

Abstract
Macrophages are known to be one of the first lines of defense against influenza virus infection. However, they may also contribute to severe disease caused by the highly pathogenic avian (HPAI) H5N1 influenza viruses. One reason for this may be the ability of certain influenza virus strains to productively replicate in macrophages. However, studies investigating the productive replication of influenza viruses in macrophages have been contradictory, and the results may depend on both the type of macrophages used and the specific viral strain. In this work, we investigated the ability of H1 to H16 viruses to productively replicate in primary murine alveolar macrophages and RAW264.7 macrophages. We show that only a subset of HPAI H5N1 viruses, those that cause high morbidity and mortality in mammals, can productively replicate in macrophages, as measured by the release of newly synthesized virus particles into the cell supernatant. Mechanistically, we found that these H5 strains can overcome a block early in the viral life cycle leading to efficient nuclear entry, viral transcription, translation, and ultimately replication. Studies with reassortant viruses demonstrated that expression of the hemagglutinin gene from an H5N1 virus rescued replication of H1N1 influenza virus in macrophages. This study is the first to characterize H5N1 influenza viruses as the only subtype of influenza virus capable of productive replication in macrophages and establishes the viral gene that is required for this characteristic. The ability to productively replicate in macrophages is unique to H5N1 influenza viruses and may contribute to their increased pathogenesis.
INTRODUCTION
According to the World Health Organization, highly pathogenic avian (HPAI) H5N1 influenza viruses have caused more than 600 human infections with a 60% mortality rate since 2003. These viruses remain a serious public health threat because they are endemic in domestic poultry populations on three continents (1), increasing the likelihood of continued epidemic outbreaks leading to human infection as well as the opportunity for the virus to continue its adaptation to mammals. Thus, it is imperative that we gain a better understanding of how H5N1 influenza viruses cause severe disease.
The exacerbated disease severity and high mortality rates associated with human H5N1 infection correlates with high viral load, tropism for alveolar epithelium, and dysregulation of the host cytokine response (2–5). A number of molecular determinants of H5N1-induced pathogenesis have been described. For example, specific amino acid residues in the hemagglutinin (HA) protein of avian influenza viruses have been correlated with increased pathogenicity (6–9). Much less is known, however, about how H5N1 influenza viruses interact with specific components of the host immune response.
Macrophages are a critical component of the host response to infection, playing an important role in the phagocytosis of pathogenic agents and interaction with cells of the adaptive immune response (10, 11). Particularly for respiratory pathogens, alveolar macrophages represent an early point of contact at the host-pathogen interface. Pathogen recognition by alveolar macrophages initiates the host response to infection and the quantitative and qualitative nature of this response is important in determining the outcome of infection. During influenza virus infection, macrophages are an important source of antiviral and proinflammatory cytokines, which serve to control early virus replication and regulate the progression of an effective antiviral response (12). The importance of macrophages for protection against influenza virus infection is clear from several studies wherein clodronate liposome-mediated depletion of macrophages resulted in greater virus replication in the lungs, systemic dissemination of the virus, and exacerbated disease severity (12, 13).
Despite this demonstrated role for macrophages in preventing severe influenza virus-mediated disease, an excessive cytokine response is thought to be one of the causes of death in patients experiencing an H5N1 infection, and macrophages have been implicated in this response (3–5). H5N1 influenza virus infection results in an early infiltration of excessive numbers of macrophages into the lungs, which correlates with increased expression of proinflammatory cytokines (5). Further, infection of macrophages in vitro with H5N1 influenza viruses results in the induction of greater levels of proinflammatory cytokines compared to seasonal influenza viruses (14–16).
Taken together, these studies suggest a fundamental difference in the interaction of macrophages with highly pathogenic and seasonal influenza viruses. Human autopsy studies and ex vivo infections demonstrate that the primary targets of influenza virus infection are the respiratory epithelium and alveolar macrophages (17). However, infection of alveolar macrophages with influenza viruses is believed to be abortive, failing to result in the release of infectious virus progeny (18–20). Recent investigation into the nature of H5N1 infection of macrophages with regard to virus replication has produced inconsistent results (21–24). While van Riel et al. demonstrated a failure of HPAI H5N1 viruses to productively replicate in alveolar macrophages, work by Yu et al. demonstrated that productive replication of H5N1 influenza viruses in human alveolar macrophages correlated with increased expression of various proinflammatory cytokines, highlighting the impact that replication of H5N1 influenza viruses may have on the course of infection (23, 24).
To address these seemingly contradictory reports and to determine which viral gene(s) contributes to the ability of H5N1 influenza viruses to productively infect macrophages and the cellular mechanism for replication, we used primary murine alveolar macrophages and RAW264.7 immortalized murine macrophages to study the interaction of seasonal and H5N1 influenza viruses with macrophages in vitro. Our results demonstrate that productive replication in macrophages is unique to a subset of HPAI H5N1 avian influenza viruses. These viruses overcome a block early in the virus life cycle, allowing entry of the viral ribonucleoprotein (vRNP) into the nucleus followed by transcription, translation, and replication of the viral genes. Further, we demonstrate that the ability to overcome this block to productive replication maps to the HA gene. Expressing the HA of the H5N1 influenza virus, A/Hong Kong/483/97 (HK/483), on the background of A/California/04/09 (CA/09) H1N1 conferred the ability of the virus to replicate in macrophages. Our studies demonstrate that the HA gene from the HPAI H5N1 viruses supports the productive replication of influenza viruses in macrophages.
MATERIALS AND METHODS
Ethics statement.
All procedures were approved by the St. Jude Children's Research Hospital Institutional Biosafety Committee and Institutional Animal Care and Use Committee and were in compliance with the Guide for the Care and Use of Laboratory Animals. These guidelines were established by the Institute of Laboratory Animal Resources and approved by the Governing Board of the U.S. National Research Council.
Laboratory facilities.
All experiments using parental HPAI H5N1 and CA/09 H1N1 containing the H5 HA gene were conducted in a biosafety level 3 enhanced containment laboratory (25). Investigators were required to wear appropriate respirator equipment (RACAL Health and Safety, Inc., Frederick, MD). Mice were housed in HEPA-filtered, negative pressure, vented isolation containers. All other viruses were used under enhanced biosafety level 2 conditions by vaccinated personnel.
