

Abstract
The innate immune system is responsible for recognizing invading pathogens and initiating a protective response. In particular, the retinoic acid-inducible gene 1 protein (RIG-I) participates in the recognition of single- and double-stranded RNA viruses. RIG-I activation leads to the production of an appropriate cytokine and chemokine cocktail that stimulates an antiviral state and drives the adaptive immune system toward an efficient and specific response against the ongoing infection. One of the best-characterized natural RIG-I agonists is the defective interfering (DI) RNA produced by Sendai virus strain Cantell. This 546-nucleotide RNA is a well-known activator of the innate immune system and an extremely potent inducer of type I interferon. We designed an in vitro-transcribed RNA that retains the type I interferon stimulatory properties, and the RIG-I affinity of the Sendai virus produced DI RNA both in vitro and in vivo. This in vitro-synthesized RNA is capable of enhancing the production of anti-influenza virus hemagglutinin (HA)-specific IgG after intramuscular or intranasal coadministration with inactivated H1N1 2009 pandemic vaccine. Furthermore, our adjuvant is equally effective at increasing the efficiency of an influenza A/Puerto Rico/8/34 virus inactivated vaccine as a poly(I·C)- or a squalene-based adjuvant. Our in vitro-transcribed DI RNA represents an excellent tool for the study of RIG-I agonists as vaccine adjuvants and a starting point in the development of such a vaccine.
INTRODUCTION
Cellular innate immunity constitutes the first barrier against pathogen invasion. It represents a nonspecific defense present in all eukaryotic cells. The system has evolved from RNA-based immunity (still present in plants and invertebrates) to protein-based immunity (1). In both cases, the activation of the innate system relies on a rapid and efficient recognition of the invading pathogen. In protein-based immunity, there are a wide variety of receptors involved in the recognition of different types of molecular patterns (pathogen-associated molecular pattern, or PAMP) generated during a pathogen's invasion (2). These pathogen sensors are known as pattern recognition receptors (PRRs). Once a PRR detects the presence of a PAMP, it will trigger a signal cascade that leads to the production of an appropriate set of cytokines and chemokines which, in most cases, includes interferon (IFN). This protein cocktail will lead to a partial or total block of the infection in the infected cell (3) or to the acquisition of a refractory state to pathogen invasion in uninfected cells. The efficiency of the system is such that most viruses, in order to establish a successful infection, must encode a mechanism to evade the innate immune system (4).
Among the different groups of PRRs, Toll-like receptors (TLRs) can be found in the plasma membrane and endosomal vesicles, while retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLRs), the nucleotide oligomerization domain-like receptors (NLRs), and cytosolic DNA sensors (like the AIM2 family) are responsible for the detection of PAMPs within the cytoplasm (5). The localization of the PRR and its binding specificity will determine the pathogen being recognized and, therefore, the type of response.
The RLR family is composed of three members, RIG-I, the melanoma differentiation-associated protein 5 (MDA5), and the laboratory of genetics and physiology protein 2 (LGP2). RIG-I recognizes RNA molecules with a 5′-triphosphate (5′ppp) group and double-stranded structure (6). It contains an amino-terminal region consisting of a tandem caspase activation and recruitment domain (CARD), a central RNA helicase domain capable of binding and possibly unwinding RNA, and a carboxy-terminal repressor domain involved in autoregulation (7). RIG-I is crucial for the recognition of many double- and single-stranded RNA viruses, including Newcastle disease virus, hepatitis C virus, Sendai virus (SeV), vesicular stomatitis virus, Japanese encephalitis virus, and influenza A virus (8). Once RIG-I is activated, it will stimulate an immune reaction appropriate for the nature of the infection. This innate immune response plays a role not only in the direct inhibition of an invading pathogen but also in the modulation of the adaptive immune response (9). Type I IFN, for example, participates in NK cell activation (10), regulation of effector and memory T cells (11), and B-cell activation (12); IL-15 modulates T-cell and NK cell activation and function (13); and IL-12 stimulates NK-cell activity, CD8+ cytotoxic T-cell activity, and development of a CD4+ Th1 responses (14). These and many other studies (see reference 15 for a full review) highlight the important role of the innate immune system in controlling and enhancing the subsequent adaptive immune response. In fact, many of the vaccine adjuvants developed so far exploit the PRR function to enhance vaccine efficiency. For example, poly(I·C) (pIC) activates TLR3 and the RLR MDA5, the synthetic oligodeoxynucleotide CpG is a TLR9 agonist, and the monophosphoryl lipid A stimulates TLR4 signaling (16). A RIG-I-based vaccine adjuvant will potentially mimic the PAMP generated during a natural infection and subsequently stimulate an innate immune reaction suitable for those viruses recognized by RIG-I. This type of adjuvant may be the most appropriate for several viral vaccines, including influenza A and B virus vaccines, hepatitis C virus vaccine, and Japanese encephalitis virus vaccine.
SeV is a parainfluenza virus from the Paramyxoviridae family. Some strains (e.g., the Cantell strain) can produce large amounts of defective interfering RNA particles (DI) (17). The DI RNAs are replicative RNA species smaller in length (∼550 bp) than wild-type infectious RNA. SeV DI RNA is generated during vRNA synthesis from a cRNA template. In most cases, the viral polymerase copies the cRNA into a full complementary copy corresponding to the vRNA. However, in some instances, the polymerase leaves the cRNA template and back copies the 5′ end of the vRNA product before complete synthesis of this vRNA is completed, giving rise to a copy-back DI. These copy-back DIs therefore display perfect complementarity of their 5′ and 3′ ends, which correspond to the sequences of the cRNA promoter, and become templates for further amplification by the viral polymerase. The SeV Cantell DI particles are well-known inducers of IFN (18, 19) and can, independently of type I IFN, stimulate dendritic cell maturation (20). Their RNAs contain a 5′ triphosphate (ppp) and a partially double-stranded structure (17). It has been shown recently that SeV DI RNAs are excellent RIG-I agonists (21). In the current manuscript, we explore the use of RIG-I ligands, particularly SeV RNA (containing mostly DI RNA), and an in vitro-transcribed SeV DI RNA (IVT DI) as adjuvants for inactivated influenza A virus vaccines.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee at Mount Sinai School of Medicine. Mice were euthanized according to these guidelines, and all efforts were made to minimize suffering of the animals.
Cell lines and antibodies.
Madin-Darby canine kidney (MDCK), 293T, and Vero cells were obtained from the ATCC and were maintained in minimal essential medium (MEM) or Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone) and penicillin-streptomycin (Gibco). LA-4 cells were obtained from the ATCC and maintained in F-12K medium (Gibco) supplemented with 15% FBS (HyClone) and penicillin-streptomycin (Gibco). For the generation of a stable 293T cell line expressing firefly luciferase under the control of the IFN-β promoter (293T-FF), a plasmid carrying a cassette with the firefly luciferase gene under the control of the murine IFN-β promoter (22) was transfected into 293T-ISRE-mRFP cells (23). Cells were selected in the presence of Geneticin (Invitrogen), and individual clones were isolated and tested for reporter induction upon Sendai virus infection. The selected clone was maintained in DMEM supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 2 mg/ml of Geneticin.
