

Abstract
The Vif protein of human immunodeficiency virus type 1 (HIV-1) promotes viral replication by downregulation of the cell-encoded, antiviral APOBEC3 proteins. These proteins exert their suppressive effects through the inhibition of viral reverse transcription as well as the induction of cytidine deamination within nascent viral cDNA. Importantly, these two effects have not been characterized in detail in human CD4+ T cells, leading to controversies over their possible contributions to viral inhibition in the natural cell targets of HIV-1 replication. Here we use wild-type and Vif-deficient viruses derived from the CD4+ T cells of multiple donors to examine the consequences of APOBEC3 protein function at natural levels of expression. We demonstrate that APOBEC3 proteins impart a profound deficiency to reverse transcription from the initial stages of cDNA synthesis, as well as excessive cytidine deamination (hypermutation) of the DNAs that are synthesized. Experiments using viruses from transfected cells and a novel method for mapping the 3′ termini of cDNAs indicate that the inhibition of reverse transcription is not limited to a few specific sites, arguing that APOBEC3 proteins impede enzymatic processivity. Detailed analyses of mutation spectra in viral cDNA strongly imply that one particular APOBEC3 protein, APOBEC3G, provides the bulk of the antiviral phenotype in CD4+ T cells, with the effects of APOBEC3F and APOBEC3D being less significant. Taken together, we conclude that the dual mechanisms of action of APOBEC3 proteins combine to deliver more effective restriction of HIV-1 than either function would by itself.
INTRODUCTION
The apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3) proteins are cellular cytidine deaminases that have potent anti(retro)viral and antiretrotransposon functions (1–4). The first family member to be recognized for this activity, APOBEC3G (A3G), was identified on account of being the cellular target for the human immunodeficiency virus type 1 (HIV-1) regulatory protein Vif (5), a feature that is also shared by APOBEC3D (A3D) (6, 7), APOBEC3F (A3F) (8–11) and haplotypes II, V, and VII of APOBEC3H (A3H) (12–14). In the context of HIV-1 infection, the current weight of evidence indicates that A3G may exert the more potent antiviral effect, with A3D, A3F, and A3H eliciting less pronounced effects, though this continues to be debated. HIV-1 Vif, together with its cellular cofactor, core binding factor β (15, 16), binds to these APOBEC3 proteins and recruits them to a cullin5-elonginB/C ubiquitin ligase complex, leading to their polyubiquitylation and proteasomal degradation (17–22). As a consequence, these APOBEC3 proteins are largely excluded from wild-type HIV-1 particles, their antiviral properties are essentially averted, and viral infectivity is preserved.
When Vif is absent, A3G, A3D, A3F, and A3H are incorporated into progeny HIV-1 particles and transferred to target cells in association with the viral capsid, where they exert their antiviral effects. Most prominently, because these enzymes are polynucleotide cytidine deaminases, they can postsynthetically convert cytidine residues to uridines in nascent (mostly single-stranded negative-sense) viral cDNA. Upon fixation, such changes then register as guanosine-to-adenosine (G-to-A) mutations in the complementary positive strand of viral cDNA (23–25). Since mutational frequencies can exceed 10% of all G residues, this phenomenon is known as hypermutation. Indeed, mutational loads of this extent are lethal and preclude the subsequent generation of functional virus components, a process that therefore shares analogies with error catastrophe (26).
While it is indisputable that APOBEC3 proteins can impose a destructive mutational burden upon HIV-1 in the absence of Vif, it has also been clear that lower levels of cDNA accumulate during HIV-1 infections in the absence of Vif but in the presence of APOBEC3 proteins (24, 27–34). It was initially suggested that U-containing reverse transcripts were recognized by cellular uracil DNA glycosylases (UDGs), leading to uracil removal and DNA degradation following recognition of abasic sites by cellular endonucleases (35). This mechanism is now considered unlikely for a variety of reasons. (i) It has been shown that even in the absence of the cellular UDGs UNG2 and SMUG1, A3G-induced decreases in viral cDNA accumulation remain (31, 36, 37). (ii) Transfection-based experiments with catalytically inactive A3G and A3F mutant proteins demonstrate that cytidine deamination is not necessarily required to register an antiviral phenotype (30, 38, 39). (iii) The extents of mutagenesis detected with HIV-1 stocks generated in the presence of a panel of A3F/A3G chimeric proteins do not correlate well with the degrees of viral inhibition (28). (iv) Little or no DNA editing is associated with the APOBEC3 protein-mediated inhibition of hepatitis B virus, adeno-associated virus, mouse mammary tumor virus, or retrotransposons, under some experimental conditions (3, 40–45).
