

Abstract
Both entry and cell-to-cell spread of herpes simplex virus (HSV) involve a cascade of cooperative interactions among the essential glycoproteins D, B, and H/L (gD, gB, and gH/gL, respectively) initiated by the binding of gD to a cognate HSV entry receptor. We previously reported that a variant (D285N/A549T) of glycoprotein B (gB:NT) enabled primary virus entry into cells that were devoid of typical HSV entry receptors. Here, we compared the activities of the gB:NT variant with those of a newly selected variant of glycoprotein H (gH:KV) and a frequently coselected gB variant (gB:S668N). In combination, gH:KV and gB:S668N enabled primary virus entry into cells that lacked established HSV entry receptors as efficiently as did gB:NT, but separately, each variant enabled only limited entry. Remarkably, gH:KV uniquely facilitated secondary virus spread between cells that lacked canonical entry receptors. Transient expression of the four essential entry glycoproteins revealed that gH:KV, but not gB:NT, induced fusion between cells lacking the standard receptors. Because the involvement of gD remained essential for virus spread and cell fusion, we propose that gH:KV mimics a transition state of gH that responds efficiently to weak signals from gD to reach the active state. Computational modeling of the structures of wild-type gH and gH:KV revealed relatively subtle differences that may have accounted for our experimental findings. Our study shows that (i) the dependence of HSV-1 entry and spread on specific gD receptors can be reduced by sequence changes in the downstream effectors gB and gH, and (ii) the relative roles of gB and gH are different in entry and spread.
INTRODUCTION
The entry of free herpes simplex virus 1 (HSV-1) into susceptible cells requires the coordinated activities of 4 viral envelope glycoproteins (1–4). After virion adsorption to cell surface heparan sulfates, mediated mainly by a fifth envelope glycoprotein, glycoprotein C (gC) (5, 6), gD binds to one of its specific receptors, herpesvirus entry mediator (HVEM or HveA), nectin-1 (HveC), or 3-O-sulfated heparan sulfate (7–9). Receptor binding results in a conformational change in gD, which in turn activates the fusion mechanism mediated by gB and the gH/gL heterodimer; fusion merges the viral envelope with the cell surface or endosomal membrane, resulting in capsid release into the cytoplasm (10–15).
HSV entry by direct cell-to-cell (lateral) spread involves a similar fusion step and requires the same 4 entry glycoproteins along with a receptor for gD (1–4, 16–18). However, evidence exists to indicate that the two entry processes are not mechanistically identical. For example, gD mutants that allow gD receptor-independent cell-to-cell spread, but not receptor-independent entry, have been isolated (19). Likewise, gD is required for entry of the related alphaherpesvirus pseudorabies virus (PRV) but not for PRV lateral spread (20–23). Two additional glycoproteins, gE and gI, are important for HSV-1 spread but are dispensable for entry of free virus (reviewed in reference 24). However, it is unknown whether the roles of gB and gH/gL are different in entry and spread.
The crystal structure of the HSV-1 gB ectodomain shows unexpected homology to the postfusion form of glycoprotein G from vesicular stomatitis virus, a well-characterized fusion protein (25), suggesting direct participation of gB in the fusion reaction. Although previous studies reported that gH displays hallmarks of fusion proteins (26–29), the crystal structure of the HSV-2 gH ectodomain bound to gL bears no similarity to any known viral fusogen (30). Nonetheless, both gB and gH may possess fusogenic capabilities, as each alone can promote membrane fusion during nuclear egress (31). Previous evidence indicated that gH/gL could carry out hemifusion, an intermediate state in which the outer membrane leaflets of the target and viral membranes mix, whereas both gB and gH/gL were required for complete fusion (32), but these findings were recently called into question (33). Recent studies suggested that gH/gL activates the dormant fusogenic activity of gB in response to signaling by receptor-bound gD (30, 34, 35), indicating that gH/gL performs an intermediate regulatory function in the steps leading to membrane fusion. However, since these studies were performed in virus-free cell fusion systems, it is unclear whether they apply equally to virus entry and spread. Moreover, it remains uncertain whether gH/gL also has a role as cofusogen in either event.
By taking advantage of the ability of HSV to adapt to constraints on receptor usage for entry, we previously identified gain-of-function derivatives of a gD mutant virus that was impaired for its ability to use nectin-1 as an entry receptor. This approach identified new mutations in gD as well as revertants (36). In an attempt to isolate a broader spectrum of adaptive mutations, including mutations in other essential glycoproteins, we further disabled the gD–nectin-1 interaction by (i) creating mutations in both binding partners or (ii) introducing further changes in nectin-1 without mutations in gD. We previously reported that the first strategy consistently identified a pair of substitution mutations in gB (D285N/A549T, referred to here as NT) that allowed virus entry in the absence of canonical gD receptors, enhanced entry through unconventional receptors, and an increased rate of entry into canonical receptor-bearing cells (37). Here, we describe the results of the second strategy and our analyses to identify key changes allowing plaque formation despite the absence of known functional receptors for gD.
MATERIALS AND METHODS
Cells.
Baby hamster kidney J1.1-2 (provided by Gabriella Campadelli-Fiume, University of Bologna), HVEM-transduced J/A (38), nectin-1-transduced J/C (36), TMCΔC-transduced J/TMCΔC (37), murine melanoma B78H1 (provided by Gary Cohen, University of Pennsylvania), HVEM-transduced B78/A (36), nectin-1-transduced B78/C (36), TMC-transduced B78/TMC (37), Chinese hamster ovary CHO-K1 (ATCC CCL-61), African green monkey kidney Vero (ATCC CCL-81), gD-complementing VD60 (4) (provided by David Johnson, Oregon Health and Science University), and gH-complementing F6 (39) (provided by Anthony Minson, University of Cambridge) cells were described previously. B78/0G and CHO/0G cells were established by cotransfection of B78H1 or CHO-K1 cells with a plasmid expressing green fluorescent protein (GFP) from the HSV-1 immediate early (IE) ICP0 promoter (pO7G) and a puromycin resistance plasmid, followed by selection for resistance to 2.5 μg/ml or 10 μg/ml puromycin, respectively. Clonal lines were screened for VP16-dependent GFP expression by transfection of VP16 expression plasmid pCAVP16.
Viruses.
K26GFP (40) (provided by Prashant Desai, Johns Hopkins University), QOZHG (41), KΔT (42) (provided by Stanley Person, Johns Hopkins University), and K-gB:NT (K-gB:N/T in reference 37) were described previously. K-gB:668N was established by cotransfection of Vero cells with KΔT viral DNA and plasmid pgB1:S668N, followed by plaque purification through three rounds of limiting dilution on Vero cells.
K-gH:KV and K-gB:668N-gH:KV were established in two steps. First, KΔgH and K-gB:668NΔgH were established by cotransfection of F6 cells with plasmid pΔgH-EGFP and viral DNA from KOS or K-gB:668N, respectively, and purification of green plaques on F6 cells. K-gH:KV and K-gB:668N-gH:KV were then established by cotransfection of Vero cells with plasmid pgH1:N753K/A778V and viral DNA from KΔgH or K-gB:668NΔgH, respectively, and plaque purification on Vero cells. Wild-type KOS, K-gB:668N, K-gB:NT, K-gH:KV, and K-gB:668N-gH:KV were all propagated and titered on Vero cells simultaneously. Genome titers of these stocks were determined by real-time quantitative PCR (qPCR) for the IE gene ICP47, as described previously (43). Table 1 lists the genome and PFU titers and their ratios.
Table 1.