Viruses.
The H1N1 virus strains influenza A/New Caledonia/20/99 (New Caledonia) and A/Mallard/Wisconsin/8/76 (Mal/WI), the H2N3 virus strain influenza A/Chicken/Ohio/494832/07 (Ck/OH), the H3N2 virus strains influenza A/Aichi/2/68 (Aichi), A/Wyoming/3/03 (Wyoming), A/Fujian/411/02 (Fujian), and A/Brisbane/10/07 (Brisbane), the H5N1 virus strains influenza A/Hong Kong/483/97 (HK/483), A/Vietnam/1194/04 (VN/1194), A/Vietnam/1203/04 (VN/1203), and A/Hong Kong/156/97 (HK/156), and the H5 virus strains influenza A/Mallard/Alberta/85/76 (H5N2; Mallard/Alb), A/Duck/Hong Kong/820/80 (H5N3; Duck/HK), A/Duck/Potsdam/2216-4/84 (H5N6; Duck/Potsdam), and A/Shorebird/Delaware/35/98 (H5N8; Shorebird/DE) were all propagated in Madin-Darby canine kidney (MDCK) cells as described previously (26, 27). The influenza viruses H1N1 A/Puerto Rico/8/34 (PR8) and A/California/04/09 (CA/09), H4N4 A/Gray Teal/Australia/2/79, H6N1 A/Teal/Hong Kong/W312/97, H7N3 A/Duck/Alberta/48/76, H8N4 A/Turkey/Ontario/6118/68, H9N2 A/Chicken/Bangladesh/659/08, H10N7 Chicken/Germany/N/49, H11N6 A/Duck/Memphis/546/74, H12N5 A/Duck/Alberta/60/76, H13N6 A/Gull/Maryland/704/77, H14N5 A/Mallard/Astrakhan/263/82, H15N8 A/Shearwater/Australia/2576/79, and H16N3 A/shorebird/Delaware/172/06 were propagated in 10-day-old specific-pathogen-free embryonated chicken eggs at 37°C. Allantoic fluid was harvested, clarified by centrifugation, and stored at −70°C.
Reverse genetics.
The CA/09 viruses expressing genes from A/HK/483/97, A/Turkey/Egypt/06, A/Duck/Hunan/02, or A/VN/1203/04 were generated by using the eight-plasmid system as described previously (28), and the viruses were confirmed by sequence analysis. Virus titers were determined by 50% tissue culture infectious dose (TCID50) analysis in MDCK cells as described previously (29). The limit of detection for the TCID50 assay is 100 TCID50/ml. All in vitro experiments were performed with at least two different preparations of the reassortant virus (30).
Cells and culture media.
MDCK cells were cultured in Eagle minimum essential medium (MediaTech, Manassas, VA) supplemented with 2 mM glutamine and 10% fetal bovine serum (FBS; Gemini BioProducts, West Sacramento, CA). A549 cells were cultured in Dulbecco minimum essential medium (DMEM; Lonza, Walkersville, MD) supplemented with 4.5 g of glutamine/liter and 10% FBS. RAW264.7 murine macrophages were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 4.5 g of glutamine/liter and 10% FBS. All cells were grown at 37°C under 5% CO2.
Alveolar macrophage isolation and culture.
Six-week-old C57BL/6 mice were euthanized by CO2 asphyxiation, and the lungs were gently infused three times with 1 ml of phosphate-buffered saline (PBS) supplemented with 1% FBS and 0.5 mM EDTA. Cells from multiple mice were pooled and pelleted by centrifugation for 10 min at 400 × g and 4°C and then resuspended in DMEM, and 3 × 105 cells/well were plated into a 24-well cell culture plate (Corning, Corning, NY). Purity was determined by the Quik-Dip differential staining kit (Mercedes Medical, Sarasota, FL) to be >95%. At 24 h after plating, the cells were recounted and used for subsequent experiments.
In vitro infections.
RAW264.7 or primary murine alveolar macrophages were infected in triplicate at the indicated multiplicity of infection for 1 h at 37°C. Unbound virus was removed, and the cells were washed in PBS and maintained in RPMI 1640 medium containing 0.075% bovine serum albumin (BSA) in the presence (non-H5 viruses and low-pathogenicity H5 viruses) or absence (HPAI H5 viruses) of 1 μg of TPCK (tolylsulfonyl phenylalanyl chloromethyl ketone)-treated trypsin (Pierce, Rockford, IL)/ml. Cell culture medium was removed at the indicated times and stored at −80°C for the determination of virus titers by TCID50 analysis on MDCK cells as described previously (30). All TCID50 titers are normalized to background levels of residual virus remaining in the culture wells after the inoculum was washed off.
Quantitation of vRNA, cRNA, and mRNA by real-time RT-PCR.
Total RNA was isolated from RAW264.7 macrophages and A549 cells at the indicated time points by TRIzol extraction (Ambion, Carlsbad, CA) according to the manufacturer's instructions. cDNA complementary to the three species of viral RNA were synthesized by a procedure similar to that described by Kawakami et al. (31). Primers specific to the viral NP gene segment and containing a nucleotide tag that is unrelated to the viral sequence were used for cDNA synthesis (Table 1). A 13-μl mixture containing 200 ng of total RNA, 10 pmol of tagged primer, 1 μl of 10 mM deoxynucleoside triphosphate mix (Invitrogen), and 8 μl of RNase-free water was heated to 65°C for 5 min and then returned to ice. After 1 min, cDNA synthesis was carried out using the SuperScript III First-strand synthesis system for reverse transcription-PCR (RT-PCR) kit (Invitrogen) according to the manufacturer's instructions.
Table 1.
