

Abstract
The hepatitis C virus (HCV) viroporin p7 is crucial for production of infectious viral progeny. However, its role in the viral replication cycle remains incompletely understood, in part due to the poor availability of p7-specific antibodies. To circumvent this obstacle, we inserted two consecutive hemagglutinin (HA) epitope tags at its N terminus. HA-tagged p7 reduced peak virus titers ca. 10-fold and decreased kinetics of virus production compared to the wild-type virus. However, HA-tagged p7 rescued virus production of a mutant virus lacking p7, thus providing formal proof that the tag does not disrupt p7 function. In HCV-producing cells, p7 displayed a reticular staining pattern which colocalized with the HCV envelope glycoprotein 2 (E2) but also partially with viral nonstructural proteins 2, 3, and 5A. Using coimmunoprecipitation, we confirmed a specific interaction between p7 and NS2, whereas we did not detect a stable interaction with core, E2, or NS5A. Moreover, we did not observe p7 incorporation into affinity-purified virus particles. Consistently, there was no evidence supporting a role of p7 in viral entry, as an anti-HA antibody was not able to neutralize Jc1 virus produced from an HA-p7-tagged genome. Collectively, these findings highlight a stable interaction between p7 and NS2 which is likely crucial for production of infectious HCV particles. Use of this functional epitope-tagged p7 variant should facilitate the analysis of the final steps of the HCV replication cycle.
INTRODUCTION
Viroporins are small viral proteins able to form ion channels into membranes upon multimerization (1). They are encoded by a range of enveloped and nonenveloped viruses, encompassing members of the families Togaviridae, Retroviridae, Flaviviridae, Coronaviridae, Picornaviridae, Polyomaviridae, Papillomaviridae, Orthomyxoviridae, and Paramyxoviridae. Viroporins typically play an important role in virus production, although an involvement in virus entry, genome replication, or pathogenesis has been reported in certain cases. Recently, much interest was raised by the assignment of viroporin function to HCV p7 protein (2–5) and the relative new possibility of studying its role in the whole viral replication cycle (6–10).
HCV is an enveloped, positive-stranded RNA virus belonging to the family Flaviviridae. Its genome encodes a polyprotein that is cleaved into 10 functional proteins. Structural proteins, that is to say the capsid (core) and envelope (E1 and E2) proteins, are located in the N-terminal part of the polyprotein, whereas the replication machinery (nonstructural [NS] proteins 3 to 5B) is contained in its C terminus. The p7 and NS2 proteins, located in between, are also classified as nonstructural proteins but are dispensable for viral RNA replication. However, it is now clear that both proteins are essential for virus production (6, 8, 11).
HCV p7 is a 63-amino-acid membrane-associated polypeptide that is able to oligomerize, as has been shown in vitro or in cells (2, 12–14). Notably, the precise oligomeric state of p7 is still debated, with both hexameric (2, 13, 15) and heptameric (12, 15) species having been reported. Each p7 monomer consists of two transmembrane segments separated by a hydrophilic loop orientated toward the cytosol. This hairpin-like topology is stabilized by two fully conserved basic residues at positions 33 and 35 of the p7 coding region. These residues are part of the cytoplasmic loop of p7, and they are essential for ion channel activity in vitro (16) as well as for production of infectious progeny in cell culture (8) and infectivity in vivo (11). Interestingly, there is evidence that HCV p7 has different functions in HCV production, including a contribution to assembly of viral progeny as well as release of virus particles from infected cells (8, 17). Moreover, interactions of p7 with other viral proteins have been reported, suggesting that p7 ion channel activity and its functions during virus production may be regulated via specific protein-protein interactions (18, 19). Notably, the p7 ion channeling function can be (at least partially) rescued in trans by another viroporin (for instance, the influenza virus M2 viroporin) (17). In contrast, it was shown by using chimeric HCV constructs that at least some functions of p7 are highly virus and genotype specific, because virus genomes carrying p7 variants from other isolates were strongly attenuated in virus production (20, 21).
Regarding the ion-channeling activity of p7, the ion specificity has not been fully established (15), although a preference for the channeling of cations has been reported (5). Recently, p7-mediated transfer of protons across intracellular membranes was observed (17). This property of p7 may preserve newly assembled virions from a premature conformational change of the glycoproteins during virus secretion (17). Currently, it is unclear if and how p7 protein interactions, like for instance between p7 and NS2 (18, 19) impact HCV assembly, ion channel activity, and release of viral progeny. Interestingly, genetic evidence (22) and localization studies (23) also suggested a possible interaction between core and p7, but so far, no physical interaction has been demonstrated.
Epitope-tagged p7 variants have been used to establish the topology of p7 (24, 25) and its subcellular localization. Using these constructs, a complex localization of p7 was revealed with prominent staining of the endoplasmic reticulum (ER) (24, 26, 27) but also labeling of mitochondria (26) and the plasma membrane (24). These observations suggested that p7-containing protein complexes may influence virus replication at various sites within infected cells. However, some caution is warranted, since the function of these epitope-tagged p7 variants was not confirmed and localization studies of virus-producing cells with functional p7 are still lacking. Therefore, to facilitate subcellular localization of p7 in virus-producing cells and to explore the role of p7-containing viral complexes during HCV assembly and release, we created a functional, epitope-tagged p7 and used this protein to assess subcellular localization, protein interaction, and its incorporation into progeny particles.
MATERIALS AND METHODS
Antibodies.
Mouse and rabbit anti-HA antibodies were purchased from Covance (Emeryville, CA; product MMS-101P) and Sigma (Steinheim, Germany; product H6908), respectively. Mouse anti-β-actin and anti-Flag M2 antibodies were obtained from Sigma (A2228 and F1804), rabbit anti-GM130 antibody from Epitomics (Burlingame, CA; product 1837-1), and rabbit anti-calnexin antibody from Enzo Life Sciences (Lörrach, Germany; product ADI-SPA-860). The mouse antibodies C7-50 (anti-core [28]) and 9E10 (anti-NS5A [7]), the human anti-E2 antibody CBH23 (29), and sheep anti-ADRP antiserum (30) were generous gifts from D. Moradpour (University of Lausanne), C. M. Rice (Rockefeller University), S. Foung (Stanford University), and J. McLauchlan (Glasgow University), respectively. The mouse anti-NS2 6H6 antibody and rabbit anti-NS3 4949 antiserum were previously reported (31, 32) and were kindly provided by C. M. Rice and R. Bartenschlager, respectively.
The IgG1 control isotype antibody used in the immunoprecipitation assays was directed against a neuronal antigen and kindly provided by C. Erck (HZI, Braunschweig, Germany). All fluorescent secondary antibodies were supplied by Invitrogen (Karlsruhe, Germany). The peroxidase-conjugated secondary anti-mouse antibody used for Western blots was obtained from Sigma. The antibody dilutions used for Western blotting or immunofluorescence assays are specified in the relevant sections below.
Plasmids.
Plasmids pFK-Jc1 (33), pFK-Jc1Δp7half, pFK-Jc1/KR33,35QQ (8), and pFK-Jc1/FlagE2 (34) were reported previously and served as parental constructs for all other monocistronic full-length constructs. Note that a silent XbaI restriction site had been introduced at the beginning of the p7-coding region of each construct so as to facilitate further cloning (8). For trans-complementation assays, bicistronic helper replicons were based on the construct pFK PI-EI-NS3-5B/JFH1, which was described previously (20). The coding sequences for HA-p7, HA-L-p7, Sp-HA-p7, Sp-HA-L-p7, and Sp-HA-HA-L-p7 were engineered into the first cistron as previously described for the G-Luc, p7, and Sp-p7 controls (20). In all cases, individual mutations were introduced by standard PCR-based techniques and verified by sequencing. Further sequence information is available upon request.
Cell and virus culture.
Huh-7.5 and Huh7-Lunet cells (35, 36) were grown at 37°C and with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 10% fetal calf serum. The stable Huh-7.5/shApoE/HA-ApoE cell line (34) was cultivated in the same medium but with the addition of 5 μg/ml blasticidin and 0.75 mg/ml G418.
In vitro transcription and electroporation of HCV RNA into Huh-7.5 cells were performed as previously described (37). Viral supernatants were filtered through 0.45-μm-pore-size membranes. Titers of infectious viruses were determined by a limiting dilution assay as described elsewhere (50% tissue culture infective dose [TCID50] method [38]).
For trans-complementation assays, Huh7-Lunet or Huh-7.5 cells were electroporated with 5 μg of Jc1Δp7half RNA and 5 μg of RNA corresponding to one of the helper replicons (see Fig. 1A). Cell culture supernatants were harvested at different time points postelectroporation, and titers of infectious particles were determined on naïve Huh-7.5 cells.