Physical and biological titers of recombinant viruses
| Name | No. of gca/ml | No. of PFU/mlb | No. of gc/PFU |
|---|---|---|---|
| KOS | 8.5 × 1010 | 3.3 × 109 | 25.8 |
| K-gB:668N | 1.9 × 1011 | 5.9 × 109 | 32.2 |
| K-gB:NT | 1.4 × 1011 | 3.9 × 109 | 35.9 |
| K-gH:KV | 2.4 × 1011 | 7.3 × 109 | 32.8 |
| K-gB:668N-gH:KV | 2.0 × 1011 | 2.7 × 109 | 74.1 |
gc, genome copies determined by qPCR.
Biological titers on Vero cells.
K-gH:KVΔgD and K-gB:668N-gH:KVΔgD were produced by cotransfection of VD60 cells with plasmid pΔgD-EGFP and viral DNA from K-gH:KV or K-gB:668N-gH:KV, respectively, followed by purification of green plaques on VD60 cells. The recombinants were propagated and titered on VD60 cells.
All recombinant viruses were confirmed by PCR and DNA sequencing through the relevant glycoprotein genes or deletions.
Virus titration.
Standard virus titrations were performed by incubating Vero cells with serial 10-fold dilutions of virus for 2 h at 37°C with intermittent agitation. Medium containing methylcellulose was then added, and the cultures were incubated at 37°C for 3 days prior to staining with crystal violet and counting the number of plaques.
Plasmids.
Plasmid pgB1:S668N was created by substituting a sequenced S668N-containing gB fragment amplified on DNA from one of the isolates for the corresponding fragment of pgB1, a plasmid containing the gB open reading frame (ORF) and flanking regulatory sequences from K26GFP (37). The gH-null recombination plasmid pΔgH-EGFP was created by replacing the sequence of the gH ectodomain and transmembrane region in pgH1:wt, a plasmid that contains the gH ORF and flanking regulatory sequences from KOS, with the enhanced GFP (EGFP) ORF from pEGFP-C1 (Clontech). Plasmid pgH1:N753K/A778V was created by the substitution of a sequenced N753K/A778V-containing gH fragment amplified on DNA from one of the isolates for the corresponding fragment of pgH1:wt; the transferred gH fragment did not include a highly GC-rich region that was difficult to sequence (positions 2,079 through 2,102 of GenBank accession number X03896), so this GC-rich region in the recombinant was derived from the wild-type (wt) gH gene. The gD-null recombination plasmid pΔgD-EGFP was described previously (37). Expression plasmids for gB, gD, gH, and gL under the control of the chicken β-actin promoter and human cytomegalovirus (CMV) IE enhancer, pPEP98, pPEP99, pPEP100, and pPEP101, respectively (44), were provided by Patricia Spear (Northwestern University). Plasmid pCAgB:wt was created by replacing the gB ORF of pPEP98, which diverged from the wild-type KOS gB sequence (44), with that of K26GFP. To create pCAgB:NT, a fragment containing the NT mutations was isolated from plasmid pgB1:D285N/A549T (37) and substituted for the corresponding fragment of pPEP98. Plasmid pCAgH:KV was created by replacing the gH ORF of pPEP100 with that of pgH1:N753K/A778V. Plasmids pCAgH:N753K and pCAgH:A778V were created by substituting a gH fragment containing either N753K or A778V for the corresponding fragment of pPEP100. All plasmid constructs were confirmed by DNA sequencing. Plasmid pO7G was provided by David Krisky (Diamyd, Inc., Pittsburgh, PA). Plasmid pCAVP16 expresses HSV-1 VP16 from the chicken β-actin promoter and human CMV IE enhancer.
Selection of virus isolates.
J/TMCΔC cells (in twenty 10-cm dishes) were inoculated with K26GFP at an approximate multiplicity of infection (MOI) of 1,000 and rinsed with 0.1 M glycine (pH 3.0) (referred to hereafter as acidic wash) at 24 h postinfection (hpi). Combined intracellular and extracellular virus harvested at 72 hpi (first-round product) was expanded on J/A cells for a second round of infection of J/TMCΔC cells (in twenty 10-cm dishes) at an MOI of ∼1,000 and acidic wash at 24 hpi. Progeny virus was again harvested and expanded (second-round product). After two more rounds of selection at the same MOI and one round at an MOI of ∼300, plaques were purified by limiting dilution on B78/TMC cells expressing the full-length version of TMCΔC (37). Selected isolates were analyzed by selective sequencing. All mutant sequences reported here were unambiguous, confirming the purity of the isolates and the absence of wild-type virus.
Entry assay.
Entry assays were performed as described previously (37). Briefly, cells were infected for 6 or 8 h and immunostained using monoclonal mouse anti-ICP4 (1:300) (Santa Cruz Biotechnology) as the primary antibody and Cy3-conjugated sheep anti-mouse IgG (1:400) (Sigma) as the secondary antibody. Images were obtained with a Nikon Diaphot fluorescence microscope.
Infectious center assay.
Infectious center assays were performed as described previously (36). Briefly, donor cells (Vero or VD60) were infected at an MOI of 10 at 37°C for 2 h followed by an acidic wash. The cells were incubated at 37°C for 1 h, trypsinized, and suspended in culture medium. Equal numbers of infected (donor) cells were seeded onto monolayers of uninfected (acceptor) cells in a 48-well plate. After a 3-h incubation at 37°C, the cells were overlaid with methylcellulose-containing medium. Two or 3 days later, the overlay was removed, and the cells were fixed with 100% methanol and immunostained with monoclonal mouse anti-VP16 antibody (1:400) (Santa Cruz Biotechnology) and Cy3-conjugated sheep anti-mouse IgG (1:400). Images were obtained with a Nikon Diaphot fluorescence microscope.
Virus growth assay.
Vero and B78/C cells were infected at an MOI of 3 for 1 h followed by an acidic wash. The cells were incubated for 4, 8, or 24 h at 37°C, the media and cells were separated by centrifugation, and cell lysates were prepared by three rounds of sonication. The titers of the cell lysates and media were determined separately on Vero cells.
Cell fusion assay.
B78H1 or B78/C cells were seeded at a density of 4 × 104 cells per well in a 48-well plate and cotransfected the following day with 1 μg each of pPEP99, pCAgB:wt, pPEP101, and either pPEP100, pCAgH:KV, pCAgH:N753K, pCAgH:A778V, or pCAgB:NT using Lipofectamine 2000 (Invitrogen). An empty pSP72 vector was substituted where plasmids expressing gD, gB, or gL were omitted. B78/C cells were fixed with 100% methanol and stained with Giemsa solution (Invitrogen) at either 20 h posttransfection (all glycoproteins included) or 72 h posttransfection (gD, gB, or gL omitted). To quantify the number of syncytia formed in the absence of nectin-1, transfected B78H1 cells were trypsinized at 6 h posttransfection, diluted 1:1 (vol/vol) with untransfected B78H1 cells, and replated in 48-well plates. At 72 h posttransfection, cells were fixed with 100% methanol and stained with Giemsa solution, and syncytia with more than 5 nuclei were counted. The average number of syncytia per well (± the standard error of the mean [SEM]) was scored for two separate experiments, with each transfection performed in triplicate.
Molecular dynamics simulations.