Influenza virus strains tested for productive replication in macrophages
| HA subtype | Virus |
|---|---|
| H1 | A/California/04/2009 |
| H2 | A/Chicken/Ohio/494832/2007 |
| H3 | A/Brisbane/10/2007 |
| H4 | A/Gray Teal/Australia/2/1979 |
| H5 | A/Hong Kong/483/1997 |
| H6 | A/Teal/Hong Kong/W312/1997 |
| H7 | A/Duck/Alberta/48/1976 |
| H8 | A/Turkey/Ontario/6118/1968 |
| H9 | A/Chicken/Bangladesh/659/2008 |
| H10 | A/Chicken/Germany/N/1949 |
| H11 | A/Duck/Memphis/546/1974 |
| H12 | A/Duck/Alberta/60/1976 |
| H13 | A/Gull/Maryland/704/1977 |
| H14 | A/Mallard/Astrakhan/263/1982 |
| H15 | A/Shearwater/Australia/2576/1979 |
| H16 | A/shorebird/Delaware/172/2006 |
Real-time PCR was performed with SsoFast EvaGreen Supermix on a CFX96 real-time system (Bio-Rad, Hercules, CA) by a method similar to that described by Kawakami et al. (31). Four microliters of a 1:10 dilution of cDNA was added to a master mix containing 10 μl of 2× SsoFast EvaGreen Supermix, 1.5 μl of forward primer (10 μM), 1.5 μl of reverse primer (10 μM), and 3 μl of sterile water. Human or mouse GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels were also measured using GAPDH control reagents (Invitrogen or Applied Biosystems [Foster City, CA]). Viral gene levels were normalized to GAPDH levels. The primers used are listed in Table 2.
Table 2.
Primer sets for strand-specific RT-PCR
| Viral target | Reaction | Primer |
|
|---|---|---|---|
| Name | Sequence (5′–3′) | ||
| HK/483 NP | |||
| vRNA | RT | vRNA_483_RT | GCCGTCATGGTGGCGAATGAATGGACGAACAAGGATTGC |
| RT-PCR | vRNA_483F | GGCCGTCATGGTGGCGAAT | |
| vRNA_483R | CTCAGGATGAGTGCAGACCGTGCC | ||
| cRNA | RT | cRNA_483_RT | GCTAGCTTCAGCTAGGCATCAGTAGAAACAAGGGTATTTTTCTTC |
| RT-PCR | cRNA_483F | GCTAGCTTCAGCTAGGCATC | |
| cRNA_483R | CGATCGTGCCTTCCTTTG | ||
| mRNA | RT | mRNA_483_RT | CCAGATCGTTCGAGTCGTTTTTTTTTTTTTTTTCTTCAATTGTC |
| RT-PCR | mRNA_483F | CCAGATCGTTCGAGTCGT | |
| mRNA_483R | CGATCGTGCCTTCCTTTG | ||
| CA/09 NP | |||
| vRNA | RT | vRNA_ca09_RT | GGCCGTCATGGTGGCGAATAAATGGACGAAGGACAAGGGTTGC |
| RT-PCR | vRNA_ca09F | GGCCGTCATGGTGGCGAAT | |
| vRNA_ca09R | CTCAGAATGAGTGCTGACCGTGCC | ||
| cRNA | RT | cRNA_ca09_RT | GCTAGCTTCAGCTAGGCATCAGTAGAAACAAGGGTATTTTTCTTC |
| RT-PCR | cRNA_ca09F | GCTAGCTTCAGCTAGGCATC | |
| cRNA_ca09R | CGATCGTGCCTTCCTTTG | ||
| mRNA | RT | mRNA_ca09_RT | CCAGATCGTTCGAGTCGTTTTTTTTTTTTTTTTTCTTCAACTGTC |
| RT-PCR | mRNA_ca09F | CCAGATCGTTCGAGTCGT | |
| mRNA_ca09R | CGATCGTGCCTTCCTTTG | ||
Preparation of cell lysates.
Mock- or influenza virus-infected RAW264.7 or MDCK cells were disrupted in RIPA buffer (150uM NaCl, 1% Triton X-100, 0.5% sodium dodecyl sulfate [SDS], 0.5% deoxycholate in PBS) supplemented with Halt Protease and Phophastase Inhibitor Cocktail (Thermo Scientific, Rockford, IL). Lysates were incubated on ice for 15 min, followed by centrifugation at 12,000 rpm for 10 min at 4°C and frozen at −80°C until further use.
Western blot.
Cell lysates were quantitated using the Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL). Ten micrograms of total cell lysate was separated on a 4%–20% SDS-PAGE gel under reducing conditions. After a transfer to nitrocellulose, blots were blocked in 5% nonfat dry milk in Tris-buffered saline plus 1% Tween 20 (TTBS) overnight at 4°C and probed for the influenza virus nonstructural protein 1 (NS1) with mouse anti-NS1 (1:1,000; a generous gift from Robert Webster) in TTBS for 1 h at room temperature. Blots were washed and incubated with goat anti-mouse-HRP (1:10,000; Jackson Laboratories, Bar Harbor, ME). The blots were stripped and reprobed for actin with goat anti-actin (1:500) in TTBS for 1 h at room temperature. The blots were washed and incubated with donkey anti-goat HRP-conjugated antibody (Jackson Laboratories, Bar Harbor, ME).
Immunofluorescence.
RAW264.7 macrophages (3 × 105 cells) or MDCK cells (1.5 × 105 cells) seeded onto sterile glass coverslips were inoculated with medium alone, HK/483, or CA/09 (multiplicity of infection [MOI] = 3.0 and 1.0 for RAW264.7 cells and MDCK cells, respectively) for 1 h at 4°C and then washed with cold PBS to remove unbound virus. The cells were shifted to 37°C for 30 min, 90 min, 2 h, or 4 h, fixed with 4% paraformaldehyde, and permeabilized in 0.1% Triton X-100 in PBS for 10 min at room temperature. After permeabilization, the cells were blocked in 1% BSA in PBS for 30 min at room temperature and stained for nucleoprotein (clone HB-65; American Type Culture Collection, Manassas, VA) and DNA (4′,6′-diamidino-2-phenylindole [DAPI], 1:1,000; Sigma). The secondary antibody was anti-mouse IgG-Alexa 488 (Invitrogen) diluted 1:200 in 1% BSA-PBS overnight at 4°C. Coverslips were mounted in ProLong Gold antifade reagent (Molecular Probes, Eugene, OR), and fluorescence was examined on a Nikon TE2000 E2 microscope equipped with a Nikon C1Si confocal scanhead. Excitation was performed with 404-nm and 488-nm DPSS lasers, and the emission was collected through 450/35 and 515/60 band-pass filters. Images were acquired with a Nikon ×40 1.3 NA Plan Fluor objective lens using Nikon EZC1 software. All images were acquired under the same condition.
Statistics.
