

Abstract
The hepatitis B virus (HBV) surface proteins not only are incorporated into the virion envelope but in addition form subviral particles (SVP) consisting solely of surface proteins and lipids. Heterologous expression of the small HBV envelope protein S produces secreted spherical SVP 20 nm in diameter, with approximately 100 S molecules per particle. The pathway leading from the initial S translation product as a multispanning transmembrane protein to the final SVP is largely unknown. To investigate the role of the four transmembrane domains (TM) of S in this process, we introduced mutations in these regions and characterized their effects on SVP formation in transfected Huh7 cells. We found that the insertion of one amino acid in the center of the α-helix of TM1 or the exchange of TM1 with a heterologous TM blocked SVP release and SVP formation by coexpressed wild-type S chains in a transdominant negative fashion. Surprisingly, this effect was partially neutralized when the mutations were expressed in the background of the HBV surface protein M, suggesting that mutations in TM1 could partially be complemented by the pre-S2 domain. The exchange of TM2 with heterologous TMs that form α-helices of the same lengths was also incompatible with SVP formation. However, these mutants no longer blocked SVP formation by coexpressed wild-type S. We conclude that TM2 is essential for the stable assembly of S chains by establishing intramembrane interactions.
INTRODUCTION
Hepatitis B virus (HBV) causes chronic infections of the liver in more than 350 million people worldwide and an increased risk for the development of hepatocellular carcinoma in these individuals (1). Today's options for the treatment of persistent HBV infections are unsatisfying. Therefore, the prevention of HBV infections is particularly important and can be achieved by active vaccination. The most common vaccine consists of the major HBV envelope protein S expressed in yeast. This protein is peculiar as it not only is incorporated into the envelope of virions which have a diameter of 42 nm but, together with host lipids, also assembles into spherical or filamentous lipoprotein particles of 20 nm in diameter (2). Such subviral particles (SVP) are secreted in great excess over virions from infected hepatocytes. Spherical SVP containing approximately 100 protein molecules are also formed by cells expressing the S protein in the absence of any other HBV-encoded protein and represent the antigenic component of HBV vaccines.
The gene encoding the S protein constitutes the 3′ end of a longer continuous open reading frame that is divided into the regions pre-S1, pre-S2, and S. Two further HBV envelope proteins, M and L, are encoded by the pre-S2 plus S and by the pre-S1 plus pre-S2 plus S sequences, respectively. Therefore, all three proteins share the S domain at their C termini. Virion envelopes contain the three proteins S, M, and L at a ratio of approximately 4:1:1. Natural spherical SVP contain mainly S proteins and some M proteins but only small amounts of L, while filamentous SVP carry more L proteins (3).
Our knowledge of the pathway leading from S protein synthesis to SVP formation is fragmentary. First, the 226-amino-acid (aa)-long S protein is translated as a transmembrane protein at the rough endoplasmic reticulum (ER). Its transmembrane topology is determined by an N-terminal type I signal which is not cleaved by signal peptidase and causes the translocation of the N terminus across the ER membrane (Fig. 1A). The hydrophobic region of this signal forms the first transmembrane domain (TM1, aa 8 to 23) of the S protein. In addition the protein carries a central type II signal causing the anchorage of the peptide chain in the membrane by the second transmembrane domain, TM2 (aa 80 to 98), and the translocation of downstream parts of the protein into the ER lumen (4). The C-terminal part of S (aa 179 to 226) is relatively hydrophobic and is assumed to be inserted in the ER membrane in most of the existing models, with some experiments supporting this assumption (5). The S chain is cotranslationally modified by partial N-linked glycosylation at asparagine residue 146 and by disulfide bridge formation. However, neither N-glycosylation (6) nor S-S bridge formation (7) is required for SVP formation and release. Early disulfide bridge formation leads to covalently linked S homodimers (7).
Fig 1.
