

Abstract
The extracellular virion form (EV) of vaccinia virus (VACV) is essential for viral pathogenesis and is difficult to neutralize with antibodies. Why this is the case and how the smallpox vaccine overcomes this challenge remain incompletely understood. We previously showed that high concentrations of anti-B5 antibodies are insufficient to directly neutralize EV (M. R. Benhnia, et al., J. Virol. 83:1201–1215, 2009). This allowed for at least two possible interpretations: covering the EV surface is insufficient for neutralization, or there are insufficient copies of B5 to allow anti-B5 IgG to cover the whole surface of EV and another viral receptor protein remains active. We endeavored to test these possibilities, focusing on the antibody responses elicited by immunization against smallpox. We tested whether human monoclonal antibodies (MAbs) against the three major EV antigens, B5, A33, and A56, could individually or together neutralize EV. While anti-B5 or anti-A33 (but not anti-A56) MAbs of appropriate isotypes were capable of neutralizing EV in the presence of complement, a mixture of anti-B5, anti-A33, and anti-A56 MAbs was incapable of directly neutralizing EV, even at high concentrations. This remained true when neutralizing the IHD-J strain, which lacks a functional version of the fourth and final known EV surface protein, A34. These immunological data are consistent with the possibility that viral proteins may not be the active component of the EV surface for target cell binding and infectivity. We conclude that the protection afforded by the smallpox vaccine anti-EV response is predominantly mediated not by direct neutralization but by isotype-dependent effector functions, such as complement recruitment for antibodies targeting B5 and A33.
INTRODUCTION
The smallpox vaccine, live vaccinia virus (VACV), has been outstanding in controlling smallpox disease, since it has led to the complete eradication of wild smallpox (variola virus) from the world via massive vaccination campaigns of the World Health Organization 40 years ago (1). VACV is considered a gold standard of human vaccinology and one of the greatest successes of modern medicine (1, 2). Unfortunately, smallpox is still considered a danger due to the potential use of smallpox as a biological weapon (3–7), which could spread rapidly through a susceptible population. In addition, related pathogenic poxviruses are a concern, highlighted by the monkeypox outbreak that occurred in the United States (8, 9). The smallpox vaccine affords substantial protection against monkeypox as well as smallpox (10–13).
Orthopoxviruses (vaccinia, variola/smallpox, and monkeypox), which are large, double-stranded DNA viruses with overall dimensions of approximately 360 by 270 by 250 nm, are unusual because their morphogenesis consists of the generation of at least two distinct virion forms with distinct biology: intracellular mature virions (IMV; referred to as MV) and extracellular enveloped virions (EEV; referred to as EV) (14–17). The most abundant viral particles are MV, which accumulate in infected cells until lysis and are released as free virus (18–20). Some MV particles are wrapped in additional membrane and leave the infected cells by exocytosis as a double membraned EV form. EV particles, while less abundant, are critical for virulence in vivo due to their importance in viral dissemination and spread within the host (17, 19–23). Importantly, EV possess nonoverlapping antigens with MV (14, 17, 19, 20, 24). EV contain 5 known outer surface antigens: A33 (25), A34 (26), A36 (27), A56 (28), and B5 (29). A36 extrudes only 2 amino acids (aa) out of the membrane and instead has a large luminal domain that is important for actin tail formation (17, 30, 31). Early expression of proteins A33 and A36 affects virion repulsion and rapid spread (22). Deletion of genes needed for actin tail formation results in decreased virus spread or infectivity (16). Inhibition of F13, a membrane-bound luminal protein of EV involved in EV morphogenesis, is highly effective at stopping orthopoxvirus infections in vivo in an animal model (32–34).
The lipid membrane composition also contributes to the infectivity of VACV MV and is well characterized (15, 35–39). The lipid components of EV outer membrane are mostly sphingomyelin and phosphatidylserine (PS), with smaller amounts of phosphatidylinositol (15). However, the role of the EV lipid membrane in the infectivity of EV is not well characterized due to the fragility of the EV. Recently, it was shown that serum protein Gas6 binding to PS enhances the infectivity and entry of EV particles (40).
The detailed immunologic mechanisms through which smallpox vaccination mediates efficient protection remain unclear and need to be understood in order to consolidate principles that can be applied to future vaccine development against other infectious diseases (41). Neutralizing antibodies (Abs) elicited by immunization with the smallpox vaccine provide protection to humans (20, 42–46). B cell responses against both EV and MV forms of VACV can be important components of protective immunity in animal models and likely contribute to the protection of immunized humans against orthopoxviruses (20, 42, 47). Neutralizing Abs against EV have been of particular interest, given the essential role of EV for viral pathogenesis (47–51). Animals receiving the smallpox vaccine raise Abs against B5 and/or A33 surface antigens (10, 49, 52, 53). Humans also make Abs against B5 and/or A33 after vaccination (43, 54–57).
Previous findings regarding EV neutralization have shown the difficulty of EV neutralization. Early studies showed that protection in vivo in mouse models could be mediated by anti-B5 or anti-A33 Abs, but it was difficult to correlate those results with in vitro activities of the Abs against EV (20, 42, 58). It was later shown that polyclonal antibodies (PAbs) against A33 in the presence of anti-MV Abs and complement reduced the infectivity of EV (59).
The basic coating/occupancy model of Ab-mediated neutralization states that any Ab that binds to a viral surface antigen with sufficient affinity and high occupancy of available surface sites on the virion can neutralize viral infectivity by coating the entire viral surface and thereby blocking viral attachment to host cells (60). As this model applies to a range of viral pathogens (61–66), we initially presumed that it would also be an accurate model for VACV EV particles. However, this model does not apply to VACV EV even at saturating Ab levels against B5 antigens (48, 49). Instead, protective efficacy of anti-B5 MAbs strongly correlated with complement-dependent neutralization of EV by opsonization and the ability to kill VACV-infected cells in the presence of complement (48, 49, 51). Virolysis is a second mechanism of EV particles in the presence of anti-A33 Abs and complement (67). In addition, several studies of the serological responses induced by smallpox vaccine have shown human Ab responses against other EV proteins (55, 57, 68), but the functional value of those responses has not been well defined. We therefore endeavored to more deeply examine the human and murine Ab responses against multiple EV antigens and assess their functionality to understand the underlying immunobiological and virological parameters that determine protective immunity.
MATERIALS AND METHODS
Ethics statement.
Informed consent was obtained for all human work, and the human studies were institutional review board (IRB) approved. All human samples were identified prior to use.
Sera.
Human plasma samples from a nonimmune donor and nine Dryvax-immunized donors from 1 to 10 months postvaccination were stored at −80°C. Rabbit-anti L1 sera were generated by immunizing rabbits with recombinant L1 protein as described in reference 44. Rabbit anti-A33 PAb (NR-628) was produced by immunization of rabbits with recombinant A33R protein (rA33) and was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH (BEI Resources, Manassas, VA) from R. J. Eisenberg and G. H. Cohen (69). Human plasma and rabbit sera were heat inactivated prior to use to eliminate complement activity (56°C for 30 and 60 min, respectively).
Viruses.
MV of the Western Reserve strain (VACVWR), the International Health Department-J (VACVIHDJ) strain, or VACVWR B5-green fluorescent protein (VACV/B5-GFP) stocks were grown on HeLa cells in D-10 (Dulbecco's modified Eagle medium [DMEM], 10% fetal calf serum [FCS], penicillin, streptomycin, and glutamine) as described previously (49). FCS used in all experiments was heat inactivated (56°C, 30 min) prior to use to eliminate complement activity. Purified VACVWR, VACVIHDJ, or VACV/B5-GFP stocks were made via centrifugation through a sucrose cushion as described previously (44). Virus was stored at −80°C. VACVWR was used unless otherwise stated.
EV stocks of VACV WR or IHDJ strains were prepared using HeLa cells grown in D-10, and the medium containing EV was harvested at 2 days after infection as previously described (49). Clarified supernatant was used immediately or stored at 4 to 8°C for a maximum of 3 to 4 weeks (70). The titers of EV VACVWR stocks (∼5 × 105 PFU/ml) and EV VACVIHDJ stocks (6 × 105 PFU/ml) were determined on Vero E6 cells in the presence of rabbit anti-L1 Abs to block contaminating MV or damaged EV present in the EV stock as previously described (49). The percentage of undamaged EV was, on average, 52%.
Recombinant VACV proteins.
Recombinant B5 protein (rB5) was produced using Trichoplusia ni High-Five BTI-TN-5b1-4 (Tn5) insect cells infected with the B5 recombinant baculovirus and purified as established previously (49). B5 is also known as B5R or WR187. rA33 protein was produced and purified comparably to methods described previously in the literature (71).
Human monoclonal antibodies.
Human anti-B5 MAbs (h101 and h102) were generated as described previously (48). Human anti-A33 MAbs were generated from modified vaccinia Ankara (MVA)-vaccinated donors using memory B cell electrofusion methods (72). Acquisition of human blood samples was approved by the Vanderbilt University IRB. Peripheral blood mononuclear cells (PBMCs) were isolated with Histopaque 1077 (Sigma) and Epstein-Barr virus (EBV) transformed in 384-well plates (Nunc) in the presence of 2.5 μg/ml CpG ODN 2006 (Invivogen), 10 μM Chk2 inhibitor II (Sigma C3742), and 1 μg/ml cyclosporine (Sigma). Supernatants from wells containing EBV-transformed lymphoblastoid cell lines were screened for binding activity by enzyme-linked immunosorbent assay (ELISA) against rA33 protein. Positive wells were fused with HMMA2.5 myeloma cells. Human hybridomas secreting antibodies specific for rA33 protein were selected, and the purified human anti-A33 MAbs were produced in culture. We generated two fully human anti-A33 MAbs of the IgG1 isotype, designated VV80 and VV22.
Human anti-A56 MAbs were selected from a human Fab phage display library and subsequently cloned into expression vectors and expressed in stable CHO cells (73).
Murine monoclonal antibodies.
Murine anti-B5 MAbs (clones B126 and B96) were generated as described previously (49).
Mouse anti-A33 MAb NR-565 (similar to VMC-34) was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH (BEI Resources, Manassas, VA) from R. J. Eisenberg and G. H. Cohen. The mouse B cell hybridoma was generated by the fusion of the SP2/0 myeloma cell line with splenocytes, and NR-565 was purified from B cell hybridoma using ammonium sulfate precipitation and size-exclusion chromatography.
Murine anti-A33 MAb (KA10 IgG2a isotype) was generated from a mouse immunized with VACV as described previously (74). Additional unpublished VACV-specific MAbs, including 12D4.1 (IgG1), were generated similarly. A33 specificity was first determined by the techniques described in reference 74) and then by rA33 ELISA.
Gene sequences of heavy- and light-chain variable regions of MAbs.
