

Abstract
Assembly of the poxvirus immature virion (IV) membrane is a poorly understood event that occurs within the cytoplasm. At least eight viral proteins participate in formation of the viral membrane. Of these, A14, A17, and D13 are structural components whereas A6, A11, F10, H7, and L2 participate in membrane biogenesis. L2, the object of this study, is conserved in all chordopoxviruses, expressed early in infection, and associated with the endoplasmic reticulum (ER) throughout the cell and at the edges of crescent-shaped IV precursors. Previous studies with an inducible L2 mutant revealed abortive formation of the crescent membrane. However, possible low-level L2 synthesis under nonpermissive conditions led to ambiguity in interpretation. Here, we constructed a cell line that expresses L2, which allowed the creation of an L2-deletion mutant. In noncomplementing cells, replication was aborted prior to formation of mature virions and two types of aberrant structures were recognized. One consisted of short crescents, at the surface of dense masses of viroplasm, which were labeled with antibodies to the A11, A14, A17, and D13 proteins. The other structure consisted of “empty” IV-like membranes, also labeled with antibodies to the viral proteins, which appeared to be derived from adjacent calnexin-containing ER. A subset of 25 proteins examined, exemplified by components of the entry-fusion complex, were greatly diminished in amount. The primary role of L2 may be to recruit ER and modulate its transformation to viral membranes in juxtaposition with the viroplasm, simultaneously preventing the degradation of viral proteins dependent on viral membranes for stability.
INTRODUCTION
Poxviruses are double-stranded DNA viruses that carry out their replication cycle in the cytoplasm. Vaccinia virus (VACV), the prototype of the family, has a genome of nearly 200 kbp that encodes approximately 200 proteins involved in virus entry, RNA and DNA synthesis, assembly, and host defense. A poorly understood and controversial aspect of poxvirus replication concerns the origin and formation of the viral lipoprotein membrane, which first appears in the cytoplasm as a crescent-shaped, single lipid bilayer with an external honeycomb lattice consisting of trimers of the VACV D13 protein (1–3). As the crescent enlarges to form the spherical immature virion (IV), electron-dense material containing core proteins and DNA is engulfed (4, 5). Subsequently, the IV undergoes a dramatic change involving the loss of the D13 scaffold following processing of the A17 membrane protein (6), cleavage of the major core proteins (7), and formation of intramolecular disulfide bonds within the membrane entry proteins (8, 9), culminating in the dense, brick-shaped infectious mature virion (MV) (10). Some MVs are wrapped with a double membrane from the trans-Golgi network or endosomal cisternae (11–13) and transported via microtubules to the periphery of the cell (14, 15), where exocytosis and loss of the outer membrane occur to liberate extracellular enveloped virions (EVs) (16).
Several alternative origins for the crescent viral membrane, based primarily on transmission electron microscopy (TEM) images, have been proposed. The absence of a clear physical connection between viral and cellular membranes suggested a de novo origin (17). Subsequently, the intermediate compartment between the endoplasmic reticulum (ER) and Golgi membrane was considered the source of the crescent membrane based on localization of viral proteins in that organelle (18, 19). However, further studies showed that viral proteins could traffic from the ER to the crescent membrane and that blockage of the secretory pathway from the ER to the Golgi apparatus did not perturb IV or MV formation, although the subsequent wrapping step failed to occur and EVs were not produced (20, 21). In addition, TEM images showed the A17 membrane protein associated with ER adjacent to crescent membranes (21). Nevertheless, additional evidence is needed to support the ER origin of the crescent membrane.
To facilitate an understanding of the initial steps of morphogenesis, attempts have been made to identify the viral components that are necessary for the formation of crescent membranes. Three major constituents of the crescent membrane have been identified: the two transmembrane proteins A17 and A14 and the scaffold protein D13. When D13 is repressed, or the infected cells are treated with the drug rifampin, irregular viral membranes form adjacent to masses of viroplasm (22–24). Upon drug reversal, the membranes acquire D13 and adopt curvature, forming bona fide IVs that develop into MVs. Repression of A17 (25, 26) or A14 (27, 28) also results in the accumulation of masses of viroplasm, but numerous small vesicles or tubules are formed. Interestingly, repression or mutation of several other viral proteins that are not components of the viral membrane or present at a low level also interrupts crescent and IV formation. These include the protein kinase F10 (29–31) and A11 (32, 33), H7 (34), L2 (35), and A6 (36) proteins.
Of the second group of proteins, L2 has unique features: it is the only one that is expressed early in infection before factory formation and that colocalizes with the ER throughout the cytoplasm (37). In addition, L2 associates with the ER near the growing edge of the crescent membrane in the factory and some L2 can be detected in purified MV preparations. Although the formation of typical crescents and IVs was inhibited when L2 was repressed, short membrane arcs resembling abortive crescents were at the periphery of masses of viroplasm (35). However, at times later than 8 h or at multiplicities of 5 PFU per cell or greater, immature and more mature viral forms were detected in some cell sections, suggesting incomplete repression of L2 (35). Therefore, we were uncertain whether the formation of the short membrane arcs also could be due to leaky L2 expression. To resolve this question, we have now generated an L2-complementing cell line that enabled the construction of the VACV L2-deletion mutant vΔL2R. In noncomplementing cells, VACV replication was completely inhibited and no MVs formed regardless of the virus multiplicity or time after infection. Nevertheless, very short crescent-like arcs still appeared at the periphery of dense cytoplasmic masses. In addition, we detected aberrant IV-like membranes containing the viral A17, A14, and D13 proteins that appeared to be derived from adjacent calnexin-containing ER membranes.
MATERIALS AND METHODS
Cells and virus.
BS-C-1, HeLa, RK-13, and RK-L2-HA cells were grown in minimum essential medium with Earle's salts (E-MEM) and Dulbecco's minimum essential medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 100 units of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). RK-L2-HA cells were supplemented with 300 μg/ml Zeocin. The VACV Western Reserve (WR) strain and the recombinant virus vL2-HA were propagated as described previously (38). The L2-deletion mutant vΔL2R was propagated in RK-L2-HA cells. MVs were purified by sedimentation twice through 36% sucrose cushions and once in 25 to 40% sucrose gradients as described previously (38).
Antibodies.
Anti-HA.11 mouse monoclonal antibody (MAb) (Covance, Denver, PA), anti-FLAG M2 mouse MAb (Sigma-Aldrich, St. Louis, MO), and SV5-Pk1 (Abcam, Cambridge, MA) recognize the influenza virus hemagglutinin (HA), the FLAG epitope, and the V5 epitope, respectively. The viral proteins were detected with the rabbit antisera to L2 (35), A17-N (39), A3 (R. Doms and B. Moss, unpublished data), A21 (40), L5 (41), A11 (32), D13 (42), A16 (43), H2 (44), G3 (45), F13 (46), H3 (47), A27 (48), F10 (49), A14 (28), G7 (51), L1 (52), F9 (53), and A10 C-terminal peptide (R. Doms and B. Moss, unpublished data) and mouse MAb to A14 (50), A13 (67), and D8 (54). The cellular proteins were detected with the following antibodies: anti-protein disulfide isomerase (anti-PDI) goat polyclonal IgG (Santa Cruz Biotechnology), anticalnexin rabbit polyclonal antibody, and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) MAb (Covance, Emeryville, CA).
Construction of the RK-L2-HA cell line.
The Zeocin (Invitrogen, Carlsbad, CA) sensitivity of RK-13 cells was determined by following the manufacturer's instructions. Zeocin at 750 μg/ml was found to kill RK-13 cells within 10 days. The L2R open reading frame (ORF) with an HA tag at the 5′ end was cloned in pcDNA 3.1/Zeo(+) plasmid (Invitrogen). The plasmid was transfected into RK-13 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 48 h, the transfected cells were distributed to new flasks at approximately 25% confluence with fresh medium containing 750 μg/ml Zeocin. The cells were fed with selective medium every 3 days until cell foci were identified on day 10. The individual colonies were isolated with cloning discs (Sigma-Aldrich), transferred to 96-well plates, and screened for HA epitope expression by Western blotting. The positive colonies were put through a second phase of selection with 750 μg/ml Zeocin. The established recombinant RK-L2-HA cell line was grown as described above and supplemented with 300 μg/ml Zeocin to maintain the selection pressure.