The statistical significance of the data was determined by using analysis of variance or the Student t test on GraphPad Prism (GraphPad, San Diego, CA). All assays were run in triplicate and are representative of at least two separate experiments. Error bars represent standard deviations, and statistical significance was defined as a P value of <0.05.
RESULTS
Highly pathogenic H5N1 influenza viruses replicate productively in macrophages.
Influenza virus infection of macrophages was thought to be an abortive process in mammalian cells, failing to result in the release of progeny virus (12, 20). However, recent studies on alveolar macrophages infected with HPAI H5N1 influenza viruses have been contradictory, with one group showing productive replication (24) and another group demonstrating abortive infection (23). To address this issue and to get a complete picture of which influenza virus HA subtypes are capable of productive replication in macrophages, RAW264.7 murine macrophages were infected with a panel of influenza viruses representative of the 16 known HA subtypes (Table 1), and virus titers were determined in the cell culture supernatant by TCID50 assay at 24 h postinfection (hpi). In support of previous findings (12, 20), the majority of influenza virus subtypes failed to productively replicate and yield infectious virus into the supernatant (Fig. 1A). However, infectious virus could be detected in the supernatants of macrophages infected with HK/483, a representative HPAI H5 virus. To rule out the possibility that our results in Fig. 1A were due to the specific viral strains that we chose, we infected macrophages with a broader panel of human and avian H1, H3, and H5 influenza viruses. Similar to our observations shown in Fig. 1A, none of the H1 or H3 viruses that we tested productively replicated in macrophages (Fig. 1B). In contrast, a subset of H5N1 influenza viruses replicated in macrophages and, with one exception (HK/156), replication was either sustained or continued to increase as late as 72 hpi (Fig. 1B).
Fig 1.

H5N1 influenza viruses productively infect RAW cells. RAW264.7 cells were infected in triplicate with a panel of influenza viruses representing all 16 HA subtypes (A [the viruses are listed in Table 1]) or with the indicated viruses (B) at an MOI of 0.01. At the indicated times postinfection, the medium was collected, and virus titers were determined by TCID50 analysis in triplicate. (C) RAW cells were infected with the indicated influenza viruses (MOI = 3). The medium was collected at 24 and 48 hpi, and virus titers were determined by TCID50 analysis. (D) Primary murine alveolar macrophages were infected with the indicated viruses (MOI = 3), cell supernatants were collected at 24 and 48 h postinfection, and virus titers were determined as described above (limit of detection = 102 TCID50/ml). The data are representative of duplicate experiments. Error bars represent the mean TCID50 value ± the standard deviation. Titers determined at 1 hpi were below the limit of detection for the H5 viruses and ranged from below the limit of detection to 102 TCID50/ml for H1 and H3 viruses.
All strains of influenza virus except the HPAI influenza H5 and H7 viruses require exogenous TPCK-trypsin to productively replicate in vitro. To rule out an inhibitory effect of TPCK-trypsin on the replication of non-H5 influenza viruses in macrophages, macrophages were infected with HPAI H5N1 influenza viruses in the presence or absence of TPCK-trypsin, and the virus titers were determined as described above. We observed no inhibition of H5N1 virus replication in the presence of trypsin (data not shown), demonstrating that the trypsin is not inhibiting non-H5 viruses from replicating in macrophages.
To address the possibility that non-H5 influenza viruses complete a productive replication cycle in the cells that are initially infected but are limited in their ability to spread and initiate a subsequent round of infection, macrophages were infected at a higher MOI (MOI = 3), and titers were measured over time. Similar to the low-dose infection, non-H5 viruses failed to productively replicate in macrophages (Fig. 1C). Finally, to confirm our findings and rule out the possibility that our results are unique to RAW264.7 cells, primary alveolar macrophages were isolated from C57BL/6 mice and infected with CA/09 H1N1 virus or the HPAI H5N1 viruses HK/483 or A/Vietnam/1203/2004 (VN/1203). Similar to our observations in RAW264.7 cells, only the H5N1 influenza viruses productively replicated in primary alveolar macrophages (Fig. 1D). Based on these findings, we used RAW264.7 cells in all subsequent experiments to investigate the mechanism of differential replicative capacity. In summary, our results support those of Yu et al. (24) demonstrating that macrophages support productive replication of H5N1 influenza viruses. Intriguingly, we further show that only a subset of the H5 influenza viruses can productively replicate in macrophages, specifically those associated with high pathogenicity in mammals (32, 33).
H5N1 influenza viruses overcome a block early in the viral life cycle.
Influenza viruses must gain access to the cellular replication machinery in the nucleus in order for productive infection to occur. Previously published reports have demonstrated that influenza viruses differ in their ability to gain entry into macrophages (18). Thus, to determine whether macrophages restrict the uptake of non-H5 influenza viruses, RAW264.7 cells were incubated with CA/09 or HK/483, strains that represent viruses that fail to replicate or productively replicate in macrophages, respectively, at an MOI = 5 for 1 h at 4°C, followed by incubation at 37°C for infection to proceed. At the indicated time points, the cells were fixed, stained for the viral nucleoprotein (NP), and visualized by confocal microscopy. At 30 min postinfection, both viruses were internalized with equal efficiency (98.5% and 97.4% NP+ for CA/09 and HK/483, respectively), and NP was localized to the cytoplasm (Fig. 2A, left panels), indicating that the initial stage of virus entry is not blocked during H1N1 influenza virus infection. At 90 min postinfection ∼89% of the HK/483-infected cells were positive for NP (Fig. 2A, lower middle panel), and by 4 hpi ∼60% of HK/483-infected cells were NP-positive with staining localizing to the nucleus (Fig. 2A lower right panel). In contrast, at 90 min postinfection only 43% of the CA/09-infected macrophages remained NP-positive (Fig. 2A, upper middle panel). This continued to decrease, and by 4 hpi only 14% of the cells had nuclear NP staining while viral antigen was not detected at all in the remaining cells (Fig. 2A, upper right panel). Differences in NP staining in infected macrophages were not due to a failure of the NP antibody to recognize the CA/09 NP protein since equivalent staining was observed in CA/09- and HK/483-infected MDCK cells at all time points (Fig. 2B). Thus, abortive infection of macrophages with an H1N1 influenza virus is not due to restricted entry but may be associated with a rapid loss of viral antigen in infected cells upstream of nuclear entry.