Total RNA from 5 × 106 murine hybridoma cells (12D4.1 and KA10) and CHO cell lines specific for A56 (WR2 and ES1) was isolated using an RNAeasy Minikit according to the manufacturer's instructions (Qiagen). First-strand cDNA and PCR amplification were produced using a OneStep reverse transcription-PCR (RT-PCR) kit (Qiagen). The cycling profile used for the first-strand PCR was 1 cycle of 30 min at 50°C and 15 min at 95°C; 40 cycles of 30 s at 94°C, 30 s at 56 to 60°C (depending on specific primers), and 55 s at 72°C; and 1 cycle of 10 min at 72°C with a cool down at 4°C. Second-strand PCR was performed using heavy-chain primer 5′MsVHE, 3′Cy1 (IgG1) outer, 3′Cy2c (IgG2a) outer, or 3′Cy3 (IgG3) outer according to the isotype. Light-chain kappa primers used were 5′mVkappa and 3′mCk (75). PCR products were verified by gel electrophoresis, with ∼500-bp products for heavy chains and ∼450-bp products for light chains. PCR fragments were purified using the QIAquick PCR purification kit (Qiagen) and sent for sequencing (5′-primer extension using light- and heavy-chain-specific primers). Sequences include only V-D-J regions for heavy chains and V-J regions for light chains. Ab germ line origin was inferred by analyzing sequences using the V-Quest service of the ImMunoGeneTics information system (http://www.imgt.org) (76).
The antibody genes for the human hybridomas VV22 and VV80 were analyzed similarly. The antibody genes were cloned molecularly from mRNA isolated from the biologically cloned hybridoma cell lines using previously described primer sets (77) into pGEM-T Easy vector (Promega), sequenced by Sanger technique using vector-specific primers, and analyzed using IMGT.
EV neutralization.
Briefly, Vero E6 cells were seeded at 1.5 × 105 cells/well into 24-well Costar plates (Corning Inc., Corning, NY) and used the following day for EV VACVWR or EV VACVIHDJ neutralization assay as described previously (49), using diluted Ab, human plasma, or treated human plasma samples (as described below for the blockade assay) in the absence or the presence of sterile baby rabbit complement (Cedarlane Laboratories, Ontario, Canada) as described previously (49). Ten percent complement (final concentration) was used for all experiments with the exception of the combination Ab experiments, where 1% complement (final concentration) was used instead. Rabbit anti-L1 (1:25 to 1:100, final concentration) was used to block the MV present in the EV stock (58, 70, 78) unless explicitly stated otherwise (e.g., see Fig. 2C and 3F). EV alone or supplemented with anti-L1 Ab was regularly used in each assay with or without complement as negative controls.
Fig 2.

Complement and isotype dependence of murine anti-A33 MAb neutralization of VACV EV. (A to C) VACV EV neutralization by anti-A33 rabbit PAbs is dependent on complement. (A) VACV EV neutralization activity of rabbit PAbs against A33 (NR628) at 1/100 dilution in the absence or the presence of complement. VACV EV alone and a naïve rabbit serum (rabbit serum) were negative controls. (B) Titrated VACV EV neutralization activity of NR628 in the absence (closed circle) or the presence (open circles) of complement. Negative-control samples are shown at single dilutions: naïve rabbit serum alone (closed squares) or plus complement (open squares). The dashed line indicates the plaque numbers of VACV EV in the presence of complement without antibody. (C) VACV EV neutralization by anti-A33 PAb is independent of anti-MV Abs. VACV EV neutralization activity of NR628 at 1/100 dilution with or without complement in the absence of anti-L1 Ab. VACV EV with and without complement (—) were negative controls. (D) Rabbit anti-A33 PAb exhibited comet tail inhibition activity in vitro. Absence of Ab, no treatment; N628, anti-A33 PAb; naïve rabbit serum, rabbit serum. Data in panels B and D are representative of two independent experiments. (E) Sequence analysis of heavy- and light-chain variable regions of murine anti-A33 MAbs (12D4.1 and KA10). Framework (FR) and CDR regions are shown. (F and G) VACV EV neutralization is dependent on complement and anti-A33 MAb isotype. (F) Neutralization activity of purified murine anti-A33 MAbs (12D4.1, KA10, and NR565) in the presence of anti-L1 Abs and in the absence (left) or the presence (right) of complement. Murine anti-B5 MAbs B126 (IgG2a) and B96 (IgG1) were used as controls for neutralization activity in each panel. Anti-DNP (IgG1) and VACV EV with or without complement (—) were negative controls. (G) VACV EV neutralization activity of mouse anti-A33 MAb IgG1 isotype (NR565) in the absence (left) or the presence (middle) of complement-fixing anti-mouse PAb (2° Ab IgG) with or without complement. Murine anti-B5 MAb B126 (IgG2a) was used as a positive control. Human anti-DNP MAb (isotype control) at 10 μg/ml with or without 2° Ab IgG with or without complement was used as a negative control (right). 2° Ab IgG with or without complement also was used as a negative control. Error bars indicate SEM under each condition. The dashed line indicates 50% of the plaque numbers of VACV EV with or without complement in panels A, C, F, and G. All data are representative of three or more experiments.
Fig 3.

Neutralization of EV by human anti-A33 MAb with effector function. (A) Titration of fully human anti-A33 IgG1 MAbs (VV22 and VV80) by rA33 ELISA. Human anti-DNP was used as a negative control. OD, optical density. (B) Cell surface expression of A33 on VACV-B5-GFP-infected cells detected by human anti-A33 MAbs VV22 (red line curve) and VV80 (green line curve) using flow cytometry. Anti-DNP MAb was the negative control (gray line curve). Data are representative of three independent experiments. (C) Sequence analysis of heavy-chain variable regions of human anti-A33 MAbs (VV22 and VV80). Framework (FR) and CDR regions are shown. (D and E) VACV EV virion neutralization by fully human anti-A33 MAbs. (D) VACV EV neutralization activity of human anti-A33 isotype IgG1 MAbs (VV22 and VV80 clones) in the presence of anti-L1 Abs with or without complement. (E) Titrated VACV EV neutralization activity of human anti-A33 MAbs VV22 (closed squares) and VV80 (open circles) in the presence of anti-L1 Abs with or without complement. Human anti-DNP MAb (open squares) and EV VACV (closed diamonds) with or without complement were used as negative controls. Data in panel D are representative of two experiments, each of which was done in the presence of anti-L1. Data are represented as plaque numbers. (F) VACV EV neutralization activity of human anti-A33 MAbs with or without complement and in the absence of anti-L1 Abs. Human anti-B5 MAb clone h101 (IgG1) was used as the positive control in each experiment. Human anti-DNP MAb (IgG1) and VACV EV with or without complement (—) were used as a negative control. The dashed line indicates 50% of the plaque numbers of VACV EV with or without complement in panels D and F. Error bars indicate SEM under each condition. All data in panels D and F are representative of three or more experiments.
MAb or PAb samples were used at 10 μg/ml (final concentration) or 1/100 (final dilution), respectively, unless otherwise stated. Ab samples with or without complement (10% final concentration) then were incubated at 37°C in an equal volume (50 μl) of EV VACVWR (1:100 to 1:400 dilution from stock) unless otherwise indicated and supplemented with rabbit anti-L1 Abs for 60 min, with the exception of the experiment shown in Fig. 2C and 3F. In Fig. 2G or 5G, mouse or human MAb samples (10 μg/ml, final concentration) with or without anti-mouse or anti-human IgG Ab samples, respectively (secondary antibody [2° Ab] at 10 μg/ml final concentration), and with or without complement (10% final concentration) were incubated with an equal volume (50 μl) of EV VACVWR. In all experiments with serial dilutions, 2-fold dilutions of PAb (1/10 starting final dilution) or MAb (40 μg/ml, starting final concentration) samples were used. In titration experiments, 50% EV neutralization was calculated based on sigmoidal dose-response nonlinear regression (Prism 5.0).
Fig 5.

Human anti-A56 MAbs have no effect on EV neutralization. (A to C) Vero E6 monolayers cell were infected with VACV-B5-GFP (green), and surface expression of A56 (red) was determined 12 h postinfection by surface staining with human anti-A56 MAb ES1 or WR2 and performing immunofluorescence (A) or flow cytometry (B and C). Surface expression of A56 was tested after infection with VACV-B5-GFP by surface staining infected cells with human anti-A56 MAb ES1 (red curve) or anti-DNP MAb (control; gray curve). (C) MFI of cell-based ELISA, quantitating surface-bound anti-A56 MAbs to VACV infected cells. Data are representative of three independent experiments. Irrelevant human MAb (anti-DNP, IgG1) was used as a negative control. (D) Sequence analysis of heavy-chain variable regions of human anti-A56 MAbs (WR2 and ES1). Framework (FR) and CDR regions are shown. (E) Human anti-A56 MAbs do not neutralize VACV EV. VACV EV neutralization activity of human anti-A56 MAbs (ES1 and WR2) with or without complement in the presence of anti-L1 Abs. Complement-fixing human anti-B5 MAb h101 (IgG1) was used as a positive control in each experiment. (F) Comet tail plaque inhibition. Shown are the absence of Ab (termed no treatment) or presence of anti-A56 MAbs (ES1 and WR2) or irrelevant MAb (control IgG1). Data are representative of two independent experiments. (G) Addition of complement-fixing anti-human IgG at 10 μg/ml (left) to human anti-A56 MAbs does not improve the neutralization of VACV EV. VACV EV neutralization activity of mouse anti-A33 MAb (NR565) in the absence or presence of complement-fixing anti-mouse IgG at 10 μg/ml with and without complement were control Abs (right). Irrelevant human MAb (anti-DNP, IgG1) was used as a negative control (right). The dashed line indicates 50% of the plaque numbers of VACV EV with or without complement. Error bars indicate SEM under each condition. All data in panels E and G are representative of three or more experiments.
To assess complement-independent VACV EV neutralization in the presence of secondary antibodies (see Fig. 6), VACV EV samples (plus anti-L1) were added to MAb samples with or without goat anti-mouse or anti-human IgG Ab samples (2° Ab at 10 μg/ml final concentration) and with or without donkey anti-goat IgG Ab samples (3° Ab at 10 μg/ml final concentration) at 37°C and 5% CO2.
Fig 6.