Construction of vΔL2R.
PCR was used to prepare DNA encoding green fluorescent protein (GFP) regulated by the VACV P11 promoter and flanked with the 500-bp sequences flanking the L2R ORF. BS-C-1 cells were infected with VACV WR and transfected with the PCR product using Lipofectamine 2000 (Invitrogen Life Technologies). The lysate was diluted and applied to RK-L2-HA cells, and plaques that fluoresced green were picked and plaque purified 5 times. The mutant virus vΔL2R was grown in RK-L2-HA cells and purified by sucrose gradient centrifugation. The deletion of the L2R ORF was verified by PCR and sequencing.
Confocal microscopy.
HeLa cells grown on coverslips were infected for 16 h and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature (RT) and washed with PBS. The cells were permeabilized for 15 min with 0.1% Triton X-100 in PBS at RT and blocked with 10% FBS for 30 min. After blocking, the cells were incubated with the primary antibody in PBS containing 10% FBS for 1 h at RT. Cells were washed and incubated with the secondary antibody conjugated to dye (Molecular Probes, Eugene, OR) for 1 h. The coverslips were washed and mounted on a glass slide by using Prolong Gold (Invitrogen).
Transfection studies.
HeLa cells were grown in 6-well plates and infected. After 1 h, the cells were washed and transfected with plasmids using Lipofectamine 2000. The cells were replenished with fresh medium at 6 h posttransfection. At 16 or 24 h after infection, the cells were harvested, the virus titer was determined by plaque assay, and protein expression was determined by Western blotting.
Plaque assay and virus yield determination.
For plaque assays, BS-C-1 or RK-L2-HA cell monolayers in 6-well tissue culture plates were infected with 10-fold dilutions of virus. After 1 h of absorption, the virus inoculum was removed, the cells were washed with E-MEM containing 2.5% FBS, and medium containing 0.5% methylcellulose was added. The infection was allowed to proceed for 48 h at 37°C, the cells were stained with crystal violet or immunostained with anti-VACV polyclonal antibody, and the plaques were counted.
For virus yield determinations, cell monolayers in 12-well tissue culture plates were infected with 3 PFU/cell of vΔL2R, vL2-HA, or VACV WR. After 1 h of absorption, the supernatant was removed and the cells were washed with E-MEM containing 2% FBS. The infected cells were incubated with medium and harvested at several times. The cells were lysed by three freeze-thaw cycles and sonicated. Virus titers were determined by plaque assay in RK-L2-HA cells.
Western blotting.
Cells were lysed with SDS and reducing agent, and the proteins were resolved by electrophoresis on 4 to 12% Novex NuPAGE acrylamide gels and transferred to nitrocellulose membranes using Mini-iBlot gel transfer stacks (Invitrogen). Membranes were blocked for 1 h at RT with 5% nonfat dried milk in PBS containing 0.05% Tween 20 and incubated with the primary antibody for 1 h at RT. The membranes were washed four times with PBS containing 0.05% Tween and then incubated with the secondary antibody for 1 h at RT. The secondary antibodies used were species-specific, horseradish peroxidase-conjugated antibodies (Pierce, Rockford, IL). The membranes were washed and developed using Dura or Femto chemiluminescent substrate (Pierce).
TEM.
For conventional TEM, infected BS-C-1 or RK-13 cells stably expressing calnexin with a V5 epitope tag in 60-mm-diameter wells were fixed with 2% glutaraldehyde and embedded in EmBed-182 resin (Electron Microscopy Sciences, Hatfield, PA). Procedures for cryosectioning and immunogold labeling were described previously (55). Cryosections were picked up on grids, thawed, washed free of sucrose, and stained with anti-A14, anti-A11, anti-A17, and anti-D13 polyclonal antibodies or a mouse MAb to the V5 epitope followed by rabbit anti-mouse IgG from Cappel-ICN Pharmaceuticals (Aurora, OH) and then protein A conjugated to 10-nm gold spheres (University Medical Center, Utrecht, Netherlands). Specimens were viewed with an FEI Tecnai Spirit transmission electron microscope (FEI, Hillsboro, OR).
RESULTS
Construction of a stable cell line expressing L2.
To critically evaluate the role of L2 in viral membrane biogenesis, we decided to make a deletion mutant. However, our previous inducible mutant displayed a conditional lethal phenotype, indicating that a complementing cell line would be required. The similar ER localizations of L2 synthesized in infected cells and in uninfected cells that were transiently transfected with an L2-expressing plasmid (37) encouraged us to attempt the production of a cell line that stably expresses L2. We obtained a chemically synthesized, mammalian codon-optimized version of the L2 ORF with a cytomegalovirus (CMV) promoter that is recognized by the cellular transcription system and an N-terminal HA epitope tag. Previous studies had shown that the tag does not alter the function or localization of L2 (37). A plasmid containing the L2-HA DNA was transfected into RK-13 cells. Stably transfected cells were selected with the antibiotic Zeocin, and individual clones were picked, expanded, and screened for L2-HA expression by Western blotting. Of 25 colonies tested, 15 expressed L2-HA and two were subcloned to obtain homogeneous populations. Subclones 1 and 2 were expanded and maintained in 300 μg of Zeocin, and those of clone 1 were used for further studies reported here. Encouragingly, Western blotting (Fig. 1A) showed that the amount of L2 in the RK-L2-HA cells was similar to that in RK-13 cells infected with 1 to 5 PFU of vL2-HA, a VACV encoding L2-HA (37).
Fig 1.

Expression of L2 by RK-L2-HA cells. (A) Expression of L2-HA in an RK-L2-HA stable cell line and RK-13 cells infected with VACV. Lysates of uninfected RK-L2-HA cells and equivalent amounts of RK-13 cells infected with a multiplicity of infection (MOI) of 1 to 5 PFU of vL2-HA for 24 h were resolved by SDS-polyacrylamide gel electrophoresis. The expression of L2 was determined by Western blotting using HA antibody for the detection of L2 and GAPDH as a loading control. (B) Confocal microscopy of the RK-L2-HA stable cell line. Confluent cells were fixed, permeabilized, and stained with the mouse anti-HA MAb and the polyclonal primary antibodies to calnexin and protein disulfide isomerase (PDI), followed by goat anti-mouse and goat anti-rabbit IgG coupled to Alexa Fluor 488 and 594, respectively. DNA was detected by staining with 4′,6-diamidino-2-phenylindole (DAPI).
Confocal microscopy was used to determine the intracellular location of L2-HA and evaluate the consistency of L2-HA expression in the RK-L2-HA cell population. RK-L2-HA cells were fixed and costained with anti-HA MAb to detect L2 and antibodies against calnexin and protein disulfide isomerase, which are membrane and luminal ER proteins, respectively. L2 colocalized with the ER in all cells examined, although the staining intensity varied (Fig. 1B). There was no obvious difference in the appearance of the ER due to L2 expression.
Construction of a VACV L2R-deletion mutant.
Having successfully produced a stable cell line expressing L2 that localized in the ER, we anticipated that it could complement a VACV L2R deletion mutant. The deletion mutant was constructed by transfecting DNA, containing the GFP open reading frame (ORF) regulated by the VACV P11 late promoter and flanked by sequences preceding and following the L2R ORF (Fig. 2A), into BS-C-1 cells infected with VACV to allow homologous recombination. The lysate was diluted and applied to a monolayer of RK-L2-HA cells, which were subsequently screened by fluorescence microscopy. Virus was extracted from fluorescent plaques and clonally purified by repeated plaque isolations. The replacement of the L2R ORF by GFP was confirmed by DNA sequencing, and the deletion mutant was named vΔL2R.
Fig 2.