Anti-mouse IgG horseradish peroxidase (HRP) antibody was purchased from GE Healthcare, and anti-mouse IgG1 and anti-mouse IgG2a were from Southern Biotech. For the IgA determination, we used a goat anti-mouse IgA from Sigma and a phosphatase alkaline-conjugated anti-goat antibody from Sigma. Enzyme-linked immunosorbent assay (ELISA) was done with Costar 3797 plates coated with either influenza A/Cal/04/2009 virus hemagglutinin (HA) protein from BEI Resources or influenza A/Puerto Rico/8/34 virus purified in house.
Adjuvants and vaccines.
Vaccine-grade poly(I·C) and MF59 equivalent emulsion (AddaVax) were obtained from InvivoGen. SeV Cantell RNA enriched in DI RNA was obtained by growing the virus in 10-day-old chicken eggs (18). The virus was then purified by ultracentrifugation through a sucrose cushion, and the RNA was extracted using TRIzol (Invitrogen) according to the manufacturer's protocol. The RNA was further cleaned up using the RNeasy kit from Qiagen.
The plasmid expressing the SeV DI RNA was constructed by PCR amplification of the SeV DI sequence from A549 SeV-infected cells using a 5′ primer containing the T7 promoter and a 3′ primer containing the hepatitis delta virus genomic ribozyme site followed by the T7 terminator. The resulting DNA was then cloned between EcoRI/HindIII sites in the pUC19 plasmid. The sequence of the plasmid was confirmed by Sanger sequencing. IVT DI and T7 control RNA were synthesized from the respective DNA plasmids using the HiScribe T7 in vitro transcription kit (New England BioLabs) by following the manufacturer's instructions. Control RNA was produced from the Litmus 28iMal control plasmid provided by the manufacturer. After in vitro transcription, the RNA was cleaned up using the RNeasy kit. To test for endotoxin contamination in the IVT DI, we used the QCL-1000 endpoint chromogenic LAL assay from Lonza. The sequence of the IVT DI, including the T7 promoter, hepatitis delta virus ribozyme, and the T7 terminator, is TAATACGACTCACTATAACCAGACAAGAGTTTAAGAGATATGTATCCTTTTAAATTTTCTTGTCTTCTTGTAAGTTTTTCTTACTATTGTCATATGGATAAGTCCAAGACTTCCAGGTACCGCGGAGCTTCGATCGTTCTGCACGATAGGGACTAATTATTACGAGCTGTCATATGGCTCGATATCACCCAGTGATCCATCATCAATCACGGTCGTGTATTCATTTTGCCTGGCCCCGAACATCTTGACTGCCCCTAAAATCTTCATCAAAATCTTTATTTCTTTGGTGAGGAATCTATACGTTATACTATGTATAATATCCTCAAACCTGTCTAATAAAGTTTTTGTGATAACCCTCAGGTTCCTGATTTCACGGGATGATAATGAAACTATTCCCAATTGAAGTCTTGCTTCAAACTTCTGGTCAGGGAATGACCCAGTTACCAATCTTGTGGACATAGATAAAGATAGTCTTGGACTTATCCATATGACAATAGTAAGAAAAACTTACAAGAAGACAAGAAAATTTAAAAGGATACATATCTCTTAAACTCTTGTCTGGTGGCCGGCATGGTCCCAGCCTCCTCGCTGGCGCCGGCTGGGCAACATTCCGAGGGGACCGTCCCCTCGGTAATGGCGAATAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTG. The sequence of the SeV DI is highlighted in boldface.
Mouse vaccines (both intramuscular [i.m.] and intranasal [i.n.]) consisted of (i) 25 μl of phosphate-buffered saline (PBS) containing the indicated vaccine dose plus (ii) 25 μl of PBS and 25 μl of PBS with 4 μg of pIC, SeV RNA, or IVT DI or AddaVax at a 1:1 (vol/vol) ratio.
In vitro, transfection of pIC, SeV RNA, and IVT RNA was done with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and using the indicated amount of RNA. RNase A was obtained from Qiagen, and enzymatic reactions were performed as indicated by the manufacturer.
Generation of inactivated PR8 vaccine.
One hundred PFU of influenza A/PuertoRico/8/34 virus was injected into 8-day-old embryonated chicken eggs, and allantoic fluid was collected after 48 h. The allantoic fluid was clarified by centrifugation at 1,000 × g for 30 min, and the virus was subsequently pelleted through a cushion (30% sucrose in PBS) by ultracentrifugation with an SW28 rotor at ∼100,000 × g for 2 h. The virus was resuspended in PBS, adjusted to a protein concentration of 0.5 mg/ml, and incubated on ice. A 10× inactivation solution was prepared by diluting 100 μl of 37% formaldehyde with 3.9 ml of PBS. The solution was passed through a 0.22-μm filter, chilled on ice, and added dropwise to the virus to yield a final ratio of 1:10. The reaction proceeded on a rotator at 4°C for 3 days, and the vaccine was stored at −80°C. Inactivation of the PR8 virus was confirmed by plaque assay.
RNA tissue extraction.
RNA from mammalian cell culture was obtained using RNeasy from Qiagen. For in vivo RNA extraction, mouse calf muscles or lungs were extracted and homogenized in 1 ml of TRIzol-LS (Ambion) using a FastPrep-24 instrument (MP). Homogenized samples then were passed through a QIAshredder column (Qiagen), and the RNA was isolated according to the manufacturer's protocol. Residual DNA in the samples was removed using DNA-free (Ambion). To ensure optimal RNA quality, the samples were cleaned up using an RNeasy kit from Qiagen.
qRT-PCR.
cDNA was synthesized using SuperScript III reverse transcriptase (Invitrogen) by following the manufacturer's instructions. Real-time PCR quantification was performed using SYBR green mix (Roche) and a LightCycler 480 Instrument (Roche). Samples were normalized to β-actin. Primer sequences for quantitative reverse transcription-PCR (qRT-PCR) were the following: mouse β-actin forward, TTTGCAAGCTCCTTCGTTGC; reverse, TCGTCATCCATGGCGAACT; mouse IFN-β forward, CCAGCTCCAAGAAAGGACGA; reverse, CGCCCTGTAGGTGAGGTTGAT; mouse Mx forward, GACCAATAGGGGTCTTGACCAA; reverse, AGACTTGCTCTTTCTGAAAAGCC; mouse IP-10 forward, GTGTTGAGATCATTGCCACGA; reverse, GCGTGGCTTCACTCCAGTTAA.
In vivo analysis of the antiviral activity of the stimulatory RNAs.