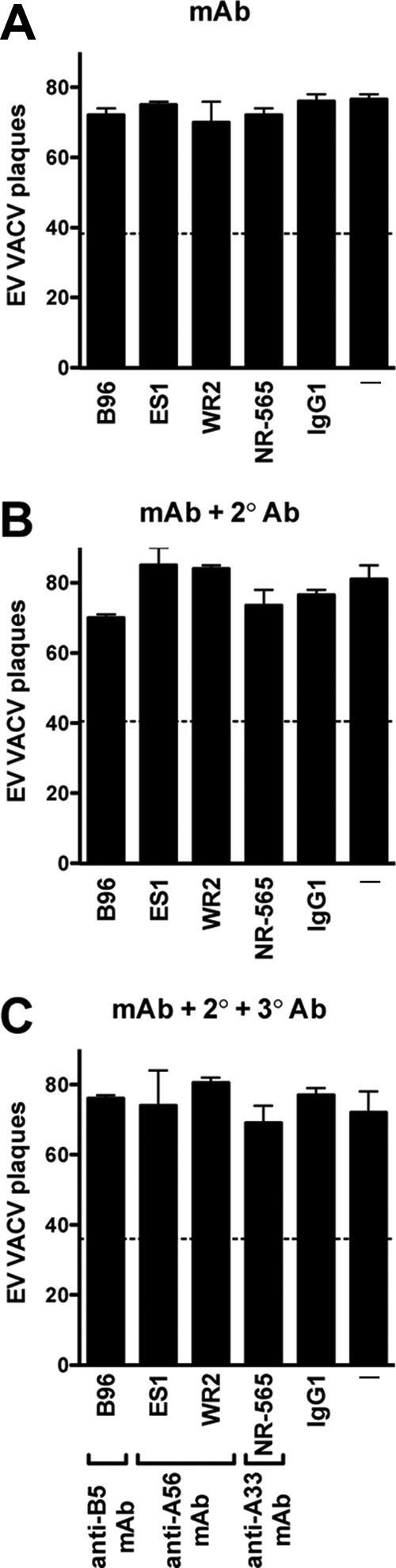
Expanding the footprint of single-specificity MAb does not result in EV neutralization. (A to C) VACV EV neutralization activity of mouse anti-B5 MAbs (B96, IgG1), mouse anti-A33 MAbs (NR565, IgG1), or human anti-A56 MAbs (ES1 or WR2, IgG1) at 10 μg/ml alone (A), in the presence of species-specific anti-Ig secondary antibodies (B), or in the presence of secondary and tertiary anti-Ig antibodies (C). EV alone (—) and human anti-DNP IgG1 (“IgG1”) were negative controls. Data are representative of two independent experiments. The dashed line indicates 50% of plaque numbers of VACV EV with or without complement. Error bars indicate SEM under each condition.
Comet tail inhibition assay.
Vero E6 cells were seeded at 5 × 105 cells/well into 6-well Costar plates and infected the following day with VACVIHDJ (20 to 60 PFU/well) for 60 min at 37°C and 5% CO2. Human anti-A56, mouse anti-A33, or irrelevant IgG1 MAbs at 20 μg/ml or naïve rabbit serum or rabbit anti-A33 Abs at 1/100 dilution, which was present in the medium throughout the culture, was used to inhibit the comet tail formation as described previously (79, 80).
ELISA.
rA33 ELISA or rB5 ELISA was done as described previously (49). For the blockade experiment, human plasma samples and rB5, rA33, or rB5 plus rA33-treated plasma samples were screened for B5 or A33 reactivity using rB5 ELISA or rA33 ELISA. The secondary Ab was streptavidin-horseradish peroxidase-conjugated mouse anti-human IgG (Hybridoma Reagent Laboratory, Baltimore, MD). The EC50, the half-saturation binding concentration of the Ab corresponding to 50% maximum optical density, for each individual anti-A33 MAb was determined by sigmoidal dose-response nonlinear regression with a variable slope (Prism 5.0).
Blockade of anti-EV-specific antibodies.
Human plasma samples (1/10 final dilution) or anti-B5 MAb clone h101 (10 μg/ml final concentration) in a volume of 10 μl was preincubated with rB5 protein (10 μg) for 2 h at room temperature in a 50-μl total volume. Samples were then used to test the efficacy and specificity of the blockade of anti-B5 Abs in a VACV EV neutralization assay. The plaque numbers were quantified, and 50% plaque reduction was calculated from VACV EV with anti-L1 alone, with or without rB5, or with or without complement. A33 was assayed with the same strategy. The method used to quantify the percentage of VACV EV neutralization at a single plasma/Ab sample dilution before and after the blockade of Abs against B5, A33, or both proteins was according to the formula (PNV − PNS)/PNV × 100, where PNV is the average number of plaques by VACVWR with or without complement and PNS is the average number of plaques in the presence of each experiment sample with or without complement. Treated and untreated samples in the presence of complement were then compared to obtain the percent reduction in neutralization activity.
Protein synthesis and proteome arrays.
VACV protein microarrays were produced and used as described previously (54). Secondary Ab was Cy3-conjugated goat anti-human IgG gamma chain Fc region-specific immunoglobulin (Jackson ImmunoResearch). Signal strength was quantified as the total fluorescence intensity at 532 nm (TFI532) of each spot. Background signal was subtracted using relevant matched control samples (e.g., rapid translation system [RTS] translation reaction without plasmid or with buffer alone), and background-subtracted signal was converted to the final IgG relative units (RU) via 10−6 transformation.
Surface staining of VACV-infected cells.
Vero E6 cell monolayers were infected with VACV/B5-GFP for 12 h at a multiplicity of infection (MOI) of 5 at 37°C and 5% CO2. A33 or A56 expression then was tested with human anti-A33 (clones VV22 and VV80) or anti-A56 (clones ES1 and WR2) MAbs at 20 μg/ml by flow cytometry as described previously (48). An irrelevant IgG1 Ab was used as a negative control of EV surface expression. The secondary Ab was donkey anti-human IgG (H+L) allophycocyanin (APC; Jackson ImmunoResearch, West Grove, PA) used at a 1:100 dilution. To determine the relative affinity of human anti-A56 MAbs, a fluorescence-activated cell sorting (FACS)-based ELISA strategy was used as described previously (49). The EC50, the half-saturation binding concentration of the Ab corresponding to 50% maximum mean fluorescence intensity (MFI) for each individual anti-A56 MAb, was determined using Prism.
Immunofluorescence (IF) assay. Confluent monolayers of Vero E6 cells seeded in 8-well chambered cover glass slides (Lab-Tek; Nalge Nunc International, Rochester, NY) were infected overnight with 8 PFU/cell of VACV/B5-GFP. A56 surface expression was detected with human anti-A56 Abs and using donkey anti-human IgG Ab conjugated to APC (Jackson ImmunoResearch, West Grove, PA) at 1:200 dilution as described in reference 48. Slides were examined using a Nikon Eclipse E1000 IF microscope (Mariana).
Mapping the epitope for KA10.
The plasmids for expressing the fusion of glutathione S-transferase (GST) and A13 were constructed by PCR amplifying the viral gene from WR DNA and cloning the PCR fragment into pGEX6P-1 (GE Healthcare Life Sciences). The expression of the fusion proteins in Escherichia coli BL21 was induced with isopropyl-beta-d-thiogalactoside (IPTG; Invitrogen). For Western blot analysis, the bacteria were lysed via sonication in SDS-PAGE sample buffer, and the clarified cell lysates were directly used in Western blot analysis.
Virion surface area calculations.
A vaccinia virus virion is a cylinder with approximate dimensions of 360-nm height by 130-nm radius (20, 81–84); this gives a surface area of 400,239 nm2. The virion mass is 5 × 10−15 g and is 90% protein, therefore 4.5 × 10−15g/virion. H3 is approximately 3% of total mass (85), thus 4.5 × 10−15g × 0.03 = 1.4 × 10−16g H3/virion. The total, given the size of H3, comes to 2,276 H3 molecules per virion. IgG at 1 μg/ml is 6.25 nM.
Statistical analysis.
Tests were performed using Prism 4.0 or 5.0 (GraphPad, San Diego, CA). Statistics were done using two-tailed, unpaired t tests with 95% confidence bounds unless otherwise indicated. For Fig. 4C, a Newman-Keuls test was used for multiple comparisons. Bar graph error bars are ±1 standard error of the means (SEM).
Fig 4.
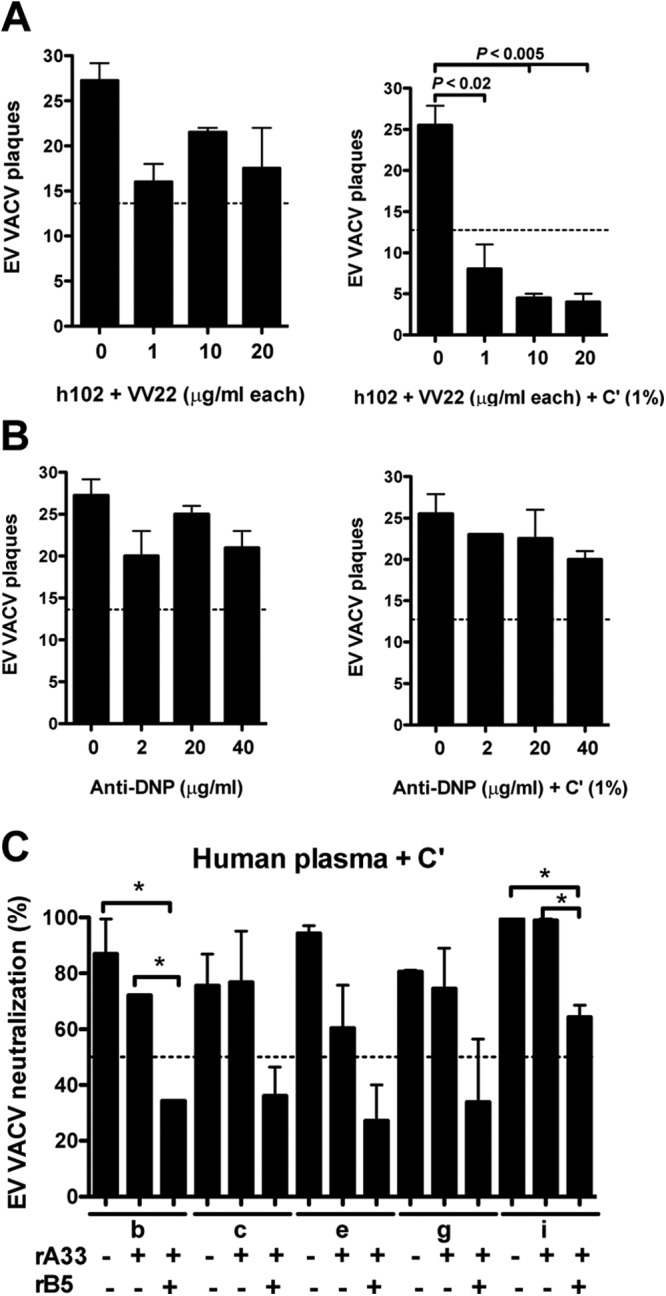
Combined effect of human A33 and B5 against EV virion and the impact of anti-A33 Ab in smallpox-vaccinated humans. (A) VACV EV neutralization activity of the fully human MAbs against B5 (h102) and A33 (VV22) at 20, 10, or 1 μg/ml of each MAb in the absence or presence of 1% complement. (B) Irrelevant human IgG1 MAb and EV (anti-DNP) at 40, 20, or 2 μg/ml were negative controls with or without complement. Data are represented as plaque numbers. The dashed line indicates 50% of the plaque numbers of VACV EV with or without complement. (C) VACV EV neutralization activity (%) of plasma from donors (b, c, e, g, and i) in the presence of complement and after the blockade of anti-A33 or anti-B5/A33 Abs with 10 μg of rA33 or rB5/rA33 proteins. *, P < 0.05. The dashed line indicates 50% of neutralization activity. Error bars indicate SEM under each condition. All data are representative of two independent experiments.
