell lysates were subjected to Western blot analysis. As shown in Fig. 7B, wild-type IRF7 was cleaved when coexpressed with GFP-3C, producing a 45-kDa band (Fig. 7B, lanes 1 and 2). However, the Q189A mutant is resistant to the 3C cleavage (Fig. 7B, lanes 11 and 12). In contrast, Q151A (Fig. 7B, lanes 3 and 4), Q167A (Fig. 7B, lanes 5 and 6), Q185A (Fig. 7B, lanes 7 and 8), Q188A (Fig. 7B, lanes 9 and 10), and Q215A (Fig. 7B, lanes 13 and 14) were cleaved in the presence of 3C. To further analyze IRF7 cleavage, we carried out in vitro assays. As shown in Fig. 7C, the recombinant 3C cleaved wild-type IRF7 supplied in the cell lysates, producing a 25-kDa amino-terminal fragment and a 45-kDa carboxyl-terminal fragment, respectively. However, the Q189A substitution in IRF7 abolished this activity. Furthermore, the 3C protein cleaved purified IRF7 but not IRF3 (Fig. 7D and E). These results indicate that the Q189-S190 pair in IRF7 is a cleavage site for EV71 3C.
Fig 7.
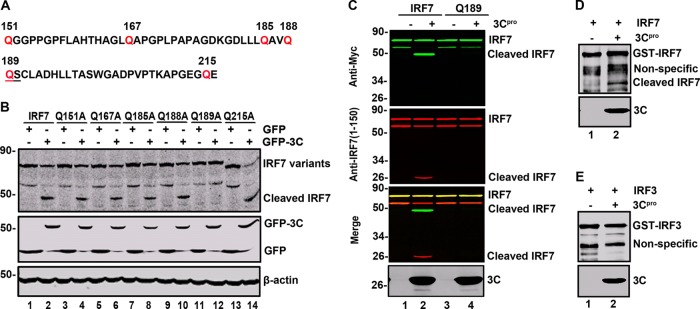
The Q189-S190 pair is the site of IRF7 cleavage. (A) Primary sequences of amino acids 151 to 216 within IRF7. In this region, glutamine was substituted with alanine. (B) IRF7 variant cleavage with substituting amino acids indicated above the lanes. 293T cells were transfected with wild-type IRF7 or IRF7 mutants along with GFP (lines 1, 3, 5, 7, 9, 11, and 13) or GFP-3C (lines 2, 4, 6, 8, 10, 12, and 14) as indicated. At 24 h after transfection, cell lysates were subjected to Western blot analysis with antibodies against Myc, GFP, and β-actin. (C) EV71 3C cleaves wild-type IRF7 but not the Q189A mutant in vitro. 293T cells were transfected with wild-type IRF7 or Q189A. At 24 h after transfection, cell lysates were incubated with the recombinant 3C protease. Samples were processed for Western blot analysis with antibodies using the LI-COR Odyssey dual-color system. Two antibodies that recognize Myc-IRF7 (Myc, C-terminal of IRF7, 800 nm, green; IRF7, 1 to 150 amino acids, 700 nm, red) were used. (D and E) EV71 3C cleaves recombinant IRF7 in vitro. The purified GST-IRF7 or GST-IRF3 was incubated with the 3C protease. Samples were analyzed by Western blotting using anti-GST antibody.
To test the consequence of IRF7 cleavage, we generated two IRF7 mutants that mimic cleaved products. As illustrated in Fig. 8A, 189N represents the amino-terminal fragment whereas 189C represents the carboxyl-terminal fragment. In reporter assays, wild-type IRF7 activated the IFN-β promoter (Fig. 8B). The addition of GFP-3C reduced its activity. However, neither 189N nor 189C was able to activate the IFN-β promoter in the presence or absence of GFP-3C. A similar phenotype was also observed for the IFN-α4 promoter activation (Fig. 8C). Western blot analysis revealed that IRF7 variants were expressed at comparable levels (Fig. 8D). Importantly, GFP-3C induced cleavage of wild-type IRF7 but had no effect on 189N or 189C. These results suggest that EV71 3C mediates IRF7 cleavage at the Q189-S190 site, resulting in two fragments which are unable to stimulate IFN expression.
Fig 8.