In addition to APOBEC3 proteins reducing the levels of nascent HIV-1 reverse transcripts that accumulate in target cells, measurements of endogenous reverse transcription (ERT) activity using purified virus particles (29, 46, 47), as well as reverse transcriptase (RT) activity in reconstituted in vitro systems (48), have each indicated that A3G can inhibit the process of reverse transcription directly. Together with the aforementioned instances of discordance between DNA editing and antiviral phenotypes, these observations provide further support for the notion that APOBEC3 proteins can exert deaminase-independent effects that contribute to the inhibition of viral infection.
Almost all of the previous work defining the antiviral activities of APOBEC3 proteins has been carried out using virions generated from transfected cell lines, raising the objection that the results may not accurately reflect the physiological situation. Moreover, studies of editing-deficient mutant APOBEC3 proteins continue to court controversy, with objections being raised over the potentially broad consequences of the mutations on protein function, as well as the levels of protein expression being assessed (30, 39, 49, 50). What has been lacking, therefore, is a detailed analysis of the editing and nonediting effects of APOBEC3 proteins on HIV-1 virions produced from infected primary human CD4+ T cells. To address this important deficiency, we have utilized the natural, Vif-mediated control of APOBEC3 proteins to produce virus stocks from primary T cells whose infectivity is or is not severely inhibited by these proteins. Here, we report the phenotypic characterization of these viruses, examining cDNA accumulation and G-to-A editing patterns during virus infection of primary CD4+ T cells and cDNA synthesis in ERT reactions. We further characterize the effects of A3G on reverse transcription using ERT reactions by means of a novel assay that identifies the 3′ termini of nascent cDNA. Our findings demonstrate that both cytidine deamination and the inhibition of reverse transcription contribute to the antiviral activity of endogenous APOBEC3 proteins in CD4+ T cells.
MATERIALS AND METHODS
Plasmid constructs.
Expression vectors for wild-type and vif-deficient HIV-1 (pIIIB and pIIIB Δvif, respectively) and the G protein from vesicular stomatitis virus G (VSV-G) have been described before (5, 51). A3G was expressed using pcDNA3.1 (38).
Cells and virus production.
All cell lines and primary cells were maintained under standard conditions. Pseudotyped wild-type and ΔVif HIV-1 stocks were initially prepared by cotransfection of 293T cells with the proviral plasmid and VSV-G expression plasmid at a ratio of 20:1. For the A3G titration experiment in 293T cells, 50% confluent cultures of 293T cells in 10-cm plates were cotransfected with 10 μg of pIIIB Δvif and various amounts of pcDNA3.1/A3G plasmid, as indicated (ratios refer to DNA quantities, and transfection cocktails were balanced with pcDNA3.1). After ∼18 h, the cells were washed three times with phosphate-buffered saline (PBS), and fresh medium was added for a further 24 h, before virus-containing supernatants were harvested.
To generate stocks of wild-type and ΔVif virus from primary T cells, CD4+ T cells were isolated from 100 ml of fresh blood collected from healthy volunteer donors by negative selection using magnetic activated cell sorting (CD4+ T-cell isolation kit II; product number 130-091-155; Miltenyi Biotec). The A3H genotypes of the donors were determined by a combination of exon and cDNA sequencing: donors 1 and 2 are I/IV, and donor 3 is I/III. T cells were then activated by incubation with 20 U/ml interleukin-2 (IL-2; BD Bioscience) and 5 μg/ml phytohemagglutinin L (PHA-L; Oxoid) for 48 h. Approximately 20 × 106 cells were challenged with filtered stocks of pseudotyped virus produced in 293T cells (virus corresponding to 80 ng p24Gag per 106 cells) by spin infection at 2,000 × g for 2.5 h at 37°C. The cells were extensively washed and incubated for 24 h before the medium was changed again, and virus-containing supernatants were harvested 24 h later. Virus stocks were quantified by an enzyme-linked immunosorbent assay (ELISA) for p24Gag content.
Immunoblot analysis.