RESULTS
To understand the immunobiology of the smallpox vaccine, we have been studying the mechanisms of poxvirus neutralization. In our recent studies on EV virion neutralization, we showed that high concentrations of anti-B5 Abs are insufficient on their own to neutralize VACV EV (48, 49). This allowed for two possible interpretations: covering the virion surface is insufficient for neutralization, or B5 is insufficiently abundant to allow anti-B5 IgG to cover the full EV surface, leaving another viral receptor protein exposed and active. These possibilities have central implications for the physiological mechanisms of poxvirus neutralization in vivo and likely have major implications for other infectious agents. Vaccinia virus is a large virus with an approximate surface area of 400,000 nm2, and it is not clear if Abs against a given antigen or multiple antigens could cover sufficient surface area of the virion to neutralize the virus by coating. An Ig molecule has a footprint of approximately 300 nm3 (86). It would take ∼1,300 IgG molecules to cover the virion surface (see Materials and Methods for detailed calculations). Is this reasonable? In vitro, under normal neutralizing Ab assay conditions and with a MAb with a reasonable affinity of 1 nM, approximately 90% occupancy is obtained at 1 μg/ml. Therefore, if the antigen is present at sufficient density, the criteria for neutralization by occupancy will be fulfilled. We do not know the abundance of B5 or other proteins on the surface of EV, because the fragility of the virions precludes biochemical purification. However, MV particles are durable and the abundance of MV surface proteins has been calculated based on mass spectrophotometry (85, 87), and MV and EV are of comparable size. H3 is an abundant MV surface protein, as determined by mass spectrometry, and calculations provide a rough estimate of 2,300 H3 molecules on the surface of each MV virion. We estimate that it takes ∼1,300 IgG molecules to cover more than half of the surface area of a vaccinia virion. If B5 protein is present at fewer than 1,300 copies per virion (or less than ∼60% of the level of the highly abundant H3 protein), then the criteria for neutralization by coating will not be fulfilled and the virus may escape direct neutralization. Conceptually, it is also possible that the EV surface antigens are not uniformly distributed, and “patchiness” of different receptors in different regions of the surface enhances resistance to neutralization. Therefore, the occupancy model of neutralization may not apply to EV particles. To test these possibilities, we examined antibodies against multiple EV virion proteins and combinations of antibodies.
Human anti-B5 antibodies are not the only specificity mediating EV neutralization.
B5 is a known target of human Ab responses, and anti-B5 Ab responses are known to be protective in animal models (20, 42). Plasma samples from nine human donors who had received the smallpox vaccine were tested for EV neutralizing Abs. Untreated human plasma samples alone exhibited minimal EV neutralization activity in the absence of complement (Fig. 1A). Addition of complement to mimic physiological conditions (48, 49) resulted in efficient EV neutralization by plasma from all donors (Fig. 1B). All donors had anti-B5 IgG responses (Fig. 1C and data not shown). To determine whether B5 is the only EV neutralizing Ab target in immunized humans, we blocked anti-B5 Ig in the human plasma samples using an excess of rB5. B5 Abs were completely blocked after rB5 adsorption of human plasma (93 to 100%) (Fig. 1D and E and data not shown), but other VACV Ab specificities were unaffected (Fig. 1E and data not shown). As a control, the impact of rB5 blocking on the human anti-B5 MAb h101 was tested. One hundred percent loss of neutralization was observed (P < 0.0001 for h101, rB5, and complement versus h101 and complement; Fig. 1F). From the panel of nine donors, blockade of anti-B5 IgG resulted in near-complete loss of EV neutralization activity in plasma from four donors (donors a, d, f, and h) (Fig. 1G), demonstrating that anti-B5 neutralizing Abs were the immunodominant EV neutralizing Ab response in these vaccinees. Somewhat surprisingly, blockade of B5 Abs had only a moderate effect on VACV EV neutralization activity of plasma from four other donors (b, c, g, and i), resulting in only 28, 23, 15, and 18% loss of virus neutralization activity, respectively (P < 0.0001 for donors i and g and P < 0.0004 for donors b and c) (Fig. 1B and G), suggesting these donors primarily had complement fixing EV neutralizing Abs against other EV antigens. Donor e had an intermediate phenotype, with 48% loss of EV neutralization activity after B5 blockade (P < 0.005).
Fig 1.

Human anti-B5 antibodies are not the only specificities mediating EV neutralization. VACV EV neutralization activity of plasma samples in the absence of complement (A) or the presence of complement (C′) (B). (C and D) Quantitation of anti-B5 IgG in untreated plasma (C) or in plasma after blocking with 10 μg of rB5 protein (D) from vaccinated human donors (lanes a to i) or a nonimmune human donor (Non imm). An IgG signal level of 2 relative units (RU) was selected as a stringent cutoff (dashed line), establishing 98% specificity (see Materials and Methods). (E) VACV viral protein microarrays. B5 protein spots (red box) in untreated plasma from an individual immunized with the smallpox vaccine (human plasma, left) and B5-treated plasma (human plasma + rB5, right). B5 protein is also present in row 12, spot 4. The C3L spot was not printed on the left panel, row 11, spot 8. (F) VACV EV neutralization activity after rB5 blockade of anti-B5 MAb h101 at 10 μg/ml. (G) VACV EV neutralization activity of plasma samples after the blockade of anti-B5 antibodies in the presence of complement. VACV EV alone (A) or plus complement (B and G) were negative controls (—). Dashed lines in panels A, B, F, and G indicate 50% of the plaque numbers of VACV EV with or without complement. *, P < 0.005; **, P < 0.0004; ***, P < 0.0001. Error bars indicate SEM under each condition. All data are representative of two or more experiments. (H) Quantitation of anti-A33 binding IgG in plasma from donors by recombinant A33 protein (rA33) ELISA. Data are representative of two independent experiments. OD, optical density. An IgG signal level of 0.2 was selected as the cutoff (dashed line).
A33 is another prominent EV antigen known to be a target of protective Ab responses in mouse models (50, 58, 88). We therefore screened each donor for the presence of anti-A33 IgG using rA33 protein ELISA. Most vaccinees had a substantial Ab response to A33, with the exception of donor a (Fig. 1H). There was not a direct correlation between the magnitude of the anti-A33 IgG and the B5-independent EV neutralization activity, leaving the role of the human anti-A33 IgG uncertain in complement-mediated EV neutralization. Furthermore, in most individuals both anti-B5 and anti-A33 IgG are present. This raised the question of why EV was not neutralized in the absence of complement.
Complement and isotype-dependent murine anti-A33 MAb neutralization of EV.
As a first step to examining if the human anti-A33 Ab response is functional for EV neutralization, we studied the capacity of rabbit and murine Abs to neutralize EV by complement-dependent and -independent processes. We examined rabbit anti-A33 PAbs (NR628) using direct and complement EV neutralization assays. NR628 exhibited weak direct EV neutralizing activity in vitro (PAb with EV VACV) (Fig. 2A and B) but showed strong neutralization in the presence of the complement (P < 0.002 for NR628 versus NR628 and complement) (Fig. 2A) in a dose-dependent manner (Fig. 2B). EV neutralization assays are regularly performed in the presence of anti-MV IgG to eliminate contaminating MV and damaged EV (EV/MV) particles in EV stocks (58, 70, 78). Experiments shown in Fig. 2A, B, F, and G were all performed in the presence of anti-L1 IgG to eliminate MV and damaged EV virions. Previously, we demonstrated that anti-MV IgG has no effect on EV neutralization measured by anti-B5 MAbs when using fresh, undamaged EV stocks (48, 49). To determine whether the anti-MV IgG was influencing the EV neutralization being measured for anti-A33 PAbs, neutralization assays were performed in the absence of anti-L1 IgG, with or without complement (Fig. 2C). The neutralization activity was comparable in the absence of anti-MV IgG (98%) (Fig. 2C). This differs from a previous study (59). Rabbit anti-A33 PAbs were efficient at inhibiting comet tail formation in the absence of exogenous complement (Fig. 2D).
Sequence analysis of murine anti-A33 MAbs to the germ line showed that 12D4.1 and KA10 clones originate from the same inferred immunoglobulin heavy-chain variable (IGHV) precursor gene allele, IGHV7-3*02 (Fig. 2E). The variable light-chain sequence for 12D4.1 and KA10 is IGKV1-110*01. Murine anti-A33 MAbs NR565, 12D4.1, and KA10 were tested for functional activity against EV. IgG1 anti-A33 MAbs NR565 and 12D4.1 exhibited weak complement-mediated neutralization activity (Fig. 2F). This was consistent with the well-characterized lack of complement recruitment by murine IgG1 Abs. Anti-A33 MAb KA10 belongs to the IgG2a isotype. However, epitope mapping revealed that KA10 binds to A33 at aa 2 to 24, which is contained within the luminal domain of A33 (data not shown). In summary, the anti-A33 MAb tested, which was able to fix complement (KA10), is not able to neutralize because it binds an epitope on the luminal domain, while the remaining antibodies have diverse specificities but are not able to fix complement.
To determine if the murine anti-A33 MAbs specific to the ectodomain of A33 failed to neutralize only due to their inability to recruit complement, we tested EV neutralization activity of NR565 in the presence of complement-fixing anti-murine IgG Abs. As shown in Fig. 2G, NR565 MAb exhibited complement-mediated EV neutralization in the presence of complement-recruiting Fc regions (P < 0.01 for NR565 and complement versus NR565 with 2° Ab IgG and complement) (Fig. 2G). An isotype control had no effect, as expected (the P value was not significant). These data indicate that anti-A33 MAb NR565 binds to A33 on the EV surface but is incapable of neutralizing the virus without complement recruitment by the Fc domain.
Neutralization of EV by human anti-A33 MAbs.
Human MAbs were developed by electrofusion generation of hybridomas using memory B cells from MVA-vaccinated donors. Two hybridomas, VV80 and VV22, were identified that produced IgG1 specific for rA33 protein by ELISA (Fig. 3A) (IgG1 is the most abundant human IgG isotype and is complement fixing). Both human MAbs bound A33 on the surface of VACV-infected cells (Fig. 3B), indicating that they are specific for the ectodomain of native A33 on the EV surface. Clone VV22 had a higher rA33 relative binding affinity (EC50 of 0.11 nM) than clone VV80 (EC50 of 83 nM) (Fig. 3A). Sequence analysis of human anti-A33 VV22 and VV80 clones revealed that they originated from separate germ lines, IGHV3-33*01 and IGHV3-74*02, respectively (Fig. 3C).