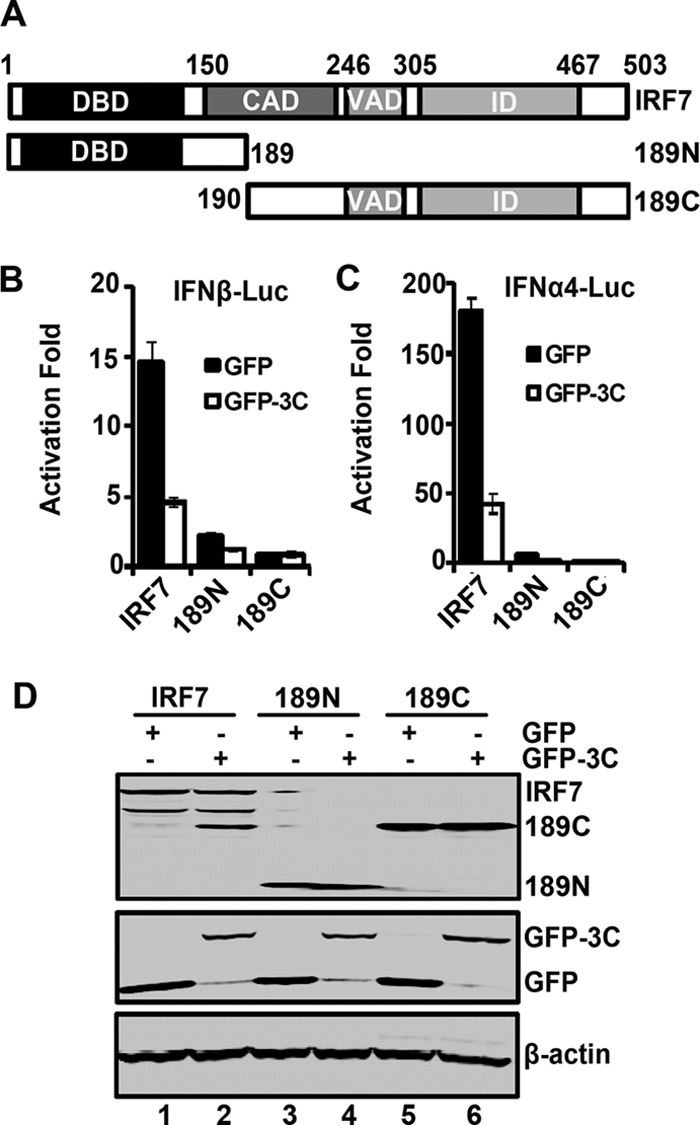
(A) Schematic diagrams of IRF7 deletion mutants. The following different domains are shown: the DNA binding domain (DBD), the constitutive activation domain (CAD), the virus-activated domain (VAD), the inhibitory domain (ID). 189N and 189C represent cleaved fragments of IRF7 by 3C. (B) The effect of 3C on IFN-β promoter activation induced by IRF7 and its mutants. 293T cells were transfected with IRF7, 189N, or 189C and GFP-3C along with IFN-β-Luc. GFP or pRL-SV40 was used as a control. At 24 h after transfection, cell lysates were assayed for luciferase activities. Data are representative of three independent experiments with triplicate samples. (C) The effect of 3C on IFN-α4 promoter activation induced by IRF7 and its mutants. 293T cells were transfected with IRF7, 189N, or 189C and GFP-3C along with IFN-α4-Luc. Cell lysates were detected as indicated in panel B. (D) Expression of IRF7 mutants. Cell lysates in panel B were detected by Western blotting with antibodies against Myc, GFP, and β-actin.
As IRF7 acts to stimulate IFN expression by forming a complex with IRF3 (19), we asked whether cleaved IRF7 fragments negatively regulated IRF3, and we carried out report assays. As illustrated in Fig. 9A, when expressed alone in 293T cells, IRF3 or IRF7 stimulated ISRE promoter activation. Coexpression of IRF3 and IRF7 had a synergistic effect. However, the addition of 189N suppressed IRF3-mediated activation whereas 189C had little effect. Western blot analysis detected the expression of IRF3, IRF7, 189N, and 189C (Fig. 9B). Interestingly, both 189N and 189C associated with IRF3. As shown in Fig. 9C, IRF7, 189N, and 189C coprecipitated with GFP-IRF3. Conversely, IRF3 was also coprecipitated with IRF7, 189N, and 189C. Apparently, 189N functioned as a dominant negative inhibitor of IRF3.
Fig 9.