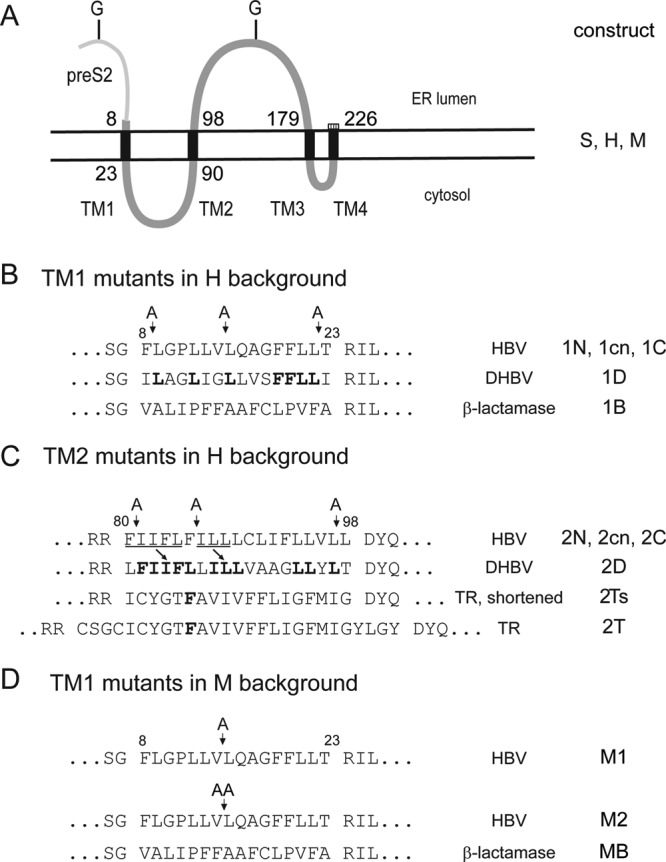
Derivatives of the S and M envelope proteins used in this work. (A) Transmembrane topologies of the S (thick gray line) and M (thin plus thick gray line) proteins in the ER membrane (double horizontal lines). The numbers denote the suggested N- and C-terminal amino acid positions of the transmembrane domains (TM1 to -4). H, S derivative carrying a C-terminal addition of 11 aa (hatched box). All S mutants except mutant 2cn were generated in the H background. G, N-glycosylation sites. (B) Mutations in TM1 in the H background. The 1N (N-terminal), 1cn (central), and 1C (C-terminal) mutants carry one additional alanine residue at the positions indicated by arrows. 1D has a substitution of TM1 with the corresponding TM1 sequence of the DHBV S protein. Mutant 1B bears a substitution of TM1 with the type I secretion signal of β-lactamase. Amino acid residues identical in the TM1 of DHBV and HBV are in boldface. (C) Mutations in TM2 in the H background (except mutant 2cn). 2N, 2cn, and 2C carry one additional alanine residue at the positions indicated by arrows. 2cn is in the S background. 2D contains a substitution of TM2 with the homologous region from the DHBV S protein. 2T contains a substitution of TM2 with the type II signal of the human transferrin receptor. 2Ts is the same as 2T except that the four N- and four C-terminal amino acids of the transmembrane domain have been removed, such that the length of the replacement transmembrane domain is identical to the length of TM2. The N-terminal half of DHBV TM2 has high homology to the N-terminal half of HBV TM2 shifted by one amino acid (arrows). (D) Mutations in TM1 in the M background. Numbers above the sequence in panels B, C, and D indicate amino acid positions in the S protein or S domain.
How the S proteins mature further on and especially how approximately 100 S proteins assemble together with lipids and bud from the host membrane to form a soluble SVP in the luminal compartment of the secretory pathway of the cell are unknown. In recent years it has become more and more evident that transmembrane domains can serve as major initiators of protein-protein interactions (8, 9). We investigated the role of the TMs of S in SVP biogenesis by introducing mutations in these regions or replacing them with foreign transmembrane domains and by characterizing these S variants in transiently transfected cells. The aim of this work was to clarify whether the transmembrane domains of S are merely components of signal sequences determining the transmembrane topology of the peptide chain or whether they have additional functions during SVP morphogenesis, e.g., by establishing lateral interactions in the membrane bilayer.
MATERIALS AND METHODS
Plasmids.
All plasmids for the expression of S variants were derived from plasmid pSVBX24H (10), which contains the S gene (HBsAg subtype adw2) of a genotype A HBV strain (11) under the transcriptional control of the simian virus 40 early promoter. Plasmids for the expression of M variants were derived from plasmid pSV33H (12), corresponding to plasmid pSVBX24H but containing in addition the pre-S2 region of the same HBV strain in front of the S gene. For expression of variant H, 11 codons encoding the influenza virus hemagglutinin tag YPYDVPDYASL were inserted between the last codon of the S gene and the stop codon using PCRs. Alanine insertions into TM1 and TM2 (Fig. 1B and C) were generated by using the Transformer site-directed mutagenesis kit (Clontech, Saint-Germain-en-Laye, France). Substitutions of TM1 or TM2 were produced by overlapping PCR. Those parts of all constructs generated in vitro by PCR or by primer elongation were sequenced to exclude unintentional mutations.
Cells, transfection, Western blotting, and HBsAg assay.
Huh7 cells were transiently transfected with Fugene 6/HD/X-treme (Roche, Penzberg, Germany) in 6-well plates using 1 μg of plasmid DNA per well. At 2 days posttransfection, the culture supernatant was harvested and centrifuged for 10 min at 13,000 rpm, and 38 μl of the supernatant was reduced and denatured with dithiothreitol and sodium dodecyl sulfate and used for polyacrylamide gel electrophoresis. Cells were washed once with 2 ml of phosphate-buffered saline and lysed by incubation for 10 min with 0.5 ml of 50 mM Tris-Cl (pH 7.5)–100 mM NaCl–20 mM EDTA–0.5% (vol/vol) Nonidet P-40 on ice. The lysate was collected and centrifuged for 10 min at 13,000 rpm, and 30 μl of the supernatant was used for polyacrylamide gel electrophoresis after reduction and denaturation. When samples from lysates and culture supernatants from one transfection were loaded side by side on one gel (see Fig. 3B and 5A and B), 19 μl of lysate and 70 μl of culture supernatant were used for comparison. After electrophoresis, proteins were transferred onto nitrocellulose membranes by blotting and detected using the monoclonal antibody HB1 against S (kindly provided by Aurelia Zvirbliene, Vilnius University, Lithuania), which detects a linear epitope at aa 120 to 125.