The higher-affinity MAb VV22 exhibited complement-dependent EV neutralization (Fig. 3D). The lower-affinity MAb VV80 exhibited no complement-mediated EV neutralization (Fig. 3D). This is consistent with a requirement for high affinity to successfully neutralize the virus in vitro and for protection in vivo, as was seen for human anti-B5 MAbs (48). Dose titrations showed that high-affinity MAb VV22 had a 50% plaque neutralization titer (PRNT50) of 4.55 μg/ml (Fig. 3E). Those assays were done in the presence of anti-MV IgG to eliminate contaminating MV particles or damaged EV particles in EV stocks. Given our previous work with anti-B5 MAbs (48, 49) and the anti-A33 PAb results discussed above (Fig. 2), anti-A33 EV neutralization assays were performed in the absence of anti-L1 IgG (Fig. 3F). The complement-dependent EV neutralization activity of human anti-A33 MAb VV22 was similar irrespective of the presence or absence of anti-MV IgG (80 and 89%, respectively) (Fig. 3D and F). Human anti-B5 MAb h102 shows neutralization activity similar to that of human anti-A33 MAb (VV22) in the presence or absence of anti-MV IgG (Fig. 3D and F). These data support a model where human anti-A33 or anti-B5 Abs can neutralize EV in the presence of complement via opsonization of the EV particle surface.
The combined effects of human A33 and B5 MAbs against EV.
Saturating amounts of anti-B5 MAb are insufficient to neutralize EV in the absence of complement (Fig. 1 and references 48 and 49). We postulated that the coating model fails and EV escape direct neutralization by anti-B5 IgG, because direct occupancy of all B5 binding sites is insufficient to fully cover the surface of the virus, and alternative receptors are sufficient for infectivity when B5 is blocked. The data in Fig. 2 and 3 and references 50 and 59 indicate that A33 is also dispensable for infectivity of EV when blocked by anti-A33 Abs. Are B5 and A33 compensating for each other as EV receptor proteins? We tested this hypothesis by attempting to directly neutralize EV with a combination of human MAbs against both A33 and B5 (h102 plus VV22). EV neutralization by the combination was modest, with maximal neutralization of 36%, even at high Ab concentrations (20 μg/ml each MAb) (Fig. 4A). The neutralization observed with the combination of the anti-A33 and anti-B5 Abs may be due to agglutination of EV, aggregating the particles. Irrelevant IgG1 anti-DNP MAbs at the same concentration showed no effect (Fig. 4B). The efficiency of anti-B5 plus anti-A33 MAb neutralization was substantially increased upon adding a low concentration of complement (Fig. 4A).
The role of polyclonal anti-A33 Ig in vaccinated humans.
Anti-B5 Abs were the primary EV neutralizing Ab in donors a, d, f, and h (Fig. 1). However, plasma from vaccinees b, c, e, g, and i showed a less dominant role for anti-B5 Abs. We therefore tested the role of anti-A33 Abs from those donors in complement-dependent EV neutralization. Anti-A33 Ig was blocked by preincubation of plasma samples with an excess of free rA33 protein. The blockade of anti-A33 Abs was specific and effective (80, 68, 61, 47, and 52% for donors b, c, e, g, and i, respectively). Blockade of anti-A33 Abs alone resulted in undetectable or modest reductions in the EV neutralization activity of the plasma from the five donors (17, 0, 36, 7.5, and 0.5%, respectively) (Fig. 4C). Blockade of B5 Abs alone again had a moderate effect on the EV neutralization activity, in agreement with Fig. 1G. We then blocked the plasma with a combination of rA33 and rB5 (Fig. 4C). Donor i gave results comparable to those for blocking anti-B5 Abs alone, which was consistent with the lack of an effect of blocking anti-A33 Abs alone in this donor. Samples from donors b, c, e, and g exhibited an additive effect of blocking both anti-A33 and anti-B5 Ig (61, 52, 71, and 58%, respectively). Therefore, anti-A33 Abs do significantly contribute, in combination with anti-B5 Abs, to complement-mediated EV neutralization in a number of vaccinees. Curiously, we were unable to completely abrogate EV neutralization by blocking anti-A33 and anti-B5 Abs together. This finding suggested that either the blockade was incomplete or that additional EV neutralization targets exist.
Effect of human anti-A56 MAbs on EV neutralization.
We hypothesized that the anti-B5- and anti-A33-independent EV neutralization activity observed in approximately half of the vaccinees was due to anti-A56 or anti-A34 Abs. Several studies of the serological responses induced by smallpox vaccine have shown human Ab responses against A56 antigen (55, 57, 68). However, no evidence is available that Abs against A56 can neutralize EV or provide protection in vivo. We identified two human anti-A56 MAbs (ES1 and WR2; see Materials and Methods) and examined their properties. Native A56 expression was detected on VACV-infected cells by IF staining (Fig. 5A) and by flow cytometry (Fig. 5B and C). WR2 exhibited good relative binding affinity to native A56 (EC50 of 1.23 nM), while ES1 possessed lower binding capacity (70.94 nM) (Fig. 5C). Sequence analysis of human anti-A56 MAbs, ES1 and WR2 clones, revealed that they originated from the different but related germ line V genes IGHV1-69*02 and IGHV1-69*10, respectively (Fig. 5D).
We tested the EV neutralizing capacity of human anti-A56 MAbs. Both clones failed to neutralize EV at 10 or 20 μg/ml in the absence or presence of complement (Fig. 5E). The two human anti-A56 MAbs exhibited moderate reduction of comet formation at 20 μg/ml (Fig. 5F). Given that anti-A56 MAb WR2 recognized native A56, was a complement binding isotype, and had high-affinity binding, why was EV neutralization not observed? It has been reported that A56 binds the complement control protein VCP, and VCP can protect cells from complement-mediated lysis (89, 90). It is possible that VCP also affects complement recruitment to EV (91, 92). The inability of anti-A56 MAb WR2 to neutralize EV therefore may be due to competition with VCP on the virion and inability to fix and activate complement. We reasoned that coating the EV virion with anti-A56 MAb and then a second layer of anti-human IgG PAb would restore complement sensitivity (Fig. 5G). EV neutralization with human anti-A56 MAbs plus 2° Ab and complement was poor, with maximum neutralization of ∼29% (Fig. 5G). As a positive control, anti-A33 MAb plus secondary PAbs exhibited good neutralization activity in the presence of complement (P < 0.003 for anti-A33 plus 2° Ab versus anti-A33 plus 2° Ab and complement) (Fig. 5G). Therefore, while vaccinated humans do produce Ab responses against A56, it does not appear that anti-A56 Abs are significant contributors to EV neutralization in the human immune response to the smallpox vaccine. The lack of activity may be due to anti-A56 Abs (i) recognizing epitopes located such that complement cannot be recruited appropriately, (ii) binding A56 on the surface of VACV-infected cells but not A56 on fully mature released EV, or (iii) being inhibited by proximity to VCP.
Testing the impact of increasing Ig footprints on EV.
The coating model of neutralization posits that a virion is neutralized at the point at which the surface of the virion is sufficiently covered to block accessibility to the target cell. We have found that high-affinity MAbs of any single specificity, B5, A33, or A56, are unable to directly neutralize EV in the absence of complement. We reasoned that perhaps all three proteins serve as redundant viral receptors. We further reasoned, as a corollary of this hypothesis, that each of these antigens must be at a low enough density on the surface of EV that saturating amounts of Ab against one specificity (e.g., A33) do not sterically block the accessibility of the other EV surface proteins (e.g., B5 and A56). If this hypothesis is correct, expanding the footprint made by anti-EV Ig of a given specificity may block accessibility of all EV surface proteins and result in neutralization of EV. Amplification of the footprint of anti-A56 or anti-A33 MAbs with secondary anti-Ig Ab (2° Ab) did not result in EV neutralization (Fig. 6A and B). Amplification of the footprint of anti-B5 MAb gave the same result (Fig. 6A and B). Given that complement component C1q has three times the molecular mass of an Ig molecule and has an extended footprint, we then tested whether the additional mass of a tertiary anti-Ig (3° Ab) would facilitate direct neutralization of EV by anti-A33, -A56, or -B5 MAbs. No EV neutralization was observed even in the presence of 3° Ig (Fig. 6C).
Fig 7.

Simultaneously coating EV virion with human MAbs specific to multiple antigens, B5, A33, and A56, is insufficient for neutralization of EV from the WR or IHD-J strain. VACV EV neutralization activity of human anti-A33 (VV22), A56 (WR2), and B5 (h101) IgG1 MAbs, starting at 20 μg/ml of each MAb and then using 2-fold serial dilutions, against VACV EV of the WR (A) or IHD-J strain (I) with or without a small amount of complement (1%) was determined. (B and J) Irrelevant human MAb (anti-DNP, control IgG1), starting at 60 μg/ml and then 2-fold serial dilution, in the presence of VACV EV WR (B) or IHD-J strain (J) with or without complement (1%). All data are representative of three or more experiments. (C to F) VACV EV neutralization activity of human anti-A33 (VV22) and B5 (h101) MAbs IgG1 (C) in combination with human anti-A56 (WR2) (E), starting at 40 μg/ml of each MAb, against VACV EV of WR with or without complement (1%). (D and F) Irrelevant human MAb (anti-DNP; control IgG1), starting at 80 or 120 μg/ml, in the presence of VACV EV with or without complement (1%). The data are representative of one independent experiment. (G and H) VACV EV neutralization activity of human anti-A56 MAbs in the presence of anti-L1 Abs against the VACVWR (G) or VACVIHDJ strain (H) with or without complement (10%) in each experiment. Mouse anti-B5 MAb B126 (IgG2a) and rabbit anti-A33 PAbs (NR628) were used as positive controls. VACV EV alone (—) and an irrelevant human IgG1 MAb plus EV (IgG1) were negative controls. The dashed line indicates 50% of the plaque numbers of VACV EV with or without complement. Error bars indicate SEM under each condition. All data are representative of two independent experiments.
Effects of Abs against multiple EV antigens simultaneously.
As a complementary approach, we hypothesized that simultaneously coating EV with Abs specific to multiple antigens may accomplish direct neutralization if each of the three receptors are functionally redundant. Saturating concentrations of Abs against A33 and B5 together were unable to neutralize EV (Fig. 4). We therefore tested a combination of MAbs against all three targets. Saturating amounts of anti-A33, -A56, and -B5 together exhibited no consistent neutralization of EV (Fig. 7A and B). In contrast, the combination of Abs can neutralize EV at 1.2 μg/ml of each MAb upon the addition of only a small amount of exogenous complement (1%) (Fig. 7A). Anti-A56 Abs do not contribute in combination with anti-B5 and -A33 Abs to mediated EV neutralization even in the presence of 1% complement (Fig. 7C and E).