To test the antiviral properties of the different RNAs in vivo, mice (n = 3) were treated intranasally with 1 μg of pIC, SeV RNA, or pIC in 50 μl of PBS. As a negative control, we used 50 μl of PBS. Inoculation was done 1, 5, or 10 days prior to the infection (day 0) with 3,000 PFU of influenza A/WSN/33 virus. Five days postinfection, mice were euthanized and their lungs extracted. The viral load (PFU/ml) was then measured by standard plaque assay in MDCK cells. To monitor weight loss and survival, animals (n = 5) were treated 24 h before the infection with PBS, pIC, or IVT DI as described previously. After infection (day 0) with influenza A/WSN/33 virus, the weight was recorded daily until the animals reached 75% of the original weight.
RESULTS
The in vitro-transcribed SeV DI RNA is a potent inducer of IFN-β and IFN-stimulated genes (ISGs).
Sendai virus (SeV) defective interfering particles (DI) are natural RIG-I agonists capable of inducing large amounts of interferon (21, 24). One can obtain a high percentage of SeV DI RNA by growing SeV Cantell, an SeV strain with a propensity to produce DI RNA, under the appropriate conditions (18 and see Materials and Methods for details). We sought to imitate the characteristics of the natural SeV DI with an in vitro transcript. To synthesize the SeV DI in vitro (IVT DI), the DI sequence was first cloned from human lung carcinoma A549 cells infected with SeV Cantell and inserted into a pUC19 plasmid between the T7 promoter and the hepatitis delta virus genomic ribozyme site followed by the T7 terminator (Fig. 1A). With this design, we rely on the T7 polymerase activity to in vitro transcribe the SeV DI RNA and incorporate the 5′ppp required for RIG-I recognition (25, 26) and on the viral ribozyme to create the appropriate 3′ end. Like the natural DI RNA, this in vitro-transcribed RNA is predicted to contain a 5′ppp and a double-stranded region (94 bp) composed of the complementary 5′ and 3′ ends flanking a 358-nucleotide loop.
Fig 1.

Type I interferon induction by the IVT DI RNA. (A) Schematic representation of the IVT DI RNA design and RNA structure (top) and sequence of the SeV DI RNA (bottom). (B) Titration of pIC in 293T-FF cells. The IFN-β reporter cells were transfected with increasing amounts of pIC, and 24 h later the luciferase signal was measured. (C) To compare their IFN-inducing abilities, equivalent amounts of the following RNAs (50 ng) were transfected into the 293T IFN-β reporter cells: pIC (orange bar; size, 1.5 to 8 kb), SeV RNA (green bar; size, 546 bp) (0.39 pM), in vitro-transcribed DI (IVT DI; blue bar; size, 546 bp) (0.39 pM), and a control RNA produced in vitro by the T7 polymerase (IVT C; gray bar; size, ∼800 bp). After 24 h, the luciferase signal was measured and is represented as fold induction above the level of the mock-transfected cells. (D) Comparison of the IFN-β induction activity of in vitro-transcribed DI RNA (IVT DI; blue bar; size, 546 bp; 50 ng) and low molecular size pIC (LMW pIC; pale orange bar; size, 0.2 to 1 kb; 50 and 200 ng). (E) RNase treatment of SeV RNA and the IVT DI RNA. The RNA was digested (+) or mock treated (−) and transfected into the 293T IFN-β reporter cell line to monitor the stimulatory activity. The fold induction of the mock-treated RNA was set to 100%. Error bars represent standard deviations from three replicates. *, P < 0.5; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
To assess the ability of the IVT DI to induce IFN-β, we utilized a reporter 293T cell line expressing firefly luciferase under the control of the IFN-β promoter (293T-FF) (27). It is important to note that 293-T cells do not express most TLRs; therefore, the results obtained will be biased toward the ability of this RNA to stimulate RLRs (28).
First, 293T-FF cells were transfected with increasing amounts of pIC to find a concentration that produces a signal in the linear range (Fig. 1B). Based on these results, 50 ng of each of the following RNAs was transfected into 293T-FF cells to compare their stimulatory properties: (i) SeV RNA, isolated from virions and enriched in DI RNA (SeV RNA), (ii) IVT DI, (iii) in vitro-transcribed T7 control RNA (IVT C), and (iv) pIC as a standard stimulatory nucleic acid (Fig. 1C). Cells transfected with SeV RNA or IVT DI showed similar levels of luciferase induction, indicating that IVT DI RNA retains the type I IFN stimulatory potency of the DI RNA extracted from SeV virions. Furthermore, the fold induction over mock-transfected cells was significantly higher for the IVT DI than for pIC or control T7 RNA (Fig. 1C). The superior activity of the IVT DI over the control T7 RNA suggests that the IVT RNA contains other stimulatory characteristics besides the 5′ppp incorporated by the T7 polymerase.
There is a considerable difference between the molecular size of our IVT DI RNA (546 bp) and that of pIC (1.5 to 8 kb). To exclude the possibility that the observed differences in reporter stimulation were simply due to the use of different molar concentrations for pIC and the IVT DI, we compared the induction of luciferase by the SeV-derived RNA (546 bp) to that of low-molecular-size pIC (with an estimated size of 0.2 to 1 kb) (Fig. 1D). Once more, the fold induction above the level of mock-transfected cells was significantly higher for the IVT DI RNA than for pIC (low molecular size)-transfected cells even when we used a 4 times higher concentration of low-molecular-size pIC.
To rule out the possibility that contamination during the production of RNA was responsible for the observed effect, we treated both SeV RNA and the IVT RNA with RNase A before transfecting them into 293T-FF cells. Complete removal of the IFN-inducing activity in the RNase-treated samples confirmed that the stimulation was RNA dependent (Fig. 1E). We also tested our IVT DI RNA for endotoxin contamination. The levels of endotoxin in the purified RNA were below the limit of detection (data not shown).
The role of RIG-I in the induction of IFN-β by the IVT DI was explored using mouse embryonic fibroblasts (MEFs) from RIG-I knockout mice and their wild-type counterparts. Stimulation of IFN-β and myxovirus resistance gene (Mx) mRNA production after transfection with pIC was comparable in RIG-I−/− and RIG-I+/+ MEFs (Fig. 2). On the other hand, the levels of IFN-β mRNA obtained in RIG-I knockout MEFs after stimulation with the IVT DI were significantly lower than those obtained in wild-type cells. A similar trend was observed for the Mx gene mRNA, although the difference between wild-type and knockout cells was not significant in this case. Together, these results indicate that the IVT DI is primarily recognized by RIG-I in MEFs.
Fig 2.

Induction of IFN-β in RIG-I−/− versus RIG-I+/+ cells. Equivalent amounts of pIC (A) and IVT DI RNA (B) were transfected into wild-type MEFs (RIG-I+/+; empty bars) and RIG-I knockout MEFs (RIG-I−/−; hatched bars). Twenty-four hours after transfection, total RNA was extracted, and mRNA for IFN-β and Mx was measured by qRT-PCR. The fold induction for each gene in the RIG-I+/+ cells was set to 100%. Error bars represent standard deviations from three replicates. *, P < 0.5.