Fig 2.
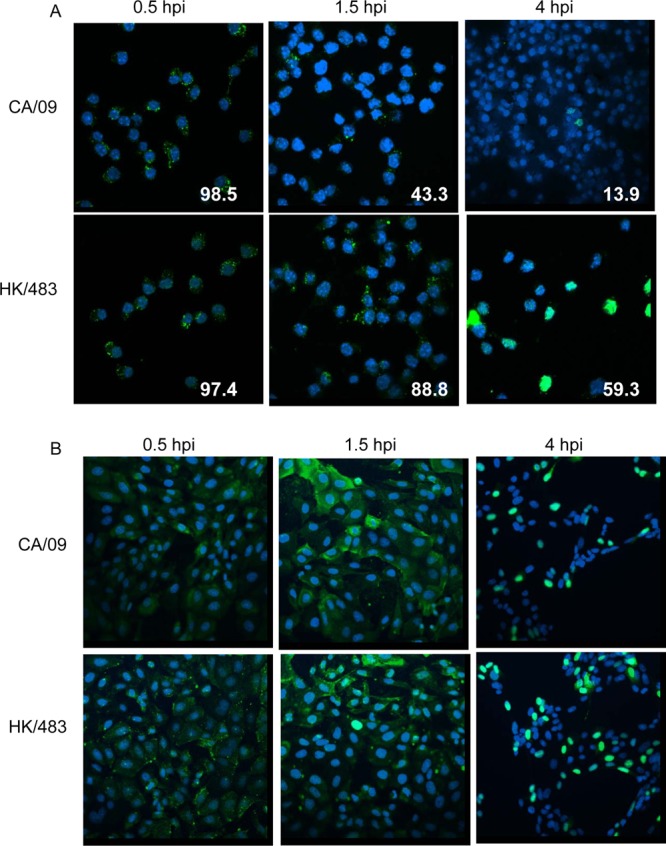
NP staining decreases in macrophages infected with CA/09. RAW264.7 macrophages (A) or MDCK cells (B) seeded onto coverslips were incubated with CA/09 or HK/483 (MOI = 5) on ice for 1 h. At time zero, warm infection medium was added, and the cells were incubated at 37°C. At the indicated time points, the cells were fixed and processed for immunofluorescent staining to detect viral NP. The slides were viewed by confocal microscopy as described in Materials and Methods. The mean percent NP+ macrophages was quantified from three independent images and is indicated in the lower right corner of each panel (A). Nuclei were visualized by DAPI staining, and representative images from two independent experiments are shown.
RNA synthesis is disrupted during H1N1 influenza virus infection of macrophages.
We hypothesized that the decrease in nuclear NP localization in CA/09-infected macrophages would be associated with less viral transcription and ultimately replication. To test this hypothesis, we infected macrophages with CA/09 or HK/483 viruses at an MOI = 5, synchronizing the infections at 4°C as described above, and isolated total RNA at 30 min and 3, 6, or 12 hpi. The viral NP gene levels were then quantitated using a strand-specific, real-time RT-PCR assay to distinguish viral RNA (vRNA), mRNA, and cRNA in infected cells as described by Kawakami et al. (31). The oligonucleotide sequences of the primers are provided in Table 2. Human respiratory epithelial A549 cells were infected in parallel as a positive control.
As shown in Fig. 3A, all three RNA species could be detected in A549 cells infected with either the CA/09 or HK/483 viruses, a finding consistent with the fact that A549 cells support productive viral infection. Synthesis of new vRNA and cRNA was detected by 12 hpi with both viruses, whereas synthesis of mRNA occurred earlier at 3 hpi. Higher levels of vRNA and cRNA were detected in HK/483-infected A549 cells (Fig. 3A). In HK/483-infected macrophages, synthesis of new vRNA and cRNA was detected as early as 6 hpi, and mRNA synthesis had begun by 3 hpi. In contrast, there was no synthesis of cRNA or new vRNA in CA/09-infected macrophages as late as 24 hpi (Fig. 3B and data not shown), which is consistent with a lack of productive replication (Fig. 1). Synthesis of mRNA in CA/09-infected macrophages was detected, but to lower levels and with delayed kinetics relative to HK/483-infected macrophages and CA/09-infected A549 cells, a finding consistent with decreased NP in the nucleus in the CA/09-infected macrophages (Fig. 2). In summary, while NP protein is detected in a minority of CA/09-infected macrophages, the synthesis of new vRNA and cRNA is not detected, a result suggestive of a block downstream of entry of the vRNP into the nucleus in addition to the block that causes decreased NP staining in the nucleus relative to HK/483-infected macrophages.
Fig 3.

RNA synthesis is inhibited in CA/09-infected macrophages. A549 (A) or RAW (B) cells were incubated with HK/483 or CA/09 at 4°C for 1 h. At time zero, warm medium was added, and the cells were incubated at 37°C. At the indicated time points, total RNA was isolated, and the levels of vRNA, mRNA, and cRNA were determined in triplicate using primers that amplify the NP gene as described in Materials and Methods. The RNA level in HK/483-infected cells at 12 hpi was set as a value of 1.0, and all other samples are presented relative to that amount. Viral RNA levels were normalized to GAPDH and to the amount of RNA present at 30 min postinfection. The data are representative of two independent experiments. The primer sequences are presented in Table 2.
Replication of CA/09 H1N1 influenza virus in macrophages is blocked upstream of translation.
Finally, to determine whether the decrease in RNA synthesis in CA/09-infected macrophages was associated with less viral protein synthesis, we monitored the level of the viral nonstructural protein (NS1) by Western blotting. Briefly, macrophages (MOI = 5) or MDCK cells (MOI = 1) were incubated in triplicate with the CA/09 or HK/483 viruses on ice for 1 h, followed by incubation at 37°C. A lower MOI was used in MDCK cells to avoid destruction of the monolayer that could affect interpretation of the results. Total cell lysates were prepared at the indicated time points, and the NS1 levels were determined by Western blotting. We could not detect NS1 expression in MDCK or macrophage lysates infected with either virus at 30 min or 6 h postinfection, a result consistent with the fact that NS1 is not a structural component of the virus and must be synthesized de novo in the infected cell (Fig. 4). By 24 hpi, NS1 was detected in MDCK cells infected with both viruses, consistent with the fact that MDCK cells support productive replication of influenza viruses. NS1 was also detected in macrophages infected with HK/483 at 24 hpi. In contrast, NS1 protein was not detected in CA/09-infected macrophages at any time postinfection (Fig. 4). Actin protein levels were monitored in parallel as a loading control (Fig. 4, lower panels). In summary, our studies demonstrate that certain HPAI H5 influenza viruses can productively replicate in macrophages by overcoming a block early in the viral life cycle leading to efficient nuclear entry and to viral transcription and translation.