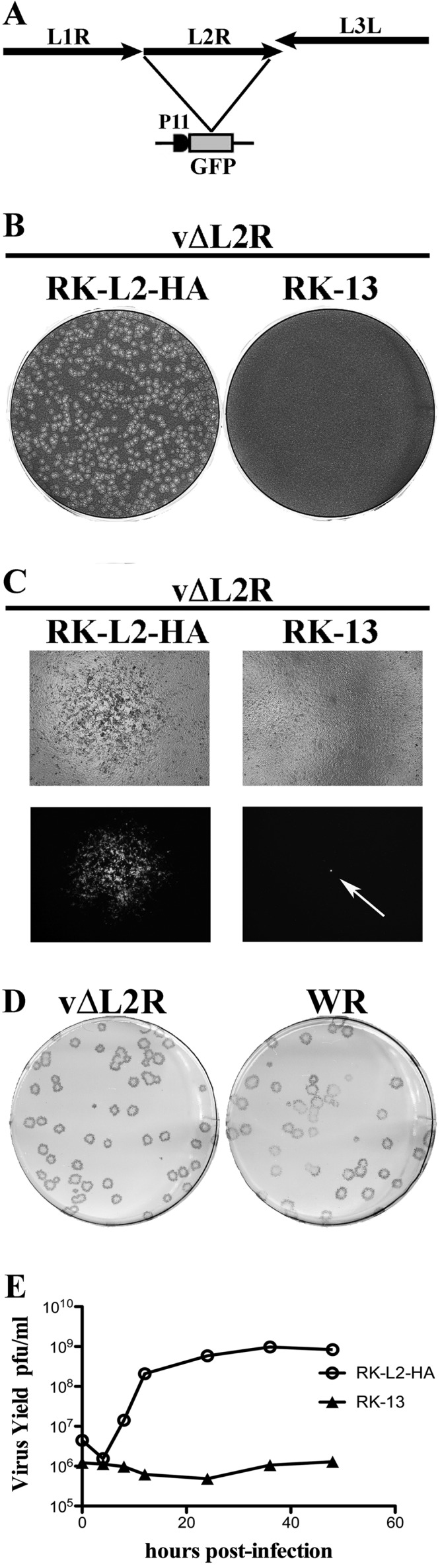
Construction, plaque phenotype, and one-step growth curve of vΔL2R. (A) Schematic representation of the genome structure of vΔL2R. The L2R ORF was replaced with the GFP ORF regulated by the VACV P11 late promoter. (B) Plaque phenotype of vΔL2R. RK-L2-HA and RK-13 cells were infected with vΔL2R. After 48 h, the cells were stained with crystal violet. (C) Cells infected as in panel B were visualized with a fluorescence microscope. The bright field (upper) and the fluorescent field (lower) are shown with a single plaque on the left and a single fluorescent cell on the right shown by the pointing arrow. (D) Plaques formed by vΔL2R and wild-type VACV strain WR on RK-L2-HA cells. After 72 h, plaques were visualized by staining with antibody to VACV followed by anti-rabbit IgG conjugated to alkaline phosphatase. (E) One-step growth curve of vΔL2R. RK-L2-HA and RK-13 cells were infected with 3 PFU per cell of vΔL2R. After 0, 6, 9, 12, 24, 36, and 48 h, the infected cells were harvested and virus titers were determined by plaque assay on RK-L2-HA cells.
vΔL2R formed plaques in RK-L2-HA cells but not in the parental RK-13 cells (Fig. 2B) or HeLa or BS-C-1 cells (data not shown). At higher magnification, individual RK-13 cells infected with vΔL2R were discerned by GFP fluorescence, but spread to neighboring cells failed to occur, confirming the defect in formation of infectious progeny (Fig. 2C). Plaques formed by vΔL2R in RK-L2-HA cells were indistinguishable in size and shape from wild-type (WT) VACV strain WR plaques (Fig. 2D). Furthermore, vΔL2R replicated with normal kinetics in RK-L2-HA cells but not in RK-13 cells as determined by virus yields (Fig. 2E). The yields of vΔL2R in RK-L2-HA cells were similar to that of WT virus, allowing preparation of large mutant virus stocks for infection and purification.
Effect of L2 deletion on viral morphogenesis.
The principal reason for making the L2-deletion mutant was to ascertain whether abortive membrane structures, seen previously with a conditional lethal mutant, represented a true null phenotype or formed because of incomplete repression of L2 synthesis. In order to distinguish between these possibilities, BS-C-1 cells were infected with 5 PFU per cell of vΔL2R and samples were fixed at 8, 12, and 19 h postinfection for TEM. In an infection with the parental virus, all stages of VACV morphogenesis, including MVs and EVs, were seen (data not shown). However, in cells infected with vΔL2R the most prominent feature was the presence of large spherical masses of dense viroplasm (Fig. 3A to F), indistinguishable from those previously shown by immunogold labeling to contain core proteins in cells infected with conditional lethal A11, A14, and H7 mutants (32, 34). Short “spicule”-coated membrane arcs resembling crescent segments were detected around the large dense aggregates at 12 and 19 h after infection (Fig. 3C to F) as with the L2-inducible mutant (35). Additional, less dense inclusions (Fig. 3A and B), previously shown to contain D13 and ER membranes with A17 in cells infected with the A11, A14, and H7 conditional lethal mutants (6, 32, 34), were also present. However, typical large crescents, IVs, or MVs were not detected even with high-multiplicity infection at late times.
Fig 3.
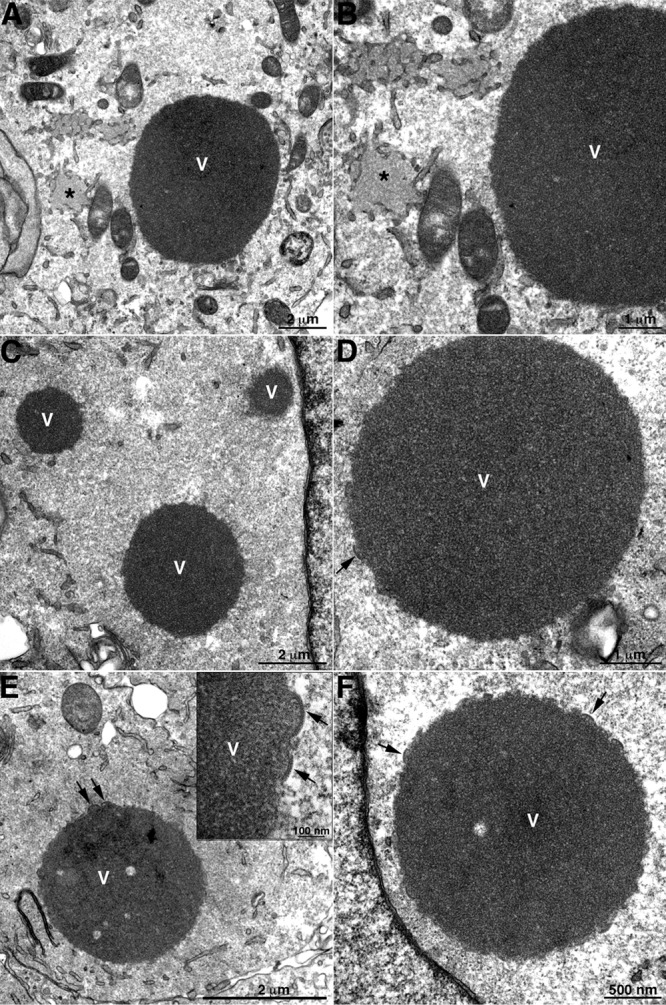
TEM of cells infected with vΔL2R showing dense masses of viroplasm and short crescent-like structures. BS-C-1 cells were infected with vΔL2R at 5 PFU per cell. The cells were fixed and prepared for TEM at 8 (A and B), 12 (C and D), and 19 (E and F) h after infection. V, dense masses of viroplasm; asterisk, intermediate-density D13 accumulation sites. The arrows point to short membrane arcs. The scale bar at the bottom of each panel indicates magnification.
Formation of aberrant IV-like membranes in the absence of L2.