Fig 8.

Mechanism of EV neutralization. (A) Models of VACV EV neutralization. Schematic diagrams of potential virion neutralization pathways. B5, A33, and A56 are drawn in green, blue, and gray, respectively. An unknown hypothetical EV surface antigen is drawn in cyan. Abs in model I are anti-B5, anti-A33, and anti-A56. Abs in model II are anti-B5 and anti-A33. Complement (C′) components include C1q and C3. Model I is the basic occupancy model. MAbs against three exposed EV antigens, B5, A33, and A56, could completely coat the functional antigens on the virion surface and subsequently neutralize the virus. This model failed. Direct occupancy of B5, A33, and A56 with Abs is insufficient to block infection of targets cells. VACV EV escape of neutralization by anti-B5, anti-A33, and anti-A56 Ab binding may be due to limited abundance of each antigen on the surface of EV, resulting in a lack of blocking of an unknown EV surface protein still able to facilitate infection. Alternatively, the infectious component of the virion surface is lipid and there is insufficient viral protein on the surface for the anti-VACV Abs to cover the surface, leaving the fusogenic membrane accessible. Model II shows complement-assisted single-specificity or two-specificity anti-EV MAbs coating VACV EV via opsonization. Antibody-mediated protection against VACV EV is dependent on activation of complement C1q via the Fc domain of the immunoglobulin and covalent attachment of C3 to the lipid outer membrane of the virus. (B and C) The presence of anti-MV Abs in EV stock is not necessary for EV neutralization. Schematic diagrams of potential virion neutralization pathways. EV and damaged EV (EV/MV) particles are presented in EV stock. B5 and A33 are drawn in cyan and gray, respectively. L1 or H3, as an MV protein, is drawn in green. The Abs were anti-B5 and anti-A33. Complement (C′) components included C1q and C3. (B) MAbs against one or two exposed EV antigens, B5 and/or A33, could completely coat the functional antigens on EV and EV/MV and subsequently neutralize EV in the presence of complement components C1q and C3. (C) MAbs against one or two exposed EV antigens, B5 and/or A33, could completely coat the specific antigens on EV and EV/MV, and anti-MV Abs coat a specific antigen on the surface of EV/MV particles. Subsequently, the addition of the complement enhances the neutralization of EV and EV/MV particles.
A34 is the fourth and final known EV transmembrane protein with an ectodomain. Is A34 functioning as a cell binding receptor in the absence of available B5, A33, and A56? To test this possibility, we made use of VACVIHDJ. The IHDJ strain of VACV has a mutation in the A34 gene resulting in defective A34 (93). Abs against A33 and B5 give comparable neutralization activity using both strains of VACV (Fig. 7G and H). Anti-A56 MAbs failed to neutralize EV virion independent of the strain of VACV (Fig. 7G and H). We tested whether a combination of MAbs against A33, B5, and A56 antigens could neutralize EV in the absence of a functional A34. No direct neutralization of VACVIHDJ in the presence of anti-A33, -B5, and -A56 MAbs was found (Fig. 7I and J). The combination of MAbs exhibited neutralization of VACVIHDJ upon the addition of a small amount of exogenous complement, comparable to the results with VACVWR, which possesses an intact A34 gene (Fig. 7I). In summary, we were not able to demonstrate that the coating model of neutralization holds for EV by targeting all known EV protein surface antigens.
DISCUSSION
We have been studying the mechanism of poxvirus neutralization in order to understand how the smallpox vaccine provides such effective protection to immunized humans. The EV virion of VACV is essential for viral pathogenesis and is difficult to neutralize. Given the general agreement in virology of the basic coating/occupancy model for neutralization of a range of viral pathogens (60, 64), this model was expected to be accurate for neutralization of VACV EV particles. However, the published literature on anti-B5 Abs does not easily fit the model. We suggested at least two possible interpretations: (i) the occupancy model is fundamentally unsound for larger pathogens, and (ii) the surface of an EV particle has restricted coverage by B5 and other functionally redundant receptors are present. In this study, we therefore hypothesized that blocking one or more additional EV proteins would result in direct neutralization of EV. We explored this problem by asking a series of questions. Do high-affinity MAbs of any single specificity directly neutralize EV? Does a combination of MAbs against B5 and A33 inhibit EV infectivity? Does expanding the footprint made by anti-EV Ig of a given specificity, with 2° or 3° Abs, result in neutralization of the EV? Does simultaneously coating EV with saturating amounts of Abs specific to multiple antigens accomplish direct neutralization of EV? Direct neutralization of EV was not observed in any instance.
Previously, we found that comet tail inhibition occurs with human anti-B5 Abs (48) and with both mouse IgG1 and IgG2a anti-B5 MAbs irrespective of isotype (49). Our data show strong inhibition of comet tail formation using antibodies against A33 and moderate inhibition with antibodies against A56. However, the comet tail assay is not predictive of in vivo efficacy. The comet tail inhibition assay measures the reduction of VACV cell-to-cell spread mediated by EV, but probably only EV before it has been released from the surface of a cell (i.e., cell-associated enveloped virus [CEV]). The comet tail inhibition assay predicts that MAbs of any single specificity (B5, A33, or A56) would be protective in vivo as the MAbs were effective or moderately effective at comet tail inhibition in vitro. However, it has been shown that mouse IgG1 comet tail-inhibiting MAbs against B5 do not protect in vivo against VACV (49) and, in contrast, that human IgG3 protective MAbs against B5 do not inhibit or partially inhibit comet tail formation (48). Given that both mouse IgG1 and IgG2a MAbs can function in comet tail inhibition, as do anti-A56 MAbs, it seems likely that the comet tail assay predominantly functions by an aggregation-type cross-linking mechanism, whereby the Abs bind to target antigen on neighboring CEVs on the surface of infected cells and reduce the release of CEV as EV. We therefore consider that the comet tail assay has minimal relevance, whereas the EV plaque reduction assay that measures the complement-mediated EV neutralization activity of free EV is highly predictive of in vivo protection.
It was surprising to find that the combination of anti-A33, -A56, and -B5 together exhibited no consistent direct neutralization of EV (Fig. 8A, model I). Combined with our findings here, the observation that C1q with C3 is required but C5 is not (48, 67) indicates that blocking the EV proteins is insufficient to neutralize EV, and that additional activities are required. C3 is covalently attached to targets and is reactive to a wide range of chemical moieties, including lipids. The fact that anti-A33, -A56, or -B5 Abs individually were unable to neutralize EV even in the presence of 2° or 3° Igs appears conceptually similar to the finding that C1q plus anti-B5 MAb was insufficient to neutralize EV (48, 49). Our findings support a model where anti-A33 or anti-B5 Abs can neutralize EV in the presence of complement, independent of anti-MV Abs, via opsonization of the EV particle surface (Fig. 8). These findings provide indirect evidence that surface proteins are not the required infectious component of EV, leaving open the possibility that EV membrane lipids directly mediate adhesion and the initial events leading to VACV fusion with the target cell. Full fusion with the host cell requires revealing the intricate VACV entry-fusion complex contained in the MV membrane (15, 18, 19). The fragility of the outer lipid envelope of EV particles may be due to limited abundance of proteins in the membrane, the biophysical properties of the lipid components, like sphingomyelin and PS, the fact that the two lipid membranes (the outer membrane and the internal MV membrane) are in direct apposition, or a combination of all three factors. Sparse proteins on the exterior of the EV particle could result in large areas of naked membrane and a fluid surface similar to that of HIV, making it very difficult for antibodies to neutralize the virus (94–96). In addition, the macropinocytic mechanism of endocytosis also may play an important role in EV infectivity (97).
Alternatively, it is possible that the lack of direct neutralization of EV observed here is because A34 is still functional and plays a role in infectivity, even though the IHDJ strain contains a mutated A34 with no known function. Another possibility is that the occupancy model fails because orthopoxviruses have somehow evolved to successfully infect target cells even when only a very small percentage of viral protein receptor molecules are available. The viruses may also possess an unknown evasion mechanism.
Without direct neutralization, Ab-mediated protection against the EV form of VACV depends on effector functions of the immunoglobulins. Anti-EV antibodies can utilize a range of effector functions to accomplish protection. Our data support a prominent role for complement-mediated opsonization that is dependent on C3 and C1q (48, 49). Complement-mediated opsonization, without virolysis, has also been shown to be the predominant complement-mediated mechanism of neutralization for other viruses, including ectromelia virus, influenza virus, West Nile virus, mumps virus, paramyxovirus simian virus 5, and vesicular stomatitis virus (98–105). Isaacs and colleagues have shown that anti-A33 in combination with complement (C1q and C5) efficiently mediates EV destruction (67). Complement-dependent virolysis is also effective against other families of viruses (106–110). It is also possible for complement to disrupt the EV outer membrane but not inner membrane and allow access of anti-MV Abs to neutralize the virus, as shown by experiments in which EV neutralization was dependent on the concurrent presence of anti-MV Abs (59). However, our data show that under the complement conditions used here, anti-B5 or anti-A33 Abs can mediate neutralization in the absence of anti-MV Abs. Together these data show that EV opsonization, stepwise neutralization (release of MV and then neutralization of MV), and EV virolysis are all available mechanisms of EV neutralization. It is possible that differences in the density of B5 and A33 on the surface of EV result under conditions where antibodies against A33 may not be able to recruit sufficient C1q and C3 to completely opsonize an EV particle (58), revealing the value of C5-dependent membrane attack complexes as a secondary mechanism of EV neutralization. There is also clear evidence that anti-EV antibodies can be protective through additional effector mechanisms in vivo, including complement-mediated killing (CDC) of vaccinia virus-infected cells (48, 49) and binding to Fcγ receptors (67).
These findings indicate that vaccines based on protective antibodies do not necessarily need to elicit antibodies that can accomplish direct neutralization of the virus. Furthermore, these findings suggest that our understanding of EV biology is insufficient to explain how EV binds to and initiates infection of target cells.
ACKNOWLEDGMENTS
This work was supported in part by NIH NIAID AI63107 and NIH NIAID AI077953 to S.C., by NIH NIAID HHSN272200900048C to B.P., and by HHSN272200900047C to J.E.C.
Multiple reagents were obtained through the NIH BEI Resources center. We thank Lindsay Crickard for technical assistance, Ravi Kolla for management assistance, and Howard Gray for his advice. We thank Bernard Moss, Alessandro Sette, and David N. Garboczi for virus strains, VACV protein, and reagents.
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. 1988. Smallpox and its eradication. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Reference deleted.