Fig 1.
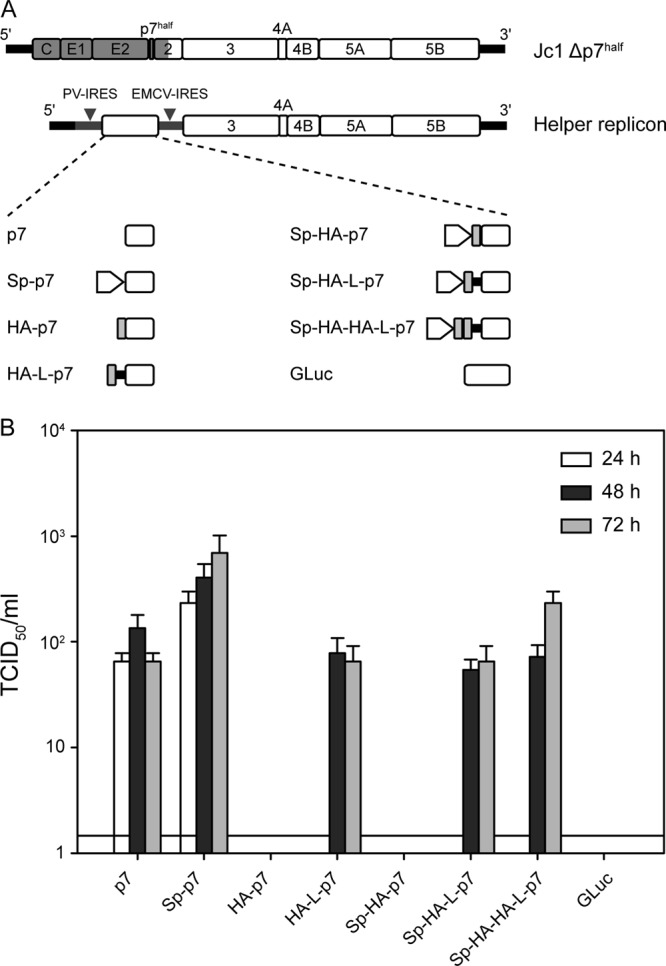
HA-tagged p7 can rescue virus production of a p7-defective virus by trans-complementation. (A) Schematic representation of the HCV assembly-deficient p7 mutant genome (Jc1Δp7half) and the helper replicons used in this study. J6-derived genome segments are depicted in gray, JFH1 portions are shown as open boxes. The 5′ and 3′ nontranslated regions are given as black bars. (B) Titers of infectious particles released by cotransfection of the Jc1Δp7half and the helper replicon in Huh-7.5 cells. Titration was performed using the TCID50 method at 24, 48, and 72 h postelectroporation. Representative results of three independent experiments with error bars representing standard deviations are shown. The background level of the limiting dilution assay is indicated by a black line.
Neutralization assays.
The different virus preparations were obtained by electroporation of Huh-7.5 or Huh-7.5/shApoE/HA-ApoE cells with the relevant HCV RNA. Target Huh-7.5 cells were seeded at 104 cells per well in 96-well dishes 1 day before infection. Virus (around 30 focus-forming units [FFU]/well) were mixed with appropriate amounts of antibody (mouse anti-Flag M2 or rabbit anti-HA antibody) and incubated for 1 h at 37°C prior to infection of target cells. Infections were performed in 35 μl/well for 3 h at 37°C with gentle agitation. Cell supernatants were then replaced with fresh medium (100 μl/well), and the cells were further incubated for 40 h before fixation. Cell fixation, immunostaining, and focus counting were performed as described elsewhere (38).
SDS-PAGE and Western blotting.
SDS-PAGE (12% acrylamide) and Western blotting were performed as previously described (37). Note that we routinely treat our protein samples in 1× Laemmli buffer (75 mM Tris-HCl [pH 6.8], 0.6% SDS, 15% glycerol, 0.001% bromophenol blue, 7.5% ß-mercaptoethanol) for 5 min at 98°C. However, we observed that this heating step impaired the detection of HA-tagged p7. As a consequence, samples intended for p7 detection were heated only to 37°C for 15 min, conditions that did not alter the detection of the other viral proteins tested. For Fig. 2B, Huh-7.5 cells were electroporated with the relevant RNA as described above and seeded in 6-well dishes. At different time points postelectroporation, the confluent cells from one well were lysed in 100 μl Laemmli buffer and treated with Benzonase (Novagen) (1 μl per sample) for 15 min at 37°C, and 15 μl was analyzed by SDS-PAGE for protein expression.
Fig 2.
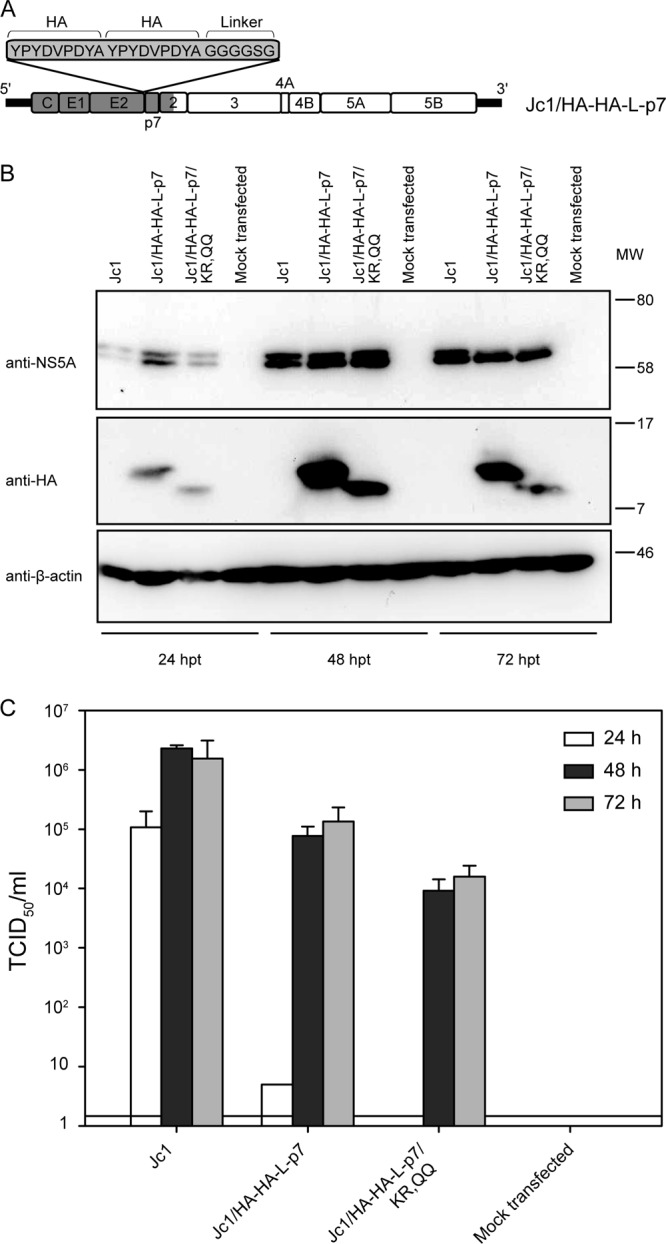
HA-tagged p7 is functional in the full-length infectious Jc1 virus. (A) Schematic representation of the Jc1/HA-HA-L-p7 construct. (B) Western blot analysis of NS5A, p7 (HA antibody), and actin expression in cell lysates at 24, 48, or 72 h postelectroporation with the indicated construct. (C) Titers of infectious particles released by transfection of the mentioned constructs. Titration was performed using the TCID50 method at 24, 48, and 72 h postelectroporation. Representative results of three independent experiments with error bars representing standard deviations are shown. The background level of the limiting dilution assay is indicated by a black line. MW, molecular weight (in thousands).
Antibodies used for immunostaining were diluted as follows: anti-β-actin, 1/1,000; anti-NS5A, 1/1,000; mouse anti-HA, 1/1,000; anti-NS2, 1/1,000; and anti-core, 1/1,000. As a secondary antibody, the peroxidase-conjugated anti-mouse antibody was diluted 1/20,000 except after 6H6 treatment (1/2,000 dilution), and the peroxidase-conjugated anti-rabbit antibody was diluted 1/25,000.
Immunoprecipitation of cell lysates.