Fig 3.
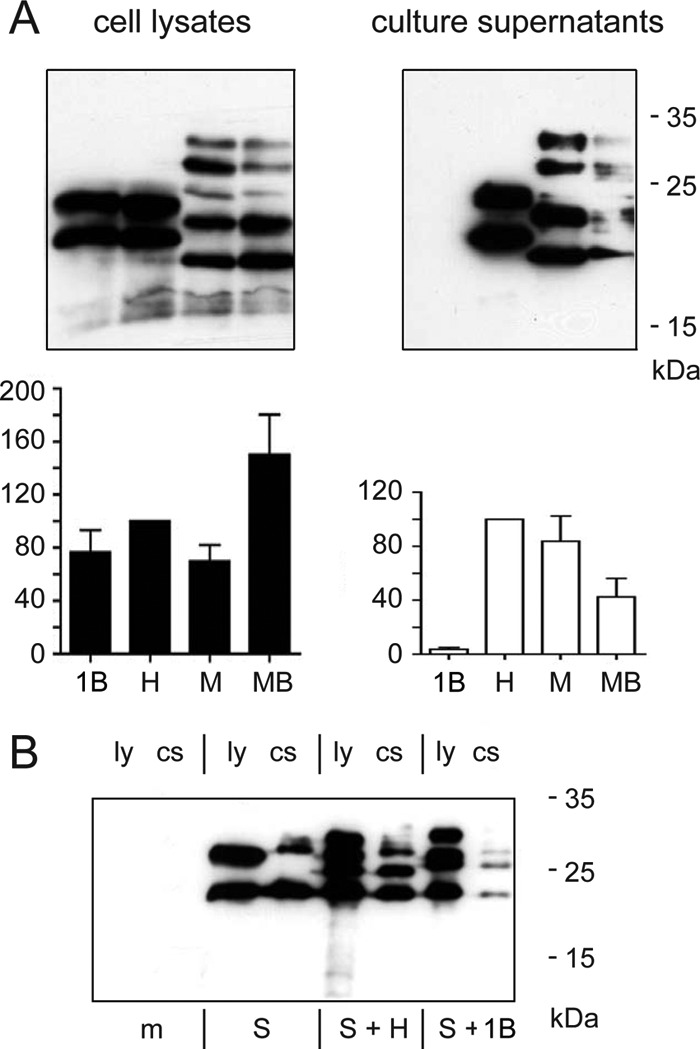
Effects of substitutions of TM1. (A) Substitution of TM1 with the type I signal from β-lactamase in the H background (mutant 1B) and M background (mutant MB). (B) Marked transdominant negative effect of mutant 1B on the release of coexpressed S protein. ly, cell lysate; cs, culture supernatant; m, mock transfected.
Fig 5.
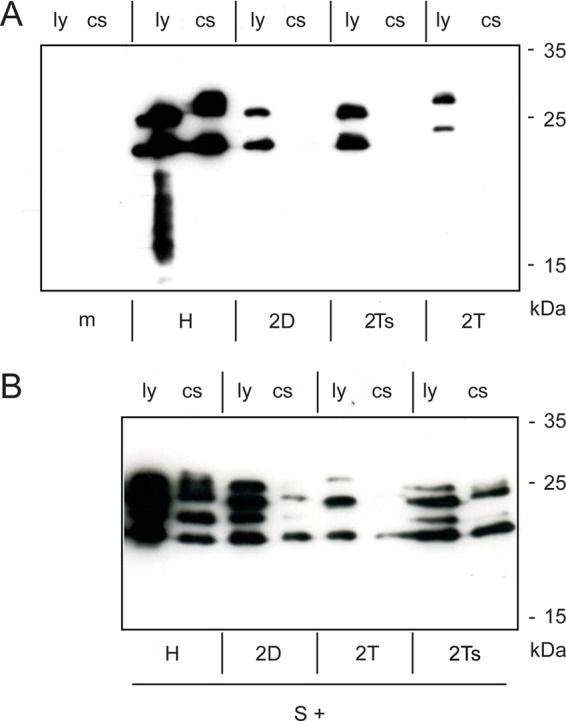
Effects of substitutions of TM2. (A) Phenotypic characterization of mutants with substitutions of TM2 with the homologous region from DHBV (2D), with the type II translocation signal of the human transferrin receptor (2T), or with a shortened version of this signal (2Ts). (B) No or weak transdominant negative effect on the release of coexpressed S protein. ly, cell lysate; cs, culture supernatant.