The simulation system in this study consisted of domains H2 and H3 of HSV-1 glycoprotein H (gH1). In the absence of structural X-ray data for gH1, we used Swiss-Model (45) (http://swissmodel.expasy.org) to build a homology model for gH1 based on the structure of HSV-2 gH (gH2) (Protein Data Bank identification no. 3MIC) (30); the sequences of gH1 and gH2 are very similar (∼92% identical residues). The H2-H3 portion of the homology model, encompassing residues N332 to P797, was solvated using explicit single-point charge water molecules (46), leading to a simulation system consisting of a total of ∼47,926 atoms. The system was initially energy minimized using the steepest-descent algorithm, followed by an equilibrium simulation for 1 ns. During the equilibrium simulation, the backbone atoms of the protein were fixed by a harmonic restraint, allowing the water molecules and side chain atoms to relax. This was followed by a production run of 100 ns during which all restraints were removed. The simulation was performed at 310 K, with temperature coupling using Berendsen's method (47). Electrostatic potentials were calculated using the particle mesh Ewald method (48). The bond lengths were restrained using the LINCS algorithm (49), and an integration time step of 2 fs was employed. The GROMOS 43a1 force field (50) was applied, and the GROMACS program (51), version 4.1, was used to generate the simulation trajectories. A second trajectory was generated with the same protocol, except that residues N753 and A778 in the H3 domain were changed to K753 and V778, respectively.
RESULTS
Selection of virus mutants.
Cells that lack functional forms of the canonical HSV-1 entry receptors HVEM, nectin-1, and 3-OS HS are referred to here as gD receptor-deficient cells, although they may express minor receptors for wild-type gD (CHO cells) (52, 53) or receptors that recognize mutated versions of gD (nectin-2 or nectin-3) (54, 55).
J/TMCΔC cells are gD receptor-deficient J1.1-2 baby hamster kidney cells that stably express a severely debilitated version of the HSV entry receptor nectin-1 (37). The defective receptor, TMCΔC, has mutations in the nectin-1 variable (V) domain that reduce gD binding and lacks the two constant (C) domains of nectin-1 to further limit its entry-mediating activity (56). After several rounds of high-MOI infection with K26GFP, a recombinant HSV-1 strain-KOS virus that expresses VP26-GFP as a fusion product (40), and progeny amplification on HVEM-transduced J1.1-2 (J/A) cells, green plaques were observed on J/TMCΔC cells, and a number of them were purified. Sequencing of the 4 essential glycoprotein genes of one of these isolates identified 2 substitution mutations in the gH gene, N753K and A778V, without mutations in gD, gB, or gL. We characterized the gH genes of another 6 isolates that yielded large plaques on J/TMCΔC cells and found the same pair of gH substitutions, referred to hereafter as KV, in all of them (Fig. 1). We also identified a frequent substitution in gB, gB:S668N, that created a new HincII restriction site [Fig. 1, Hinc(+)]. Sequencing or HincII digestion demonstrated the presence of this substitution in 4 of the 7 gH:KV isolates and a majority of incompletely characterized isolates (77% overall, n = 30) (Fig. 1). Several of the gH:KV isolates also had one of three missense mutations in gD, but the frequency of gD mutations in the entire set of isolates was relatively low (35%, n = 46) (Fig. 1). These results suggested that the KV mutations in gH were essential for the acquired ability of the isolates to infect and form plaques on J/TMCΔC cells and that additional mutations in gB and/or gD could facilitate these new activities.
Fig 1.

Selective characterization of virus isolates. A total of 46 isolates were initially characterized by complete sequencing of their gD genes and grouped according to the results; the number of isolates in each group is shown. Sequencing of the gB genes of selected isolates identified the existence of 2 alleles, wt and S668N. Additional isolates were screened for the presence of the S668N-diagnostic HincII restriction site, and the number of isolates scoring positive [Hinc(+)] or negative [Hinc(-)] is indicated. Several isolates representing similar or different combinations of gD/gB alleles were further characterized by nearly complete or localized (*) gH sequencing, and the complete sequence of the gL gene of one isolate was determined.
The gB and gH mutations enhance virus entry into cells that lack canonical gD receptors.
To evaluate the suggestion that the KV mutations in gH were sufficient for virus growth in the absence of known functional gD receptors, we transferred these mutations into a wild-type virus background; we also separately introduced the gB:S668N mutation into a wild-type virus backbone to explore its effects on entry and spread. The resulting recombinant viruses are referred to as K-gH:KV and K-gB:668N, respectively. In addition, we established a double-recombinant virus, K-gB:668N–gH:KV, to identify potential combinatorial effects of the mutant gB and gH alleles. Each recombinant virus was confirmed by DNA sequencing, and stocks were propagated simultaneously and their titers were determined on Vero cells. Stocks of wild-type HSV-1 KOS and K-gB:NT, containing the previously described entry-accelerating NT mutations in gB (37), were prepared at the same time. Determination of the genome titers of these various stocks, expressed as the number of genome copies (gc)/ml (43), demonstrated <3-fold differences in their particle-to-PFU ratios (Table 1).
Since our previous work (37) showed that K-gB:NT has the ability to enter CHO-K1 cells, another HSV-1-resistant cell line that lacks canonical gD receptors (8), we first asked whether the new recombinant viruses shared this ability. As shown in Fig. 2A, entry of K-gB:NT into CHO-K1 cells was readily detectable at an MOI of 30, whereas essentially no entry was seen for wild-type KOS at an MOI of 300. K-gH:KV reproducibly showed a higher level of entry than wild-type KOS but an approximately 10-fold lower level than K-gB:NT, while K-gB:668N showed only a slightly increased entry level relative to KOS. Interestingly, the double-recombinant K-gB:668N–gH:KV yielded an entry level that was at least 10-fold higher than that of K-gB:668N or K-gH:KV, comparable to the level observed for K-gB:NT. As expected, all of the mutant viruses and wild-type KOS entered Vero cells to similar degrees (Fig. 2A), confirming equal input of functional virus particles. Comparison of the new viruses with KOS on B78H1 cells that also lack canonical gD receptors showed a similar pattern (Fig. 2B), demonstrating that the observed effects of the mutant alleles were not unique to CHO-K1 cells. Together, these results indicate that the gH:KV and gB:S668N mutant alleles possessed limited and differing abilities to compensate for the absence of canonical gD receptors in virus entry but could act cooperatively to enhance this ability.
Fig 2.
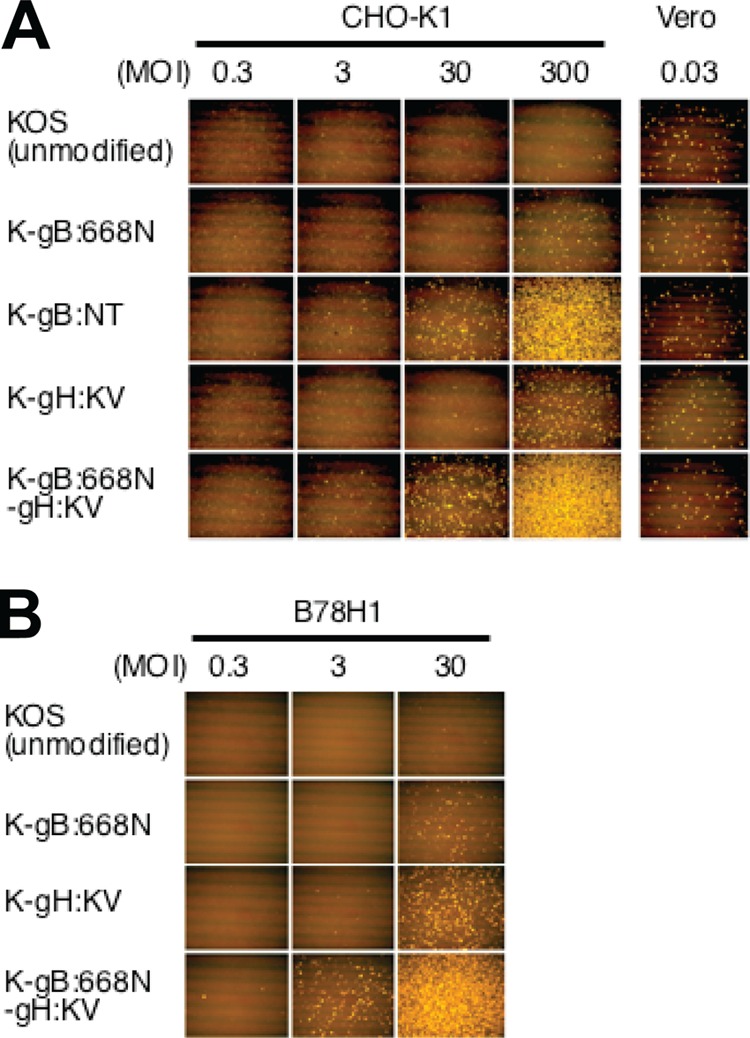
Effects of gB and gH mutations on virus entry into gD receptor-deficient cells. CHO-K1 and Vero cells (A) and B78H1 cells (B) were infected for 6 h at the MOIs shown at the top of the columns and immunostained for ICP4. Images are representative of 3 independent experiments.