Production of IFN should stimulate an antiviral state in the cells. To confirm the IFN production shown by the previous results, 293T, mouse lung adenoma (LA4), and Vero cells were stimulated with either pIC, SeV RNA, or IVT DI or were mock treated with PBS. Twenty-four hours later, cells were infected with vesicular stomatitis virus (a highly IFN-sensitive virus) expressing green fluorescent protein (VSV-GFP). After 18 to 20 h of incubation, the GFP signal was measured as an indirect readout of viral replication. The IFN produced should stimulate an antiviral state and subsequently inhibit viral growth. Therefore, the levels of IFN will be inversely proportional to viral replication and, in this case, the levels of GFP. The outcome of this experiment closely recapitulated the previous results, showing the enhanced potency of SeV DI (both virion purified and in vitro transcribed) over pIC in 293T cells (Fig. 3A). In LA4 cells, both SeV and IVT DI again were better at inhibiting VSV-GFP than pIC, indicating a stronger IFN induction by the IVT DI in mouse lung-derived cells. As a control, we included interferon-incompetent Vero cells (29, 30). The lack of protection against VSV-GFP infection in Vero cells (despite the use of a higher dose of pIC, SeV RNA, or IVT DI) indicates that the type I IFN produced in response to the stimulatory RNAs is responsible for the antiviral state observed in 293T and LA4 cells.
Fig 3.

In vitro antiviral activity of SeV RNA and IVT DI. 293T, LA4, and Vero cells were transfected with equivalent amounts (50 ng) of pIC (orange bars), SeV RNA (green bars), and IVT DI RNA (blue bars) or were mock transfected (gray bars). Twenty-four hours later, cells were infected with VSV-GFP. After 18 to 20 h, GFP was measured as an indirect readout of viral replication. The fluorescence levels in the mock-transfected cells were set to 100%, and the results are expressed as a percentage of GFP. (B) Influenza A virus minigenome activity in 293T cells transfected with the influenza virus firefly luciferase minigenome reporter, PB1, PB2, PA, and NP protein-expressing plasmids. A GFP-expressing plasmid was included to normalize for transfection efficiency. Luciferase activity and GFP signal were assayed 24 h posttransfection. Error bars represent standard deviations from three replicates. ****, P < 0.0001; **, P < 0.01; ns, not significant.
In addition, and to rule out the possibility that the SeV DI RNA could stimulate the synthesis of the FF-luciferase reporter and alter our results in the IFN-β reporter cell line, we performed an influenza virus minigenome assay which measures the activity of the viral polymerase complex in the context of reconstituted viral ribonucleoprotein complexes (Fig. 3B). 293T cells were cotransfected with expression plasmids for the influenza A/WSN/33 virus polymerase proteins (PB1, PB2, and PA), the nucleoprotein (NP), and an influenza virus-specific FF-luciferase reporter plasmid. The IVT DI RNA strongly reduced the activity of the influenza minigenome, confirming the antiviral effect of our SeV-derived RNA.
The IVT DI can induce the synthesis of IFN-β and Mx in vivo.
We decided to explore the induction of IFN by the IVT DI RNA in vivo. For this purpose, pIC, SeV RNA, IVT DI, and PBS (as mock treatment) were administered to mice either i.m. or i.n. We decided to use these two methods because they are the most common routes for vaccine delivery. Six hours after administration, the calf muscles (in the case of i.m. administration) or the lungs (for the i.n. delivery) were removed, and the RNA present in the tissue was extracted for measuring induction of IFN-β, Mx, and IP-10. In this case, the IVT DI, SeV RNA, and pIC treatments induced, in both muscle and lungs, equivalent levels of IFN-β, Mx, and IP-10 mRNA (Fig. 4A and B), which demonstrates that both SeV RNA and, more importantly, the IVT DI can stimulate the innate immune system in vivo. Once more, to rule out the possibility that contamination during the production of RNA was responsible for the observed effect, we treated both SeV RNA and the IVT RNA with RNase A. Complete removal of the IFN-inducing activity in the RNase-treated samples confirmed that the stimulation in vivo was also RNA dependent (Fig. 4C).
Fig 4.
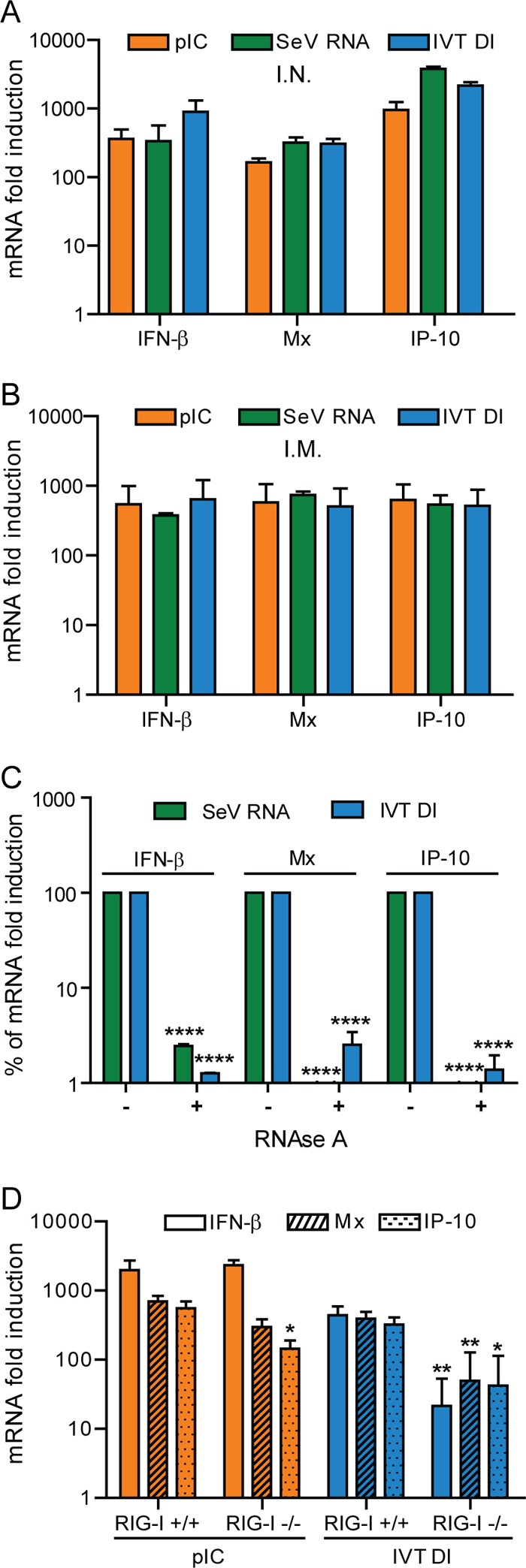
Induction of IFN and interferon-stimulated genes in vivo. BALB/c mice were treated intramuscularly (A) or intranasally (B) with 50 μl of PBS containing 4 μg of pIC (orange bars), SeV RNA (green bars), IVT DI RNA (blue bars), or PBS alone. Six hours later the calf muscles (A) or the lungs (B) were extracted and total RNA isolated. The levels of mRNA for IFN-β, Mx, and IP-10 then were measured by qRT-PCR. Data represent the mRNA fold induction for individual genes above the level for the PBS-treated mice. (C) RNase treatment of SeV RNA and the IVT DI RNA. The RNAs were digested (+) or mock treated (−) and injected i.m. into the calf muscle of mice to monitor their stimulatory activity. The fold induction of the mock-treated RNA was set to 100%. (D) Induction of IFN and ISGs in RIG-I−/− versus RIG-I+/+ mice. Mice were injected with pIC (4 μg), IVT DI (4 μg), and PBS via the i.m. route. Six hours later, the mRNA from the muscle was extracted and analyzed by qRT-PCR. Results represent the fold induction above the level of the PBS-treated samples. Error bars represent standard deviations from three replicates. *, P < 0.5; **, P < 0.01.