HBsAg was measured using the Axsym HBsAg kit from Abbott (Wiesbaden, Germany). The signal from the mock control was subtracted from the signals from the sample and from the wild-type control. The signal of the sample was then expressed as percentage of that of the wild-type control. The experiments were repeated four times. Standard deviations were calculated. The Western blots shown in the figures are representatives from one out of the four repeated experiments.
RESULTS
All mutations (except that for mutant 2cn) were introduced in a derivative of S, variant H, that carries 11 additional amino acids at the C terminus (Fig. 1A). This addition did not alter the behavior of H relative to S with respect to SVP formation and release (compare, e.g., Fig. 2A, lanes H, with 2C, left lanes; see also reference 13). The increase in molecular mass of approximately 2 kDa allowed distinction of the H protein with the mutation of interest from the smaller wild-type S protein on Western blots in cotransfection experiments. The S-derived proteins were detected by Western blotting using the monoclonal antibody HB1 against a linear epitope at aa 120 to 125. We presume that the appearance of S mutants in the culture supernatant of transfected cells is indicative of SVP formation and release, although the particulate nature of the protein/lipid complex is formally not proven by Western blotting of the material directly from the culture supernatant.
Fig 2.
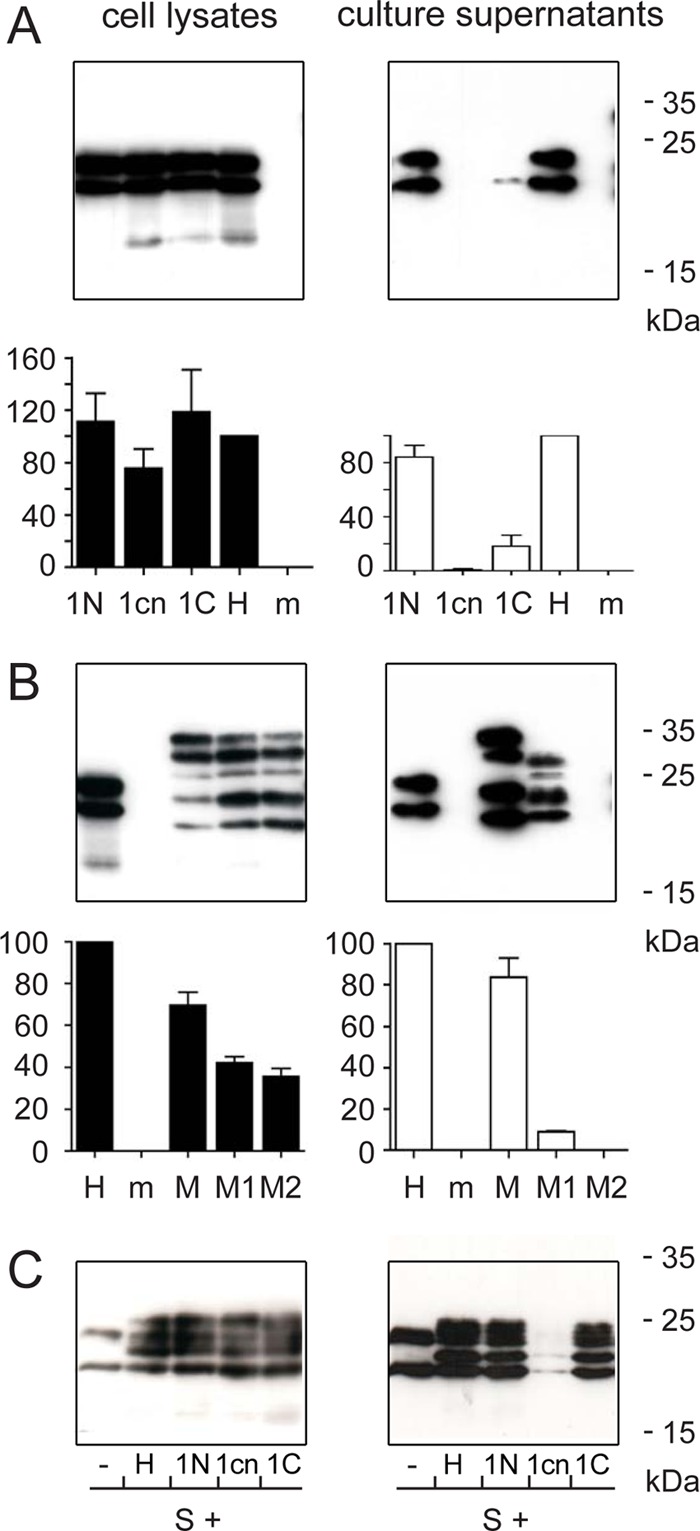
Effects of insertions in TM1. Huh7 cells were transiently transfected with the indicated constructs (Fig. 1) or mock transfected (m). HBsAg levels from cell lysates (left panels) and culture supernatants (right panels) are indicated as percentage of the S protein with hemagglutinin (HA) tag (H) (standard deviations from 4 experiments are indicated). Western blots from cell lysates and culture supernatants from one of the four experiments are shown. (A) Effect of insertion of one amino acid in TM1 on intracellular expression levels and protein release into the culture supernatant. (B) Phenotypic characterization of mutants M1 and M2. (C) Transdominant negative effect of the central insertion in TM1 on the release of coexpressed S protein. Numbers at the right indicate the positions and molecular masses of size marker proteins.