Aliquots (1 ml) of virions corresponding to 10 ng p24Gag were concentrated by centrifugation at 20,000 × g for 1 h at 4°C in a 2-ml Eppendorf tube through a 300-μl 20% (wt/vol) sucrose cushion before immunoblot analysis. A3G or viral p24Gag was detected in virion lysates using a polyclonal rabbit serum to A3G (38) or 24-2 (52), a p24Gag-specific monoclonal antibody, respectively, secondary antibody IRDye800CW goat anti-rabbit or IRDye800CW anti-mouse (LI-COR Biosciences UK Ltd.), and LI-COR Odyssey infrared imaging and quantitation.
Viral infections and DNA isolation.
Virus infectivity was determined in single-cycle assays by challenging 105 TZM-bl β-galactosidase (β-Gal) indicator cells with viruses corresponding to 5 ng p24Gag and measuring the induced expression of β-Gal activity in cell lysates after ∼24 h, using a Galacto-Star system (Applied Biosystems). The average counts of β-galactosidase activity from two uninfected control cell lysates were subtracted from each sample.
Primary CD4+ T-cell infections used for detailed DNA analyses were initiated by spin infecting 20 × 106 PHA/IL-2-treated cells, obtained from the same three donors used for viral stock preparation, with stocks corresponding to 40 ng p24Gag, at 2,000 × g for 2.5 h at 4°C. Cells were extensively washed with cold PBS before virus entry was initiated by resuspension in warm medium. Since viruses produced from the cells of three independent donors were used to infect fresh cells harvested from the same three donors, there were nine wild-type and nine ΔVif infections.
For quantitative real-time PCR (qPCR) analysis of viral cDNA in target cells, total DNA was purified using a DNeasy kit from Qiagen and eluted in a total volume of 50 μl. DNA (2 μl) was analyzed by qPCR, and the data for each infection were normalized by setting the maximum level of cDNA to 1. Mean values are provided for all time points for each source of target cell by averaging values obtained with each of the three challenge stocks.
Endogenous reverse transcription assays.
Viral preparations containing 25 ng of p24Gag were pelleted by centrifugation for 1 h at 20,000 × g at 4°C through a 300-μl 20% (wt/vol) sucrose cushion. Virions were resuspended in PBS, 2.5 mM MgCl2, 15 μg/ml melittin, and 1 mM deoxynucleoside triphosphates (dNTPs) and incubated at 37°C. At the specified time points, aliquots were removed and an equal volume of 2× cell-to-signal lysis buffer (Ambion) was added. Control reactions without dNTPs were incubated for the amount of time equivalent to the final time point. Samples were diluted 1:10 in water before analyzing 2 μl by qPCR (46).
qPCR.
Strong stop reverse transcription products were detected using primers that amplify the region between nucleotides 500 and 635 of the provirus: oHC64 (5′-TAACTAGGGAACCCACTGC) and oHC65 (5′-GCTAGAGATTTTCCACACTG) with probe oHC66 (5′-FAM-ACACAACAGACGGGCACACACTA-TAMRA, where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine) (28). A later product formed after first-strand transfer was detected using primers that amplify the region between nucleotides 411 and 635 of the provirus: HIV-FST-F1 (5′-GAGCCCTCAGATCCTGCATAT) and oHC65 with probe oHC66 (53).
Reactions were performed in triplicate in TaqMan universal PCR master mix using 900 nM each primer and 250 nM probe. After 10 min at 95°C, reactions were cycled through 15 s at 95°C, followed by 1 min at 60°C for 40 repeats, carried out on an ABI Prism model 7900HT thermal cycler (Applied Biosystems). The pIIIB Δvif vector was diluted into purified SupT1 cellular DNA to create a series of control samples that were used to calculate cDNA copy numbers and confirm the linearity of the assays.
Single-molecule amplification, analysis of mutations in cDNA, and statistics.
A 500-bp region between nucleotides 9114 and 9613 of the provirus (nef-R fragment) was sequenced (forward and reverse) following single-molecule amplification, using two-step limiting-dilution PCR, from DNA isolated from infected cells in a 96-well plate format (54). The DNA samples were diluted so that no more than 33% of wells gave a product (0.4 molecule per well). Initial amplification used outer primers EKf_Nef_HIV1_IIIB (5′-CAGGTACCTTTAAGACCAATGACT) and EKr_U5_HIV1_IIIB (5′-TGCTAGAGATTTTCCACACTGACT); 1 μl of these reaction mixtures was then transferred into a second reaction mixture using primers oKG82 (5′-AGGCAGCTGTAGATCTTAGCCACTT) and oKG81 (5′-GGTCTGAGGGATCTCTAGTTAC). Products were analyzed by agarose gel electrophoresis, and the products in wells containing a PCR product were sequenced; any sections of imperfect sequence reads were eliminated prior to further analysis.