We have previously shown that the IVT DI requires RIG-I to induce IFN-β in MEFs. Since the IVT DI can stimulate IFN production in vivo, we decided to investigate the role of RIG-I in the production of this cytokine in vivo. In this case, RIG-I knockout (RIG-I−/−) and RIG-I wild-type (RIG-I+/+) mice received the IVT DI or pIC via the i.m. route. At 6 h posttreatment with the IVT DI, RIG-I−/− mice showed significantly reduced mRNA levels for IFN-β, Mx, and IP-10 compared to RIG-I+/+ mice (Fig. 4D). On the other hand, treatment with pIC was equally effective at inducing IFN-β and Mx mRNA in both RIG-I−/− and RIG-I+/+ mice. Surprisingly, after pIC stimulation, the mRNA levels for IP-10 were significantly reduced in RIG-I−/− mice. This experiment corroborates our results in tissue culture and confirms that the IVT DI is an RIG-I agonist, which allows us to explore its use in vivo as an antiviral and as a vaccine adjuvant.
Treatment with the IVT DI reduces influenza A virus lung titers in mice.
pIC has been reported to protect mice from influenza A virus infection due to its immune-stimulatory properties (30–32). To determine whether our in vitro-transcribed RIG-I agonist (IVT DI) could also be used as an antiviral, 6-week-old BALB/c mice were inoculated i.n. with 50 μl of PBS containing 1 μg (μg) of pIC, SeV RNA, IVT DI, or PBS alone either 1 or 5 days before infection. Mice were then challenged i.n. with 3,000 PFU (∼100 50% lethal infectious doses [LD50]) of influenza A/WSN/33 virus. Five days postinfection, lungs were collected and viral titers measured by standard plaque assay in Madin-Darby canine kidney cells. A stronger reduction of the viral load was detected when animals were treated with pIC or the SeV RNA than with pIC 1 day before the infection (Fig. 5A). Equivalent levels of viral inhibition for pIC, IVT DI, or SeV RNA were observed when the animals where treated 5 days before challenge (Fig. 5B). Treatment 10 days prior to the infection, however, failed to reduce the viral load in the lungs in all cases (data not shown).
Fig 5.

In vivo antiviral activity of the SeV DI. (A) Mice were treated intranasally with PBS (gray dots), 1 μg of pIC (orange dots), 1 μg of SeV RNA (green dots), or 1 μg of IVT DI RNA (blue dots) at 1 (A) or 5 (B) days before infection with 3,000 PFU of influenza A/WSN/33 virus. Five days postinfection, the viral load in the lungs was determined by plaque assay in MDCK cells. Error bars represent the standard errors of the means from three replicates.
The IVT DI enhances IgG production and vaccine efficiency.
We next examined the potential of the IVT DI to act as a vaccine adjuvant. For this purpose, we used the inactivated, monovalent, influenza H1N1 vaccine that was administered during the 2009 pandemic response. For i.m. delivery, 190 ng of vaccine in 50 μl of PBS was administered together with 4 μg of SeV RNA or IVT DI. Controls included AddaVax (MF59 equivalent, at a 1:1 [vol/vol] ratio), pIC (4 μg), and vaccine with no adjuvant. At 21 days postvaccination, the levels of HA-specific IgG antibodies in the sera were analyzed by ELISA (Fig. 6A and B). The mice receiving AddaVax and pIC had the highest anti-HA titers, but mice receiving SeV RNA or IVT DI also showed specific antibody levels above those in animals that received the vaccine alone.
Fig 6.

SeV RNA and IVT DI RNA increase IgG responses to inactivated influenza vaccine. Mice were vaccinated i.m. (A and B) or i.n. (C and D) with the 2009 H1N1 monovalent influenza virus vaccine supplemented with PBS (gray line and dots), pIC (4 μg; orange), the squalene emulsion AddaVax (1:1 [vol/vol]; purple), SeV RNA (4 μg; green), or the IVT DI RNA (4 μg; blue). Panels A and C show the average values of the HA-ELISA data from the sera of i.m. and i.n. vaccinated animals, respectively. Panels B and D show the endpoint titers from individual animals for the same experiment. Error bars represent the standard errors of the means from five mice. O.D. 405nm, optical density at 405 nm.
Noticeably, an IgG1-biased response was observed in mice receiving the AddaVax adjuvant, consistent with previous reports indicating that MF59 elicits a Th2 response (33, 34). However, an IgG1/IgG2a ratio indicative of a Th1 response was observed in mice receiving pIC or SeV IVT DI via the i.m. route (Fig. 7). Type I IFN is thought to drive a Th1 antibody response (35); therefore, these results correlate with the IFN-inducing ability of these adjuvants. Due to the amount of vaccine delivered, low levels of IgG1 and IgG2a antibodies were detected. To confirm the observed IgG1 versus IgG2a trend, we decided to repeat the experiment but this time using 750 ng of the 2009 vaccine (data not shown). Once again, the results showed a higher presence of HA-specific IgG1 antibodies in the case of the AddaVax-adjuvanted vaccine and a Th1-like antibody response for the pIC- and IVT DI-supplemented vaccines.
Fig 7.

IgG1 and IgG2a levels in vaccinated mice. Levels of HA-specific IgG1 (red line) and IgG2a (black line) from intramuscularly (i.m., top) and intranasally (i.n., bottom) vaccinated animals measured by ELISA. Equivalent amounts of sera from 5 animals treated with the same adjuvant were pooled together before the antibody quantification.