For each HCV RNA construct, 6 × 106 cells were electroporated with the relevant RNA, and two-thirds of the cells were seeded into one 10-cm-diameter tissue culture dish (the rest of the cells was used to control the electroporation efficiency and the virus titers released). The cells were harvested 48 h posttransfection by trypsinization, and cell pellets were lysed in 230 μl lysis buffer (phosphate-buffered saline [PBS] supplemented with 1% Triton X-100 and a protease inhibitor cocktail [Complete; Roche]). From these mixtures, 200 μl was used for immunoprecipitation with the mouse anti-HA or control isotype antibodies (100 μl each) and 20 μl was kept for the inputs (1/5 of the input). For each immunoprecipitation, 25 μl of protein G-agarose beads (Roche) were washed 3 times in PBS. Subsequently, washed beads were incubated with 3.3 μg antibody diluted in PBS for 2 h at 4°C with rotation. In parallel, cell lysates were precleared on protein G-agarose in the absence of antibody for 2 h at 4°C. Afterwards, precleared cell lysates were added onto the washed antibody-bound beads and incubated overnight at 4°C with constant rotation, in PBS–1% Triton X-100. Beads were then washed 5 times in PBS–1% Triton X-100 (three times quickly and twice with a 5-min incubation time with rotation) and finally once quickly in water before protein elution in Laemmli buffer and SDS-PAGE analysis.
Immunoprecipitation of native secreted viral particles.
Viral supernatants were produced as mentioned above for the neutralization assays. Cleared supernatants were concentrated by ultracentrifugation over a 20% sucrose cushion in sterile PBS in a Sorvall TH-641 rotor for 4 h at 110,000 × g (ThermoScientific Sorvall WX 80 ultracentrifuge). The supernatant was carefully decanted by inverting the ultracentrifuge tube and pipetting out the last drops at its rim. Subsequently, pelleted virus was recovered by addition of ca. 150 μl PBS per tube, incubation at 4°C for several hours or overnight, and subsequent resuspension by pipetting. Concentrated virus stocks were stored at −80°C or used fresh. On average, virus stocks were concentrated 200 times in volume. A 100-μl sample of concentrated virus stocks was used per immunoprecipitation, and 20 μl was kept for the inputs (1/5 inputs). For the anti-Flag immunoprecipitations, the M2 anti-Flag affinity matrix purchased from Sigma (A2220) was used. For the anti-HA immunoprecipitations, we used protein G-agarose matrix linked with the mouse anti-HA antibody or the IgG1 control isotype (in each case, 3.3 μg antibody/immunoprecipitation, as indicated above). For each condition, 30 μl beads were used. Concentrated virus supernatants were incubated directly with the antibody-bound beads overnight at 4°C under rotation, in PBS. Beads were washed 6 times in PBS (four times quickly and twice with a 5-min incubation with rotation), and proteins were eluted in 1× Laemmli buffer and analyzed by SDS-PAGE and Western blot. Alternatively, core protein in the culture fluid of transfected cells or after immunoprecipitation was quantified by using the Architect HCV Core AG test (Abbott, Wiesbaden, Germany) according to the instructions of the manufacturer.
Immunofluorescence.
Huh-7.5 cells were seeded on coverslips and infected for 4 h at 37°C with undiluted supernatants harvested from Jc1/HA-HA-L-p7-transfected cells. Forty-eight hours postinfection, cells were washed in PBS and fixed in PBS–1% paraformaldehyde for 30 min at room temperature (RT). Cells were then permeabilized for 15 min in PBS–0.05% Triton X-100. Afterwards, cells were incubated in blocking buffer (PBS complemented with 5% goat serum [Sigma]) for 1 h at RT. Primary antibodies were diluted in blocking buffer (see above) and incubated with the cells overnight at RT. Relevant species-specific secondary antibodies (A488 or A467-conjugated anti-mouse IgG, anti-rabbit IgG–A488 or –A647, anti-human IgG–A546, and anti-sheep IgG–A447) were diluted 1/1,000 in blocking buffer and added to the cells for 6 h at RT in the dark. Finally, cell nuclei were stained with DAPI (Invitrogen) diluted 1/3,000 for 1 min at RT, and coverslips were mounted on glass slides using Fluoromount-G (product 100-01; Southern Biotech, Birmingham, AL). Between steps, cells were washed three times in PBS. For lipid droplet staining, BODIPY 495/503 (Invitrogen) was diluted 1/1,000 and added together with the secondary anti-mouse antibody (for detection of HA-tagged p7). Mitochondria were stained by incubating live cells with the dye Mitotracker RedCMXRos (Invitrogen), diluted at 100 nM in unsupplemented DMEM, for 45 min just before cell fixation. In this particular case, cells were fixed in PBS–3% paraformaldehyde, for 10 min at RT.
Primary antibody dilutions were as follows: mouse anti-HA, rabbit anti-HA, mouse anti-core C7-50 (28), mouse anti-NS2 6H6 (32), and mouse anti-NS5A 9E10 (7) were diluted 1/1,000; human anti-E2 CBH-23 (29) hybridoma supernatant was diluted 1/250, rabbit anti-NS3 4949 was diluted 1/400 (31); sheep anti-ADRP (30) and rabbit anti-GM130 were diluted 1/500; and rabbit anti-calnexin was diluted 1/300. Note that the mouse anti-HA antibody yielded better staining than the rabbit anti-HA antibody. However, we observed that both staining patterns colocalized similarly with E2 (data not shown).
Pictures were taken with an inversed confocal laser-scanning microscope (Olympus Fluoview 1000), using a ×100 magnification lens. The three channels (blue, green, and red) were read in a sequential acquisition mode, with an average of 3 frames for each picture (Kalman n = 3). The pixel size of the original pictures was between 70 and 100 nm.
Colocalization analysis.
Intensity profiles were generated with the Fluoview 1000 viewer software (Olympus). Frequency scatter plots and Pearson's colocalization coefficients (39) were obtained with the Mander's coefficient plugin (Tony Collins and Wayne Rasban, Wright Cell Imaging Facility, Toronto, Canada) in ImageJ (National Institutes of Health Bethesda, MD [http://rsb.info.nih.gov/ij/]). Note that Pearson's coefficients close to 1 are indicative of reliable colocalization.
RESULTS
HA epitope-tagged p7 rescues virus production of p7-deleted HCV genome.
Studies of the HCV p7 ion channel have been limited by the restricted availability of efficient specific antibodies and the difficulties inherent to the small size of the protein. Various tagged p7 versions have previously been reported (19, 24, 26, 27) and have allowed p7 detection, but none of these constructs proved to be functional when incorporated into the HCV genome, limiting the studies to artificial heterologous expression systems. The first aim of this study was to generate an epitope-tagged p7 that would be functional for HCV production. Importantly, while p7 is crucial for virus production (6, 8), its sequence also affects HCV polyprotein processing (8, 40). Therefore, genetic modification, and in particular epitope tagging of p7, can have pleiotropic effects on the HCV replication cycle. We recently described a trans-complementation system allowing the production of single-round infectious particles based on HCV subgenomic replicons packaged in helper cell lines that encode the remaining HCV proteins (41). Of particular interest, it is possible to restore HCV production of mutant HCV genomes harboring a deletion of half the p7 protein (Jc1Δp7half) by expressing the complete p7 protein together with its signal peptide in trans from a helper replicon (Fig. 1A) (20, 41). This system can be used to perform genetic modifications of p7; it also allows the contribution of p7 in HCV production to be specifically studied, thereby circumventing possible polyprotein processing defects due to genetic modification of the p7 sequence. We therefore used the trans-complementation system to test the capacity of tagged p7 variants to rescue virus production of the Jc1Δp7half mutant.
Several p7-encoding helper replicons were designed incorporating a single or double influenza hemagglutinin (HA) epitope at the N terminus of p7 (Fig. 1A). We additionally tested the insertion of a short linker (L) sequence (GGGGSG) between the tag and the p7 sequence, as well as the addition of the p7 signal peptide (Sp). The influenza hemagglutinin A (HA) epitope (YPYDVPDYA) (42, 43) was chosen for its small size (1.1 kDa), broad use, and precise characterization and for the availability of characterized HA-specific tools (antibodies and peptides). As was previously reported, Jc1Δp7half was unable to produce infectious particles (8, 20), but virus production was restored by providing, via a helper replicon, p7 alone or together with its minimal signal peptide (the 17 C-terminal residues of the E2 glycoprotein), with a slightly higher efficiency in the latter case (Fig. 1B) (20). Expression of the Gaussia luciferase instead of p7 served as a negative control for the assay and did not rescue virus release. Interestingly, addition of a HA tag at the N terminus of p7 abrogated the trans-complementation, unless a linker sequence was inserted between the tag and p7. The HA-L-p7 and Sp-HA-L-p7 constructs trans-complemented the Jc1Δp7half genome with an efficiency comparable to that of the constructs encoding nontagged p7, although with delayed kinetics in virus production. Finally, we tested the addition of a double-HA tag at the N terminus of p7, in order to enhance the affinity and specificity of HA-specific antibodies for the tagged protein. This double tag was inserted in combination with the linker peptide described above and the p7 signal peptide, as both features seemed to contribute to the optimal function of the single tagged or wild-type (WT) p7, respectively. Importantly, the double-tagged construct was as efficient as the single-tagged construct in restoring virus production of Jc1Δp7half. Collectively, these results provide formal proof that HA-epitope-tagged p7 preserves the function of the protein in the course of virus production.