In addition to the short crescent-like membranes at the periphery of the dense viroplasm, we noted IV-like membranes in about 10% of the thin cell sections. These structures appeared in clusters within the viral factories but distant from the dense inclusions and surrounded by ER membranes (Fig. 4A and C). The IV-like membranes had the characteristic spicule surface representing the D13 scaffold but appeared “empty” of electron-dense material, and some were less spherical than typical IVs (Fig. 4B and D). The ends of IV-like structures frequently appeared continuous with membranes that lacked the spicule coat (Fig. 4B and D). In representative examples, we could not discern discontinuity between the spicule-coated IV-like structures and the naked membranes by tilting thicker sections (data not shown), supporting membrane continuity.
Fig 4.
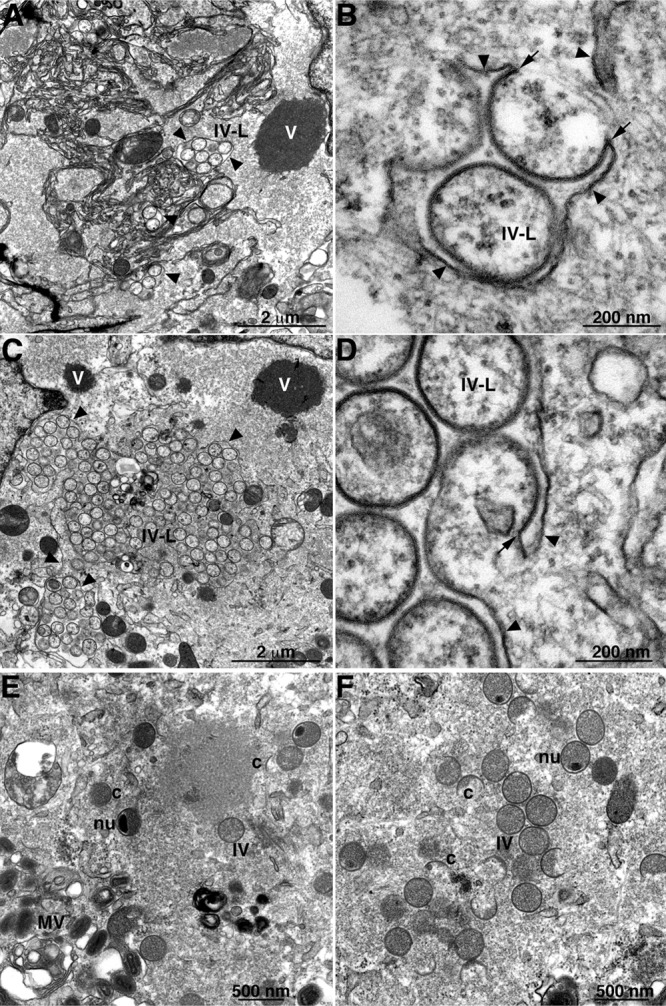
TEM of cells infected with vΔL2R showing IV-like structures. (A to D) BS-C-1 cells were infected with vΔL2R at a multiplicity of 1 PFU (A and B) and 5 PFU (C and D) per cell. (E and F) Cells were infected with 1 PFU per cell of vΔL2R and transfected 1 h later with a plasmid that expresses L2-HA under the L2R natural promoter. At 20 h after infection, the cells were fixed and prepared for TEM. Abbreviations: c, crescents; IV, immature virion; nu, IV that contains a nucleoid; MV, mature virion; V, dense masses of viroplasm; IV-L, IV-like structures. Arrowheads point to smooth ER membranes; arrows point to the apparent continuity of spicule-coated IV-like structures with smooth ER membranes. The scale bar at the bottom of each panel indicates magnification.
To ensure that that our mutant had no other defects contributing to the phenotype, we carried out a transcomplementation experiment. At 1 h after infection of HeLa cells with vΔL2R, the cells were transfected with a plasmid containing the L2R ORF with the natural promoter. TEM images of cells at 20 h postinfection revealed all stages of morphogenesis, including crescents, IVs, and MVs (Fig. 4E and F).
Identification of viral proteins in the aberrant membranes formed in the absence of L2.
Further experiments were performed to identify viral proteins associated with the aberrant membranes made in the absence of L2. BS-C-1 cells were infected with vΔL2R, and thawed cryosections were probed with rabbit antibodies to the A14 (Fig. 5A and B), A11 (Fig. 5C and D), A17 (Fig. 5E and F), and D13 (Fig. 5G and H) proteins followed by protein A conjugated to 10-nm gold. Note that membranes appear somewhat different than in the Epon-imbedded samples in Fig. 4. The short crescent membranes adjacent to dense viroplasm are shown in Fig. 5A, C, E, and G. In general, the labeling was relatively sparse, as only cut surfaces can bind antibody and protein A gold by this procedure. In addition, some proteins such as A14 were reduced in amount in the absence of L2, as will be shown below. The two principal components of the IV membrane, A14 (Fig. 5A) and A17 (Fig. 5E), were associated with the body of the short crescent membranes, whereas A11 (Fig. 5C) was predominantly at the ends, similar to the location of L2 during a normal infection (37). Some D13 was associated with the short crescent membranes (Fig. 5G), but the majority was in the intermediate-density inclusions (Fig. 5G), as had previously been found with A11 and H7 mutants (32, 34). A14 (Fig. 5B), A11 (Fig. 5D), A17 (Fig. 5F), and D13 (Fig. 5H) were also associated with the IV-like membranes.
Fig 5.
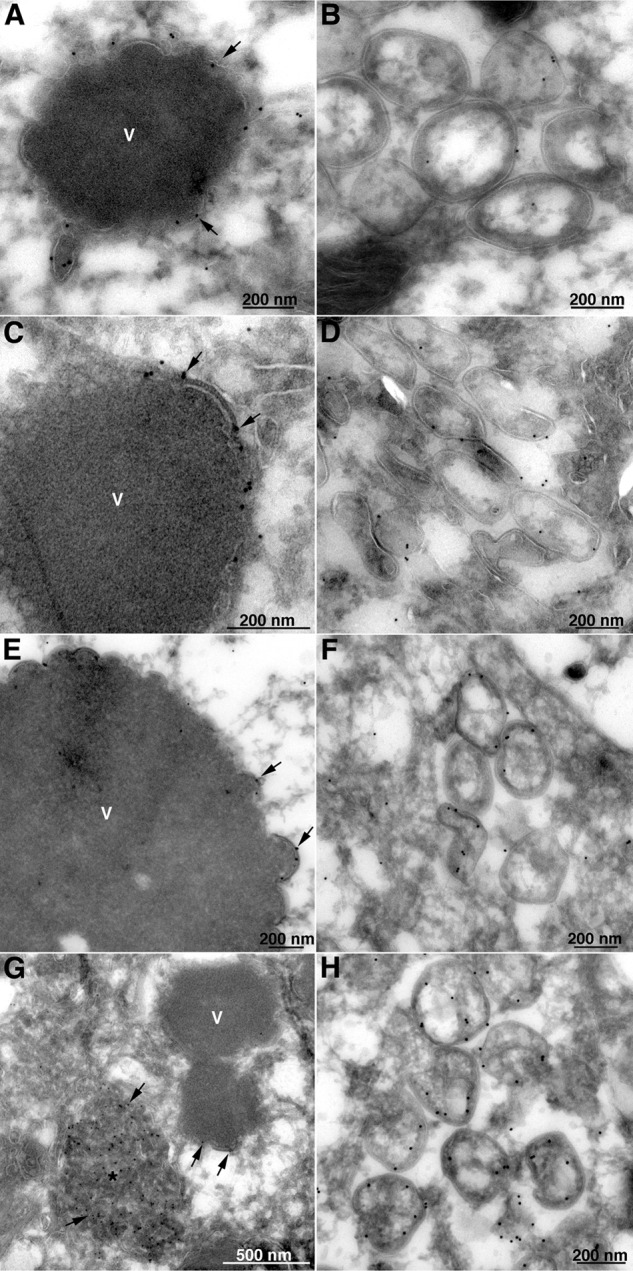
Localization of A11, A14, A17, and D13 by immunogold TEM in the absence of L2. BS-C-1 cells were infected with vΔL2R. After 20 h, the cells were fixed, cryosectioned, and stained with the primary polyclonal antibodies to A14 (A and B), A11 (C and D), A17 (E and F), and D13 (G and H), followed by protein A conjugated to 10-nm gold spheres. V, dense masses of viroplasm; asterisk, D13 accumulation sites. Arrows point to the gold particles that are labeling the viral proteins identified close to the short membrane arcs. The scale bar at the bottom of each panel indicates magnification.