Fig 4.
Viral protein synthesis is blocked during CA/09 infection of macrophages. RAW264.7 (left panels) or MDCK (right panels) cells were infected with HK/483 or CA/09, and the cells were lysed at the indicated times postinfection. Proteins were separated by SDS-PAGE under reducing conditions, transferred to nitrocellulose, and probed with anti-NS1 (top panels) or anti-actin (bottom panels) by Western blotting.
The HA protein confers the ability of influenza viruses to productively infect macrophages.
Our results indicate that the primary restriction to replication of non-H5 influenza viruses in macrophages occurs between 30 and 90 min postinfection. At this early stage of infection, the viral HA protein has an important role in mediating the escape of internalized virions from the uptake vesicle in order for the viral RNP to traffic to the nucleus.
To directly determine which viral protein(s) are important for the productive replication in macrophages, reverse genetics reassortant CA/09 viruses expressing individual HK/483 viral genes were generated (30). The reassortant viruses all replicated to similar levels in MDCK cells (30). Macrophages were infected with the reassortant or reverse genetics parental CA/09 and HK/483 viruses (MOI = 0.01), and virus titers were measured at 24 hpi. As shown in Fig. 5, the reverse genetics-derived parental CA/09 and HK/483 viruses replicated similarly to the wild-type viruses (compare Fig. 5A and Fig. 1). Of the eight reassortant viruses, only the CA/09 virus expressing the HA gene of HK/483 (CA/09-HK/483HA) productively replicated in macrophages (Fig. 5A). A replication kinetics assay showed that, although the CA/09-HK/483HA virus productively replicated in macrophages, at 24 and 48 hpi it was not as efficient as the reverse genetics-derived HK/483 parental virus (Fig. 5B). However, at 72 hpi, the titers of the CA/09-HK/483HA virus were still increasing, whereas those of the HK/483 control were beginning to decrease (Fig. 5B). These data demonstrate that the HA gene of an HPAI H5N1 influenza virus is sufficient to rescue productive replication of the CA/09 virus in macrophages.
Fig 5.
The HA gene mediates replication of influenza viruses in macrophages. (A) RAW264.7 cells were infected with the parental reverse genetics (rg) viruses or with rgCA/09 expressing individual genes from HK/483 (MOI = 0.1). Cell culture supernatants were collected at 24 hpi, and virus titers were determined by TCID50 analysis. (B) RAW264.7 cells were infected with the indicated viruses, and the virus titers determined as described for panel A. (C) RAW264.7 cells were infected with the indicated reverse genetics viruses as described above. The medium was collected 24 hpi, and virus titers were determined by TCID50 analysis on MDCK cells. Error bars represent the mean value ± the SD.
The HK/483 virus was isolated from a fatal human infection during the initial outbreak of avian H5N1 influenza virus in humans in 1997, and this clade (clade 0) no longer appears to be circulating in nature. To determine whether the HA protein of contemporary H5N1 influenza viruses also promotes replication in macrophages, we generated CA/09 viruses expressing the HA of A/Duck/Hunan/795/2002 (clade 2.1), A/Vietnam/1203/2004 (clade 1), and A/Turkey/Egypt/2006 (clade 2.2.1). All of these viruses replicated in macrophages (Fig. 5C). Overall, these data confirm the role of the HA gene of H5N1 influenza viruses in mediating productive replication of influenza virus in macrophages.
DISCUSSION
Early investigation into the interaction of influenza viruses with mammalian alveolar and peritoneal macrophages demonstrated that infection is abortive, failing to yield infectious virus into the cellular supernatant (19, 20, 34). More recent studies, however, demonstrate that the fate of viral infection may be dependent on the viral strain and the source of the macrophages, but all studies are consistent in demonstrating that alveolar macrophages do not support productive replication of seasonal influenza viruses or the 2009 pandemic H1N1 virus (5, 21, 23, 24). The potential contribution of macrophages to severe H5N1 disease is now recognized, leading to several recent investigations into the replicative capacity of H5N1 influenza viruses in this cell type. The resulting studies present conflicting evidence, with some groups suggesting that H5N1 influenza viruses productively replicate in alveolar macrophages (5, 24), while other groups report abortive infection (21–23). In the studies reported here, we address these conflicting results and demonstrate that certain H5 influenza viruses are unique among the 16 known HA subtypes of influenza virus in their capacity to replicate productively in macrophages. Further, our work extends that of others by investigating the nature of the restriction of influenza virus replication in macrophages and demonstrating which gene from H5N1 influenza viruses is required for productive replication. We show that the replication of most influenza virus strains is blocked early in the course of the viral life cycle, leading to decreased NP levels in the nucleus, less viral transcription, translation, and ultimately viral replication, as determined by monitoring the release of infectious virus. However, the exact step in the process that is blocked and the role of cellular host proteins remains under investigation.
Previously published studies suggest that influenza viruses are differentially capable of infecting macrophages (18, 23). In contrast, and in support of the work of others (21, 24), we demonstrate here using the A/CA/04/2009, A/Puerto Rico/8/1934 H1N1 viruses and the A/Aichi/2/1968 H3N2 virus that non-H5 influenza viruses are not restricted in their ability to be internalized by macrophages (Fig. 2 and data not shown). Although the source of macrophages or choice of viral strain may, in some cases, explain the disparity between these results, other work that observes differential infection based on visualization of the viral NP at 8 to 10 hpi (18) is unlikely to be representative of how well the initial uptake of the virus occurs since we observed a rapid decrease in NP levels by 90 min postinfection following efficient uptake.