Of note, we also designed p7 constructs fused to the green fluorescent protein (GFP) and tested their ability to rescue virus production of the Jc1Δp7half mutant, in the same trans-complementation assay (data not shown). Our constructs contained a glycine-serine linker (GGGGSGGGGST) between the GFP and p7 and, in the case of a C-terminal tag, a point mutation (A63I, terminal p7 residue) intended to prevent the natural signal peptide cleavage between p7 and NS2. These constructs failed either to rescue virus production (N-terminal tag) or to maintain the GFP tag (C-terminal tag). Given these circumstances, we did not pursue p7 localization with the described GFP-p7 fusion proteins and rather focused on the HA-tagged p7 constructs.
A double-HA-tagged p7 is functional in the full-length Jc1 system.
Both single- and double-HA-tagged p7 constructs were able to trans-complement, with comparable efficiencies, the virus production of a p7-deficient HCV mutant (Fig. 1). We next aimed to validate this result in the context of the full-length infectious Jc1 clone. A Jc1 genome incorporating a double HA tag followed by a linker sequence at the N terminus of p7 was created as illustrated in Fig. 2A. This construct efficiently replicated after transfection in Huh-7.5 cells, as indicated by NS5A detection in the cell lysates (Fig. 2B). Moreover, the tagged p7 could be readily detected in transfected cells, at the expected molecular size of around 10 kDa (7 kDa [p7] + [2 × 1.1] kDa [double HA tag] + 0.4 kDa [linker]), with an HA-specific antibody (Fig. 2B). p7 detection was optimal 48 h posttransfection and decreased thereafter, together with detection of the NS5A signal, probably due to the cells' reaching confluence in this assay setup. Last, the cell supernatant of transfected Huh-7.5 cells was titrated for infectious particles on naïve cells. High-titer infectious particles, reaching 105 TCID50/ml, were produced by the HCV genome carrying the double HA tag, confirming the functionality of this p7 variant as first deduced from the trans-complementation assay. However, a ca. 10-fold decrease in titer and a slower kinetics of virus production compared to those of WT Jc1 were observed, indicating that the double HA tag did affect p7 function in the HCV replication cycle to some extent. To verify that the p7 function could still be investigated in the context of a tagged-p7 virus, a derived construct harboring two extra point mutations (KR33,35QQ) was also generated as a control. This double mutation has been described as preventing the ion channel activity of p7 and therefore hindering the p7 function in virus production (8, 11, 16, 17). Replication of the genome was verified by NS5A detection in the transfected cells (Fig. 2B). Interestingly, the double mutation resulted in an increased electrophoretic mobility of the tagged p7 protein (Fig. 2B, bottom), which could be explained by a change in the local charge of the protein and in the subsequent interaction with SDS in the gel. More importantly, as observed in the WT construct background (6, 8), the KR33,35QQ mutation resulted in a decrease in titers of infectious particles (10- or 30-fold decrease on average at 48 and 72 h postelectroporation) in the context of the Jc1/HA-HA-L-p7 virus (Fig. 2C). This suggested that the ion channel function of p7 was intact in the tagged p7 protein and that the Jc1/HA-HA-L-p7 construct could be used for functional studies of p7.
The possibility of tagging p7 in a system that allows the whole replication cycle of HCV has, to our knowledge, not been reported before. Therefore, we used this system to confirm previously reported observations on p7 function and to extend our knowledge of p7 localization and protein partners within the infected cells.
p7 colocalizes with E2 and nonstructural proteins in the ER of HCV-infected cells.
p7 was previously suggested to reside in the ER membrane (19, 24, 26, 27), but some reports also proposed its association with mitochondria (16, 26). We set out to investigate the localization of p7 in HCV-infected cells, using anti-HA antibodies. p7 was found in the cytoplasm of infected cells, following a typical reticular pattern, widespread in the cell but with an enhanced signal around the nucleus (Fig. 3 and 4, HA panels). To delineate more precisely its subcellular localization, we first stained p7 together with cellular organelles (Fig. 3 and 5). As expected, the p7 staining overlaid with an ER-marker, the calnexin chaperone. However, no obvious colocalization was observed with a mitochondrion-specific dye. Because of the proposed role of p7 for pH maintenance in the secretory pathway (17), we also stained the Golgi apparatus using GM130-specific antibodies as marker but could not detect any colocalization with p7. Finally, as lipid droplets are an important organelle for HCV assembly and concentrate several viral factors (44, 45), we stained them with either the BODIPY lipid dye or with an antibody specific for a lipid droplet-associated protein, ADRP (adipocyte differentiation-related protein). In neither case could we detect an overlay between the p7 and lipid droplet stainings. Nevertheless, a fraction of the total p7 was detected in close proximity to the lipid droplets (Fig. 3 and 5; see the enlarged images for BODIPY/p7-stained samples). The predominant ER localization of p7 was confirmed by immune gold labeling of HCV-transfected cells with an anti-HA antibody and observation by electron microscopy (data not shown).
Fig 3.

Subcellular localization of p7 relatively to cellular organelles. Huh-7.5 cells were fixed 48 h postinfection with Jc1/HA-HA-L-p7. For each staining combination, representative confocal images are shown in gray for the individual green and red channels and in color for the merged images (green, red, and blue channels, the latter representing DAPI staining of the cell nuclei). Areas highlighted with yellow boxes in the merged pictures are depicted at higher magnification in the rightmost column. Colocalization analyses of these pictures (profile intensity on a section of the picture, intensity scatter plot, and Pearson's coefficient) are presented in Fig. 5.
Fig 4.

p7 colocalizes with E2 and nonstructural proteins in infected cells. Huh-7.5 cells were fixed 48 h postinfection with Jc1/HA-HA-L-p7. For each staining combination, representative confocal images are shown in gray for the individual green and red channels and in color for the merged images (green, red, and blue channel, the latter representing the DAPI staining of the cell nuclei). Areas highlighted with yellow boxes in the merged pictures are depicted at higher magnifications in the rightmost column. Colocalization analysis of these pictures (profile intensity on a section of the picture, intensity scatter plot, and Pearson's coefficient) are presented in Fig. 6.
Fig 5.

Colocalization analysis of tagged p7 with cellular markers. Analysis was performed on the pictures showed in Fig. 3. The middle column shows signal intensity profiles for green, red, and blue (DAPI) channels along a section of the picture that is depicted in the left column. The right column represents frequency scatter plots of the intensity registered in the red and green channels. Note that this analysis was gated on the infected cells. Moreover, the HA signal is always shown on the x axis, whereas the y axis corresponds to the intensity of the second marker (e.g., Mitotracker, BODIPY, etc.). Individual dots in the scatter plot correspond to single pixels of the original picture. The color code highlights the frequency of dots present in a certain region of the scatter plot (from blue to yellow and white with increasing frequencies). Pearson's r coefficient of colocalization is indicated in the top right corner of the plot.
In a second step, we assessed the localization of p7 respectively to other viral proteins (Fig. 4 and 6). A striking colocalization was observed between p7 and the E2 glycoprotein, as reflected by the strong correlation of the two signal intensities (Fig. 6). In contrast, p7 and core showed clearly different staining patterns, with core being enriched in one part of the cell while p7 was widely spread throughout the cytoplasm. Local spots of colocalized core-p7 signals were nevertheless detected, even though they accounted only for a fraction of the p7 protein (Fig. 4, enlarged area, and 6). Finally, p7 also colocalized with NS2, -3, and -5A, although not as strikingly as with E2.
Fig 6.

Colocalization analysis of tagged p7 with other viral proteins. Analysis was performed on the pictures showed in Fig. 5. Graphs are as described in the legend to Fig. 5, except that the scatter plot analysis and Pearson's coefficient calculation were performed on the whole picture, as both markers analyzed were detected only in infected cells.
p7 interacts with NS2 in HCV-infected cells.