The dinucleotide sequence context for each G-to-A mutation was determined, and the number of available contexts was calculated by multiplication of the proportion of each dinucleotide context within the 500-bp sequence by the number of total G residues sequenced.
To assess the influence of the nucleotide context on G-to-A mutation rates, chi-square tests on the frequency of surrounding nucleotides were performed with 3 degrees of freedom. Initially, to look at the effect of the +1 position, the expectation values were calculated by counting the frequency of each of A, C, G, and T at the +1 position of every G residue in the 500-bp sequenced region (140 in total). As the length of the sequenced fragment of DNA was relatively small, the assumption of independence of nucleotide frequencies within larger motifs is not valid; e.g., the probability of finding a 5′-GA dinucleotide is not necessarily equal to the frequency of G nucleotides multiplied by the frequency of A nucleotides. Thus, the expectation values for nucleotide frequencies at the +2 and −1 positions of 5′-GG and 5′-GA (the target nucleotide is underlined) were recalculated by counting the frequency of each of A, C, G, and T at the +2 and −1 positions solely of the relevant dinucleotide in the reference sequence.
To determine whether there was a statistically significant correlation between the percentage of mutated G residues in 5′-GG dinucleotide contexts and the total number of mutated G residues in any given sequence, the Spearman rank correlation coefficient (rS) and the corresponding P value were determined using Wessa's free statistics software.
Identification of cDNA 3′ termini in ERT reactions.
Viral cDNA (i.e., extended tRNA primer) was generated as in the ERT time course assays and purified using Qiagen minielute reaction cleanup columns following addition of 200 ng of pET28b as carrier DNA. To map viral cDNA 3′ ends, tRNA-cDNA was incubated with terminal transferase enzyme (New England BioLabs) in the presence of 1 mM CoCl2, 94 μM dGTP, and 6 μM ddGTP for 6 h at 37°C. Following heat inactivation of the enzyme, extended viral tRNA-cDNA was reverse transcribed using Tth DNA polymerase (Promega) and the primer 5′-GATCAAGCTTCGTATATCCCCCCCCCCCCCCCCX, where X is A, G, or T (in equal proportions), to generate a pure DNA molecule. This product was then used as a template for PCR, again using Tth DNA polymerase, the same 3′ primer, and 5′-TCAAGTCCCTGTTCGGGCGC (complementary to the 3′ end of tRNA-Lys3) as the 5′ primer. PCR products were cloned using a Topo-Clone system (Invitrogen), and plasmids were isolated and sequenced using an ABI 3730xl sequencer with BigDye (version 3.1) chemistry.
RESULTS
Vif-deficient HIV-1 produced in primary CD4+ T cells is less infectious than wild-type virus and yields fewer reverse transcripts.
A principal objective of this study was to clarify the antiviral activities of APOBEC3 proteins in the natural context of primary human CD4+ T cells. To address this definitively, it would be necessary to knock out individually as well as combinatorially each of the four APOBEC3 genes (A3D, A3F, A3G, and A3H) that are expressed in these cells (55, 56) and inhibit HIV-1 infection. Given that this is not technically possible in primary human T-cell cultures, we exploited the long-established capacity of the HIV-1 Vif protein to antagonize the activity of these proteins through the induction of proteolysis: for the purpose of this study, Vif-expressing virus closely approximates APOBEC3 null (a point that is addressed in greater detail below), whereas Vif-deficient (ΔVif) virus is APOBEC3 positive. We eliminated the possible contributions of antiviral A3H to these studies by using donors who do not have the II, V, or VII haplotype (data not shown). CD4+ T cells were purified from the peripheral blood of three seronegative donors, activated in culture, and challenged with high-titer pseudotyped stocks of wild-type HIV-1 or HIV-1 ΔVif. After extensive cell washing to remove the inocula and 24 h of further culture, virus stocks were harvested from each culture and stored for future analyses.