Mutations in gH, but not gB, facilitate lateral virus spread on gD receptor-deficient cells.
We performed infectious center assays (17, 36) to determine whether the new gB and gH mutations affected cell-to-cell spread. Vero cells were infected with wild-type KOS or mutant viruses at an MOI of 10 to achieve 100% infection. After inactivation of residual extracellular virions by acidic wash, equal numbers of infected (donor) cells were seeded onto monolayers of uninfected (acceptor) cells. The cultures were overlaid with methylcellulose-containing medium, and plaque formation was assessed 2 days later by VP16 immunostaining. As shown in Fig. 3A, each of the viruses formed plaques on acceptor Vero cells and gD receptor-transduced B78 cell lines (B78/A, B78/C), regardless of the nature of the gB and gH alleles. Surprisingly, however, the two viruses harboring gH:KV formed plaques on gD receptor-negative B78 cells as well (B78/0G, containing a GFP gene controlled by the virus-inducible ICP0 promoter) (Fig. 3A, top row). Observation of green fluorescence confirmed that these plaques consisted of infected acceptor cells (Fig. 3B). At higher magnifications, several layers of fluorescent cells could be seen around a vacant space in the middle, indicative of initial virus spread from a central infected Vero cell to its immediate neighbors followed by multiple rounds of spread from one gD receptor-negative cell to the next. Similar results were obtained using CHO/0G cells, another gD receptor-deficient line that expresses GFP in response to virus infection (data not shown). These observations provided compelling evidence that the gH:KV double mutation enables spread between cells that lack typical gD receptors, whereas neither of the gB mutant alleles, S668N and NT, had this ability.
Fig 3.
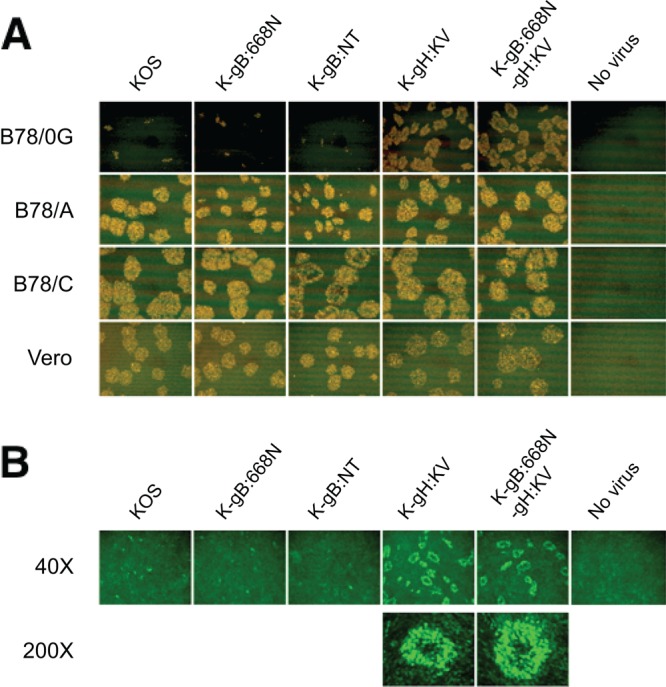
Effects of gB and gH mutations on cell-to-cell spread to and between gD receptor-deficient cells. (A) Vero cells were infected with the viruses indicated above the panels (MOI of 10). Extracellular virus was inactivated by acidic wash, and equal numbers of infected (donor) cells were added onto monolayers of the uninfected cells indicated at the left (acceptor cells). The mixed cultures were overlaid with methylcellulose-containing medium and immunostained for VP16 at 48 hpi. (B) B78/0G cells cultured for 48 h with the donor cells as described for panel A were observed under a fluorescence microscope. Magnifications are indicated at the left. Images are representative of 2 independent experiments.
Cell-to-cell spread of gH:KV mutant viruses requires gD.
To determine the requirement for gD in the spread of the two gH:KV recombinant viruses on gD receptor-deficient cells, we tested gD knockout versions of these viruses. Infectious center assays were performed as done earlier except that a gD-complementing cell line, VD60, was used for donor cells. VP16 immunostaining showed that the gD-null viruses yielded only single-cell infections or small foci on gD receptor-deficient B78H1 and receptor-positive Vero cells (Fig. 4). While the difference in signal intensities between the B78H1 and Vero cells may suggest that the gD-complemented gD-null viruses produced in the donor VD60 cells infect the immediately surrounding noncomplementing cells more efficiently when these cells express canonical gD receptors, it is clear that the subsequent spread between noncomplementing cells was minimal. Thus, it can be concluded that gD is required for the spread of gH:KV viruses regardless of the presence or absence of gD receptors.
Fig 4.
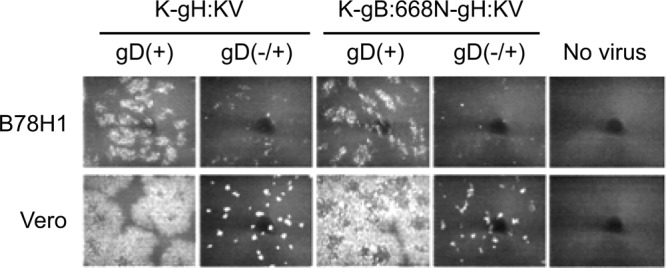
Effects of gD on cell-to-cell spread by gH:KV mutant viruses. VD60 cells were infected at an MOI of 10 with K-gH:KV, K-gB:668N-gH:KV [both gD(+)], or their gD knockout derivatives prepared on VD60 cells [gD(-/+)]. Extracellular virus was inactivated by acidic wash, and equal numbers of infected (donor) cells were added onto monolayers of uninfected B78H1 or Vero (acceptor) cells. The mixed cultures were overlaid with methylcellulose-containing medium and immunostained for VP16 at 72 hpi. Images are representative of 2 independent experiments.
gH:KV does not alter virus replication or egress.
To examine the possibility that increased virus replication, virion assembly, or transport to the cytoplasmic membrane played a role in the increased lateral spread of K-gH:KV on cells that lacked canonical gD receptors, we compared the replication and egress efficiencies of K-gH:KV with those of KOS. Vero cells were infected for 1 h at an MOI of 3, extracellular virions were inactivated by acidic wash, and viral titers in cell lysates and media at 4, 8, and 24 hpi were determined separately. As shown in Fig. 5A, the two viruses showed similar titers in both compartments at each of the selected time points. Similar results on B78/C cells (Fig. 5B) indicated that this outcome was not dependent on a specific cellular background. These observations provide evidence that the spread-enhancing activity of gH:KV could not be attributed to increases in the efficiency of virus replication or egress.