(A) The effects of IRF7 variants on the IRF3 activity. 293T cells were transfected with IRF7, 189N, 189C, or GFP-IRF3 along with ISRE-Luc as indicated. pRL-SV40 was used as a control. At 24 h after transfection, cell lysates were assayed for luciferase activities. Data are a representative of three independent experiments with triplicate samples. (B) Expression of IRF7 variants and IRF3. Cell lysates in panel A were detected by Western blotting with antibodies against Myc, GFP, and β-actin. (C) IRF7 variants interact with IRF3. 293T cells were transfected with plasmids encoding GFP-IRF3 (lanes 1, 2, 3, and 4), Myc-Flag-IRF7 (lane 2), Myc-Flag-189N (lane 3), and Myc-Flag-189C (lane 4). At 24 h after transfection, cell lysates were immunoprecipitated with anti-Myc or anti-GFP antibody. Immunoprecipitates and aliquots of cell lysates were then subjected to Western blot analysis.
IRF7 but not cleaved IRF7 fragments inhibit EV71 replication.
Finally, we assessed whether IRF7 is functionally linked to EV71 infection. Specifically, RD cells were transfected with wild-type IRF7, 189N, 189C, or IRF3. At 24 h after infection, cell lysates were processed for Western blot analysis. As illustrated in Fig. 10A, in nontransfected cells, EV71 expressed VP1, indicative of viral infection. Ectopic expression of IRF7 and IRF3 reduced VP1 production. However, unlike wild-type IRF7, neither 189N nor 189C reduced the production of VP1. Notably, expression of 189N enhanced the production of VP1. Compared to the IRF7 mutants, wild-type IRF7 and IRF3 reduced VP1 production by approximately 50% (Fig. 10B), indicating that the overexpression of IRF7 or IRF3 inhibits replication of EV71. This was reversed by 189N. These phenotypes correlated with levels of VP4 mRNA as measured by PCR analysis (Fig. 10C). On the other hand, upon infection with increasing doses of EV71, cleavage of IRF7 was seen in 293T cells (Fig. 10D).
Fig 10.

(A) IRF7 inhibits EV71 replication. RD cells were transfected with IRF7 variants and IRF3 as indicated. At 24 h after transfection, cells were treated with EV71. After 24 h, cells were harvested and resolved with 12% SDS-PAGE. Western blot analysis for VP1, Myc, or β-actin was conducted. (B) Densitometry analysis. VP1 protein bands from three independent experiments in panel A were quantified and normalized to β-actin by using the Odyssey image software. (C) The effects of IRF7 on viral RNA synthesis. RD cells were transfected with IRF7, 189N, 189C, or IRF3 for 24 h. Cells were infected with EV71, and at 24 h postinfection, the total RNA extracted from the cells were analyzed for the expression of EV71 VP4 RNA. GAPDH was used as an internal control. (D) IRF7 is cleaved in EV71-infected cells. 293T cells were transfected with IRF7 for 24 h. Cells were then mock infected or infected with an increasing dose of EV71. At 24 h after infection, cell lysates were analyzed by Western blotting using antibodies against Myc, EV71, and β-actin.
DISCUSSION
Previous studies revealed that the 3C protein of EV71 suppresses antiviral immunity (6, 7). In this complex process, EV71 3C precludes the formation of RIG-I-IPS-1 complexes. Additionally, EV71 3C mediates proteolytic cleavage of TRIF in the TLR3 pathway. Although the underlying events are not fully deciphered, these results partly explain why EV71 infection downregulates IFN-I responses (6, 7, 20). Relevant to this is the finding that EV71 3C negatively regulates IRF7, a key transcription factor to activate IFN-I expression. Remarkably, EV71 3C mediates proteolytic cleavage of IRF7, inactivating IRF7. Given the importance of IRF7 in the immune system, our work suggests that the interplay of EV71 3C and IRF7 may be critical in EV71 infection or pathogenesis.
The 3C protein of EV71 is an essential factor in viral replication (2, 3). Besides viral protein processing, the 3C protein acts to inhibit antiviral immunity (6, 7). In this respect, EV71 3C seems evolved to target IRF7 as well. When expressed, it mediated the proteolytic cleavage of IRF7. This was resistant to inhibitors of caspases, proteasomes, endocytosis, and autophagy. On the other hand, this activity was sensitive to an inhibitor of the 3C protease. A logical explanation is that EV71 3C functions as a viral protease of IRF7. Three lines of evidence support this model. First, a protease dead mutant of 3C was unable to mediate IRF7 cleavage whereas mutants defective in RNA binding worked normally. This correlated well with their capacity to modulate IRF7 activity. Second, the IRF7 cleavage site resembles a signature sequence of 3C proteases encoded by picornaviruses. Third, EV71 3C was capable of cleaving purified GST-IRF7 in vitro.