Mutations in transmembrane domain 1.
First, we introduced single alanine residues at an N-terminal, central, or C-terminal position of TM1 (mutants 1N, 1cn, and 1C, respectively) (Fig. 1B). We assume that the domain most probably forms an alpha helix and that insertions would cause a rotation of the part of the helix upstream of the insertion and of the short N-terminal sequence from aa 1 to 7 located in the ER lumen relative to the downstream portion of the protein. A sequence-specific interaction of TM1 with other transmembrane domains important for SVP maturation could potentially be disrupted by this manipulation. All three mutants and the reference H were expressed at similar levels and were produced as unglycosylated and N-glycosylated versions at a ratio of approximately 1:1 (Fig. 2A, upper left panel). Apparently, the mutations altered neither protein stability nor membrane insertion as indicated by the N-glycosylation. The N-terminal insertion 1N had no influence on SVP formation (compare lanes 1N and H in Fig. 2A, upper right panel), whereas the central insertion 1cn strongly blocked and the C-terminal insertion 1C partially blocked SVP release. The results of the Western blotting were supported by measurements of HBsAg levels in cell lysates and supernatants (Fig. 2A, lower panels).
To control whether the 1cn mutation, which blocked SVP release to undetectable levels, still supported the translocation function of signal I, we expressed this mutation in the context of the HBV M protein (mutant M1). Since the pre-S2 start codon is a weak codon for translational initiation (14), a corresponding S derivative was coexpressed from the mRNA encoding the M protein. The N-terminal pre-S2 domain of M is translocated into the ER lumen by signal I in its S domain (15), and during this translocation the N-glycosylation site at asparagine 4 of pre-S2 is modified. Therefore, a double N-glycosylated M version modified at asparagine 4 of pre-S2 and at asparagine 146 in the S domain is indicative of pre-S2 translocation across the ER membrane. Expression of the M protein resulted in five bands detected by the antibody HB1 (Fig. 2B, upper left panel, lane M): the two lower bands correspond to the coexpressed unglycosylated and N-glycosylated S protein, and the three upper bands represent the unglycosylated, monoglycosylated, and double N-glycosylated M protein. The mutant M1 also showed the same pattern, specifically including a band with a mass of approximately 36 kDa, similar to that of wild-type M protein. This band represents the double N-glycosylated protein and indicates pre-S2 translocation. Surprisingly, in the M context the central insertion in TM1 (mutant M1) allowed reduced but well-detectable formation of SVP containing both M and S derivatives (Fig. 2B, upper right panel), although the protein pattern was abnormal. We have no explanation for this behavior. Insertion of two alanines in the central position of TM1 in the M background (mutant M2), however, strongly blocked SVP release.
The importance of TM1 for SVP formation is supported by the behavior of mutant 1cn during coexpression with wild-type S protein (Fig. 2C). Coexpression of variant H, mutant 1N, or mutant 1C with S allowed the efficient release of all proteins, most probably as phenotypically mixed SVP. The presence of mutant 1cn, however, had a pronounced transdominant negative effect on S release. The most straightforward interpretation of this observation is that mutant 1cn and wild-type S form stable multimers and that the altered TM1 domains in these complexes freeze SVP biogenesis at a certain step. In the case of mixed SVP formed by mutant 1C plus coexpressed wild-type S, the efficient release of particles did not require a complete set of fully functional TM1 domains. Rather, it was apparently sufficient that approximately half of the TM1 domains were wild type and half carried the 1C mutations.
Next, we replaced TM1 with the homologous sequence of the duck hepatitis B virus (DHBV) S protein (Fig. 1B, mutant 1D) and with the hydrophobic sequence of the type I signal of β-lactamase (mutant 1B). Although in the DHBV-derived sequence, 7 out of 16 amino acid positions are occupied by the same amino acid in the HBV sequence, the chimeric protein could not be detected by Western blotting or by HBsAg enzyme-linked immunosorbent assay (ELISA) (data not shown). Apparently this chimeric protein was very unstable, for unknown reasons. Replacement of TM1 by the totally unrelated signal sequence derived from the β-lactamase (mutant 1B) generated a stable protein (Fig. 3A, left panels) which, however, was unable to generate detectable amounts of secreted SVP (Fig. 3A, right panels). Pre-S2 translocation by the altered signal I was functional as demonstrated by the double N-glycosylated version of the corresponding M derivative MB. Again, in the M context this mutation supported the appearance of the M and S mutants in the culture supernatant, although with reduced levels and additional anti-HBs-reactive bands. Apparently, the pre-S2 domain in the M derivative could complement a function of TM1 required during SVP formation. For unknown reasons, the intracellular HBsAg levels of the mutant MB were higher than those of the wild type (approximately 150%). This, however, was probably not the reason why mutant MB could be detected in the culture supernatant in contrast to mutant 1B. If the efficiency of SVP formation for mutant 1B were similar to that for mutant MB, the level of HBsAg for mutant 1B in the culture supernatant should be approximately 20% of the wild-type level. Such a signal would be clearly detectable.