Fig 5.
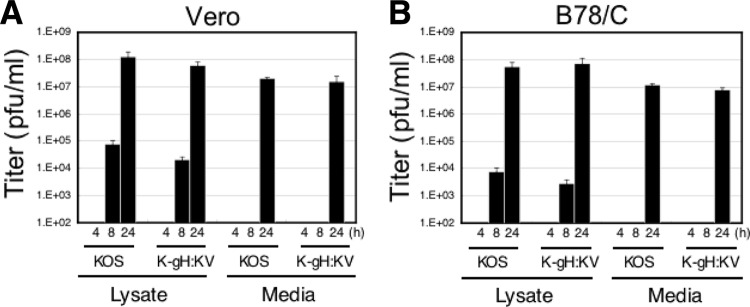
Effects of the gH:KV mutations on viral replication and egress. Vero (A) or B78/C (B) cells were infected at an MOI of 3 for 1 h followed by acid treatment. The titers of the cell lysates and media were determined following additional incubation at 37°C for 4, 8, or 24 h.
gH:KV promotes cell fusion.
Transfection of HSV-susceptible cells with the four essential glycoproteins, gB, gD, gH, and gL, causes cell-cell fusion, which is believed to reflect, at least in part, the normal functions of these glycoproteins and their receptors in HSV entry and spread (44, 57, 58). We used this cotransfection assay to determine whether gH:KV in combination with gB, gD, and gL could induce fusion of gD receptor-deficient B78H1 cells. The combined results of two separate experiments, each performed in triplicate, showed that substitution of the gH:wt gene with gH:KV increased polykaryocyte (syncytium) formation at least 10-fold (Fig. 6A). However, syncytium formation was negligible when the gD, gB, or gL gene was omitted (Fig. 6A; representative images are shown in Fig. 6B, upper panels), demonstrating that the gH:KV allele did not compensate for the absence of any of the other essential glycoprotein genes. We also tested the two gH mutations separately (gH:N753K and gH:A778V) and found that both of them increased cell-cell fusion, although less so than did the combination of them (Fig. 6A). In contrast, cotransfection of the entry-enhancing gB:NT gene with the gH:wt, gD, and gL genes did not increase syncytium formation over gB:wt in the same combination. Control experiments on receptor-bearing B78/C cells showed widespread cell-cell fusion with gH:wt and gH:KV, both of which were strictly dependent on coexpression of each of the three other glycoprotein genes (Fig. 6B, lower panels). Highly efficient B78/C syncytium formation was also observed with the other alleles tested and shown in Fig. 6A (gB:NT, gH:N753K, and gH:A778V; data not shown), demonstrating that all of them were functional in the presence of a canonical gD receptor. These results indicate that gH:KV, but not gB:NT, facilitates a rate-limiting step in the events leading to cell-cell fusion, correlating with increased viral cell-to-cell spread but only minimally with viral entry. In contrast, the lack of spread and cell fusion enhancement by gB:NT suggests that this allele acts on a rate-limiting step that is unique to viral entry.
Fig 6.

Effects of the gH:KV mutations on cell-cell fusion. (A) B78H1 cells were cotransfected with plasmids for the indicated glycoproteins, Giemsa staining was performed at 72 h posttransfection, and syncytia were counted. The gH plasmids containing either of the KV mutations alone are indicated as K (for N753K) and V (for A778V). Shown are the averages ± the SEM of data from two experiments performed in triplicate. (B) Representative images of syncytium formation by B78H1 (upper panels) and B78/C (lower panels) cells after cotransfection of different combinations of expression plasmids for wild-type gD, gB, and gL, indicated at the top, with plasmids expressing gH:wt or gH:KV, as indicated at the left. Giemsa-stained cells were photographed at either 20 h posttransfection (B78/C, first column) or 72 h posttransfection (all other panels).
Comparative structure modeling of gH:KV.
We used molecular dynamics (MD) simulations to model the possible effects of the KV mutations on the conformation and dynamic properties of gH. Since the X-ray structure of HSV-1 gH (gH1) is not yet available, we derived a homology model for the H3 domain of gH1, containing the positions of the two mutations, and the neighboring H2 domain (H2-H3, residues N332 through P797) from the published X-ray structure of HSV-2 gH (gH2) (30); gH1 and gH2 share 82.4% sequence identity in H2-H3, which is sufficient for modeling the unknown structure of these domains for gH1 (59). We first modeled a short stretch of missing H3 residues (R720 through R724) into the gH2 structure, and this structure was then used to develop a homology model for the H2-H3 domains of gH1 (Fig. 7A). The model was energy minimized in a solvated environment to remove potential steric clashes, and the dynamics of the solvated structures for gH:wt and gH:KV were accessed from 100-ns simulation trajectories generated using GROMACS (51).
Fig 7.

Global and local effects of the gH:KV mutations. (A) Ribbon diagram of the H2-H3 domains of HSV-1 gH:wt with the positions of the mutant residues in gH:KV (N753K, A778V) highlighted as space-filling models in magenta and the two halves of the flap represented as dark- and light-blue sticks. (B) Cumulative root mean square deviation (RMSD) from the respective starting structures for the H2-H3 domain of HSV-1 gH:wt and gH:KV. (C) RMSDs of two sides of the flap region, E707-K716 and R717-D726.
A measure of the stability of a simulation system is obtained from the root mean square deviation (RMSD) from the starting structure (60–62). The RMSDs for the H2-H3 region of both gH:wt and gH:KV stabilized at ∼0.5 nm after ∼30 ns (Fig. 7B), indicating that the system reached a stable equilibrium conformation at that time. We also examined the RMSD of the gH1 region E707 through D726 in domain H3 that is homologous to the “flap” identified in the crystal structure of PRV gH by Backovic et al. (63). These authors proposed that the flap is a dynamic structure with the potential to move in order to expose an H3-internal hydrophobic region for interaction with membranes. We analyzed the stability of each half of the flap separately for increased sensitivity compared to that of analysis of the complete flap. The first half (E707 through K716) (Fig. 7A) exhibited a low and stable RMSD of ∼0.22 nm for both the wt and mutant gH1 structure (Fig. 7C), suggesting that it is relatively static in HSV-1. In contrast, the RMSDs for the second half (R717 through D726) (Fig. 7A) jumped to ∼0.45 nm and further increased to ∼0.6 nm at about 50 ns for gH:KV and to ∼0.5 nm at about 70 ns for gH:wt (Fig. 7C). These observations suggest that the R717-to-D726 side of the flap is relatively mobile and that its mobility is enhanced by one or both of the mutations.
Based on the minimum distances between atoms of different residues, we examined the 6 closest interactions between pairs of residues in the H3 domain and the 3 closest interactions at the interface of H2-H3; the positions of the interacting pairs are shown in Fig. 8A (H3) and 8B (H2-H3). We monitored the time evolution of these interactions to gain a broader sense of the stability of H3, its mobility relative to H2, and the effect of the KV mutations on these parameters. An interaction was considered strong if the minimum distance between any 2 atoms of the interacting residue pair was ∼0.35 nm or less and stable if that distance was maintained for at least 20 ns (60). Figure 9 shows the time evolution of the interactions monitored within the H3 domain. The T700_G685 interactions are relatively weak (∼0.45 nm) but stable at that distance in both gH:wt and gH:KV (Fig. 9, upper panels). The T700_L680 interactions in both gH:wt and gH:KV oscillate between 0.2 nm and >0.4 nm, but in gH:wt the interaction is eventually disrupted at 80 ns. The S745_R736 and A/V778_L790 interactions (Fig. 9, middle panels) are strong (∼0.2 nm) and stable in both gH:wt and gH:KV, although the A778V substitution in gH:KV appears to cause an increase in the distance fluctuation between residues 778 and 790. The M735_V675 and G654_G708 interactions (Fig. 9, bottom panel) both settle at a steady distance, but neither is particularly strong. These results suggest that there is no dramatic difference in the overall stability of the H3 domain between gH:wt and gH:KV.