As a master regulator of IFN-I expression IRF7 normally resides in the cytosol (9). Upon activation, this transcription factor migrates to the nucleus to upregulate target gene expression. The fact that EV71 3C suppresses IRF7 activity highlights a biological link between IRF7 and EV71. Consistent with this idea, EV71 infection reduced IRF7 expression. Conversely, overexpression of IRF7 decreased EV71 replication. A relevant issue is then how EV71 3C modulates IRF7. It has been reported that besides its DNA binding domain, IRF7 possesses constitutive activation, virus activation, inhibitory, and signal response domains (21). Although the crystal structure of IRF7 is not available, a coordinated interaction among these domains is believed to regulate IRF7 in host cells (21). In this context, we noted that 3C-induced IRF7 cleavage occurred within the constitutive activation domain. One possibility is that such cleavage may uncouple the DNA binding domain at the amino terminus from the central or carboxyl-terminal region. Alternatively, IRF7 cleavage may simply destroy the constitutive activation domain, resulting in nonfunctional IRF7. These models are not necessarily mutually exclusive. Work is in progress to test these possibilities.
Our results suggest that the Q189-S190 pair within IRF7 represents a likely EV71 3C cleavage site. When expressed in mammalian cells, EV71 3C induced a 45-kDa protein band which represents the carboxyl terminus of IRF7. Among six potential cleavage sites in the region spanning amino acids 151 to 216, the Q189-S190 pair was affected by alanine substitution that blocked IRF7 cleavage. Previous studies suggest that 3C of picornaviruses typically cleaves at the Q-G pair (22, 23). Alternative sites, such as the Q-S pair, exist (22, 23). We observed that mutations in the additional potential sites, as represented by Q151A, Q167A, Q185A, Q188A, and Q215A substitutions, produced no visible effect on IRF7 cleavage. Therefore, IRF7 seems to have a single cleavage site. Intriguingly, unlike wild-type IRF7, mutants that mimic cleaved IRF7 failed to activate the IFN promoter. In addition, the cleaved IRF7 acted as a dominant inhibitor of IRF3, facilitating EV71 infection. These results support the notion that cleavage at the Q189-S190 pair by EV71 3C is an effective way to interfere with IRF7 and IRF3.
EV71 is a causative agent of hand, foot, and mouth disease and may cause neurological diseases in young children (1). While the underlying mechanisms are unclear, immature or impaired immunity is believed to contribute to viral pathogenesis (24–27). In animal models, EV71 stimulates the production of inflammatory cytokines but not IFN-I (20, 28). Concordant with this, we reported that EV71 inhibits IFN-I induction in infected cells by disrupting the RIG-I-IPS-1 complex (7). Additionally, EV71 3C inhibits the TLR3 pathway in infected cells through TRIF cleavage (6). Interestingly, a recent study shows that EV71 3C mediates IRF9 cleavage (29). The data present in this study further suggest that EV71 3C targets IRF7. Considering various functions of these host components in immunity, we suspect that EV71 3C may act as a virulence factor in vivo. Nonetheless, it is noteworthy that the 2A protease of EV71 blocks IFN signaling by reducing the level of interferon receptor I (30). These viral proteins may act cooperatively to facilitate viral pathogenesis. Additional work is required to address this issue.
ACKNOWLEDGMENTS
We thank Sheng Cui and Bei Wang for providing recombinant EV71 3C protease and technical assistance.
This work was supported by grants from the National Basic Research Program of China (973 Project; 2011CB504903), the New Century Program for Outstanding Young Talents, Chinese Ministry of Education (NCET-07-0506), the Institute of Pathogen Biology, Chinese Academy of Medical Sciences (2009IPB112), the National Natural Science Foundation of China (31270200), and the National Institute of Allergy and Infectious Diseases of the United States (AI092230).