Again, coexpression of mutant 1B with wild-type S protein had a transdominant negative effect on the levels of S released into the culture supernatant (Fig. 3B), which is in accordance with the behavior of mutant 1cn (Fig. 2C).
Mutations in transmembrane domain 2.
Insertions of one amino acid in an N-terminal (mutant 2N), central (mutant 2cn), or C-terminal (mutant 2C) position of TM2 (Fig. 1C) allowed stable protein expression and translocation of downstream domains as indicated by N-glycosylation of asparagine 146 (Fig. 4A, upper left panel). However, all three mutants were deficient in SVP formation (Fig. 4A, right panels) and blocked release of coexpressed S (or H) protein in a transdominant fashion (Fig. 4B). In the primary translation product of these mutants, the protein part C terminal of TM2 (aa 99 to 226) is expected to be rotated relative to the part N terminal of TM2 (aa 1 to 79) by 100°. This causes a relatively drastic alteration of the orientation of the cytoplasmic part of the protein relative to the luminal part and hydrophobic C terminus. Apparently, such an alteration was not compatible with SVP release.
Fig 4.
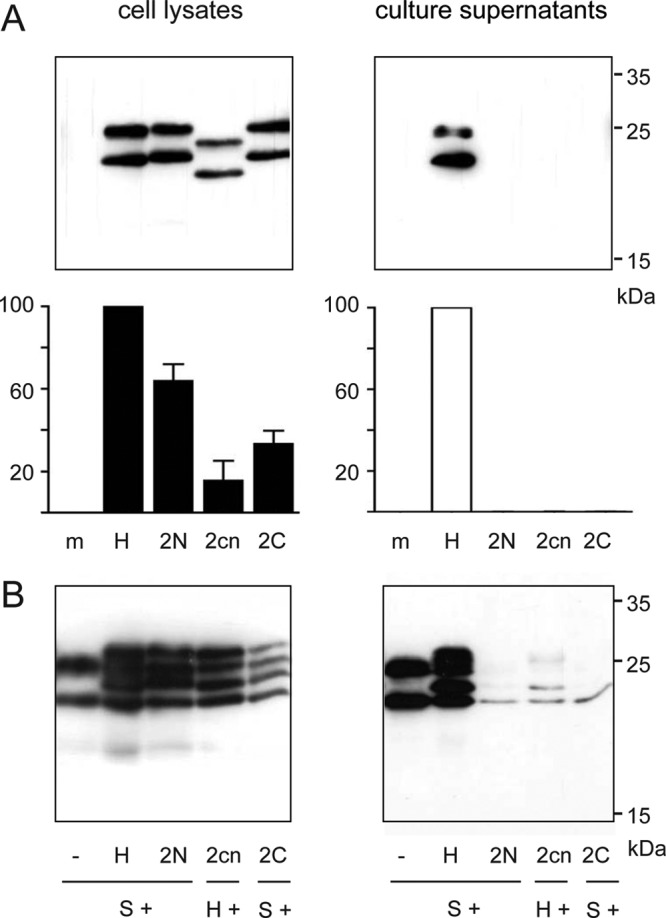
Effects of insertions in TM2. (A) Phenotypic characterization of the indicated mutants. (B) Transdominant negative effect on the release of coexpressed S protein. Mutant 2cn lacks the C-terminal 11-aa-long tag present in mutants 2N and 2C and therefore was coexpressed with variant H instead of wild-type S.
Replacement of TM2 by the homologous sequence from the DHBV S protein (mutant 2D) or by the transmembrane domain of the type II signal derived from the human transferrin receptor either in its original length (27 aa, mutant 2T) or shortened to the length of TM2 (19 aa, mutant 2Ts) resulted in stable proteins and translocation of the luminal loop, but none of the mutants were able to form secreted SVP (Fig. 5A). During coexpression with wild-type S protein, these mutants showed only a minor negative effect on the formation of SVP consisting mainly of wild-type S proteins (Fig. 5B). Apparently, the mutants did not form stable multimers with S, which is a prerequisite for retention of wild-type S within the cell. Rather, complexes of approximately 100 wild-type S chains could assemble, excluding the mutant proteins, and form nearly pure wild-type SVP. We conclude that TM2 is required as a component of the type II signal in the S protein and as a membrane anchor and that the authentic TM2 sequence is essential for the formation of stable multimers during SVP morphogenesis.