- 3. Henderson DA, Inglesby TV, Bartlett JG, Ascher MS, Eitzen E, Jahrling PB, Hauer J, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl T, Russell PK, Tonat K. 1999. Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA 281:2127–2137 [DOI] [PubMed] [Google Scholar]
- 4. LeDuc JW, Jahrling PB. 2001. Strengthening national preparedness for smallpox: an update. Emerg. Infect. Dis. 7:155–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meltzer MI, Damon I, LeDuc JW, Millar JD. 2001. Modeling potential responses to smallpox as a bioterrorist weapon. Emerg. Infect. Dis. 7:959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. NIAID 2007. NIAID high priority biodefense products. NIAID, Bethesda, MD [Google Scholar]
- 7. O'Toole T, Mair M, Inglesby TV. 2002. Shining light on “Dark Winter.” Clin. Infect. Dis. 34:972–983 [DOI] [PubMed] [Google Scholar]
- 8. Huhn GD, Bauer AM, Yorita K, Graham MB, Sejvar J, Likos A, Damon IK, Reynolds MG, Kuehnert MJ. 2005. Clinical characteristics of human monkeypox, and risk factors for severe disease. Clin. Infect. Dis. 41:1742–1751 [DOI] [PubMed] [Google Scholar]
- 9. Reynolds MG, Yorita KL, Kuehnert MJ, Davidson WB, Huhn GD, Holman RC, Damon IK. 2006. Clinical manifestations of human monkeypox influenced by route of infection. J. Infect. Dis. 194:773–780 [DOI] [PubMed] [Google Scholar]
- 10. Edghill-Smith Y, Golding H, Manischewitz J, King LR, Scott D, Bray M, Nalca A, Hooper JW, Whitehouse CA, Schmitz JE, Reimann KA, Franchini G. 2005. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat. Med. 11:740–747 [DOI] [PubMed] [Google Scholar]
- 11. Hammarlund E, Lewis MW, Carter SV, Amanna I, Hansen SG, Strelow LI, Wong SW, Yoshihara P, Hanifin JM, Slifka MK. 2005. Multiple diagnostic techniques identify previously vaccinated individuals with protective immunity against monkeypox. Nat. Med. 11:1005–1011 [DOI] [PubMed] [Google Scholar]
- 12. Karem K, Reynolds M, Hughes C, Braden Z, Nigam P, Crotty S, Glidewell J, Ahmed R, Amara R, Damon I. 2007. Monkeypox-induced immunity and failure of childhood smallpox vaccination to provide complete protection. Clin. Vaccine Immunol. 14:1318–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Likos AM, Sammons SA, Olson VA, Frace AM, Li Y, Olsen-Rasmussen M, Davidson W, Galloway R, Khristova ML, Reynolds MG, Zhao H, Carroll DS, Curns A, Formenty P, Esposito JJ, Regnery RL, Damon I. 2005. A tale of two clades: monkeypox viruses. J. Gen. Virol. 86:2661–2672 [DOI] [PubMed] [Google Scholar]
- 14. Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 15. Laliberte JP, Moss B. 2010. Lipid membranes in poxvirus replication. Viruses 2:972–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts KL, Smith GL. 2008. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 16:472–479 [DOI] [PubMed] [Google Scholar]
- 17. Smith GL, Vanderplasschen A, Law M. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915–2931 [DOI] [PubMed] [Google Scholar]
- 18. Laliberte JP, Weisberg AS, Moss B. 2011. The membrane fusion step of vaccinia virus entry is cooperatively mediated by multiple viral proteins and host cell components. PLoS Pathog. 7:e1002446 doi:10.1371/journal.ppat.1002446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moss B. 2007. Poxviridae: the viruses and their replication. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 20. Moss B. 2011. Smallpox vaccines: targets of protective immunity. Immunol. Rev. 239:8–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doceul V, Hollinshead M, Breiman A, Laval K, Smith GL. 2012. Protein B5 is required on extracellular enveloped vaccinia virus for repulsion of super-infecting virions. J. Gen. Virol. 93(Pt 9):1876–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith GL, Murphy BJ, Law M. 2003. Vaccinia virus motility. Annu. Rev. Microbiol. 57:323–342 [DOI] [PubMed] [Google Scholar]
- 24. Payne LG. 1980. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J. Gen. Virol. 50:89–100 [DOI] [PubMed] [Google Scholar]
- 25. Roper RL, Payne LG, Moss B. 1996. Extracellular vaccinia virus envelope glycoprotein encoded by the A33R gene. J. Virol. 70:3753–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duncan SA, Smith GL. 1992. Identification and characterization of an extracellular envelope glycoprotein affecting vaccinia virus egress. J. Virol. 66:1610–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parkinson JE, Smith GL. 1994. Vaccinia virus gene A36R encodes a M(r) 43–50 K protein on the surface of extracellular enveloped virus. Virology 204:376–390 [DOI] [PubMed] [Google Scholar]
- 28. Shida H. 1986. Nucleotide sequence of the vaccinia virus hemagglutinin gene. Virology 150:451–462 [DOI] [PubMed] [Google Scholar]
- 29. Isaacs SN, Wolffe EJ, Payne LG, Moss B. 1992. Characterization of a vaccinia virus-encoded 42-kilodalton class I membrane glycoprotein component of the extracellular virus envelope. J. Virol. 66:7217–7224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanderson CM, Frischknecht F, Way M, Hollinshead M, Smith GL. 1998. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol. 79(Pt 6):1415–1425 [DOI] [PubMed] [Google Scholar]
- 31. Wolffe EJ, Weisberg AS, Moss B. 1998. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology 244:20–26 [DOI] [PubMed] [Google Scholar]
- 32. Huggins J, Goff A, Hensley L, Mucker E, Shamblin J, Wlazlowski C, Johnson W, Chapman J, Larsen T, Twenhafel N, Karem K, Damon IK, Byrd CM, Bolken TC, Jordan R, Hruby D. 2009. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob. Agents Chemother. 53:2620–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jordan R, Goff A, Frimm A, Corrado ML, Hensley LE, Byrd CM, Mucker E, Shamblin J, Bolken TC, Wlazlowski C, Johnson W, Chapman J, Twenhafel N, Tyavanagimatt S, Amantana A, Chinsangaram J, Hruby DE, Huggins J. 2009. ST-246 antiviral efficacy in a nonhuman primate monkeypox model: determination of the minimal effective dose and human dose justification. Antimicrob. Agents Chemother. 53:1817–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sbrana E, Jordan R, Hruby DE, Mateo RI, Xiao SY, Siirin M, Newman PC, Da Rosa AP, Tesh RB. 2007. Efficacy of the antipoxvirus compound ST-246 for treatment of severe orthopoxvirus infection. Am. J. Trop. Med. Hyg. 76:768–773 [PubMed] [Google Scholar]
- 35. Chung CS, Huang CY, Chang W. 2005. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 79:1623–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laliberte JP, Moss B. 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. U. S. A. 106:17517–17521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535 [DOI] [PubMed] [Google Scholar]
- 38. Mercer J, Knebel S, Schmidt FI, Crouse J, Burkard C, Helenius A. 2010. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. U. S. A. 107:9346–9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oie M. 1985. Reversible inactivation and reactivation of vaccinia virus by manipulation of viral lipid composition. Virology 142:299–306 [DOI] [PubMed] [Google Scholar]
- 40. Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, Chen IS. 2011. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 9:286–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walker BD, Burton DR. 2008. Toward an AIDS vaccine. Science 320:760–764 [DOI] [PubMed] [Google Scholar]
- 42. Amanna IJ, Slifka MK, Crotty S. 2006. Immunity and immunological memory following smallpox vaccination. Immunol. Rev. 211:320–337 [DOI] [PubMed] [Google Scholar]
- 43. Bell E, Shamim M, Whitbeck JC, Sfyroera G, Lambris JD, Isaacs SN. 2004. Antibodies against the extracellular enveloped virus B5R protein are mainly responsible for the EEV neutralizing capacity of vaccinia immune globulin. Virology 325:425–431 [DOI] [PubMed] [Google Scholar]
- 44. Benhnia MR, McCausland MM, Su HP, Singh K, Hoffmann J, Davies DH, Felgner PL, Head S, Sette A, Garboczi DN, Crotty S. 2008. Redundancy and plasticity of neutralizing antibody responses are cornerstone attributes of the human immune response to the smallpox vaccine. J. Virol. 82:3751–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kempe CH, Bowles C, Meiklejohn G, Berge TO, St Vincent L, Babu BV, Govindarajan S, Ratnakannan NR, Downie AW, Murthy VR. 1961. The use of vaccinia hyperimmune gamma-globulin in the prophylaxis of smallpox. Bull. World Health Organ. 25:41–48 [PMC free article] [PubMed] [Google Scholar]
- 46. Mack TM, Noble J, Jr, Thomas DB. 1972. A prospective study of serum antibody and protection against smallpox. Am. J. Trop. Med. Hyg. 21:214–218 [DOI] [PubMed] [Google Scholar]
- 47. Moyron-Quiroz JE, McCausland MM, Kageyama R, Sette A, Crotty S. 2009. The smallpox vaccine induces an early neutralizing IgM response. Vaccine 28:140–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benhnia MR, McCausland MM, Laudenslager J, Granger SW, Rickert S, Koriazova L, Tahara T, Kubo RT, Kato S, Crotty S. 2009. Heavily isotype-dependent protective activities of human antibodies against vaccinia virus extracellular virion antigen B5. J. Virol. 83:12355–12367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benhnia MR, McCausland MM, Moyron J, Laudenslager J, Granger S, Rickert S, Koriazova L, Kubo R, Kato S, Crotty S. 2009. Vaccinia virus extracellular enveloped virion neutralization in vitro and protection in vivo depend on complement. J. Virol. 83:1201–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen Z, Earl P, Americo J, Damon I, Smith SK, Yu F, Sebrell A, Emerson S, Cohen G, Eisenberg RJ, Gorshkova I, Schuck P, Satterfield W, Moss B, Purcell R. 2007. Characterization of chimpanzee/human monoclonal antibodies to vaccinia virus A33 glycoprotein and its variola virus homolog in vitro and in a vaccinia virus mouse protection model. J. Virol. 81:8989–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McCausland MM, Benhnia MR, Crickard L, Laudenslager J, Granger SW, Tahara T, Kubo R, Koriazova L, Kato S, Crotty S. 2010. Combination therapy of vaccinia virus infection with human anti-H3 and anti-B5 monoclonal antibodies in a small animal model. Antivir. Ther. 15:661–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y, Romero KM, Nguyen TT, Kalantari-Dehaghi M, Crotty S, Baldi P, Villarreal LP, Felgner PL. 2005. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. U. S. A. 102:547–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marriott KA, Parkinson CV, Morefield SI, Davenport R, Nichols R, Monath TP. 2008. Clonal vaccinia virus grown in cell culture fully protects monkeys from lethal monkeypox challenge. Vaccine 26:581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Davies DH, Molina D, Wrammert J, Miller J, Hirst S, Mu Y, Pablo J, Unal B, Nakajima-Sasaki R, Liang X, Crotty S, Karem K, Damon I, Ahmed R, Villarreal L, Felgner P. 2007. Proteome-wide analysis of the serological response to vaccinia and smallpox. Proteomics 7:1678–1686 [DOI] [PubMed] [Google Scholar]