Footnotes
Published ahead of print 21 November 2012
REFERENCES
- 1. McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev. 26:91–107 [DOI] [PubMed] [Google Scholar]
- 2. Shih SR, Chiang C, Chen TC, Wu CN, Hsu JT, Lee JC, Hwang MJ, Li ML, Chen GW, Ho MS. 2004. Mutations at KFRDI and VGK domains of enterovirus 71 3C protease affect its RNA binding and proteolytic activities. J. Biomed. Sci. 11:239–248 [DOI] [PubMed] [Google Scholar]
- 3. Sim AC, Luhur A, Tan TM, Chow VT, Poh CL. 2005. RNA interference against enterovirus 71 infection. Virology 341:72–79 [DOI] [PubMed] [Google Scholar]
- 4. Li ML, Hsu TA, Chen TC, Chang SC, Lee JC, Chen CC, Stollar V, Shih SR. 2002. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 293:386–395 [DOI] [PubMed] [Google Scholar]
- 5. Weng KF, Li ML, Huang CT, Shih SR. 2009. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 5:e1000593 doi:10.1371/journal.ppat.1000593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85:8811–8818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lei XB, Liu XL, Ma YJ, Sun ZM, Yang YW, Jin Q, He B, Wang JW. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84:8051–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 9. Ning S, Pagano JS, Barber GN. 2011. IRF7: activation, regulation, modification and function. Genes Immun. 12:399–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U. S. A. 103:8459–8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105 [DOI] [PubMed] [Google Scholar]
- 12. Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, Akira S, Mechti N. 2009. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393:311–318 [DOI] [PubMed] [Google Scholar]
- 13. Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, Yanai H, Seko Y, Shitara H, Bishop K, Yonekawa H, Tamura T, Kaisho T, Taya C, Taniguchi T, Honda K. 2008. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A. 105:20446–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richer MJ, Lavallée DJ, Shanina I, Horwitz MS. 2009. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One 4:e4127 doi:10.1371/journal.pone.0004127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J, Zhao Y, McHenry CL, Burgens RV, Miller DJ, Sajjan U, Hershenson MB. 2009. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 183:6989–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, Racaniello VR. 2007. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 81:3677–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cui S, Wang J, Fan T, Qin B, Guo L, Lei X, Wang M, Jin Q. 2011. Crystal structure of human enterovirus 71 3C protease. J. Mol. Biol. 408:449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Au WC, Yeow WS, Pitha PM. 2001. Analysis of functional domains of interferon regulatory factor 7 and its association with IRF-3. Virology 280:273–282 [DOI] [PubMed] [Google Scholar]
- 20. Liu ML, Lee YP, Wang YF, Lei HY, Liu CC, Wang SM, Su IJ, Wang JR, Yeh TM, Chen SH, Yu CK. 2005. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 86:3263–3269 [DOI] [PubMed] [Google Scholar]
- 21. Lin R, Genin P, Mamane Y, Hiscott J. 2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 20:6342–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kundu P, Raychaudhuri S, Tsai W, Dasgupta A. 2005. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease-mediated cleavage of the TATA-binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J. Virol. 79:9702–9713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parks GD, Baker JC, Palmenberg AC. 1989. Proteolytic cleavage of encephalomyocarditis virus capsid region substrates by precursors to the 3C enzyme. J. Virol. 63:1054–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang LY, Huang LM, Gau SS, Wu YY, Hsia SH, Fan TY, Lin KL, Huang YC, Lu CY, Lin TY. 2007. Neurodevelopment and cognition in children after enterovirus 71 infection. N. Engl. J. Med. 356:1226–1234 [DOI] [PubMed] [Google Scholar]
- 25. Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shih SR. 1999. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N. Engl. J. Med. 341:929–935 [DOI] [PubMed] [Google Scholar]
- 26. Huang CC, Liu CC, Chang YC, Chen CY, Wang ST, Yeh TF. 1999. Neurologic complications in children with enterovirus 71 infection. N. Engl. J. Med. 341:936–942 [DOI] [PubMed] [Google Scholar]
- 27. Lin YW, Chang KC, Kao CM, Chang SP, Tung YY, Chen SH. 2009. Lymphocyte and antibody responses reduce enterovirus 71 lethality in mice by decreasing tissue viral loads. J. Virol. 83:6477–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khong WX, Foo DG, Trasti SL, Tan EL, Alonso S. 2011. Sustained high levels of IL-6 contribute to the pathogenesis of Enterovirus 71 in a neonate mouse model. J. Virol. 85:3067–3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP, Hsu JT. 2011. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 203:1784–1790 [DOI] [PubMed] [Google Scholar]
- 30. Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He M. 2012. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 86:3767–3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, Wang M, Jin Q, Cui S. 2011. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J. Virol. 85:10021–10030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rebsamen M, Meylan E, Curran J, Tschopp J. 2008. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 15:1804–1811 [DOI] [PubMed] [Google Scholar]
- 33. Xue Q, Zhou Z, Lei X, Liu X, He B, Wang J, Hung T. 2012. TRIM38 negatively regulates TLR3-mediated IFN-β signaling by targeting TRIF for degradation. PLoS One 7:e46825 doi:10.1371/journal.pone.0046825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. 2009. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 16:761–771 [DOI] [PubMed] [Google Scholar]