The exchange of aa 179 to 226 containing TM3 and TM4 with the homologous region (aa 125 to 167) of the DHBV S protein created a chimera which was stably expressed and secreted, although very inefficiently. However, this variant was cosecreted with coexpressed wild-type S (data not shown). Previous work showed that an S mutant lacking the hydrophobic C terminus behaved similarly (16). Apparently, this part of S is not essential for a stable interaction with S proteins and a complete set of these domains is not necessary for the biogenesis of SVP. However, a certain fraction of S chains containing the correct C-terminal domain was required for efficient particle formation.
DISCUSSION
Direct intra- and intermolecular interactions during the folding of transmembrane S proteins shortly after synthesis at the ER are constrained to domains residing in the same of three separate compartments: cytoplasm, lumen of the ER, and ER membrane. Interactions among cytoplasmic domains of the transmembrane S proteins have not yet been shown. However, interactions of luminal domains of the HBV S protein by intra- and intermolecular disulfide bridges and the formation of the conformational main “a” epitope of the hepatitis B surface antigen are well documented. On the basis of the results in this work, we propose that the transmembrane domains of S also contribute to the maturation of the protein by interactions within the membrane bilayer.
The amino acid sequence of TM1 seems to be less crucial for SVP biogenesis, as clearly demonstrated by the fact that its replacement by a totally unrelated TM from β-lactamase in the background of the M protein allowed the proteins to appear in the culture supernatant. However, mutagenesis of three hydrophobic clusters of amino acids in TM1 demonstrated that these residues were critical for S protein expression as well as for infectivity in the background of the L protein (17). On the other hand, a chimera consisting of an N-terminal hepatitis C virus E1 domain, including its transmembrane region, fused to the portion of the HBV S protein downstream of TM1 also was able to be cosecreted with wild-type S protein as SVP (18). In this construct the hepatitis C virus-derived TM replaced TM1. However, SVP formation by this chimera without coexpression of wild-type S was not possible. Berkower and coworkers (19) reported that an S chimera carrying a replacement of TM1 with the transmembrane domain of gp41 from human immunodeficiency virus type 1 was able to form SVP. However, this construct was expressed in insect cells, and SVP are not released into the culture supernatant by these cells. Rather, particles were harvested after cell lysis with detergent and sonication for analysis. Therefore, a deduction of the role of TM1 in SVP formation is limited in this experimental system. TM1 of the DHBV S protein has also been shown to play a role in SVP maturation (20).
The elongation of TM1 by one amino acid and the induced 100° rotation of aa 1 to 8 relative to TM1 was compatible with SVP biogenesis as demonstrated by mutant 1N. A 100° rotation of the N-terminal half of TM1 together with aa 1 to 7 in the luminal compartment of mutant 1cn allowed stable expression and translocation and also disulfide-linked dimer formation (data not shown), but later steps in SVP maturation were blocked. This supports the model that TM1 has further tasks during SVP morphogenesis in addition to its topological function. An initial 100° rotation of the whole transmembrane helix of TM1 relative to the rest of the protein chain was at least partially compatible with SVP formation as shown by mutant 1C (Fig. 2A). Possibly, a conformational change in the cytoplasmic loop between TM1 and TM2 can abrogate the displacement of TM1 and adjust its location in the membrane. In fact, the cytoplasmic loop between TM1 and TM2 seems to be relatively flexible, since it tolerated relatively drastic changes such as insertions of up to 40 aa without blocking SVP formation (V. Bruss, unpublished observation).
Probably the most significant result of this study can be deduced from the phenotype of mutants 2D and 2Ts (Fig. 5B), which carry a foreign transmembrane domain from the DHBV small surface protein and from the human transferrin receptor, respectively, instead of TM2. These mutants were quite strongly excluded from SVP formation by coexpressed wild-type S protein. It is very likely that all three TMs from the human and avian viruses and from the transferrin receptor form straight α-helices with identical lengths, implying that folding and location of the cytoplasmic and luminal parts of the amino acid chains are very similar if not identical to those of wild-type S in the initial translation product. The fact that 2D, 2T, and 2Ts were not or were only very inefficiently incorporated into secreted SVP when coexpressed with wild-type S protein implies that the authentic sequence of TM2 was essential for stable interaction of the peptide chain with other S molecules. At least two different plausible mechanisms could explain this behavior: (i) TM2 establishes intramolecular interactions with other TMs of the same peptide chain and these interactions are important, e.g., for the folding of other parts of the molecule so that the protein can gain competence for the intermolecular interaction with other S chains, or (ii) the mutants with TM2 substitution fold properly, but the wild-type TM2 is essential for the intermolecular interaction with other S proteins. To date, these alternatives cannot be discriminated. However, the observations that TM1 seems to be less important and TM3 and TM4 can even be deleted without blocking cosecretion with wild-type S (16) favor the second alternative.
In a model for the arrangement of HBV envelope proteins in filamentous SVP deduced from electron cryomicroscopy, the proteins form tetramers and their TMs are densely packed in a middle ring of approximately 3 nm in thickness around the helical axis of the filament (21). This size corresponds to the thickness of a lipid bilayer, and the fraction of 21% of low-density areas in the model fits well to the low content of 25% (wt/wt) of lipids in SVP (22). This tight packaging of TMs would also suggest that these domains interact with each other.