Fig 8.
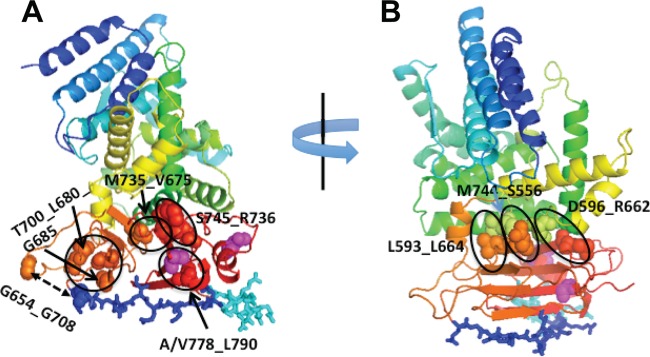
Ribbon diagrams showing the location of residues whose pairwise interactions were monitored. (A) Pairs of residues in the H3 domain; (B) pairs of residues across the H2-H3 interface. Magenta spheres, positions of the KV mutations (753 and 778); dark- and light-blue sticks, flap residues. Monitored interactions are labeled and circled or indicated by two-sided arrows.
Fig 9.

Time evolution of the interaction between pairs of residues in the H3 domain for gH:wt (A) and gH:KV (B). Top, T700_L680 and T700_G685; middle, S745_R736 and A/V778_L790; bottom, M735_V675 and G654_G708. Interactions are defined as the minimum distance between any two atoms of the pairs at a given time along the simulation trajectory.
The monitored interactions at the H2-H3 interface, L593_L664, M744_S556, and D596_R662, showed more variability between the wt and KV versions of gH1 (Fig. 10A). While the L593_L664 interactions are relatively stable in both gH:wt and gH:KV, the M744_S556 and D596_R662 interactions are not, showing considerable fluctuations, particularly in gH:KV, where both interactions are disrupted at ∼30 to 40 ns (distance, >0.5 nm) (Fig. 10A, right panel) and remained so for 30 to 35 ns. The D596_R662 interactions were disrupted once more, albeit briefly, at ∼80 ns in both the wt and the mutant form. Snapshots of gH:wt and gH:KV at 45 ns (Fig. 10B) illustrate the looser packing of the H2-H3 interface in the mutant than in wt gH1. This observation raises the possibility that the KV mutations facilitate the opening up of the interface for H2 and/or H3 interaction with other molecules, potentially gD (35).
Fig 10.

Time evolution of the interaction between pairs of residues at the H2-H3 interface and structure representations. (A) Interactions L593_L664 (black), M744_S556 (light blue), and D596_R662 (dark blue) for gH:wt (left) and gH:KV (right). (B) Ribbon diagrams of the H2-H3 domains of gH:wt (left) and gH:KV (right) at 45 ns. The pairs of interacting residues monitored in the diagrams at the top are highlighted in space-filling representation and are color coded as in panel A.
DISCUSSION
The goal of this study was to select virus mutants that had acquired the ability to enter and spread on cells that expressed only a debilitated gD receptor, TMCΔC. We have no direct evidence that any of the isolates identified here or in our previous study using slightly different starting materials (37) were selected as a consequence of compensatory mutations in gD that rescued recognition of the mutant receptor. Instead, mutations consistently found in our isolates were in the gH gene (this study) or the gB gene (37) and appeared to facilitate downstream events in the processes of virus entry or spread. Although both of these processes involve a fusion step requiring the 4 essential envelope glycoproteins, gB:NT only enhanced entry into gD receptor-deficient cells, while gH:KV mainly promoted spread. This suggests that these two events are governed by different rate-limiting steps and that the roles of gB and gH differ between entry and spread. Viruses containing gB:NT required gD for entry into cells that lacked canonical gD receptors (37), and viruses containing gH:KV required gD for spread on these cells, suggesting that both mutant proteins had acquired the ability to respond to unstimulated gD or limited activation of gD caused by binding to noncanonical receptors (37). Given our rudimentary understanding of how the 4 essential glycoproteins interact, it is premature to speculate how this potentially common trigger promotes one event but not the other depending on the particular mutant glycoprotein. While recent reports have suggested that gB is the fusogen in HSV entry and spread, whereas gH serves a regulatory function (30), much of the evidence comes from virus-free systems that do not clearly distinguish between entry and spread (30, 34, 35). In fact, the virus-free cell fusion assay in our study reflected the activities of gB:NT and gH:KV in spread but not in entry, supporting the view that this assay is limited as a model for HSV entry.
Although our results show that the gH:KV allele enhances cell fusion and lateral virus spread on gD receptor-deficient cells, our gH:KV viruses did not show a syncytial phenotype regardless of the presence or absence of canonical receptors on the cells (e.g., see Fig. 3A and 4). This suggests that while gH:KV alleviates, to a degree, a rate-limiting step in these processes in the absence of canonical entry receptors, there is another layer of control that prevents runaway virus-mediated fusion in the presence (or absence) of these receptors. Unlike gH:KV, typical syncytial mutations identified in other glycoproteins appear to breach this control. While syncytial mutations have been identified in gB (see reference 64 and references therein), we note that K-gB:NT and K-gB:668N also are not syncytial viruses. For K-gB:NT, this is consistent with our observation that the gB:NT allele has similar activity in the cell fusion assay as the wild-type gB gene; we did not test the gB:S668N gene with this assay. Thus, our gH and gB mutations differ in their mechanisms not only from one another but also from the standard syncytial mutations.
None of the three domains, H1, H2, or H3, identified in the crystal structure of the HSV-2 gH ectodomain showed any resemblance to previously characterized fusion proteins, supporting the suggestion that gH is not a viral fusogen but is instead a regulator of the fusion activity of gB (30). However, in their report of the crystal structure of a core fragment of the PRV gH ectodomain, Backovic et al. proposed fusion-related functions for the conserved flap and hydrophobic patch they identified in the H3 domains of PRV, HSV-2, and Epstein-Barr virus (EBV) (63). The authors suggested a model in which the hydrophobic patch is unmasked by movement of the flap during a conformational change in gH triggered by gD binding to a receptor, exposing the hydrophobic surface to interact with the viral membrane during the fusion process. Most recently, a targeted mutagenesis study by the same investigators provided support for aspects of this model, in particular the proposal that movement of the flap relative to the hydrophobic patch is important for gH function (65). However, based on results from a structure-function analysis of varicella-zoster virus (VZV) gH, the opposite has also been argued, i.e., that stability rather than movement of the flap is a requirement for gH functions (66).
The gH:KV mutant positions 753 and 778 are also located in the membrane-proximal H3 domain of gH. This is the most highly conserved region of gH among different herpesviruses and is therefore expected to be functionally important. It has been shown that linker insertions at amino acids 691/692, 791, or 799 within H3 significantly reduce cell fusion mediated by a syncytial strain (67). Furthermore, synthetic peptides containing amino acid 778 have been found to induce fusion of liposomal membranes in lipid-mixing assays and to inhibit HSV infection (68, 69). Amino acid 778 is adjacent to the conserved hydrophobic patch described in the PRV gH paper (63) and is close to α-helix α22 containing amino acid 753 (30). Thus, amino acids 753 and 778 may affect the proposed membrane-interacting surface of gH that is suggested to be exposed by dislocation of the flap. Alternatively, either or both N753K and A778V could change the positioning of this surface relative to the flap in such a manner that complete disengagement of these two structures requires only a weak signal, likely from gD. This second scenario is the most attractive, as it would offer an explanation for the gD dependence of gH:KV-enhanced spread between cells that lack canonical gD receptors but express minor receptors capable of limited gD activation, such as nectin-3 (36). As discussed below, our molecular dynamics simulations suggested another possibility.