It has previously been shown that Vif-induced degradation of APOBEC3 proteins results in their exclusion from newly synthesized virions, whereas the absence of Vif allows significant levels of APOBEC3 proteins to be packaged (19–22, 57). To confirm this observation in our system, the stocks from all three donors were analyzed by immunoblotting using an A3G-specific antiserum (antisera capable of recognizing endogenous levels of encapsidated A3D or A3F are not currently available) (Fig. 1A). As expected, A3G was readily detected in ΔVif virions, with good consistency in levels across the three donors. Low levels of A3G were also detected in the preparations of wild-type virions, though the amounts were 10- to 20-fold lower than those seen for the ΔVif virions. Whether this represents only residual packaged A3G or also includes a contribution from contaminating microvesicles was not determined. We also measured the relative infectivity of each stock using single-cycle infection assays and TZM-bl indicator cells (Fig. 1B). Consistent with previous analyses of viruses from primary T cells (58), the absence of Vif resulted in a 17- to 39-fold (mean, 25 ± 12) defect in virus infectivity.
Fig 1.

Comparison of CD4+ T-cell-derived wild-type and Vif-deficient HIV-1 for APOBEC3G packaging (A), single-cycle infectivity (B), and cDNA synthesis (C and D). CD4+ T cells isolated from three different donors were infected with wild-type or ΔVif HIV-1. The released viral particles were isolated, purified, and analyzed. (A) Wild-type and ΔVif HIV-1 virions corresponding to 10 ng p24Gag were analyzed by immunoblotting using antibodies specific for A3G and p24Gag (loading control). (B) Virion infectivity was determined using TZM-bl reporter cells challenged with wild-type or ΔVif viruses corresponding to 5 ng p24Gag, followed by the measurement of β-galactosidase activity in cell lysates. Background β-galactosidase activity (2,808 counts) from uninfected control cell lysates was subtracted. (C and D) Equivalent amounts of wild-type and ΔVif virions (5 ng p24Gag) were used for in vitro endogenous reverse transcription assays. Total DNA was harvested at the indicated times, and cDNA levels were measured using qPCR with primer-probe sets monitoring production of strong stop (C) or first-strand transfer (D) cDNA. wt, wild type. Statistical analysis was performed applying a 2-sample, unequal-variance t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Previous work has established that the inhibition of HIV-1 infection by APOBEC3 proteins correlates with diminished levels of cDNA accumulation and that these effects are more pronounced as later reverse transcription products are analyzed (27, 29, 30, 46). As noted above, the significance of these effects has been questioned, in part because much of the work has been performed using cell lines and ectopically expressed proteins. We therefore used qPCR to evaluate the reverse transcription competencies of the viruses derived from each donor. First, we examined the production of cDNA in ERT reactions in which dNTPs were added to permeabilized virions. Consistent with previous studies (46), the ΔVif/APOBEC3-positive (APOBEC3+) virions produced ∼2-fold (mean, 2.1 ± 0.6-fold, when averaged over all time points) less strong stop cDNA (Fig. 1C) and 2- to 10-fold (mean, 5.5 ± 3.2 for all time points) less first-strand transfer cDNA (Fig. 1D), a later reverse transcription intermediate, than wild-type virions.
We next measured reverse transcription phenotypes in infections of CD4+ T cells. Specifically, cells were purified from the same three donors whose samples had been used for viral production, and all six stocks were used to challenge cells from each (i.e., nine wild-type and nine ΔVif/APOBEC3+ infections). DNA was isolated from each culture over the following 24 h, and strong stop and first-strand transfer cDNAs were measured using qPCR (Fig. 2A and B, respectively). In keeping with both the ERT analysis (Fig. 1) and previous studies in cell lines (30, 46), the presence of APOBEC3 proteins resulted in marked decreases in cDNA levels: 2- to 10-fold less for strong stop cDNA (mean, 5.7-fold at 8 h) and 5- to 15-fold less for first-strand transfer cDNA (mean, 9.3-fold at 8 h). In sum, HIV-1 particles produced in CD4+ T cells in the presence of natural levels of APOBEC3 proteins but in the absence of Vif display severe defects in their capacity to synthesize and accumulate cDNA.
Fig 2.