Localization of the ER in relation to IV-like structures.
We were particularly interested in localizing the ER in relation to the IV-like structures formed in the absence of L2. In preliminary immunoelectron microscopy experiments, we found very poor labeling of ER with antibodies to calnexin and other ER membrane proteins in both uninfected and infected cells despite strong labeling by fluorescence microscopy. In order to enhance specific labeling, a stable RK-13 cell line that expresses calnexin with a V5 tag (to be described in detail elsewhere) was constructed. These cells were infected with vΔL2R, and thawed cryosections were incubated with a MAb to the V5 epitope tag followed by rabbit anti-mouse IgG and protein A conjugated to 10-nm gold spheres. Gold grains were present in reticular membranes throughout the cytoplasm (Fig. 6A), including the perinuclear envelope (data not shown). In addition, calnexin-stained membranes were intimately associated with the spicule-coated IV-like structures (Fig. 6B, C, and D). Calnexin staining of smooth membranes within IV-like structures was also evident (Fig. 6C and D).
Fig 6.
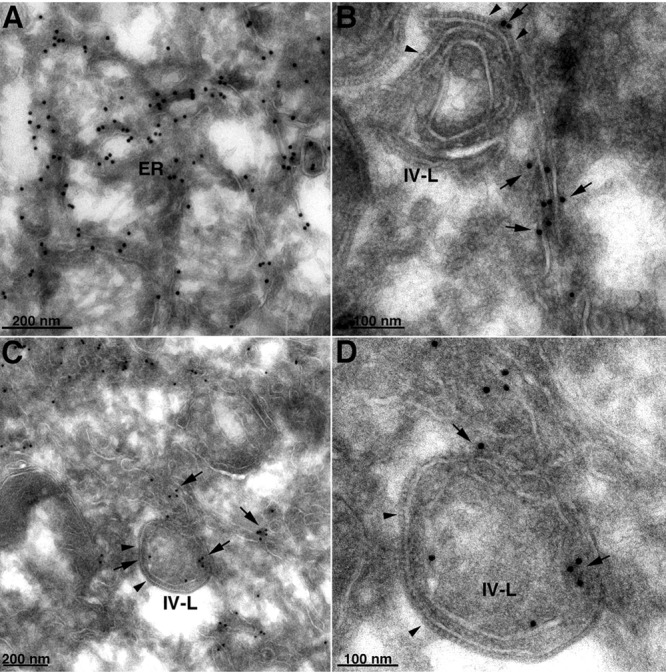
Localization of calnexin by immunogold TEM in the absence of L2. RK-13 cells stably expressing V5-tagged calnexin were infected with vΔL2R at a multiplicity of 5 PFU per cell. After 21 h, the cells were fixed, cryosectioned, and stained with anti-V5 MAb, followed by rabbit anti-mouse IgG and protein A conjugated to 10-nm gold spheres. (A) Calnexin-staining ER. (B) Image shows an IV-like structure (IV-L). Arrowheads point to the D13 scaffold spicules; arrows point to the adjacent calnexin-staining smooth membranes. (C) Similar to panel B but with calnexin staining of smooth membranes within the IV-like structure. (D) Higher magnification of panel C. The scale bar at the bottom of each panel indicates magnification.
Effect of the L2 deletion on the stability and localization of other viral proteins.
The fate of viral membrane proteins, when synthesis of viral membranes is perturbed, is an intriguing topic. In our previous studies with the conditional lethal L2 mutant vL2Ri, we found by Western blotting of cell lysates that a subset of viral membrane proteins were greatly reduced in amount or not detected in the absence of inducer due to instability as shown by pulse-chase experiments (37). Interestingly, these proteins were components of the entry-fusion complex (EFC). We repeated and extended these experiments by infecting HeLa cells with vΔL2R and analyzing a larger number of viral membrane and nonmembrane proteins with a variety of roles during the VACV replication cycle. In the experiment summarized in Fig. 7A, HeLa cells were infected with vΔL2R, vL2-HA, or VACV WR (the last two serving as L2-expressing controls). After 9 h, the cells were lysed, and the proteins were resolved by SDS-polyacrylamide gel electrophoresis and prepared for Western blotting. We assembled a panel of antibodies to 19 viral proteins classified as intermediate or late (56) in addition to L2 and the loading control cell protein GAPDH. The viral targets of these antibodies included MV membrane proteins involved in cell attachment (D8 and H3), membrane fusion (L1, L5, F9, A16, G3, A21, and H2), and crescent formation (A17, A14, and A11); the IV scaffold protein (D13); a protein indirectly associated with the MV membrane for cell attachment (A27); core proteins (A3, A10, G7, and F10); and an EV membrane protein (F13). Each protein was detected in cells infected with the control viruses, and this established the normal levels. Note that the lower mobility of L2 in cells infected with vL2-HA than in cells infected with VACV WR was due to the epitope tag. In cells infected with vΔL2R, L2 was absent and 11 of the 19 other proteins analyzed were greatly reduced in amount or not detected. The affected proteins included each of the EFC or EFC-associated MV membrane proteins probed (L1, L5, F9, A16, G3, A21, and H2), the F10 core protein and A11 putative membrane protein required for IV formation, and the H3 membrane protein involved in cell attachment. Although substantially decreased, the A11 protein could be detected using higher loading of extracts from cells infected with vΔL2R, as will be shown in a figure below. The A14 protein was detected as a major 14-kDa band and a minor glycosylated 25-kDa band in the cells infected with the control viruses as previously shown (57), but only the minor glycosylated A14 band was detected in the cells infected with vΔL2R. In contrast, the A17 MV membrane protein, which like A14 is required for IV formation, was present in substantial amounts but less processed than in the control infections. (The processing of A17 is complex, involving phosphorylation and N- and C-terminal cleavages, and the products are incompletely resolved by electrophoresis.) The A3 (p4B) and A10 (p4A) core proteins were undiminished, although processing was inhibited as expected for an assembly block (note that the 25-kDa processed form of A10 [58], detected by the antibody to the C-terminal peptide, is not shown in the blot), whereas the F10 core protein kinase was greatly reduced. On the other hand, there were normal amounts of the D13 IV scaffold protein, the D8 membrane protein involved in cell attachment, the G7 core protein, and the F13 EV membrane protein. The A27 protein, which associates with the MV membrane via attachment to A17, was only modestly reduced in amount. Thus, the effect of the L2 deletion was selective. The MV membrane proteins, with the exception of D8 and A17, were greatly reduced; the MV core proteins with the exception of F10 were stable, although processing was inhibited; and an EV membrane protein tested was stable.
Fig 7.