In our gH1 models based on the published structure of gH2 (30), the A778V mutation did not seem to alter the dynamics of the H3 domain, as indicated by our analysis of interactions within this domain close to or with residue 778 (Fig. 9). These interactions were predominantly stable in both gH:wt and gH:KV. In addition, the N-terminal half of the flap region (residues E707 through K716) was stable in both of them (Fig. 7C). However, the C-terminal half of the flap was not as stable and exhibited greater instability in gH:KV than in gH:wt. While this could facilitate partial exposure of the hydrophobic patch underneath it (63), including the proposed fusion peptide (69), it has been questioned how exposure of the patch could contribute to membrane fusion without major disruption of the H3 core (30). It is also noteworthy that the second half of the flap includes 5 residues not resolved in the gH2 structure whose positions and dynamics must thus be considered conjectural. Interactions destabilized in the KV mutant were also identified at the H2-H3 interface (M744_S556 and D596_R662) (Fig. 10A). Both interactions transiently weakened in the mutant gH (distance increases to >0.5 nm) before falling more in line with the wt pattern. This observation suggests that the KV mutations may destabilize the H2-H3 interface to facilitate interactions with other proteins involved in (pre)fusion. The gB binding site of gH has been proposed to be in a highly conserved region near the boundary between domains H1 and H2, at a distance from the H2-H3 interface (30). Thus, it seems unlikely that the KV mutations affect gB binding directly. Instead, our experimental data suggesting that gH:KV, unlike gH:wt, is responsive to unstimulated or weakly stimulated gD raise the possibility that the mutations facilitate H3 displacement by gD. For example, interaction of the membrane-proximal profusion domain of gD with the membrane-proximal face of the H2 domain of gH could mediate a conformational change in H2 necessary for gB binding, resulting in gB activation and membrane fusion; other and more complex mechanisms can also be proposed. Thus, the biological properties of the mutations we have described, along with the modeling of their effects on the structure of gH, point to the H2-H3 interface as a potential gD binding region whose role in gD-dependent HSV entry can be experimentally tested.
Aside from the KV substitutions in gH identified in all of the large-plaque isolates examined for these substitutions, many of our isolates also contained a common substitution in gB, S668N, and a smaller fraction had one of three substitutions in gD. While at least one isolate contained only gH:KV without substitutions in gB or gD, we consider it likely that the gB and/or gD mutation(s) observed in all other isolates provided some advantage in our selection protocol. For example, we showed that the S668N substitution in gB can act cooperatively with gH:KV to facilitate entry into cells that lack conventional gD receptors, thus addressing a bottleneck in the protocol. However, gB:S668N did not detectably enhance the receptor-independent spread phenotype of gH:KV; thus, it represents another glycoprotein mutation that acts differently in entry and spread. The S668N substitution was previously identified in the gB gene of an oncolytic HSV-1 mutant, HF10 (70), and has also been described as one of three gB substitutions found in an HSV-1 isolate that was selected for resistance to the virucidal activity of a phosphorothioate oligonucleotide (71). However, the effect of this substitution in the context of a wild-type virus has not been reported. The crystal structure of the HSV-1 gB ectodomain shows that amino acid 668 is located in a short outer β-strand, β36, that belongs to a neighboring protomer (25). This β-strand participates in shaping the core domain (domain III) and is close to a short α-helix, αD, that contains two of the three known gB residues (at positions 549 and 553) whose substitution can accelerate the viral rate of entry (37, 72). While this may suggest that gB:S668N and the A549T substitution of the gB:NT allele influence the same stage of the entry process, the entry-enhancing activity of gB:S668N alone was limited compared to that of gB:NT. Additional work is required to determine whether S668N and A549T act through related mechanisms.
Previous studies support the possibility that the gD substitutions identified in some of our isolates may have provided a selective advantage as well. In one report, the selection of HSV variants for growth on receptor-deficient J1.1-2 cells identified three substitutions in gD that together enabled nectin-3-mediated gD activation and virus entry (54). Two of these substitutions existed in the HSV-1 KOS strain used in our work (340H and 344R), and the third (S140N) occurred at the same position as a substitution (S140K) identified in 3 of our isolates (Fig. 1). Since the selective J/TMCΔC cells used in our study were derived from J1.1-2 cells, S140K may have promoted the selection of these 3 isolates by facilitating entry via nectin-3. The more prevalent A185T substitution (Fig. 1) was also encountered in our previous selection study, where it was one of only two gD substitutions observed in combination with gB:NT (37). A185T has been shown to facilitate lateral spread on receptor-deficient Syrian hamster US11cl19.3 cells (19, 54), consistent with the possibility that it too facilitates gD interaction with noncanonical receptors. Using reconstructed viruses containing S140K or A185T in a wild-type KOS background, we have observed small increases in entry and plaque formation by the S140K virus on J1.1-2 and B78H1 cells expressing TMC, the less debilitated predecessor of TMCΔC preserving the two nectin-1 C domains (37), but neither virus showed enhanced entry or spread on unmodified J1.1-2 or B78H1 cells compared to the KOS strain (unpublished results). These results and the observation that a majority of the selected viruses contained wild-type gD indicate that the gD substitutions identified in some of our isolates did not play a key role in the selection of these isolates, although we have not excluded the possibility that they can act cooperatively with gH:KV and/or gB:S668N.
In summary, we have identified a pair of missense mutations in the gH gene that promote lateral HSV-1 spread in the absence of canonical gD receptors. The phenotype of these mutations is different from that of the previously described entry-enhancing gB:NT mutations, despite a shared dependence on gD. The gain-of-function nature of these distinct pairs of mutations should be valuable in ongoing studies of glycoprotein interactions in HSV entry and spread.
ACKNOWLEDGMENTS
We thank David Johnson, Anthony Minson, Gary Cohen, Patricia Spear, Gabriella Campadelli-Fiume, Stanley Person, and Prashant Desai for reagents. We also thank Zhanna Hakhverdyan and David Clawson for technical assistance and Bill Goins for technical advice and helpful discussions.
This work was supported by NIH grants CA119298, NS40923, and DK044935 (to J.C.G.).