- 55. Johnson BF, Kanatani Y, Fujii T, Saito T, Yokote H, Smith GL. 2011. Serological responses in humans to the smallpox vaccine LC16m8. J. Gen. Virol. 92:2405–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lawrence SJ, Lottenbach KR, Newman FK, Buller RM, Bellone CJ, Chen JJ, Cohen GH, Eisenberg RJ, Belshe RB, Stanley SL, Jr, Frey SE. 2007. Antibody responses to vaccinia membrane proteins after smallpox vaccination. J. Infect. Dis. 196:220–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pütz MM, Midgley CM, Law M, Smith GL. 2006. Quantification of antibody responses against multiple antigens of the two infectious forms of vaccinia virus provides a benchmark for smallpox vaccination. Nat. Med. 12:1310–1315 [DOI] [PubMed] [Google Scholar]
- 58. Galmiche MC, Goenaga J, Wittek R, Rindisbacher L. 1999. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology 254:71–80 [DOI] [PubMed] [Google Scholar]
- 59. Lustig S, Fogg C, Whitbeck JC, Moss B. 2004. Synergistic neutralizing activities of antibodies to outer membrane proteins of the two infectious forms of vaccinia virus in the presence of complement. Virology 328:30–35 [DOI] [PubMed] [Google Scholar]
- 60. Parren PW, Burton DR. 2001. The antiviral activity of antibodies in vitro and in vivo. Adv. Immunol. 77:195–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dowd KA, Jost CA, Durbin AP, Whitehead SS, Pierson TC. 2011. A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog. 7:e1002111 doi:10.1371/journal.ppat.1002111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, Marx PA, Burton DR. 2007. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449:101–104 [DOI] [PubMed] [Google Scholar]
- 63. Klasse PJ. 2007. Modeling how many envelope glycoprotein trimers per virion participate in human immunodeficiency virus infectivity and its neutralization by antibody. Virology 369:245–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Klasse PJ, Burton D. 2007. Antibodies to West Nile virus: a double-edged sword. Cell Host Microbe 1:87–89 [DOI] [PubMed] [Google Scholar]
- 65. Li L, Coelingh KL, Britt WJ. 1995. Human cytomegalovirus neutralizing antibody-resistant phenotype is associated with reduced expression of glycoprotein H. J. Virol. 69:6047–6053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pierson T, Xu Q, Nelson S, Oliphant T, Nybakken G, Fremont D, Diamond M. 2007. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1:135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cohen ME, Xiao Y, Eisenberg RJ, Cohen GH, Isaacs SN. 2011. Antibody against extracellular vaccinia virus (EV) protects mice through complement and Fc receptors. PLoS One 6:e20597 doi:10.1371/journal.pone.0020597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Davies DH, Wyatt LS, Newman FK, Earl PL, Chun S, Hernandez JE, Molina D, Hirst S, Moss B, Frey SE, Felgner PL. 2008. Antibody profiling by proteome microarray reveals the immunogenicity of the attenuated smallpox vaccine modified vaccinia virus Ankara is comparable to that of Dryvax. J. Virol. 82:652–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lustig S, Fogg C, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454–13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Aldaz-Carroll L, Whitbeck JC, Ponce de Leon M, Lou H, Hirao L, Isaacs SN, Moss B, Eisenberg RJ, Cohen GH. 2005. Epitope-mapping studies define two major neutralization sites on the vaccinia virus extracellular enveloped virus glycoprotein B5R. J. Virol. 79:6260–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Su HP, Singh K, Gittis AG, Garboczi DN. 2010. The structure of the poxvirus A33 protein reveals a dimer of unique C-type lectin-like domains. J. Virol. 84:2502–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu X, McGraw PA, House FS, Crowe JE., Jr 2008. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J. Immunol. Methods 336:142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27 [DOI] [PubMed] [Google Scholar]
- 74. Meng X, Zhong Y, Embry A, Yan B, Lu S, Zhong G, Xiang Y. 2011. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409:271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tiller T, Busse CE, Wardemann H. 2009. Cloning and expression of murine Ig genes from single B cells. J. Immunol. Methods 350:183–193 [DOI] [PubMed] [Google Scholar]
- 76. Brochet X, Lefranc MP, Giudicelli V. 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J. and V-D-J. sequence analysis. Nucleic Acids Res. 36:W503–W508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. 2009. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 4:372–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vanderplasschen A, Hollinshead M, Smith GL. 1997. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J. Gen. Virol. 78(Pt 8):2041–2048 [DOI] [PubMed] [Google Scholar]
- 79. Law M, Hollinshead R, Smith GL. 2002. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J. Gen. Virol. 83:209–222 [DOI] [PubMed] [Google Scholar]
- 80. Law M, Smith GL. 2001. Antibody neutralization of the extracellular enveloped form of vaccinia virus. Virology 280:132–142 [DOI] [PubMed] [Google Scholar]
- 81. Cyrklaff M, Risco C, Fernandez JJ, Jimenez MV, Esteban M, Baumeister W, Carrascosa JL. 2005. Cryo-electron tomography of vaccinia virus. Proc. Natl. Acad. Sci. U. S. A. 102:2772–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gaylord WH, Jr, Melnick JL. 1953. Intracellular forms of pox viruses as shown by the electron microscope (Vaccinia, Ectromelia, Molluscum Contagiosum). J. Exp. Med. 98:157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morgan C, Ellison SA, Rose HM, Moore DH. 1954. Internal structure in virus particles. Nature 173:208. [DOI] [PubMed] [Google Scholar]
- 84. Morgan C, Ellison SA, Rose HM, Moore DH. 1954. Structure and development of viruses observed in the electron microscope. II. Vaccinia and fowl pox viruses. J. Exp. Med. 100:301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hulander M, Lundgren A, Berglin M, Ohrlander M, Lausmaa J, Elwing H. 2011. Immune complement activation is attenuated by surface nanotopography. Int. J. Nanomed. 6:2653–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233–247 [DOI] [PubMed] [Google Scholar]
- 88. Fogg C, Lustig S, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. 2004. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J. Virol. 78:10230–10237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. DeHaven BC, Girgis NM, Xiao Y, Hudson PN, Olson VA, Damon IK, Isaacs SN. 2010. Poxvirus complement control proteins are expressed on the cell surface through an intermolecular disulfide bridge with the viral A56 protein. J. Virol. 84:11245–11254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Girgis NM, Dehaven BC, Fan X, Viner KM, Shamim M, Isaacs SN. 2008. Cell surface expression of the vaccinia virus complement control protein is mediated by interaction with the viral A56 protein and protects infected cells from complement attack. J. Virol. 82:4205–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Isaacs SN, Argyropoulos E, Sfyroera G, Mohammad S, Lambris JD. 2003. Restoration of complement-enhanced neutralization of vaccinia virus virions by novel monoclonal antibodies raised against the vaccinia virus complement control protein. J. Virol. 77:8256–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Isaacs SN, Kotwal GJ, Moss B. 1992. Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neutralization of infectivity and contributes to virulence. Proc. Natl. Acad. Sci. U. S. A. 89:628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. McIntosh AA, Smith GL. 1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70:272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bachmann MF, Zinkernagel RM. 1997. Neutralizing antiviral B cell responses. Annu. Rev. Immunol. 15:235–270 [DOI] [PubMed] [Google Scholar]
- 95. Burton D. 2002. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2:706–713 [DOI] [PubMed] [Google Scholar]
- 96. Karlsson Hedestam G, Fouchier RA, Phogat S, Burton D, Sodroski J, Wyatt R. 2008. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat. Rev. Microbiol. 6:143–155 [DOI] [PubMed] [Google Scholar]
- 97. Schmidt FI, Bleck CK, Helenius A, Mercer J. 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J. 30:3647–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Feng JQ, Mozdzanowska K, Gerhard W. 2002. Complement component C1q enhances the biological activity of influenza virus hemagglutinin-specific antibodies depending on their fine antigen specificity and heavy-chain isotype. J. Virol. 76:1369–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Johnson JB, Capraro GA, Parks GD. 2008. Differential mechanisms of complement-mediated neutralization of the closely related paramyxoviruses simian virus 5 and mumps virus. Virology 376:112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Leddy JP, Simons RL, Douglas RG. 1977. Effect of selective complement deficiency on the rate of neutralization of enveloped viruses by human sera. J. Immunol. 118:28–34 [PubMed] [Google Scholar]
- 101. Mehlhop E, Ansarah-Sobrinho C, Johnson S, Engle M, Fremont DH, Pierson TC, Diamond MS. 2007. Complement protein C1q inhibits antibody-dependent enhancement of flavivirus infection in an IgG subclass-specific manner. Cell Host Microbe 2:417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mehlhop E, Diamond MS. 2006. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 203:1371–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mehlhop E, Fuchs A, Engle M, Diamond MS. 2009. Complement modulates pathogenesis and antibody-dependent neutralization of West Nile virus infection through a C5-independent mechanism. Virology 393:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mehlhop E, Nelson S, Jost CA, Gorlatov S, Johnson S, Fremont DH, Diamond MS, Pierson TC. 2009. Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile virus. Cell Host Microbe 6:381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Moulton EA, Atkinson JP, Buller RM. 2008. Surviving mousepox infection requires the complement system. PLoS Pathog. 4:e1000249 doi:10.1371/journal.ppat.1000249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Berry DM, Almeida JD. 1968. The morphological and biological effects of various antisera on avian infectious bronchitis virus. J. Gen. Virol. 3:97–102 [DOI] [PubMed] [Google Scholar]
- 107. Friedman HM, Wang L, Pangburn MK, Lambris JD, Lubinski J. 2000. Novel mechanism of antibody-independent complement neutralization of herpes simplex virus type 1. J. Immunol. 165:4528–4536 [DOI] [PubMed] [Google Scholar]
- 108. Radwan AI, Burger D. 1973. The complement-requiring neutralization of equine arteritis virus by late antisera. Virology 51:71–77 [DOI] [PubMed] [Google Scholar]
- 109. Stollar V. 1975. Immune lysis of Sindbis virus. Virology 66:620–624 [DOI] [PubMed] [Google Scholar]
- 110. Welsh RM, Jr, Lampert PW, Burner PA, Oldstone MB. 1976. Antibody-complement interactions with purified lymphocytic choriomeningitis virus. Virology 73:59–71 [DOI] [PubMed] [Google Scholar]