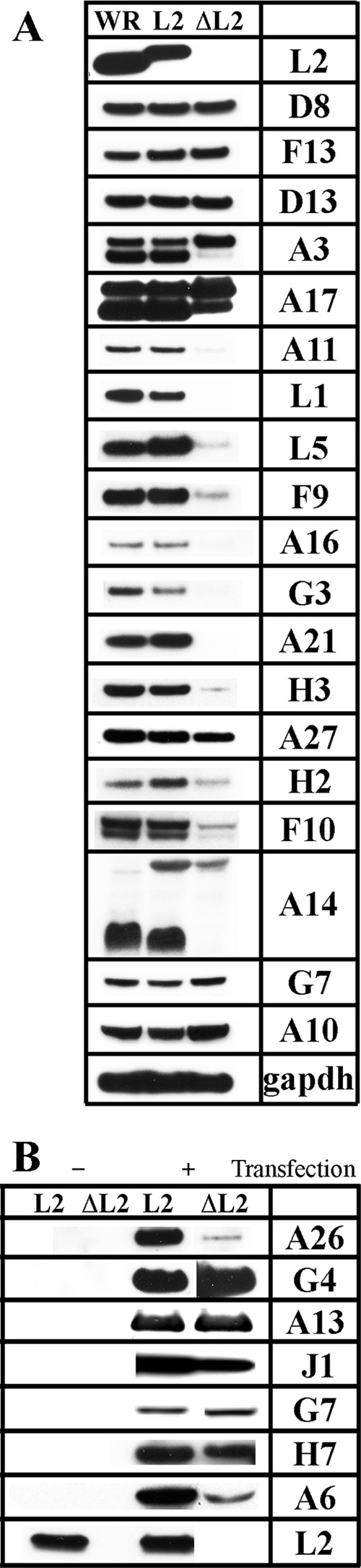
Western blotting of proteins from cells infected with L2-deletion mutant and control viruses. (A) HeLa cells were infected with 3 PFU per cell of wild type (WR), vL2-HA (L2), and vΔL2R (ΔL2). After 9 h, the cells were lysed with SDS, and the proteins were resolved by SDS-polyacrylamide gel electrophoresis and analyzed by Western blotting with antibodies for the viral proteins indicated on the right or GAPDH as a loading control. Proteins were detected by chemiluminescence. (B) HeLa cells were infected as described above with vL2-HA and vΔL2R. After 1 h, the cells were washed and transfected (+) or not (−) with a plasmid expressing a FLAG-tagged version of one of the proteins indicated on the right under their natural promoter. After 11 h, the cells were lysed with SDS, and the proteins were resolved by SDS-polyacrylamide gel electrophoresis and analyzed by Western blotting with an anti-FLAG MAb, except for L2, which was probed with a rabbit polyclonal antibody. Proteins were detected by chemiluminescence.
We were interested in checking the status of additional viral proteins for which antibodies were unavailable to us. To circumvent the need for specific antibodies, we transfected seven plasmids encoding FLAG-tagged versions of intermediate and late VACV proteins under their natural promoters (56) into HeLa cells infected with a control virus expressing L2 or with vΔL2R. By using the natural promoters, we could be assured that the proteins would be transcribed within the viral factories (59). Three of the additional proteins associate with the MV membrane: one has a transmembrane domain (A13) and two do not (G4 and A26). A13 and G4 were not diminished in the absence of L2, supporting the selectivity of the effect of the L2 deletion on a subset of membrane-associated proteins (Fig. 7B). However, A26 was reduced in the cells infected with vΔL2R. A26 is destabilized in the absence of A27 (48), but the modest reduction in A27 (Fig. 7A) did not seem sufficient to account for this effect. We probed for FLAG-tagged H7 and A6, since they, like L2, A11, and F10, are required for crescent formation. The amount of A6 was reduced in cells infected with vΔL2R, whereas H7 was only slightly affected. F10, which was decreased in the absence of L2 (Fig. 7A), is associated in a complex with six other core proteins required for the association of the viral membrane with core proteins to form IVs (60). We checked two of the proteins in the complex: G7 and J1. However, neither was reduced in amount (Fig. 7B). In summary, a total of 25 viral intermediate or late proteins were analyzed here and 13 were selectively diminished in the absence of L2. These data, and a comparison with other mutants to be discussed later, are summarized in Table 1.
Table 1.
Effects on amounts and processing of viral proteinsj
| ORF | Deleted or repressed gene |
Promotera | TM | Location | Role | |||
|---|---|---|---|---|---|---|---|---|
| L2b | A11c | A6d | A17e | |||||
| A3 | U | U | U | Intermed | − | Core | MV formation | |
| A4 | N | N | N | Early | − | Core | MV formation | |
| A6 | D | Intermed | − | Cytoplasm | IV formation | |||
| A10 | Nf | U | Late | − | MV core | MV formation | ||
| A11 | D | Ng | N | Late | + | Cytoplasm | IV formation | |
| A13 | N | Nh | Late | + | MV membrane | MV formation | ||
| A14 | D, Gi | D, Gi | Gi | Late | + | IV, MV | IV formation | |
| A16 | D | Intermed | + | MV membrane | EFC | |||
| A17 | U | U | Late | + | IV, MV membrane | IV formation | ||
| A21 | D | D | D | Late | + | MV membrane | EFC | |
| A26 | D | D | D | Late | − | MV membrane | Cell attachment | |
| A27 | N | Intermed | − | MV membrane | Cell attachment | |||
| A28 | D | D | D | Late | + | MV membrane | EFC | |
| A30 | N | N | Intermed | − | MV core | MV formation | ||
| D8 | Nh | N | Nh | N | Intermed | + | IV, MV membrane | Cell attachment |
| D13 | N | N | N | Intermed | − | IV outer | IV scaffold | |
| F9 | D | D | D | D | Late | + | MV membrane | EFC |
| F10 | D | D | Late | − | MV core | Kinase, IV formation | ||
| F13 | N | N | N | Intermed | + | EV membrane | Wrapping | |
| G3 | D | Late | + | MV membrane | EFC | |||
| G4 | N | Intermed | − | MV membrane | Redox | |||
| G7 | N | Late | − | MV core | Core complex | |||
| H2 | D | Late | + | MV membrane | EFC | |||
| H3 | D | Nh | Intermed | + | MV membrane | Cell attachment | ||
| H7 | N | Intermed | − | Cytoplasm | IV formation | |||
| J1 | N | Intermed | − | MV core | Core complex | |||
| L1 | D | D | D | D | Late | + | MV membrane | EFC |
| L2 | N | N | Early | + | ER | IV formation | ||
| L5 | D | Late | + | MV membrane | EFC | |||
Data from reference 56.
Data from present study and reference 37.
Data from reference 33.
Data from reference 37.
Precursor and processed forms not resolved.
Mislocalized to cytoplasm.
ER localization.
Glycosylation and ER localization.
Abbreviations: D, decreased; U, unprocessed; N, normal; Intermed, intermediate; TM, transmembrane domain. Shading highlights proteins that are decreased when L2 is repressed or deleted.
TEM studies (Fig. 5) showed that A14, A17, and D13 were associated with the short crescents around the periphery of the dense aggregates of viroplasm and with the IV-like membranes in the absence of L2. D13 was also present in less-dense inclusions under these conditions. We visualized these proteins, and three additional ones (A3, D8, and A13), by confocal microscopy to achieve a more global picture. Of these 6 proteins, only A14 appeared to be severely diminished in amount by Western blotting (Fig. 7A). In cells infected with an L2-expressing VACV, each protein localized predominantly within 4′,6-diamidino-2-phenylindole (DAPI)-staining viral factories presumably associated with individual immature and mature virions (Fig. 8). In cells infected with vΔL2R, the A3 protein still localized in viral factories but appeared to form ring-like structures (Fig. 8) that represent surface staining of large, dense, spherical protein aggregates seen previously by confocal microscopy (34, 35) and visualized at higher magnification by electron microscopy (Fig. 5). The inability of antibody to access the interior of the aggregates in fixed, permeabilized cells likely accounted for the decreased staining of A3. The A17 protein also appeared aggregated with some ring-like forms rather than associating with discrete virions, making it appear as if it also was diminished in amount. The D13 protein was largely near the periphery of the factories, presumably associated with the less-dense inclusions visualized by TEM in Fig. 5. The A14 protein had a punctate and ring appearance largely associated with the periphery of factories. Staining of the D8 protein was undiminished but instead of localizing in the factory was distributed throughout the cytoplasm in association with calnexin-staining ER. A13 was more widely distributed within the cytoplasm in the absence of L2, and the factory-associated protein was in particulate and ring forms, suggesting association with the periphery of dense viroplasm. A13 has greater ER localization when A6 is repressed (36). Thus, the absence of L2 and the interruption of viral morphogenesis resulted in abnormal distributions of viral proteins.
Fig 8.

Confocal microscopy of cells infected with vL2-HA and vΔL2R. HeLa cells were infected with L2-expressing vL2-HA and L2-deletion mutant vΔL2R at 5 PFU per cell. After 16 h, the infected cells were fixed, permeabilized, and stained with primary polyclonal antibodies for A3, A17, D13, and calnexin and the mouse monoclonal anti-A14, anti-D8, anti-A13, and anticalnexin antibodies followed by goat anti-rabbit IgG and goat anti-mouse coupled to Alexa Fluor 594 and 647, respectively, and DAPI. Viewing the enlarged figure on a computer screen is recommended to see detail.