In the next step, we analyzed the possible viral interaction partners of p7 within the infected cells. A physical interaction between tagged p7 and tagged NS2 was shown after heterologous expression of tagged proteins (19) or in the cell-cultured HCV (HCVcc) system (18) and is supported by genetic evidence (18, 33, 46). Moreover, genetic interactions reported between p7 and core (22) could indicate further viral interaction partners for p7. Jirasko et al. also reported the coimmunoprecipitation of p7 but also NS3, E2, and to a lesser extent NS5A together with NS2 (18), implying the formation of multimolecular assembly complexes. Finally, the strong colocalization observed between HA-HA-L-p7 and E2 or NS2 in infected cells might suggest a direct interaction between these proteins (Fig. 4 and 6). We tackled these issues by immunoprecipitating HA-HA-L-p7 from the lysates of HCV-producing cells. NS2 coprecipitated with HA-HA-L-p7 (Fig. 7A), confirming previous reports (18, 19). Interestingly, a second species corresponding to a protein of around 25 to 30 kDa was consistently coprecipitated and enriched in an anti-HA immunoprecipitation. It probably corresponded to an uncleaved p7-NS2 precursor, as this species could be detected independently by both an anti-HA and anti-NS2 antibody (data not shown).
Fig 7.

p7 interacts with NS2 in HCV-producing cells. (A) NS2 coimmunoprecipitates with HA-tagged p7. Huh-7.5 cells were electroporated with Jc1 (−) or Jc1/HA-HA-L-p7 RNA (+). At 48 h posttransfection, cells were lysed, and immunoprecipitations with an anti-HA antibody or with a control isotype were performed. HA-tagged p7 and NS2 were detected in cell lysates (inputs) and in immunoprecipitates by Western blotting. In brief, proteins were revealed with a mixture of anti-HA and anti-NS2 mouse antibodies followed by detection with an anti-mouse IgG–horseradish peroxidase conjugate. The asterisks correspond to the immunoglobulins used for the immunoprecipitations (IPs) (from top to bottom, partially dissociated immunoglobulins, heavy and light chains). To improve p7 detection, Laemmli-containing protein samples were heated only to 37°C, which probably explains why part of the immunoglobulin heavy and light chains remained undissociated. (B) The p7 F26S mutation partially compensates for the core 69-72A defect in virus production. Titers of virus supernatants were determined using the TCID50 method at 24, 48, and 72 h after electroporation with the indicated constructs. Data are averages from 4 to 7 independent experiments, with error bars showing standard deviations. The background level of the limiting dilution assay is indicated as a black line. (C) The core 69-72A mutation does not affect p7/NS2 interaction. Immunoprecipitations and Western blot were performed as described for panel A, with lysates of cells electroporated with constructs 1 to 6 (numbering corresponds to panel B). MW, molecular weight (in thousands).
Despite trying different immunoprecipitation conditions and detergents (Triton X-100 and n-dodecyl-β-maltoside), we were unable to detect a physical interaction between HA-HA-L-p7 and E2, NS5A, or core (data not shown). The possibility of an interaction between p7 and core had been supported by the ability of p7 to redirect the core protein from the lipid droplets to the ER (23). It had also been suggested by the isolation of p7 mutations (position 776 in JFH-1 polyprotein or 26 in p7 sequence) compensating for the assembly defect of core mutants (22). This compensatory effect was particularly strong for the p7 F776S rescue of the core 69-72A mutation. Because we could not detect an interaction between core and p7, we tested whether the core 69-72A mutation could affect assembly indirectly by hindering the p7/NS2 interaction and whether the p7 compensatory mutation could compensate for such a defect. We therefore engineered the core 69-72A mutation, as well as the compensatory p7 F26S mutation (corresponding to the mutation designated F776S in the original report [22]), in the Jc1/HA-HA-L-p7 construct, alone or in combination (Fig. 7B). In this background, the core 69-72A mutation resulted in a >100-fold decrease in released infectivity, a defect partially rescued by the p7 F26S mutation (3- to 7-fold increase). It is noteworthy that these phenotypes were mild compared to those described by Murray et al. (22), probably due to the slightly different parental construct used (Jc1/HA-HA-L-p7 in our case versus J6/JFH). In particular, we observed only a limited rescue of the core 69-72A phenotype by the p7 F26S mutation (3- to 7-fold increase, respectively, at 48 and 72 h posttransfection in an average of 6 [72-h time point] or 7 [48-h time point] independent experiments, among which only half showed a rescue). The moderate rescue is possibly due to the attenuating effect of the p7 F26S mutation in the context of the Jc1/HA-HA-L-p7 genome (3- to 6-fold decrease in infectivity at 48 and 72 h posttransfection, and delayed kinetics). We nevertheless tested the p7/NS2 interaction for these mutants (Fig. 7C). All mutants in the context of the tagged-p7 genome expressed NS2 and p7 at comparable levels 48 h posttransfection. Note that, similar to the p7 KR33,35QQ mutant, the p7 F26S mutant exhibited a slightly increased electrophoretic mobility. Moreover, the p7-NS2 precursor was detected for all tagged p7 constructs (see above). More importantly, the core 69-72A mutation did not significantly alter the p7/NS2 interaction, nor did the p7 rescue mutation. Therefore, the assembly defect associated with the core 69-72A mutation does not seem to be due to a decreased p7/NS2 interaction.
No evidence for p7 incorporation in HCV particles.
Finally, we addressed the controversial issues of possible p7 incorporation in the secreted viral particle and its role in entry. As described above, the HA-HA-L-p7 protein is readily expressed in HCV-producing cells (Fig. 2B) and able, albeit with a slightly lower efficiency than WT p7, to support the production of virus particles. Therefore, if p7 was incorporated into the viral particle, it is likely that epitope-tagged p7 would also be incorporated, with the N-terminal tag facing the outside of the particle.
Two approaches were therefore used to test the presence of p7 in the viral particle. First, we attempted to neutralize HCV entry with an HA-specific rabbit polyclonal antibody or with a Flag-specific antibody as a control (Fig. 8A). For this experiment, we produced viruses from different constructs: (i) untagged WT Jc1, (ii) Jc1/HA-HA-L-p7 (described above), (iii) Jc1/FlagE2, with a Flag epitope in E2 preceding HVR1 (47), or (iv) the double-tagged Jc1/FlagE2/HA-HA-L-p7. We also produced Jc1 virus in cells expressing HA-tagged ApoE (34). Because ApoE is incorporated into viral particles and plays a role in entry (47–49), we used the Jc1/HA-ApoE virus as a control for anti-HA antibody-mediated neutralization. In this setup, the anti-HA antibody was not able to neutralize the infectivity of viruses produced from the Jc1/HA-HA-L-p7 or the Jc1/FlagE2/HA-HA-L-p7 construct, while a weak but reproducible neutralization of Jc1 virus incorporating HA-tagged ApoE was achieved (Fig. 8A, left). As a control, the anti-Flag antibody efficiently neutralized virus particles incorporating a Flag-tagged E2 protein (47), including the virus particles generated from a double-tagged Jc1/FlagE2/HA-HA-L-p7 genome (Fig. 8A, right). Therefore, the anti-HA antibody was in principle able to neutralize virus entry (see Fig. 3A, left panel, Jc1/HA-ApoE) but had no effect on virus produced from the Jc1/FlagE2/HA-HA-L-p7 construct, with the latter being efficiently neutralized, however, by an anti-Flag antibody. Finally, we were unable to demonstrate a role of p7 in HCV entry or the incorporation of p7 in the virus particle by antibody-mediated neutralization.
Fig 8.

Absence of evidence for p7 incorporation in the HCV virion or role in entry. (A) The indicated viruses were incubated with anti-HA or anti-Flag antibodies prior to infection of cells. At 40 h postinfection, infectivity was evaluated by a focus-forming-unit assay. The residual infectivity relative to infectivity in the absence of antibody is shown. Means from 3 independent experiments, each containing 3 replicates, including standard deviations are shown. (B) Huh-7.5 cells were transfected with Jc1, Jc1/HA-HA-L-p7, Jc1/FlagE2 or Jc1/FlagE2/HA-HA-L-p7 RNA (respectively, lanes 1 to 4 in each part of the gel [inputs/control IgG/anti-HA/anti-Flag]). As a further control, Jc1 was prepared from Huh-7.5 cells expressing HA-ApoE (lane 5 in each part of the gel). Concentrated supernatants of the electroporated cells were either directly lysed and loaded on the gel (left) or subjected to immunoprecipitation with anti-HA antibodies or the relevant control isotype (middle) or an anti-Flag affinity matrix (right). Inputs and immunoprecipitated proteins were analyzed by Western blotting for p7 (anti-HA antibody; no signal detected), ApoE (anti-HA antibody), and core (C7-50 antibody) content. Arrowheads indicate detection of the core protein in the immunoprecipitates. The quantity of HCV core protein in each sample was also determined using a core-specific ELISA. Mean values and standard deviations from three independent experiments performed with independent virus stocks are given. MW, molecular weight (in thousands).