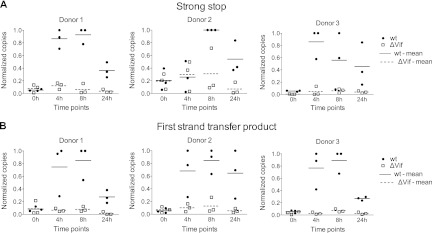
Effect of packaged APOBEC3 proteins on cDNA synthesis in infected CD4+ T cells. The six CD4+ T-cell-derived virus stocks characterized for Fig. 1 were used to infect fresh cultures of CD4+ T cells from the same three donors (corresponding to the three different graphs). Total DNA was harvested at the indicated times after infection, and the levels of strong stop (A) or first-strand transfer (B) cDNA were measured using qPCR. Levels of cDNA were normalized to the amount of total DNA extracted and are shown as proportions of the peak accumulation detected in each set of reactions, and mean values are indicated. For statistical analysis of the differences in cDNA levels between wild-type and ΔVif infections, data available from all donors were combined for each time point and a 2-sample, unequal-variance t test was carried out. For strong stop cDNA (A), P < 0.01 at 4 h, P < 0.001 at 8 h, and P < 0.01 at 24 h. For first-strand transfer cDNA (B), P < 0.001 at 4 h, P < 0.001 at 8 h, and P < 0.01 at 24 h.
Analysis of APOBEC3G's effects on reverse transcription through the mapping of 3′ termini of HIV-1 cDNAs.
There are different possibilities to explain the effects of APOBEC3 proteins on the processivity of HIV-1 reverse transcription. Specifically, (i) cDNA synthesis could be halted at particular sites, as might be provoked by the sequence- or structure-specific binding of APOBEC3 proteins to such sites and the consequent blockade of reverse transcriptase translocation along the RNA template, or (ii) APOBEC3 proteins might act with less specificity to impede processivity, perhaps via interactions with the reverse transcriptase enzyme itself, with the RNA, or with both.
To begin to address these questions, we set out to map the 3′ termini of nascent viral cDNAs formed in ERT reactions in the presence or absence of APOBEC3G. Since PCR-based methods depend on primers annealing to fixed positions on a template, these approaches are able to monitor the appearance of viral cDNA products only of a particular length or greater but are not able to provide a view of the distribution of individual DNA lengths in any given sample or at any given time point. We therefore developed a novel assay to reveal the exact lengths of cDNA molecules generated during ERT reactions, based on tailing with terminal transferase, PCR, cloning, and then sequencing (refer to Materials and Methods).
To increase cDNA recovery and to assess the profiles of measured 3′ termini in the context of titrated levels of A3G, we turned to using 293T cells cotransfected with pIIIB Δvif and escalating levels of pcDNA3.1/A3G for virus production. As shown by immunoblot analysis of the resulting virion preparations (Fig. 3, top), pA3G/provirus ratios of between 1:81 and 1:27 (lanes 4 and 5) yield viruses with an A3G content that approximates that of ΔVif virus from primary T cells (lane 2). All six 293T-derived stocks were then analyzed using ERT reactions, with samples collected over 240 min. The products were first visualized by agarose gel electrophoresis and ethidium bromide staining (see Fig. S1 in the supplemental material); as expected and consistent with previous findings (46), the smear of cDNAs from each reaction reflected progressively longer cDNA products at later time points (e.g., refer to the provirus-only samples, Fig. S1, lanes 3 to 6), and these became both shorter and less intense as the amounts of A3G were increased. The levels of strong stop cDNA were also measured by qPCR, further reconfirming that the presence of A3G in virions inhibits cDNA synthesis in a general and dose-dependent fashion (Fig. 3, bottom).
Fig 3.

Inhibition of endogenous reverse transcription by ectopically expressed APOBEC3G. (Top) Immunoblotting was used to compare wild-type and ΔVif virions derived from infected primary CD4+ T cells (lanes 1 and 2, respectively), with virions generated by transfection of 293T cells with pIIIB Δvif and increasing quantities of pcDNA3.1 A3G (lanes 3 to 8, A3G/provirus transfection ratios of 0:1, 1:81, 1:27, 1:9, 1:3, and 1:1, respectively). (Bottom) Virions analyzed in lanes 3 to 8 of panel A were assessed in ERT assays. Total DNA was harvested at the indicated times, and levels of strong stop cDNA were measured using qPCR.