DISCUSSION
Inducible conditional lethal mutants are extremely useful for studies of VACV assembly as abortive or intermediate structures may form in the absence of key proteins. Nevertheless, the difficulty in gauging whether the phenotype is moderated by low-level expression under “nonpermissive” conditions is a pervasive problem with such mutants. Thus, the detection of a few immature and mature virions in addition to mostly abortive forms at late times after infection with the L2-inducible mutant led to uncertainty regarding the actual defect (35). The purpose of the present study was to construct and characterize a true L2-null mutant and compare its phenotype with that of the previously isolated inducible mutant and wild-type virus. However, to accomplish this, we needed a complementing cell line. Until now, only two cell lines that successfully complement nonreplicating VACV deletion mutants were described. The first expresses a viral protein required for DNA replication (61), and the second expresses the two subunits of the intermediate transcription factor (62). Here, we describe a cell line that expresses the L2 membrane protein and complements a VACV deletion mutant.
The HA-tagged L2R ORF was chemically synthesized in order to optimize translation of mammalian codons and remove any sequences that might impair translocation of mRNA from the nucleus. After transfection, L2-expressing cell lines were readily obtained and confocal images verified that the protein colocalized with the ER. Importantly, the cell line allowed us to isolate an L2R deletion mutant in which the entire ORF was replaced with GFP. The deletion mutant was stable, as the absence of sequences shared with the cell line ruled out the possibility of homologous recombination. As expected, the mutant was unable to replicate in the parental RK-13 cells or other unmodified cell lines tested. Following infection of nonpermissive cells, we observed large spherical masses of electron-dense viroplasm with short crescent-like membranes on the periphery, which were indistinguishable from the abortive structures seen with the inducible mutant (35). Therefore, we could conclude that these membranes formed in the complete absence of L2 synthesis. Although we did not detect typical IVs in the absence of L2, there were clusters of spicule-coated IV-like membranes in about 10% of the 80-nm cell sections. Given the ∼5- to 10-μm thickness of an adherent cell, the 10% number probably underestimates the frequency of these structures. Interestingly, smooth membranes surrounded the IV-like structures, and in some cases, the spicule-coated and smooth membranes appeared to be continuous even when thick sections were examined with tilting. The absence of electron-dense material within the IV-like structures was reminiscent of “empty IVs” that formed with mutations of a complex of core proteins (60). However, the latter structures were much more abundant and were not intimately associated with smooth membranes.
Immunoelectron microscopy was carried out to identify proteins associated with the aberrant structures. As expected, antibodies to the viral A17 and A14 membrane proteins decorated the short crescents. We also detected the A11 protein primarily at the edges of the short crescents, as previously found for L2 during a wild-type virus infection (37), consistent with recent evidence for lipid association of A11 (33). The majority of the D13 scaffold protein was present in intermediate-density occlusions. Although fewer D13 antibody-associated gold grains localized with the short crescent membranes, the characteristic appearance of the spicule coat confirmed the presence of the D13 scaffold. The viral membrane proteins were also associated with the aberrant IV-like structures, which were well decorated with D13. We were particularly interested in the smooth membranes that appeared contiguous and often within the IV-like structures. These membranes were confirmed as ER by calnexin labeling. Several studies have shown that calnexin is retained in the ER and that little or none can be found in the ER-Golgi intermediate compartment (63–65). These data add to the growing evidence that the IV membranes are derived by modification of the ER. It will be interesting to perform similar immunoelectron microscopy with other mutants that are blocked in crescent formation.
In our previous studies with the L2-inducible mutant, we had found that certain viral membrane proteins, in particular those forming the EFC, were diminished, and pulse-chase experiments indicated that this was due to instability (37). In order to better understand the selectivity of this effect, we analyzed an expanded number of VACV proteins involved in many aspects of the VACV replication cycle. Data for 27 proteins analyzed with L2-deletion or -inducible mutants are summarized in Table 1. Fourteen of the proteins were greatly diminished in amount, confirming the severity of this effect. The decrease did not correlate with the type of promoter, e.g., both intermediate and late proteins were reduced in amount (Table 1). The group most affected was comprised of the EFC and EFC-associated membrane proteins. All eight such proteins tested were greatly diminished. Three additional proteins with transmembrane domains (A11, A14, and H3) and a few without transmembrane domains (A6, A26, and F10) were reduced, though some to a lesser extent than the EFC proteins. It is necessary to consider whether the protein instability is caused by the defect in viral membrane formation or vice versa. We favor the former because repression of the A17 membrane protein, which also prevents crescent and IV formation, also leads to selective effects on protein stability while not diminishing L2 (37). Of the 12 proteins analyzed (excluding L2) with the A17 mutant, the pattern was identical to that obtained with L2 deletion except for the stability of A11 (Table 1). Although fewer proteins were analyzed when A11 was repressed (32), the results were consistent with the data obtained with L2 deletion (Table 1). When A6 was repressed (36), both F9 and L1 were diminished; neither H3 nor A14 was decreased though the latter was glycosylated and mislocalized to the ER (Table 1). Although there are a few differences, interfering with IV formation reduced the amounts of a specific subset of proteins in each case cited above.
The protein instability data may provide a clue to the trafficking of viral membrane proteins. The exposure of hydrophobic domains in the cytoplasm is a well-known product of protein misfolding that results in rapid proteosomal degradation. We suggest that the hydrophobic domains of the EFC and some other viral membrane proteins become exposed when formation of the viral membrane is perturbed. This could mean that certain nascent viral proteins (e.g., EFC proteins) need to be inserted directly into the viral membrane to prevent degradation, whereas others (e.g., A17 and D8) can insert their hydrophobic domains into the ER and are therefore stable in the absence of L2. The instability of certain nonmembrane proteins (e.g., A26 and A6) may also result from misfolding due to altered protein associations. Treatment of cells with rifampin, in contrast to the results obtained by preventing the expression of L2, A17, A11, and A6, does not lead to protein instability (37). Although rifampin prevents IV formation, there is robust synthesis of viral membranes that lack the D13 scaffold (3, 22, 23, 66). It would be interesting to localize the EFC proteins in the presence of rifampin, although their low abundance has made this difficult.
It is risky to try organizing the existing data into a model of viral membrane formation as we are probably still missing pieces of the puzzle. Nevertheless, it may be useful to make this attempt. Increasing data suggest that the IV membrane originates from the ER and that the A17, A14, and D13 proteins are the key building blocks. The early synthesis of L2 and association with the ER and the edges of crescents imply a direct role for this protein in IV formation. In the absence of L2, the short crescent-like structures associated with the masses of viroplasm could indicate that insufficient ER membrane is available at the appropriate site for IV formation. This idea is reinforced by the presence of IV-like membranes distal from the viroplasm and near ER when L2 is not synthesized. Taken together, one role of L2 may be to recruit ER to the virus factory and ensure that the ER modification is occurring in juxtaposition with the viroplasm. A second role, which may be indirect, is to enable other membrane proteins (e.g., the EFC) to stably associate with the viral membrane. Although L2, A11, H7, and A6 seem to be involved at similar stages of viral membrane formation, their functions are not redundant. Thus far, we have been unable to demonstrate an association of L2 with these other proteins by coimmunopurification. However, a weak association of A6 and A11 was recently reported (33).
ACKNOWLEDGMENTS
We thank Catherine Cotter for preparation of cells and Zhilong Yang for plasmids. G. L. Smith, G. H. Cohen, Paula Traktman, and Y. Xiang kindly provided antibodies.
Support was provided by the Division of Intramural Research, NIAID, NIH.