We also assessed i.n. administration as a route for vaccine delivery to investigate whether our adjuvant candidates were able to enhance the mucosal immune response. Six hundred ng of the H1N1 2009 pandemic vaccine was administered i.n. together with 4 μg of SeV RNA or IVT DI. Controls consisted of the vaccine supplemented with pIC (4 μg) and the vaccine alone. Mice received an i.n. boost on day 21 with an additional 600 ng of vaccine supplemented with the same adjuvant used for the prime. On day 28 after vaccination, sera and lung lavages were collected. Equivalent anti-HA IgG titers were observed for mice that received SeV RNA, the IVT DI, or pIC as adjuvants, and all were considerably higher than those in mice that received the vaccine with no adjuvant (Fig. 6C and D). Surprisingly, in mice receiving IVT DI by the i.n. route, we observed IgG1/IgG2a ratios lower than those in pIC-treated mice (Fig. 7), indicating a stronger Th1 response for the IVT DI than for pIC. Interestingly, we also observed that SeV DI RNAs (both the virion-purified and the IVT versions), as well as pIC, induced the production of influenza virus-specific IgA antibodies in the lungs (Fig. 8).
Fig 8.

Anti-HA-specific IgA levels after i.n. vaccination. IgA levels in the lung lavages of i.n. vaccinated animals were analyzed by ELISA. The lung lavages from animals treated either with the vaccine supplemented with pIC (orange), SeV RNA (green), IVT DI (blue), or with the vaccine alone in PBS (gray) were pooled together before antibody determination.
To assess how the adjuvant-elicited immune responses alter vaccine efficiency, we generated an influenza A/Puerto Rico/8/34 virus formaldehyde-inactivated vaccine which could be assessed in a lethal challenge model. As in the previous experiment, the vaccine dose was titrated down to obtain a suboptimal vaccination regimen that could be used as a baseline (data not shown). We then vaccinated mice via the i.m. route with 50 μl of PBS containing 10 ng of influenza A/Puerto Rico/8/34 virus vaccine supplemented with either IVT DI, pIC, or AddaVax (MF59 equivalent) at a 1:1 (vol/vol) ratio. As a negative control we used the vaccine with no adjuvant. After 21 days, animals were bled and analyzed for seroconversion (data not shown). Two days later (day 23 after vaccination), mice were challenge with 5,000 PFU (∼100 LD50) of influenza A/Puerto Rico/8/34 virus. Only 20% of the animals that received the vaccine with no adjuvant survived. In contrast, a 100% survival rate was achieved when the vaccine incorporated any of the tested adjuvants, including the IVT DI (Fig. 9A). These results, together with the weight loss measurements, clearly indicate that the IVT DI enhances the efficacy of the i.m. administered vaccine, reducing both mortality and morbidity.
Fig 9.

Survival and weight loss of vaccinated animals. Mice (n = 5) were vaccinated i.m. (A, top) or i.n. (B, bottom) with inactivated influenza A/Puerto Rico/8/34 virus supplemented with squalene-based oil in water emulsion (AddaVax; 1:1 [vol/vol]; purple line; only for the i.m. vaccinated animals), pIC (4 μg; orange line), or the IVT DI (4 μg; blue line) or were mock vaccinated (black line) or vaccinated with the inactivated vaccine with no adjuvant (gray line). In the case of the i.n. vaccinations, an additional group of mice that were treated with 4 μg of the IVT DI and did not receive the vaccine (pale blue line) was included. Twenty-three days after vaccination, animals were challenged with 5,000 (i.m.) or 1,000 (i.n.) PFU of influenza A/Puerto Rico/8/34 virus. The weight of the animals was recorded over 15 days. Animals with >25% weight loss were euthanized. Left panels show the survival rates, while right panels show the percentage of initial weight throughout the course of the infection. Error bars represent standard deviations from five mice.
We also investigated whether the SeV-derived adjuvant could enhance the efficiency of our A/Puerto Rico/8/34 vaccine when an i.n. route of administration was used. The vaccine was administered with either pIC or IVT DI as vaccine adjuvant. Once more the vaccine with no adjuvant was used as a negative control. A nonvaccinated group treated only with IVT DI was included in the study to rule out the antiviral effect of the IVT DI as a source of protection. After 21 days, sera were collected and analyzed for specific influenza virus HA antibodies (data not shown). Two days later (day 23 postvaccination), they were challenged with 1,000 PFU (∼20 LD50) of influenza A/Puerto Rico/8/34 virus. No boost was administered this time. All of the nonvaccinated animals (including those treated with IVT DI) and the animals vaccinated without adjuvant succumbed to infection (Fig. 9B). On the other hand, all of the animals that received the vaccine supplemented with pIC or IVT DI survived. Furthermore, these animals did not show any weight loss or signs of infection.
We wanted to explore the requirement of RIG-I for the observed adjuvant activity of the IVT DI RNA. In this case, 50 μg of the influenza A/Puerto Rico/8/34 virus vaccine was administered i.m. (supplemented with pIC or with IVT DI RNA) to RIG-I−/− and RIG-I+/+ mice. The vaccine with no adjuvant was used as a negative control. Twenty-three days postvaccination, the animals were challenged with 2.5 × 105 PFU of influenza A/Puerto Rico/8/34 virus. This vaccination-challenge regimen with a high lethal dose of virus (∼5,000 LD50) was used to maximize the differences between the two RNA-based adjuvants. The weight and survival of the animals then was monitored throughout the course of the infection (Fig. 10). The results confirm pIC as a RIG-I-independent adjuvant, while the adjuvant effect of IVT DI RNA is clearly reduced when RIG-I is not present. Thus, the adjuvant effect of the IVT DI is RIG-I dependent. A residual activity is, nonetheless, still present in RIG-I−/− mice, consistent with the previously observed induction of IFN-β and Mx mRNA in these animals.
Fig 10.

Adjuvant activity of the IVT DI RNA in RIG-I−/− mice. RIG-I+/+ (solid lines) and RIG-I−/− (dashed lines) mice were vaccinated i.m. with 50 μg of inactivated influenza A/Puerto Rico/8/34 virus supplemented with pIC (1 μg; orange line), the IVT DI RNA (1 μg; blue line), or with no adjuvant (gray line). Twenty-three days after vaccination, animals were challenged with 2.5 × 105 PFU of influenza A/Puerto Rico/8/34 virus. (A) Percent survival (animals with >25% weight loss were euthanized). (B) Percentage of initial weight throughout the course of the infection. Error bars represent standard deviations. For vaccine in RIG-I−/− and RIG-I+/+ mice, n = 4; vaccine plus pIC in RIG-I−/− mice, n = 5; vaccine plus pIC in RIG-I+/+ mice, n = 6; vaccine plus IVT DI RNA in RIG-I−/− mice, n = 5; and vaccine plus IVT DI RNA in RIG-I+/+ mice, n = 6.