Second, we tested the direct detection of HA-HA-L-p7 in the cell culture fluid of HCV-producing cells and in affinity-purified HCV particles (Fig. 8B). To do this, we used the same viruses as for the neutralization assay, namely, untagged Jc1, Jc1 with HA-tagged p7 and/or Flag-tagged E2, and Jc1 incorporating HA-ApoE (see above). Importantly, we were never able to detect tagged p7 in concentrated infectious cell supernatants (data not shown). Furthermore, we could detect the core protein (Jc1/FlagE2 and Jc1/FlagE2/HA-HA-L-p7 viruses) but no tagged p7 (Jc1/FlagE2/HA-HA-L-p7 virus) in Flag-purified virus particles (Fig. 8B, anti-Flag immunoprecipitations and white arrowheads [left]). Finally, we attempted to purify HA-tagged proteins from the cell supernatant and investigated the coprecipitation of core protein, as a marker for the virion, using Western blots and a highly sensitive core-specific enzyme-linked immunosorbent assay (ELISA) (Fig. 8B, anti-HA immunoprecipitations). While we could coprecipitate core with a virus incorporating HA-ApoE (Fig. 8B, white arrowheads [left and right panels]), this was not the case with virus particles produced from a HA-tagged p7 genome (Jc1/HA-HA-L-p7 or Jc1/FlagE2/HA-HA-L-p7). Altogether, we had no proof of p7 secretion and could neither detect p7 in affinity-purified viral particles nor immunoprecipitate viruses produced from an HA-tagged p7 genome with an anti-HA antibody. Thus, consistently with the functional neutralization data, this biochemical approach gave no evidence of p7 incorporation in the virus particle.
DISCUSSION
HA-tagged p7, a new tool to study p7 function and HCV production.
Several tagged versions of p7 have been described (19, 24, 26, 27, 50), but none was shown to be compatible with virus production. The use of these constructs was therefore limited to single-protein (19, 24) or, at best, replicon-driven (27) expression systems. Moreover, because p7 is small and membrane integral, it is poorly immunogenic, and therefore antibodies against this polypeptide are rare. To our knowledge, the only reported anti-p7 antibodies are strain-specific polyclonal antisera raised against the N- or C-terminal residues of p7 (2, 26, 51). For these reasons, the immunological detection of p7 in a relevant HCV-infection system is still challenging (21, 52). It therefore remains extremely difficult to track p7 and p7-containing protein complexes during HCV assembly and release. Likewise, purification of p7 and its interacting partners from HCV-producing cells and, even more so, the investigation of p7 incorporation into secreted viral particles are difficult.
In this work, we created new versions of tagged p7. Our attempts to produce virus particles from constructs encoding GFP-tagged p7 were not successful (data not shown). However, addition of a double-HA tag at the N terminus of p7 permitted production of infectious virus in a trans-complementation setup but also in the full-length Jc1 infectious clone. A nearly one-log decrease in titers of infectious particles was nevertheless observed in the context of the Jc1 clone (Fig. 2C). A more detailed characterization of the tagged HCV genome would be needed to fully understand this defect. However, our data suggest that the specific role of p7 in assembly and release of HCV was, despite the tag, if not intact, at least mostly preserved. First, the tagged p7 constructs could rescue virus production in a trans-complementation system to the same extent as untagged p7, although with delayed kinetics (Fig. 1). We chose to use this system because it allows the specific investigation of HCV assembly and release (together with entry), independently of the upstream steps of HCV replication cycle. Indeed, the modified p7 sequence is provided in trans, whereas all determinants for HCV replication, polyprotein translation and processing are encoded in cis by the subgenomic replicon (in our case the Jc1Δp7half construct), and therefore stable in the assay. In a full-length Jc1 genome, p7 tagging also resulted in slower kinetics of infectious virus production but additionally yielded lower titers of infectious particles. Therefore, we hypothesize that p7 tagging might affect a step in the HCV replication cycle that was disregarded in the trans-complementation system, namely, replication, polyprotein translation, or processing. Because p7 and the neighboring E2 and NS2 proteins are not major actors in virus RNA replication (53) but p7 sequence is a determinant for polyprotein processing (8, 40), we favored this last hypothesis. Consistently, we detected by Western blotting a p7-NS2 precursor in cells transfected with the Jc1/HA-HA-L-p7 construct (Fig. 7A and C). This species was indeed stained independently by anti-HA or anti-NS2 antibodies (data not shown), was enriched in anti-HA immunoprecipitates, and had an apparent molecular size of 25 to 30 kDa, consistent with a p7-NS2 precursor (ca. 10 + 21 kDa). Importantly, such a species was not detected in Jc1-transfected cells with an anti-NS2 antibody, pointing at a specific defect of HCV polyprotein processing in our HA-tagged construct. Such a defect is intriguing, because the tag was inserted at the N terminus of p7, whereas p7-NS2 cleavage occurs at its C terminus. It could, however, explain the lower titers of infectious particles obtained with the tagged construct, either through a decreased availability of fully processed p7 or NS2 proteins (however, the latter could not be evidenced in our Western blot analysis [Fig. 7A and C]) or via a slight dominant negative effect of a nonfunctional p7-NS2 precursor. Last, we indirectly tested the ion channel function of tagged p7 by mutating the dibasic motif in the cytosolic loop of p7 (KR33,35QQ mutation) in the context of our tagged-p7 genome. These residues were previously shown in biochemical assays to be crucial for p7 ion channeling (16) but were also shown in infectious systems to be important for the role of p7 in virus production (6, 8, 11, 20) and its capacity to regulate the intravesicular pH along the secretory pathway (17). Because the KR33,35QQ mutation impaired virus production of the HA-tagged genome (Fig. 2C), we reasoned that the HA-tagged p7 maintained an ion channel activity. As shown in Fig. 7, tagged p7 also retained its ability to interact with NS2 (18), suggesting that the protein-protein interaction function of p7 was also preserved. Although we cannot exclude the possibility that the tag was lost from a fraction of the p7 proteins, the interaction of HA-tagged p7 with NS2 and its colocalization with E2 strongly support the specific involvement of HA-tagged p7, rather than a cleaved p7 form, in the HCV replication cycle.
Finally, our data showed the possibility of using the double HA tag to detect p7 by Western blotting (Fig. 2B) and immunofluorescence in HCV-producing cells (Fig. 3 to 6) and to immunoprecipitate p7 and associated proteins (Fig. 7), with a good specificity and sensitivity in all cases. Although we did not test the double-HA tag with p7 proteins derived from other isolates and genotypes, the tag position at one end of the protein and its separation from p7 by a linker sequence should increase its compatibility with a variety of p7 sequences.
Localization of p7 in HCV-infected cells and interaction with viral partners.
The localization of p7 in HCV-infected cells remains unknown (21), as previous studies were restricted to heterologous expression systems (24, 26, 27, 52). Most reports supported the ER retention of p7 (19, 24, 26, 27). This is consistent with the incorporation of p7 in assembly complexes, which are believed to gather at the ER membrane, in close proximity to lipid droplets (18, 19). However, a possible localization in the mitochondria was also proposed (16, 26). This would not be an unprecedented situation, as other viroporins or viral proteins can localize to mitochondria and alter their function (54–57). Finally, p7 might protect pH-sensitive intracellular virions from premature fusion by equilibrating the pH of acidic intracellular vesicles (17). This would suggest a possible localization of p7 along the secretory pathway, and in particular in the Golgi apparatus where the virus particle is thought to traffic before secretion. The localization of p7 in HCV-producing cells is therefore an open question and one that is crucial for understanding the function of p7.
Our data supported a cytoplasmic distribution of p7 in HCV-producing cells, with a typical reticular pattern of p7, including staining of the nuclear envelope (Fig. 3). The p7 signal colocalized with an ER marker, but little colocalization was observed with mitochondria. Also, p7 was not detected in the Golgi apparatus. Consistently, p7 colocalized nearly perfectly with the E2 envelope protein, with signal intensities of the two stainings being strongly correlated (Fig. 6). Of all cellular markers and viral proteins tested, Pearson's and Mander's coefficients were the highest for the p7/E2 colocalization (Fig. 5 and 6; also data not shown). Finally, colocalization was also observed with NS2, NS3, and NS5A proteins, while p7 and core signals overlapped only locally.