Many cell surface lipids and proteins are sialylated, and the specific molecule that serves as the primary entry receptor for influenza viruses is not known. Two C-type lectins expressed on macrophages, the macrophage mannose receptor and the macrophage galactose-type lectin, were shown to be critical for infection of macrophages by influenza viruses (35, 36). Entry through these receptors results in a nonproductive infection since none of the viruses used in these studies were able to productively replicate in macrophages. A possible explanation for our results is that a subset of H5 influenza viruses binds to a different receptor, one allowing entry through a productive pathway. Studies are under way to determine whether influenza viruses that productively infect macrophages enter the cell through a different pathway than those viruses which do not replicate productively.
In addition to binding to the target cell, HA mediates postinternalization fusion of the viral envelope with the endosomal membrane to release the viral RNP complexes into the cytoplasm. Exposure of the HA molecule to the increasingly acidic environment of the endosome triggers a conformational change that permits the fusion event to take place (37). Internalized viral particles must escape the endosome prior to its fusion with the lysosome in order to avoid degradation by the acid hydrolases present in the lysosome. The pH at which the HA molecule is triggered is virus strain specific, and variations in the pH of fusion are correlated with disease severity in animal models of influenza virus infection (38, 39). We detected a gradual decrease in the number of CA/09 virus-infected macrophages between 30 min and 4 h postinfection (Fig. 3), an observation suggestive of a possible degradation of the virus after uptake. Experiments ongoing in the laboratory are addressing the role of the pH of fusion on replication of influenza viruses in macrophages.
An alternative explanation for the restricted nuclear entry of the CA/09 virus is that the viral RNPs, rather than not being released from the uptake vesicle, are not properly shuttled to the nucleus. This hypothesis is not as attractive to us because replication of CA/09 is rescued by expression of the HK/483 HA gene and HA is not known to play a role in nuclear import of the viral RNPs. However, our data do not allow us to rule out this possibility, and future studies will differentiate between impaired fusion of the viral and endosomal membranes and impaired nuclear transport of the viral RNPs.
Previous studies have reported the importance of macrophages to the host response during influenza virus infection. Indeed, dysregulation of cytokine production by infected macrophages has been implicated in the hypercytokinemia which is linked to high mortality rates in humans infected with avian H5N1 influenza viruses (2–4). The observation, made by us and by other laboratories, that H5N1 influenza viruses productively infect macrophages is, to our knowledge, the first qualitative difference in the interaction of macrophages with influenza viruses to be reported. It is an intriguing hypothesis to consider that productive replication in macrophages may account for the virulence of HPAI H5N1 viruses by contributing to the excessive production of proinflammatory cytokines. However, unpublished results generated in our lab give no indication that live H5N1 influenza virus stimulates greater cytokine secretion from macrophages than infection with an H5N1 virus that has been UV-inactivated (data not shown). However, further investigation is needed to determine whether there are differences in the cytokine response between macrophages infected with an influenza virus that can replicate in macrophages and one that cannot.
Does productive replication of macrophages lead to more severe disease? Although this question remains under investigation, our previous work suggests this may be a possibility. We have previously demonstrated that the CA/09 virus expressing the HA of HK/483 exhibits greater pathogenicity in mice relative to the wild-type CA/09 virus (30). Although we are unsure of the mechanism of heightened disease severity during in vivo infection with CA/09-HK/483HA, the results presented here suggest that replication in macrophages may be a contributing factor to the virulence of this reassortant virus. In order to address this question, we are planning future studies in the lab to determine whether the productive replication of influenza viruses that we observe in immortalized cell lines and primary alveolar macrophages can be detected during in vivo infection.
In summary, we demonstrate that a subset of HPAI H5N1 influenza viruses is unique among influenza viruses in their capacity to replicate in a mammalian macrophage cell line and alveolar macrophages. These viruses overcome a block early in the replication cycle in an HA-dependent manner to promote transcription, translation, and replication of the viral genes, leading to the assembly and release of newly formed virus particles. Our findings may provide insight into the mechanisms of virulence of H5N1 influenza viruses.
ACKNOWLEDGMENTS
We thank Pam Freiden, Andrew Burnham, Kevin O'Brien, Hassan Zaraket, Paul Thomas, and Richard Webby for insightful discussions and critical review of the manuscript. We thank Jennifer Peters at the St. Jude Cell and Tissue Imaging Center for technical assistance with the confocal microscopy. We thank Yoshihiro Kawaoka at the University of Wisconsin and Subrata Barman at St. Jude Children's Research Hospital for the HK/483 and CA/09 reverse genetics plasmids, and we thank Scott Krauss and Robert Webster for many of the influenza viruses used in these studies.
This study was supported by National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases, contract HHSN266200700005C, NIH grant AI059049, and the American Lebanese Syrian Associated Charities.