DISCUSSION
Different pathogens show different propensities for triggering certain PRRs depending on the type of PAMP they produce. Activation of a cellular PRR triggers a specific response (36). This response is not limited to the direct inhibition of the pathogen but also participates in the generation of a protective adaptive immune response; in fact, many vaccine adjuvants take advantage of this phenomenon (e.g., pIC is a MDA5/TLR3 agonist [37], AS03 activates TLR4 [38], and CpG is recognized by TLR9 [39]). To maximize the effect of a viral vaccine, theoretically we should match the vaccine with an adjuvant that activates the appropriate PRR for that virus, i.e., mimics the natural infection. Influenza A virus and many other RNA viruses, such as Newcastle disease virus, hepatitis C virus, Sendai virus, vesicular stomatitis virus, and Japanese encephalitis virus, are recognized in a natural infection primarily by the cytosolic receptor RIG-I (8). For these viruses, a RIG-I agonist could be the most appropriate vaccine adjuvant. In this work, we explored the use of SeV DI RNA, a well-known and potent inducer of IFN via RIG-I (21), as an adjuvant for the inactivated influenza vaccine. Two approaches were used to obtain the DI RNA: (i) RNA was isolated from SeV Cantell virions, and (ii) SeV DI RNA was synthesized in vitro from a plasmid template.
As expected, the SeV-derived RNA is capable of inducing large amounts of type I IFN when transfected into cells. Both the IVT DI and the SeV RNA show a stronger induction of IFN-β than pIC and even other T7 polymerase-produced RNA. This induction, like that of the natural SeV DI RNA (21), comes primarily from RIG-I activation. This greater potency of the DI RNA over pIC to induce type I IFN was confirmed by its ability to stimulate a more robust antiviral state in 293T and LA4 cells.
In vivo, the IVT DI is capable of inducing IFN both in the lungs and in the muscle. We detected similar levels of IFN-β, Mx, and IP-10 mRNA after treatment with the IVT DI or pIC in vivo despite the expected increased stability of the pIC over the IVT DI or the SeV RNA. This observation could be attributed to a saturation of the response due to the large amounts of stimulatory RNA used (4 μg) or to the stronger ability of the SeV-derived RNAs to induce IFN.
Treatment (i.n.) of mice with SeV RNA, IVT DI, or pIC 24 h before infection with influenza A/WSN/33 virus decreased lung titers, confirming the immunostimulatory properties of these RNAs. We observed a greater reduction of viral titers when animals were treated with the SeV DI RNA (both the virion-purified and the IVT DI versions) than with pIC, as we did in tissue culture, despite the induction of similar levels of IFN-β. In this experiment, animals were treated with just 1 μg of RNA, which might favor the detection of differences in the ability to stimulate the immune system between these RNAs. This result could also be attributed to the induction of a different expression profile after pIC or IVT DI stimulation, with the IVT DI initiating a response that is more appropriate for influenza virus inhibition. However, when animals were treated at day −5, the difference was no longer detected. We expect the IVT DI to have a shorter half-life in vivo than pIC, therefore the increased stability of pIC may counteract the stronger anti-influenza properties of the IVT DI observed during the shorter time frame. The use of a molecule with a shorter half-life, however, might not be detrimental. As has been proposed previously, a molecule with properties similar to those of pIC but with a reduced half-life may offer a greater safety margin in clinical use (40).
As expected, in the case of the IVT DI and SeV RNA, RIG-I plays a key role in the stimulation. The absence of the cytosolic receptor severely decreases the induction of IFN-β mRNA. There is, however, some residual IFN being produced which is enough to stimulate the synthesis of some ISGs (e.g., Mx in Fig. 2B). On the other hand, treatment with pIC was equally effective at producing IFN-β regardless of the presence of RIG-I, most likely indicating a dependence on MDA5 and/or TLR3. We detected a significant decrease in IP-10 mRNA copies after pIC treatment of RIG-I−/− mice, which may be explained by the reported requirement of RLR signaling for IFN-γ production in myeloid dendritic cells and the fact that IP-10 is an IFN-γ-inducible protein (41, 42).
The IVT DI can increase HA-specific IgG titers when it is administered (either i.m. or i.n.) together with inactivated influenza virus split vaccine. The SeV-derived RNAs (both the IVT DI and the SeV RNA) can also enhance the efficacy of a whole inactivated influenza virus vaccine independently of the route of administration, reducing the morbidity and mortality after a homologous challenge with influenza A/Puerto Rico/8/34 virus. According to our results, the presence of RIG-I is crucial for the observed adjuvant effect of the IVT DI RNA. We should point out, however, that the differences in survival between RIG-I+/+ and RIG-I−/− mice vaccinated with the IVT DI RNA as adjuvant are not statistically significant, probably due to the small number of animals per group. We detect some residual adjuvant activity for the IVT DI in RIG-I−/− mice consistent with the mRNA levels of IFN-β observed in vivo in RIG-I-deficient animals. Therefore, partial recognition of the IVT DI RNA by other PRRs (MDA5, TLR7, and TLR8) cannot be ruled out.
As expected for a type I IFN inducer, the IVT DI induces a Th1-type response when it is delivered i.m. and i.n. Animals treated with the IVT DI via the i.n. route did not show any sign of protection despite its immunomodulatory properties. In this case the time lapse between the administration and the challenge was beyond the protective effect of the IVT. Unexpectedly, we observed a Th2-like IgG1/IgG2a ratio when pIC was administered i.n. This Th2-biased response could be due to the large amount (4 μg) of pIC used. More experiments are necessary to provide a more in-depth assessment of the type of response produced in response to pIC. However, it has been reported recently that repetitive inhalation of pIC induces a Th2-like cytokine response that results in increased production of IgG1 versus IgG2b antibodies in mice (43). This difference between pIC and IVT DI may indicate a different mechanism of action between these two stimulatory RNAs or a faster and more effective clearance of the IVT DI than pIC. The presence of IgG2a antibodies, on the other hand, correlates with clearance of virus and increased protection against lethal influenza virus challenge (44).
We showed that SeV DI RNA and, more importantly, the IVT DI RNA that we designed can be used as vaccine adjuvants for the inactivated influenza virus vaccine. The promising results obtained when the IVT DI RNA was delivered i.n. prior to infection with influenza virus and the strong IFN-inducing characteristics of this RNA in the lungs suggest a potential broad-spectrum antiviral activity against respiratory viruses when the DI RNA is delivered i.n. and for prophylactic purposes. Furthermore, the IVT DI represents an excellent tool for the study of RIG-I agonists as vaccine adjuvants and can help us to understand the role of RIG-I in modulation of the adaptive immune response.
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (grants HHSN272200900032C and U54 AI057159), the CRIP (Center for Research on Influenza Pathogenesis), and the NIAID (Center of Excellence on Influenza Research and Surveillance) (CEIRS, HHSN266200700010C).
We thank A. Baum for helpful discussion of the manuscript and assistance in the construction of the SeV DI plasmid and Michel Gale for kindly donating the RIG-I+/+ and RIG-I−/− mice. We also thank Chen Wang for excellent technical assistance.