This localization of p7 in the ER and its colocalization with E2 and NS2 reflect the position of p7 in the viral polyprotein. This also further endorses the incorporation of p7 in multimolecular assembly complexes, in particular with NS2 and the glycoproteins (18, 19). In this respect, the p7/NS2 interaction, already reported in the HCVcc and in heterologous expression systems (18, 19), was confirmed by immunoprecipitation of p7 (Fig. 7A). However, no interaction between p7 and other viral proteins, for instance E2, core, or NS5A, was detected. It was reported that p7, E2, NS3, and to a lesser extent NS5A could be copurified with NS2 in HCV-producing cells (18). Nevertheless, there was no proof that all proteins were incorporated together within the same assembly complexes. The architecture of assembly complexes might be more complicated and might gather together several types of complexes (for instance a p7/NS2 complex and an NS2/glycoprotein complex), with some proteins, such as NS2, being able to sequentially interact with one complex or another, without a need for a direct or indirect interaction between all proteins. More generally, interactions between p7 and other viral proteins might be indirect and too weak to be detected under our assay conditions, might involve only a small fraction of the total p7 protein, or might simply be too transient to be detected. It is also possible that the double-HA tag becomes poorly accessible once p7 is incorporated into a multiprotein complex. Interestingly, core mutants defective in assembly could be rescued by p7 mutations (22) (Fig. 7B). Moreover, in the absence of other viral proteins, p7 is able to relocate the core protein from the lipid droplet to the ER surface (23). Finally, we believe that p7 is important for RNA encapsidation and for capsid envelopment (J. Gentzsch and T. Pietschmann, submitted for publication). For these reasons, we were particularly interested in testing the core-p7 interaction in our system. Since we could not detect any interaction between these two partners, we investigated whether the assembly-impairing core 69-72A mutation, which is rescued by the p7 F26S mutation, could act indirectly by modulating the p7/NS2 interaction. This did not seem to be the case, as individual or double mutants exhibited comparable p7/NS2 interactions (Fig. 7C). In conclusion, it seems that NS2 is the main interacting partner of p7, while p7 colocalizes most strikingly with E2. Transient or weak interactions might occur between p7 and other proteins, or NS2 might be sufficient to pull together p7 and other assembly factors, by interacting sequentially with them.
Is p7 incorporated into the viral particle?
Finally, the tagged p7 construct was used to test the presence of p7 in the secreted viral particle, an issue that remains controversial. Most other viroporins are not incorporated into the virion (for instance, HIV Vpu, alphavirus 6K, and Jc virus agnoprotein are excluded from the virion [58, 59]). However, in the case of influenza virus, the M2 viroporin is present on the viral particle and plays an important role in entry (reviewed in references 60 and 61). What is more, the delayed and incomplete cleavage of the HCV E2-p7-NS2 precursor (8, 62–64) could theoretically favor the incorporation of p7, alone or as a precursor with E2 and eventually NS2, into the secreted HCV viral particle. Note that the related pestiviruses also encode a p7 viroporin that is crucial for virus production. However, neither p7 nor p7-containing precursors were detected in BVDV (bovine viral diarrhea virus) or CSFV (classical swine fever virus) virions (65). Furthermore, whereas an E2-p7-NS2 precursor was readily detected in JFH-1-producing cells with an anti-E2 antibody, no such precursor was found in the HCV virion (66). To our knowledge, the only indirect indication of a possible role of p7 in HCV entry comes from inhibitor studies using amantadine, rimantadine, NN-DNJ or GSK2, all of which are presumed p7 inhibitors (51). Addition of these drugs during cell entry reduced HCV infection, possibly by blocking p7 function(s) during virus entry or uncoating. However, the observed effects were moderate, and it is difficult to exclude the possibility that these drugs also perturb cellular factors, thus indirectly impeding HCV infection.
In addition to this, it is clear thanks to the HCV pseudoparticle (HCVpp) system that HCV glycoproteins can mediate virus entry by themselves (67–69). Whether HCVpp truly mimic entry of the genuine viral particle or of HCVcc is certainly disputable, however, since HCVpp are built on a retroviral backbone and typically produced in nonhepatoma cells. Undoubtedly, HCVpp cannot recapitulate the HCV uncoating, as they contain a retroviral capsid and genome, and fusion itself might be biased by the HCVpp structure and lipid composition. Therefore, the possibility that p7 might be dispensable for HCVpp entry but necessary for HCVcc entry at a late stage, to mediate fusion or uncoating, as shown for the influenza M2 protein (61), cannot be ruled out.
Further insight into the question of p7 incorporation in the virion was provided by the characterization of p7 mutants. In particular, we previously showed that the p7 KR33,35QQ mutant, although impaired in assembly, had a specific infectivity similar to that of the WT Jc1 virus (8). The report by Wozniak et al. goes along the same lines (17). In brief, the authors used the KR33,35AA mutation, which in the context of genotype 1a p7 (chimeric HJ3-5 virus) totally abrogates infectious virus production. In this context, virus particles could be rescued by bafilomycin treatment of virus-producing cells, and these particles were able to infect naïve cells. These are strong indications that p7 is crucial for assembly but not important for virus entry.
An assembly-competent virus encoding a tagged p7 protein is an interesting tool to tackle this issue from a novel and more direct angle. We therefore used several new approaches to test p7 incorporation into the virion and its possible role in entry. First, we tried to directly detect p7 in the supernatant of HCV-producing cells by using the anti-HA antibody in Western blotting. Despite a good affinity of this antibody for p7, as shown in cell lysates (Fig. 2B), we were never able to detect p7 in the concentrated infectious cell supernatant or in affinity-purified Flag-tagged HCV particles (Fig. 8B) (47). We also tried to immunoprecipitate p7 from the concentrated infectious cell supernatant using an anti-HA antibody and tested the coprecipitation of the core protein, as a marker for viral particles (Fig. 8B). In this case, the final readout is based on core protein detection. Core stoichiometry in the virion is likely to be higher than that of p7, and moreover, its detection can be performed by a highly sensitive ELISA. For these reasons, the anti-HA immunoprecipitation assay is likely to be more sensitive than the anti-Flag immunoprecipitation. However, whether we used a Western blot or core ELISA detection, we were not able to specifically detect the core protein in an anti-HA immunoprecipitate.
Finally, an anti-HA antibody failed to neutralize the infectivity of virus particles produced from a p7-tagged genome. In principle, however, the same anti-HA antibody was able to neutralize virus particle infectivity, as evidenced by the neutralization of a control virus incorporating HA-tagged ApoE. Also, a double-tagged virus, incorporating a Flag epitope at the N terminus of E2 and a double HA epitope at the N terminus of p7, could be neutralized by an anti-Flag antibody but not by an anti-HA antibody.
Obviously, these pieces of data cannot rule out the incorporation of p7 in the virus particle or its role in entry. Regarding the neutralization assay, p7 could be incorporated into the virus particle and have no role in entry, or it could have a role in entry that cannot be blocked by the anti-HA antibody used. The negative immunoprecipitation results could also be attributed to an insufficient sensitivity of the method. As a comparison, despite the important role of M2 in influenza virus entry, the virion harbors only a few copies of the viroporin (14 to 68 copies per virion), that is to say 10 to 100 times less than the HA envelope protein (500 to 1,200 copies) (70). Given these data, it is likely that production of larger stocks of affinity-purified viruses (for instance, using the Jc1/Flag E2 construct) coupled to more sensitive detection methods or attempts to immunoprecipitate HA-tagged p7 from large-scale virus preparations will be needed to answer this question more conclusively.
In conclusion, the generation of an infectious HCV clone containing an epitope-tagged p7 allowed us to analyze the subcellular localization of p7 in HCV-infected cells and to examine its interacting partners as well as its possible incorporation into the viral particle. We believe that this construct will be useful to further clarify these issues, in the context of the WT virus but also in the context of assembly mutants or pharmacological inhibition of assembly.
ACKNOWLEDGMENTS
We are grateful to Rajesh Kolli and Mathias Müsken for their advice on confocal microscopy, to Francois Penin for his advice on the choice of linker sequences, and to Regina Raupach for technical assistance. We also thank Ralf Bartenschlager, Christian Erck, Steven Foung, John McLauchlan, Darius Moradpour, and Charles M. Rice for providing us with antibodies. Finally, we acknowledge all members of the Institute of Experimental Virology at Twincore for valuable discussions.
This work was funded by an Emmy Noether fellowship to T.P. (PI 734/1-1) provided by the Deutsche Forschungsgemeinschaft and by a grant from the Initiative and Networking Fund of the Helmholtz Association (SO-024) to T.P.