ACKNOWLEDGMENTS
We thank Ruth Brack-Werner for reading of the manuscript and helpful discussions. The monoclonal anti-HBs antibody HB1 was kindly provided by Aurelia Zvirbliene, Department of Immunology and Cell Biology, Vilnius University, Lithuania. We thank Raindy Tedjokusumo, Theresa Asen, and Andrea Weicht for help with the HBsAg assays.
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118–1129 [DOI] [PubMed] [Google Scholar]
- 2. Bruss V. 2007. Hepatitis B virus morphogenesis. World J. Gastroenterol. 13:65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. 1984. Large surface proteins of hepatitis B virus containing the pre-S sequence. J. Virol. 52:396–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eble BE, MacRae DR, Lingappa VR, Ganem D. 1987. Multiple topogenic sequences determine the transmembrane orientation of the hepatitis B surface antigen. Mol. Cell. Biol. 7:3591–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Y, Simsek E, Norton P, Sinnathamby G, Philip R, Block T, Zhou T, Mehta A. 2007. The role of the downstream signal sequences in the maturation of the HBV middle surface glycoprotein: development of a novel therapeutic vaccine candidate. Virology 365:10–19 [DOI] [PubMed] [Google Scholar]
- 6. Patzer EJ, Nakamura GR, Yaffe A. 1984. Intracellular transport and secretion of hepatitis B surface antigen in mammalian cells. J. Virol. 51:346–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wunderlich G, Bruss V. 1996. Characterization of early hepatitis B virus surface protein oligomers. Arch. Virol. 141:1191–1205 [DOI] [PubMed] [Google Scholar]
- 8. Ciczora Y, Callens N, Penin F, Pecheur E-I, Dubuisson J. 2007. Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J. Virol. 81:2372–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Langosch D, Arkin IT. 2009. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 18:1343–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerhardt E, Bruss V. 1995. Phenotypic mixing of rodent but not avian hepadnavirus surface proteins into human hepatitis B virus particles. J. Virol. 69:1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valenzuela P, Quiroga M, Zaldivar J, Gray R, Rutter W. 1980. The nucleotide sequence of the hepatitis B viral genome and the identification of the major viral genes. UCLA Symp. Mol. Cell Biol. 18:57–70 [Google Scholar]
- 12. Bruss V, Lu X, Thomssen R, Gerlich WH. 1994. Post-translational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J. 13:2273–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lambert C, Prange R. 2001. Dual topology of the hepatitis B virus large envelope protein: determinants influencing post-translational pre-S translocation. J. Biol. Chem. 276:22265–22272 [DOI] [PubMed] [Google Scholar]
- 14. Sheu SY, Lo SJ. 1992. Preferential ribosomal scanning is involved in the differential synthesis of the hepatitis B viral surface antigens from subgenomic transcripts. Virology 188:353–357 [DOI] [PubMed] [Google Scholar]
- 15. Eble BE, Lingappa VR, Ganem D. 1990. The N-terminal (pre-S2) domain of a hepatitis B virus surface glycoprotein is translocated across membranes by downstream signal sequences. J. Virol. 64:1414–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bruss V, Ganem D. 1991. Mutational analysis of hepatitis B surface antigen particle assembly and secretion. J. Virol. 65:3813–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lepere-Douard C, Trotard M, Le Seyec J, Gripon P. 2009. The first transmembrane domain of the hepatitis B virus large envelope protein is crucial for infectivity. J. Virol. 83:11819–11829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patient R, Hourioux C, Vaudin P, Pages JC, Roingeard P. 2009. Chimeric hepatitis B and C viruses envelope proteins can form subviral particles: implications for the design of new vaccine strategies. Nat. Biotechnol. 25:226–234 [DOI] [PubMed] [Google Scholar]
- 19. Berkower I, Spadaccini A, Chen H, Al-Awadi D, Muller J, Gao Y, Feigelstock D, Virnik K, Ni Y. 2011. Hepatitis B virus surface antigen assembly function persists when entire transmembrane domains 1 and 3 are replaced by a heterologous transmembrane sequence. J. Virol. 85:2439–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grgacic EVL. 2002. Identification of structural determinants of the first transmembrane domain of the small envelope protein of duck hepatitis B virus essential for particle morphogenesis. J. Gen. Virol. 83:1635–1644 [DOI] [PubMed] [Google Scholar]
- 21. Short JM, Chen S, Roseman AM, Butler PJ, Crowther RA. 2009. Structure of hepatitis B surface antigen from subviral tubes determined by electron cryomicroscopy. J. Mol. Biol. 390:135–141 [DOI] [PubMed] [Google Scholar]
- 22. Gavilanes F, Gonzalez-Ros JM, Peterson DL. 1982. Structure of hepatitis B surface antigen. Characterization of the lipid components and their association with the viral proteins. J. Biol. Chem. 257:7770–7777 [PubMed] [Google Scholar]