Footnotes
Published ahead of print 21 November 2012
REFERENCES
- 1. Yan N, Chen ZJ. 2012. Intrinsic antiviral immunity. Nat. Immunol. 13:214–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30:16–34 [DOI] [PubMed] [Google Scholar]
- 3. GarcíA-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in détente. Science 312:879–882 [DOI] [PubMed] [Google Scholar]
- 4. Versteeg GA, GarcíA-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 13:508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baum A, GarcíA-Sastre A. 2011. Differential recognition of viral RNA by RIG-I. Virulence 2:166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Loo Y, Gale MJ. 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kato H, Takahasi K, Fujita T. 2011. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev. 243:91–98 [DOI] [PubMed] [Google Scholar]
- 9. González-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12:125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seya T, Kasamatsu J, Azuma M, Shime H, Matsumoto M. 2011. Natural killer cell activation secondary to innate pattern sensing. J. Innate Immun. 3:264–273 [DOI] [PubMed] [Google Scholar]
- 11. Huber JP, Farrar JD. 2011. Regulation of effector and memory T-cell functions by type I interferon. Immunology 132:466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beignon A, Skoberne M, Bhardwaj N. 2003. Type I interferons promote cross-priming: more functions for old cytokines. Nat. Immunol. 4:939–941 [DOI] [PubMed] [Google Scholar]
- 13. Bodnár A, Nizsalóczki E, Mocsár G, Szalóki N, Waldmann TA, Damjanovich S, Vámosi G. 2008. A biophysical approach to IL-2 and IL-15 receptor function: localization, conformation and interactions. Immunol. Lett. 116:117–125 [DOI] [PubMed] [Google Scholar]
- 14. Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A. 2007. Interleukin-12: biological properties and clinical application. Clin. Cancer Res. 13:4677–4685 [DOI] [PubMed] [Google Scholar]
- 15. Tovey MG, Lallemand C. 2010. Adjuvant activity of cytokines. Methods Mol. Biol. 626:287–309 [DOI] [PubMed] [Google Scholar]
- 16. Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR. 2003. Enhancement of antigen-specific immunity via the tlr4 ligands mpl adjuvant and ribi 529. Expert Rev. Vaccines 2:219–229 [DOI] [PubMed] [Google Scholar]
- 17. Kolakofsky D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555 [DOI] [PubMed] [Google Scholar]
- 18. Johnston MD. 1981. The characteristics required for a Sendai virus preparation to induce high levels of interferon in human lymphoblastoid cells. J. Gen. Virol. 56:175–184 [DOI] [PubMed] [Google Scholar]
- 19. Miller DK, Lenard J. 1982. Ultraviolet-irradiated vesicular stomatitis virus and defective-interfering particles are similar non-specific inhibitors of virus infection. J. Gen. Virol. 60:327–333 [DOI] [PubMed] [Google Scholar]
- 20. Yount JS, Kraus TA, Horvath CM, Moran TM, López CB. 2006. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J. Immunol. 177:4503–4513 [DOI] [PubMed] [Google Scholar]
- 21. Baum A, Sachidanandam R, GarcíA-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303–16308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hai R, Martínez-Sobrido L, Fraser KA, Ayllon J, GarcíA-Sastre A, Palese P. 2008. Influenza B virus ns1-truncated mutants: live-attenuated vaccine approach. J. Virol. 82:10580–10590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nguyen DN, Kim P, Martínez-Sobrido L, Beitzel B, GarcíA-Sastre A, Langer R, Anderson DG. 2009. A novel high-throughput cell-based method for integrated quantification of type I interferons and in vitro screening of immunostimulatory RNA drug delivery. Biotechnol. Bioeng. 103:664–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111 [DOI] [PubMed] [Google Scholar]
- 25. Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale MJ, Inagaki F, Fujita T. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29:428–440 [DOI] [PubMed] [Google Scholar]
- 26. Uzri D, Gehrke L. 2009. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J. Virol. 83:4174–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martínez-Sobrido L, Zúñiga EI, Rosario D, GarcíA-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 80:9192–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kirschning CJ, Wesche H, Merrill Ayres T, Rothe M. 1998. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J. Exp. Med. 188:2091–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 2:955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43:247–252 [DOI] [PubMed] [Google Scholar]
- 31. Falcoff E, Falcoff R, Cherby J, Florent J, Lunel J, Ninet L, De Ratuld Y, Tissier R, Vuillemin B, Werner GH. 1973. Double-stranded ribonucleic acid from mengo virus: production, characterization, and interferon-inducing and antiviral activities in comparison with polyriboinosinic-polyribocytidylic acid. Antimicrob. Agents Chemother. 3:590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong JP, Saravolac EG, Sabuda D, Levy HB, Kende M. 1995. Prophylactic and therapeutic efficacies of poly(IC.LC) against respiratory influenza A virus infection in mice.Antimicrob. Agents Chemother. 39:2574–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Higgins DA, Carlson JR, Van Nest G. 1996. Mf59 adjuvant enhances the immunogenicity of influenza vaccine in both young and old mice. Vaccine 14:478–484 [DOI] [PubMed] [Google Scholar]
- 34. Valensi JP, Carlson JR, Van Nest GA. 1994. Systemic cytokine profiles in BALB/c mice immunized with trivalent influenza vaccine containing mf59 oil emulsion and other advanced adjuvants. J. Immunol. 153:4029–4039 [PubMed] [Google Scholar]
- 35. Toporovski R, Morrow MP, Weiner DB. 2010. Interferons as potential adjuvants in prophylactic vaccines. Expert Opin. Biol. Ther. 10:1489–1500 [DOI] [PubMed] [Google Scholar]
- 36. Aoshi T, Koyama S, Kobiyama K, Akira S, Ishii KJ. 2011. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 1:226–232 [DOI] [PubMed] [Google Scholar]
- 37. Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. 2008. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3:352–363 [DOI] [PubMed] [Google Scholar]
- 38. Alving CR, Peachman KK, Rao M, Reed SG. 2012. Adjuvants for human vaccines. Curr. Opin. Immunol. 24:310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A toll-like receptor recognizes bacterial DNA. Nature 408:740–745 [DOI] [PubMed] [Google Scholar]
- 40. De Clercq E, Stollar BD, Thang MN. 1978. Interferon inducing activity of polyinosinic acid. J. Gen. Virol. 40:203–212 [DOI] [PubMed] [Google Scholar]
- 41. Luster AD, Ravetch JV. 1987. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J. Exp. Med. 166:1084–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luster AD, Ravetch JV. 1987. Genomic characterization of a gamma-interferon-inducible gene (IP-10) and identification of an interferon-inducible hypersensitive site. Mol. Cell. Biol. 7:3723–3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reuter S, Dehzad N, Martin H, Böhm L, Becker M, Buhl R, Stassen M, Taube C. 2012. Tlr3 but not tlr7/8 ligand induces allergic sensitization to inhaled allergen. J. Immunol. 188:5123–5131 [DOI] [PubMed] [Google Scholar]
- 44. Huber VC, McKeon RM, Brackin MN, Miller LA, Keating R, Brown SA, Makarova N, Perez DR, Macdonald GH, McCullers JA. 2006. Distinct contributions of vaccine-induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin. Vaccine Immunol. 13:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]