Footnotes
Published ahead of print 21 November 2012
REFERENCES
- 1. Nieva JL, Madan V, Carrasco L. 2012. Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10:563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. 2003. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, amantadine. FEBS Lett. 535:34–38 [DOI] [PubMed] [Google Scholar]
- 3. Montserret R, Saint N, Vanbelle C, Salvay AG, Simorre JP, Ebel C, Sapay N, Renisio JG, Bockmann A, Steinmann E, Pietschmann T, Dubuisson J, Chipot C, Penin F. 2010. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J. Biol. Chem. 285:31446–31461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pavlovic D, Neville DC, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. 2003. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl. Acad. Sci. U. S. A. 100:6104–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Premkumar A, Wilson L, Ewart GD, Gage PW. 2004. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 557:99–103 [DOI] [PubMed] [Google Scholar]
- 6. Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81:8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 8. Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3:e103 doi:10.1371/journal.ppat.0030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao ZJ, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakai A, Claire MS, Faulk K, Govindarajan S, Emerson SU, Purcell RH, Bukh J. 2003. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. U. S. A. 100:11646–11651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clarke D, Griffin S, Beales L, Gelais CS, Burgess S, Harris M, Rowlands D. 2006. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 281:37057–37068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luik P, Chew C, Aittoniemi J, Chang J, Wentworth P, Jr, Dwek RA, Biggin PC, Venien-Bryan C, Zitzmann N. 2009. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 106:12712–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patargias G, Zitzmann N, Dwek R, Fischer WB. 2006. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J. Med. Chem. 49:648–655 [DOI] [PubMed] [Google Scholar]
- 15. Chandler DE, Penin F, Schulten K, Chipot C. 2012. The p7 protein of hepatitis C virus forms structurally plastic, minimalist ion channels. PLoS Comput. Biol. 8:e1002702 doi:10.1371/journal.pcbi.1002702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Griffin SD, Harvey R, Clarke DS, Barclay WS, Harris M, Rowlands DJ. 2004. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol. 85:451–461 [DOI] [PubMed] [Google Scholar]
- 17. Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. 2010. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 6:e1001087 doi:10.1371/journal.ppat.1001087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6:e1001233 doi:10.1371/journal.ppat.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Popescu CI, Callens N, Trinel D, Roingeard P, Moradpour D, Descamps V, Duverlie G, Penin F, Heliot L, Rouille Y, Dubuisson J. 2011. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 7:e1001278 doi:10.1371/journal.ppat.1001278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brohm C, Steinmann E, Friesland M, Lorenz IC, Patel A, Penin F, Bartenschlager R, Pietschmann T. 2009. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J. Virol. 83:11682–11693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steinmann E, Pietschmann T. 2010. Hepatitis C virus p7-a viroporin crucial for virus assembly and an emerging target for antiviral therapy. Viruses 2:2078–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murray CL, Jones CT, Tassello J, Rice CM. 2007. Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J. Virol. 81:10220–10231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boson B, Granio O, Bartenschlager R, Cosset FL. 2011. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 7:e1002144 doi:10.1371/journal.ppat.1002144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carrere-Kremer S, Montpellier-Pala C, Cocquerel L, Wychowski C, Penin F, Dubuisson J. 2002. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 76:3720–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isherwood BJ, Patel AH. 2005. Analysis of the processing and transmembrane topology of the E2p7 protein of hepatitis C virus. J. Gen. Virol. 86:667–676 [DOI] [PubMed] [Google Scholar]
- 26. Griffin S, Clarke D, McCormick C, Rowlands D, Harris M. 2005. Signal peptide cleavage and internal targeting signals direct the hepatitis C virus p7 protein to distinct intracellular membranes. J. Virol. 79:15525–15536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haqshenas G, Mackenzie JM, Dong X, Gowans EJ. 2007. Hepatitis C virus p7 protein is localized in the endoplasmic reticulum when it is encoded by a replication-competent genome. J. Gen. Virol. 88:134–142 [DOI] [PubMed] [Google Scholar]
- 28. Moradpour D, Wakita T, Tokushige K, Carlson RI, Krawczynski K, Wands JR. 1996. Characterization of three novel monoclonal antibodies against hepatitis C virus core protein. J. Med. Virol. 48:234–241 [DOI] [PubMed] [Google Scholar]
- 29. Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 8:e1002653 doi:10.1371/journal.ppat.1002653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Targett-Adams P, Chambers D, Gledhill S, Hope RG, Coy JF, Girod A, McLauchlan J. 2003. Live cell analysis and targeting of the lipid droplet-binding adipocyte differentiation-related protein. J. Biol. Chem. 278:15998–16007 [DOI] [PubMed] [Google Scholar]
- 31. Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035 doi:10.1371/journal.ppat.1000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dentzer TG, Lorenz IC, Evans MJ, Rice CM. 2009. Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J. Virol. 83:12702–12713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Menzel N, Fischl W, Hueging K, Bankwitz D, Frentzen A, Haid S, Gentzsch J, Kaderali L, Bartenschlager R, Pietschmann T. 2012. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog. 8:e1002829 doi:10.1371/journal.ppat.1002829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 79:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haid S, Windisch MP, Bartenschlager R, Pietschmann T. 2010. Mouse-specific residues of claudin-1 limit hepatitis C virus genotype 2a infection in a human hepatocyte cell line. J. Virol. 84:964–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vieyres G, Pietschmann T. 2012. Entry and replication of recombinant hepatitis C viruses in cell culture. Methods http://dx.doi.org/10.1016/j.ymeth.2012.09.005 [DOI] [PubMed]
- 39. Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169:375–382 [DOI] [PubMed] [Google Scholar]
- 40. Carrere-Kremer S, Montpellier C, Lorenzo L, Brulin B, Cocquerel L, Belouzard S, Penin F, Dubuisson J. 2004. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J. Biol. Chem. 279:41384–41392 [DOI] [PubMed] [Google Scholar]
- 41. Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 82:7034–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. 1988. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 8:2159–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. 1984. The structure of an antigenic determinant in a protein. Cell 37:767–778 [DOI] [PubMed] [Google Scholar]
- 44. Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 19:95–103 [DOI] [PubMed] [Google Scholar]
- 45. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 46. Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81:629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286:3018–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783–13793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 83:12680–12691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. StGelais C, Foster TL, Verow M, Atkins E, Fishwick CW, Rowlands D, Harris M, Griffin S. 2009. Determinants of hepatitis C virus p7 ion channel function and drug sensitivity identified in vitro. J. Virol. 83:7970–7981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Griffin S, StGelais C, Owsianka AM, Patel AH, Rowlands D, Harris M. 2008. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 48:1779–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Popescu CI, Rouille Y, Dubuisson J. 2011. Hepatitis C virus assembly imaging. Viruses 3:2238–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 54. D'Agostino DM, Ranzato L, Arrigoni G, Cavallari I, Belleudi F, Torrisi MR, Silic-Benussi M, Ferro T, Petronilli V, Marin O, Chieco-Bianchi L, Bernardi P, Ciminale V. 2002. Mitochondrial alterations induced by the p13II protein of human T-cell leukemia virus type 1. Critical role of arginine residues. J. Biol. Chem. 277:34424–34433 [DOI] [PubMed] [Google Scholar]
- 55. Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. 2003. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 77:7214–7224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muthumani K, Choo AY, Premkumar A, Hwang DS, Thieu KP, Desai BM, Weiner DB. 2005. Human immunodeficiency virus type 1 (HIV-1) Vpr-regulated cell death: insights into mechanism. Cell Death Differ. 12(Suppl 1):962–970 [DOI] [PubMed] [Google Scholar]
- 57. Takada S, Shirakata Y, Kaneniwa N, Koike K. 1999. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 18:6965–6973 [DOI] [PubMed] [Google Scholar]
- 58. Gonzalez ME, Carrasco L. 2003. Viroporins. FEBS Lett. 552:28–34 [DOI] [PubMed] [Google Scholar]
- 59. Imperiale MJ, Major EO. 2007. Polyomaviruses, p 2268–2282 In Knipe DM, Howley M. (ed), Fields virology, 5th ed, vol 3 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 60. Helenius A. 1992. Unpacking the incoming influenza virus. Cell 69:577–578 [DOI] [PubMed] [Google Scholar]
- 61. Kelly ML, Cook JA, Brown-Augsburger P, Heinz BA, Smith MC, Pinto LH. 2003. Demonstrating the intrinsic ion channel activity of virally encoded proteins. FEBS Lett. 552:61–67 [DOI] [PubMed] [Google Scholar]
- 62. Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67:1385–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mizushima H, Hijikata M, Asabe S, Hirota M, Kimura K, Shimotohno K. 1994. Two hepatitis C virus glycoprotein E2 products with different C termini. J. Virol. 68:6215–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Elbers K, Tautz N, Becher P, Stoll D, Rumenapf T, Thiel HJ. 1996. Processing in the pestivirus E2-NS2 region: identification of proteins p7 and E2p7. J. Virol. 70:4131–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, Dubuisson J. 2010. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 84:10159–10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Drummer HE, Maerz A, Poumbourios P. 2003. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 546:385–390 [DOI] [PubMed] [Google Scholar]
- 69. Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zebedee SL, Lamb RA. 1988. Influenza A virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J. Virol. 62:2762–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]