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Cai WH, Gu B, Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J. Gen. Virol. 69:1147–1156 [DOI] [PubMed] [Google Scholar]
- 3. Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 66:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. 1998. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 72:6119–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 8. Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 9. Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22 [DOI] [PubMed] [Google Scholar]
- 10. Carfí A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169–179 [DOI] [PubMed] [Google Scholar]
- 11. Fusco D, Forghieri C, Campadelli-Fiume G. 2005. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc. Natl. Acad. Sci. U. S. A. 102:9323–9328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24:4144–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nicola AV, Straus SE. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508–7517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cocchi F, Menotti L, Dubreuil P, Lopez M, Campadelli-Fiume G. 2000. Cell-to-cell spread of wild-type herpes simplex virus type 1, but not of syncytial strains, is mediated by the immunoglobulin-like receptors that mediate virion entry, nectin1 (PRR1/HveC/HIgR) and nectin2 (PRR2/HveB). J. Virol. 74:3909–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roller RJ, Rauch D. 1998. Herpesvirus entry mediator HVEM mediates cell-cell spread in BHK(TK-) cell clones. J. Virol. 72:1411–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826 [DOI] [PubMed] [Google Scholar]
- 19. Rauch DA, Rodriguez N, Roller RJ. 2000. Mutations in herpes simplex virus glycoprotein D distinguish entry of free virus from cell-cell spread. J. Virol. 74:11437–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Babic N, Mettenleiter TC, Flamand A, Ugolini G. 1993. Role of essential glycoproteins gII and gp50 in transneuronal transfer of pseudorabies virus from the hypoglossal nerves of mice. J. Virol. 67:4421–4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peeters B, de Wind N, Hooisma M, Wagenaar F, Gielkens A, Moormann R. 1992. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J. Virol. 66:894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peeters B, Pol J, Gielkens A, Moormann R. 1993. Envelope glycoprotein gp50 of pseudorabies virus is essential for virus entry but is not required for viral spread in mice. J. Virol. 67:170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J. Virol. 65:5348–5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson DC, Huber MT. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 76:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 26. Galdiero S, Falanga A, Vitiello M, Browne H, Pedone C, Galdiero M. 2005. Fusogenic domains in herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 280:28632–28643 [DOI] [PubMed] [Google Scholar]
- 27. Galdiero S, Vitiello M, D'Isanto M, Falanga A, Collins C, Raieta K, Pedone C, Browne H, Galdiero M. 2006. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 87:1085–1097 [DOI] [PubMed] [Google Scholar]
- 28. Gianni T, Martelli PL, Casadio R, Campadelli-Fiume G. 2005. The ectodomain of herpes simplex virus glycoprotein H contains a membrane alpha-helix with attributes of an internal fusion peptide, positionally conserved in the Herpesviridae family. J. Virol. 79:2931–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gianni T, Menotti L, Campadelli-Fiume G. 2005. A heptad repeat in herpes simplex virus 1 gH, located downstream of the alpha-helix with attributes of a fusion peptide, is critical for virus entry and fusion. J. Virol. 79:7042–7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 104:10187–10192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian RP, Geraghty RJ. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. U. S. A. 104:2903–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jackson JO, Longnecker R. 2010. Reevaluating herpes simplex virus hemifusion. J. Virol. 84:11814–11821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazear E, Whitbeck JC, Ponce-de-Leon M, Cairns TM, Willis SH, Zuo Y, Krummenacher C, Cohen GH, Eisenberg RJ. 2012. Antibody-induced conformational changes in herpes simplex virus glycoprotein gD reveal new targets for virus neutralization. J. Virol. 86:1563–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uchida H, Shah WA, Ozuer A, Frampton AR, Jr, Goins WF, Grandi P, Cohen JB, Glorioso JC. 2009. Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J. Virol. 83:2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB, Glorioso JC. 2010. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J. Virol. 84:12200–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frampton AR, Jr, Stolz DB, Uchida H, Goins WF, Cohen JB, Glorioso JC. 2007. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J. Virol. 81:10879–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forrester A, Farrell H, Wilkinson G, Kaye J, Davis-Poynter N, Minson T. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen X, Li J, Mata M, Goss J, Wolfe D, Glorioso JC, Fink DJ. 2000. Herpes simplex virus type 1 ICP0 protein does not accumulate in the nucleus of primary neurons in culture. J. Virol. 74:10132–10141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cai WZ, Person S, Warner SC, Zhou JH, DeLuca NA. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 61:714–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jiang C, Ataai M, Ozuer A, Krisky D, Wechuck J, Pornsuwan S, Pourarian F, Glorioso JC. 2006. Inactivation of herpes simplex type 1 gene vector on immobilized metal affinity chromatography: oxidative damage by hydroxyl free radicals and its prevention. Biotechnol. Bioeng. 95:48–57 [DOI] [PubMed] [Google Scholar]
- 44. Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 45. Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. 1981. Interaction models for water in relation to protein hydration, p 331–342 In Pullman B. (ed), Intermolecular forces. Reidel Publishing, Dordrecht, Netherlands [Google Scholar]
- 47. Berendsen HJ, Van Gunsteren WF, Zwinderman HR, Geurtsen RG. 1986. Simulations of proteins in water. Ann. N. Y. Acad. Sci. 482:269–286 [DOI] [PubMed] [Google Scholar]
- 48. Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092 [Google Scholar]
- 49. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 18:1463–1472 [Google Scholar]
- 50. Lindahl E, Hess B, van der Spoel D. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317 [Google Scholar]
- 51. van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. 2005. GROMACS: fast, flexible, and free. J. Comput. Chem. 26:1701–1718 [DOI] [PubMed] [Google Scholar]
- 52. Shieh MT, WuDunn D, Montgomery RI, Esko JD, Spear PG. 1992. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 116:1273–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 77:9221–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cocchi F, Menotti L, Di Ninni V, Lopez M, Campadelli-Fiume G. 2004. The herpes simplex virus JMP mutant enters receptor-negative J cells through a novel pathway independent of the known receptors nectin1, HveA, and nectin2. J. Virol. 78:4720–4729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189 [DOI] [PubMed] [Google Scholar]
- 56. Struyf F, Plate AE, Spear PG. 2005. Deletion of the second immunoglobulin-like domain of nectin-1 alters its intracellular processing and localization and ability to mediate entry of herpes simplex virus. J. Virol. 79:3841–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Muggeridge MI. 2000. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J. Gen. Virol. 81:2017–2027 [DOI] [PubMed] [Google Scholar]
- 58. Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leach AR. 2001. Molecular modelling: principles and applications, 2nd ed Pearson Education, New York, NY [Google Scholar]
- 60. Shrivastava I, LaLonde JM. 2011. Enhanced dynamics of HIV gp120 glycoprotein by small molecule binding. Biochemistry 50:4173–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shrivastava IH, Jiang J, Amara SG, Bahar I. 2008. Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J. Biol. Chem. 283:28680–28690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shrivastava IH, Sansom MS. 2000. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 78:557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. U. S. A. 107:22635–22640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J. Virol. 86:8171–8184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fuchs W, Backovic M, Klupp BG, Rey FA, Mettenleiter TC. 2012. Structure-based mutational analysis of the highly conserved domain IV of glycoprotein H of pseudorabies virus. J. Virol. 86:8002–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc. Natl. Acad. Sci. U. S. A. 108:18412–18417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Galdiero M, Whiteley A, Bruun B, Bell S, Minson T, Browne H. 1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J. Virol. 71:2163–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Galdiero S, Falanga A, Vitiello G, Vitiello M, Pedone C, D'Errico G, Galdiero M. 2010. Role of membranotropic sequences from herpes simplex virus type I glycoproteins B and H in the fusion process. Biochim. Biophys. Acta 1798:579–591 [DOI] [PubMed] [Google Scholar]
- 69. Galdiero S, Falanga A, Vitiello M, D'Isanto M, Collins C, Orrei V, Browne H, Pedone C, Galdiero M. 2007. Evidence for a role of the membrane-proximal region of herpes simplex virus Type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem 8:885–895 [DOI] [PubMed] [Google Scholar]
- 70. Ushijima Y, Luo C, Goshima F, Yamauchi Y, Kimura H, Nishiyama Y. 2007. Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect. 9:142–149 [DOI] [PubMed] [Google Scholar]
- 71. Shogan B, Kruse L, Mulamba GB, Hu A, Coen DM. 2006. Virucidal activity of a GT-rich oligonucleotide against herpes simplex virus mediated by glycoprotein B. J. Virol. 80:4740–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bzik DJ, Fox BA, DeLuca NA, Person S. 1984. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology 137:185–190 [DOI] [PubMed] [Google Scholar]