Footnotes
Published ahead of print 28 November 2012
REFERENCES
- 1. Heuser J. 2005. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 169:269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hollinshead M, Vanderplasschen A, Smith GL, Vaux DJ. 1999. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 73:1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dales S, Siminovitch L. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morgan C. 1976. The insertion of DNA into vaccinia virus. Science 193:591–592 [DOI] [PubMed] [Google Scholar]
- 6. Bisht H, Weisberg AS, Szajner P, Moss B. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moss B, Rosenblum EN. 1973. Protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J. Mol. Biol. 81:267–269 [DOI] [PubMed] [Google Scholar]
- 8. Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. U. S. A. 102:18572–18577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Senkevich TG, White CL, Koonin EV, Moss B. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. U. S. A. 99:6667–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 11. Hiller G, Weber K. 1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmelz M, Sodeik B, Ericsson M, Wolffe EJ, Shida H, Hiller G, Griffiths G. 1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J. Virol. 68:130–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tooze J, Hollinshead M, Reis B, Radsak K, Kern H. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 60:163–178 [PubMed] [Google Scholar]
- 14. Hollinshead M, Rodger G, Van Eijl H, Law M, Hollinshead R, Vaux DJ, Smith GL. 2001. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 154:389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ward BM, Moss B. 2001. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 75:11651–11663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith GL, Law M. 2004. The exit of vaccinia virus from infected cells. Virus Res. 106:189–197 [DOI] [PubMed] [Google Scholar]
- 17. Dales S, Mosbach EH. 1968. Vaccinia as a model for membrane biogenesis. Virology 35:564–583 [DOI] [PubMed] [Google Scholar]
- 18. Risco C, Rodriguez JR, Lopez-Iglesias C, Carrascosa JL, Esteban M, Rodriguez D. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van 't Hof W, van Meer G, Moss B, Griffiths G. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 121:521–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Husain M, Moss B. 2003. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 77:11754–11766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Husain M, Weisberg AS, Moss B. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. U. S. A. 103:19506–19511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moss B, Rosenblum EN, Katz E, Grimley PM. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280–1284 [DOI] [PubMed] [Google Scholar]
- 23. Nagayama A, Pogo BGT, Dales S. 1970. Biogenesis of vaccinia: separation of early stages from maturation by means of rifampicin. Virology 40:1039–1051 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Y, Moss B. 1992. Immature viral envelope formation is interrupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology 187:643–653 [DOI] [PubMed] [Google Scholar]
- 25. Rodriguez JR, Risco C, Carrascosa JL, Esteban M, Rodriguez D. 1997. Characterization of early stages in vaccinia virus membrane biogenesis: implications of the 21-kilodalton protein and a newly identified 15-kilodalton envelope protein. J. Virol. 71:1821–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wolffe EJ, Moore DM, Peters PJ, Moss B. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodriguez JR, Risco C, Carrascosa JL, Esteban M, Rodriguez D. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Traktman P, Liu K, DeMasi J, Rollins R, Jesty S, Unger B. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Punjabi A, Traktman P. 2005. Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J. Virol. 79:2171–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szajner P, Weisberg AS, Moss B. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Traktman P, Caligiuri A, Jesty SA, Sankar U. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of morphogenesis. J. Virol. 69:6581–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu X, Meng X, Yan B, Rose L, Deng J, Xiang Y. 2012. Vaccinia virus virion membrane biogenesis protein A11 associates with viral membranes in a manner that requires the expression of another membrane biogenesis protein, a6. J. Virol. 86:11276–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Satheshkumar PS, Weisberg A, Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maruri-Avidal L, Domi A, Weisberg AS, Moss B. 2011. Participation of vaccinia virus L2 protein in the formation of crescent membranes and immature virions. J. Virol. 85:2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meng X, Embry A, Rose L, Yan B, Xu C, Xiang Y. 2012. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J. Virol. 86:5603–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maruri-Avidal L, Weisberg AS, Moss B. 2011. Vaccinia virus L2 protein associates with the endoplasmic reticulum near the growing edge of crescent precursors of immature virions and stabilizes a subset of viral membrane proteins. J. Virol. 85:12431–12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. 1998. Preparation of cell cultures and vaccinia virus stocks, p 16.16.1–16.16.3 In Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (ed), Current protocols in molecular biology, vol 2 John Wiley and Sons, New York, NY [Google Scholar]
- 39. Betakova T, Wolffe EJ, Moss B. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L protein kinase. J. Virol. 73:3534–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Townsley A, Senkevich TG, Moss B. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Townsley A, Senkevich TG, Moss B. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 79:10988–10998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ojeda S, Senkevich TG, Moss B. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 80:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nelson GE, Wagenaar TR, Moss B. 2008. A conserved sequence within the H2 subunit of the vaccinia virus entry/fusion complex is important for interaction with the A28 subunit and infectivity. J. Virol. 82:6244–6250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wagenaar TR, Ojeda S, Moss B. 2008. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J. Virol. 82:5153–5160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Husain M, Moss B. 2005. Role of receptor-mediated endocytosis in the formation of vaccinia virus extracellular enveloped particles. J. Virol. 79:4080–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. da Fonseca FG, Weisberg A, Wolffe EJ, Moss B. 2000. Characterization of the vaccinia virus H3L envelope protein: topology and post-translational membrane insertion via the C-terminal hydrophobic tail. J. Virol. 74:7508–7517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Howard AR, Senkevich TG, Moss B. 2008. Vaccinia virus A26 and A27 proteins form a stable complex tethered to mature virions by association with the A17 transmembrane protein. J. Virol. 82:12384–12391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szajner P, Weisberg AS, Moss B. 2004. Physical and functional interactions between vaccinia virus F10 protein kinase and virion assembly proteins A30 and G7. J. Virol. 78:266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meng X, Zhong Y, Embry A, Yan B, Lu S, Zhong G, Xiang Y. 2011. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409:271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szajner P, Jaffe H, Weisberg AS, Moss B. 2003. Vaccinia virus G7L protein interacts with the A30L protein and is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 77:3418–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lustig S, Fogg C, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454–13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brown E, Senkevich TG, Moss B. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related l1 protein. J. Virol. 80:9455–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vanderplasschen A, Hollinshead M, Smith GL. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79:877–887 [DOI] [PubMed] [Google Scholar]
- 55. Senkevich TG, Wyatt LS, Weisberg AS, Koonin EV, Moss B. 2008. A conserved poxvirus NlpC/P60 superfamily protein contributes to vaccinia virus virulence in mice but not to replication in cell culture. Virology 374:506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mercer J, Traktman P. 2003. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J. Virol. 77:8857–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vanslyke JK, Whitehead SS, Wilson EM, Hruby DE. 1991. The multistep proteolytic maturation pathway utilized by vaccinia virus P4a protein: a degenerate conserved cleavage motif within core proteins. Virology 183:467–478 [DOI] [PubMed] [Google Scholar]
- 59. Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szajner P, Jaffe H, Weisberg AS, Moss B. 2004. A complex of seven vaccinia virus proteins conserved in all chordopoxviruses is required for the association of membranes and viroplasm to form immature virions. Virology 330:447–459 [DOI] [PubMed] [Google Scholar]
- 61. Holzer G, Falkner FG. 1997. Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol. 71:4997–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Warren RD, Cotter C, Moss B. 2012. Reverse genetic analysis of poxvirus intermediate transcription factors. J. Virol. 86:9514–9519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Butler J, Watson HR, Lee AG, Schuppe HJ, East JM. 2011. Retrieval from the ER-Golgi intermediate compartment is key to the targeting of C-terminally anchored ER-resident proteins. J. Cell. Biochem. 112:3543–3548 [DOI] [PubMed] [Google Scholar]
- 64. Greenfield JJ, High S. 1999. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 112:1477–1486 [DOI] [PubMed] [Google Scholar]
- 65. Hammond C, Helenius A. 1994. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 126:41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grimley PM, Rosenblum EN, Mims SJ, Moss B. 1970. Interruption by rifampin of an early stage in vaccinia virus morphogenesis: accumulation of membranes which are precursors of virus envelopes. J. Virol. 6:519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu CG, Meng XZ, Yan B, Crotty S, Deng JP, Xiang Y. 2011. An epitope conserved in orthopoxvirus A13 envelope protein is the target of neutralizing and protective antibodies. Virology 418:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]