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Chen H, Smith GJ, Li KS, Wang J, Fan XH, Rayner JM, Vijaykrishna D, Zhang JX, Zhang LJ, Guo CT, Cheung CL, Xu KM, Duan L, Huang K, Qin K, Leung YH, Wu WL, Lu HR, Chen Y, Xia NS, Naipospos TS, Yuen KY, Hassan SS, Bahri S, Nguyen TD, Webster RG, Peiris JS, Guan Y. 2006. Establishment of multiple sublineages of H5N1 influenza virus in Asia: implications for pandemic control. Proc. Natl. Acad. Sci. U. S. A. 103:2845–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, Garcia-Sastre A, Tolnay AE, Albrecht R, Pyles JA, Olson PH, Aicher LD, Rosenzweig ER, Murali-Krishna K, Clark EA, Kotur MS, Fornek JL, Proll S, Palermo RE, Sabourin CL, Katze MG. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JS. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837 [DOI] [PubMed] [Google Scholar]
- 4. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12:1203–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4:e1000115 doi:10.1371/journal.ppat.1000115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bosch FX, Orlich M, Klenk HD, Rott R. 1979. The structure of the hemagglutinin, a determinant for the pathogenicity of influenza viruses. Virology 95:197–207 [DOI] [PubMed] [Google Scholar]
- 7. Horimoto T, Kawaoka Y. 1994. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J. Virol. 68:3120–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawaoka Y, Webster RG. 1988. Sequence requirements for cleavage activation of influenza virus hemagglutinin expressed in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 85:324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z, Jiang Y, Jiao P, Wang A, Zhao F, Tian G, Wang X, Yu K, Bu Z, Chen H. 2006. The NS1 gene contributes to the virulence of H5N1 avian influenza viruses. J. Virol. 80:11115–11123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kreijtz JH, Fouchier RA, Rimmelzwaan GF. 2011. Immune responses to influenza virus infection. Virus Res. 162:19–30 [DOI] [PubMed] [Google Scholar]
- 11. Wijburg OL, DiNatale S, Vadolas J, van Rooijen N, Strugnell RA. 1997. Alveolar macrophages regulate the induction of primary cytotoxic T-lymphocyte responses during influenza virus infection. J. Virol. 71:9450–9457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC. 2010. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J. Virol. 84:7569–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim HM, Lee YW, Lee KJ, Kim HS, Cho SW, van Rooijen N, Guan Y, Seo SH. 2008. Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J. Virol. 82:4265–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hui KP, Lee SM, Cheung CY, Ng IH, Poon LL, Guan Y, Ip NY, Lau AS, Peiris JS. 2009. Induction of proinflammatory cytokines in primary human macrophages by influenza A virus (H5N1) is selectively regulated by IFN regulatory factor 3 and p38 MAPK. J. Immunol. 182:1088–1098 [DOI] [PubMed] [Google Scholar]
- 15. Lee SM, Gardy JL, Cheung CY, Cheung TK, Hui KP, Ip NY, Guan Y, Hancock RE, Peiris JS. 2009. Systems-level comparison of host-responses elicited by avian H5N1 and seasonal H1N1 influenza viruses in primary human macrophages. PLoS One 4:e8072 doi:10.1371/journal.pone.0008072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mok KP, Wong CH, Cheung CY, Chan MC, Lee SM, Nicholls JM, Guan Y, Peiris JS. 2009. Viral genetic determinants of H5N1 influenza viruses that contribute to cytokine dysregulation. J. Infect. Dis. 200:1104–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, Wong MP, Chui WH, Poon LL, Tsao SW, Guan Y, Peiris JS. 2007. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat. Med. 13:147–149 [DOI] [PubMed] [Google Scholar]
- 18. Reading PC, Whitney PG, Pickett DL, Tate MD, Brooks AG. 2010. Influenza viruses differ in ability to infect macrophages and to induce a local inflammatory response following intraperitoneal injection of mice. Immunol. Cell Biol. 88:641–650 [DOI] [PubMed] [Google Scholar]
- 19. Rodgers B, Mims CA. 1981. Interaction of influenza virus with mouse macrophages. Infect. Immun. 31:751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wells MA, Albrecht P, Daniel S, Ennis FA. 1978. Host defense mechanisms against influenza virus: interaction of influenza virus with murine macrophages in vitro. Infect. Immun. 22:758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friesenhagen J, Boergeling Y, Hrincius E, Ludwig S, Roth J, Viemann D. 2012. Highly pathogenic avian influenza viruses inhibit effective immune responses of human blood-derived macrophages. J. Leukoc. Biol. 92:11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sakabe S, Iwatsuki-Horimoto K, Takano R, Nidom CA, Le M, Nagamura-Inoue T, Horimoto T, Yamashita N, Kawaoka Y. 2011. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J. Gen. Virol. 92:1428–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Riel D, Leijten LM, van der Eerden M, Hoogsteden HC, Boven LA, Lambrecht BN, Osterhaus AD, Kuiken T. 2011. Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog. 7:e1002099 doi:10.1371/journal.ppat.1002099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu WC, Chan RW, Wang J, Travanty EA, Nicholls JM, Peiris JS, Mason RJ, Chan MC. 2011. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J. Virol. 85:6844–6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Richmond JY, McKinney RW., III 1993. Biosafety in microbiological and biomedical laboratories, p 26–36 U.S. Department of Health and Human Services, Centers for Disease Control, Atlanta, GA [Google Scholar]
- 26. Carlson CM, Turpin EA, Moser LA, O'Brien KB, Cline TD, Jones JC, Tumpey TM, Katz JM, Kelley LA, Gauldie J, Schultz-Cherry S. 2010. Transforming growth factor-beta: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 6:e1001136 doi:10.1371/journal.ppat.1001136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones JC, Turpin EA, Bultmann H, Brandt CR, Schultz-Cherry S. 2006. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol. 80:11960–11967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. 2002. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20:3165–3170 [DOI] [PubMed] [Google Scholar]
- 29. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. (Lond.) 1938:493–497 [Google Scholar]
- 30. Cline TD, Karlsson EA, Freiden P, Seufzer BJ, Rehg JE, Webby RJ, Schultz-Cherry S. 2011. Increased pathogenicity of a reassortant 2009 pandemic H1N1 influenza virus containing an H5N1 hemagglutinin. J. Virol. 85:12262–12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods 173:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu X, Tumpey TM, Morken T, Zaki SR, Cox NJ, Katz JM. 1999. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J. Virol. 73:5903–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yen HL, Aldridge JR, Boon AC, Ilyushina NA, Salomon R, Hulse-Post DJ, Marjuki H, Franks J, Boltz DA, Bush D, Lipatov AS, Webby RJ, Rehg JE, Webster RG. 2009. Changes in H5N1 influenza virus hemagglutinin receptor binding domain affect systemic spread. Proc. Natl. Acad. Sci. U. S. A. 106:286–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bird RA, Sweet C, Husseini RH, Smith H. 1983. The similar interaction of ferret alveolar macrophages with influenza virus strains of differing virulence at normal and pyrexial temperatures. J. Gen. Virol. 64(Pt 8):1807–1810 [DOI] [PubMed] [Google Scholar]
- 35. Reading PC, Miller JL, Anders EM. 2000. Involvement of the mannose receptor in infection of macrophages by influenza virus. J. Virol. 74:5190–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Upham JP, Pickett D, Irimura T, Anders EM, Reading PC. 2010. Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry. J. Virol. 84:3730–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569 [DOI] [PubMed] [Google Scholar]
- 38. DuBois RM, Zaraket H, Reddivari M, Heath RJ, White SW, Russell CJ. 2011. Acid stability of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity. PLoS Pathog. 7:e1002398 doi:10.1371/journal.ppat.1002398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reed ML, Bridges OA, Seiler P, Kim JK, Yen HL, Salomon R, Govorkova EA, Webster RG, Russell CJ. 2010. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity and transmissibility in ducks. J. Virol. 84:1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]