We next used sequencing to determine the exact 3′ termini of >20 cDNAs from all four time points of the provirus-only, 1:27, and 1:1 sample sets. The results are displayed in Fig. 4, with the length of each cDNA plotted as the ordinate and its ranking by length within a sample plotted as the abscissa and with the shortest cDNA from each sample being the first point (e.g., for the 240-min sample from the provirus-only reaction, four cDNAs were between 179 and 181 nucleotides [nt] in length). The results for the provirus-only samples were as expected and matched those of the gel-based analysis, in that there were progressively longer reverse transcripts at later times. Within this sampling, no specific stop sites for reverse transcription were apparent (which would be seen as discrete plateaux in these plots). Both of these trends were also evident within the 1:27 and 1:1 sample sets, and increasing A3G levels further corresponded with the detection of shorter cDNAs. Moreover, the presence of A3G (at either dose) failed to induce the appearance of cDNAs with obviously preferred stop sites, indicating that the A3G-mediated inhibition of reverse transcription may be attributable to a general reduction in processivity, as opposed to termination or pausing at a low number of specific or selected sites.
Fig 4.

Impact of APOBEC3G on length of HIV-1 cDNA synthesized by endogenous reverse transcription. Virions characterized for Fig. 3 and generated by transfection of 293T cells with pcDNA3.1 A3G and pIIIB Δvif (0:1, 1:27, and 1:1 ratios) were purified and subjected to ERT reactions. Nucleic acids were extracted at the indicated times, and cDNA products were tailed, amplified, cloned, and sequenced. The lengths of between 22 and 46 reverse transcripts per time point and A3G concentration are shown. cDNA length is plotted on the ordinate, with the abscissa corresponding to individual cDNAs sorted by length. Each graph corresponds to one A3G/provirus ratio and demonstrates progression of reverse transcription over four time points (30, 60, 120, and 240 min).
Mutational analysis.
As summarized above, a significant body of previous work has described APOBEC3 protein-mediated C-to-U hypermutation of nascent viral cDNA in infections with ΔVif virions (1–4, 7–11, 23–25, 59). Previous work has also established that different APOBEC3 proteins exhibit various local sequence preferences for deamination. Specifically, A3G has a unique and strong preference for substrates with 5′-CC (minus-strand sequence, with the edited nucleotide underlined), which equates to 5′-GG on the plus strand (8, 23, 60, 61), whereas A3F, A3D, and A3H preferentially target 5′-TC dinucleotides (5′-GA on the plus strand) (7–10, 62). Accordingly, examination of local sequence contexts of G-to-A mutations can help identify the causative APOBEC3 protein(s). We therefore undertook an extensive sequencing analysis of nascent cDNAs in our primary cell system (i.e., using the DNA samples analyzed for Fig. 2).
We utilized PCR-mediated single-molecule amplification of individual cDNAs followed by cloning and sequencing to ensure that recovered sequences reflected independent viral cDNAs. Sequences were obtained from all nine ΔVif/APOBEC3+ infections at the 4-, 8-, and 24-h time points, from the wild-type infections at the 24-h time point, and from the wild-type infections of donor 1's cells at 4 h. In total, the numbers of cDNAs analyzed were 117, 115, and 131 for the 4-, 8-, and 24-h ΔVif samples, respectively, and 36 and 101 from the wild-type virus infections at 4 and 24 h, respectively.
As expected, G-to-A plus-strand transition mutations were by far the most predominant mutation seen in the ΔVif and wild-type infections (Fig. 5A). These changes were distributed throughout the analyzed 500-bp region, though some clear preferences for particular sites were evident, as has been previously described (not shown). At least for the ΔVif infections, a significant number of C-to-T changes were also observed. We attribute this to the relatively infrequent deamination events on plus-strand reverse transcripts, but it could also be caused by editing of viral RNA (63). Noticeably, the mutational spectrum seen in cDNAs recovered from wild-type samples was skewed toward G-to-A changes and was therefore very different from the error bias typically reported for HIV-1 reverse transcriptase (64). Given that the majority of these changes (156/171) occurred at A3G consensus target sites (see below), we conclude that the low levels of APOBEC3 protein that were detected in these virion preparations (Fig. 1A) must have evaded Vif-mediated suppression and are functional following entry into target cells.
Fig 5.

Analysis of mutational load imposed on HIV-1 cDNA by endogenous APOBEC3 proteins in CD4+ T cells. A 500-nt nef-R fragment was amplified by limiting dilution, followed by PCR from DNA samples extracted from the 18 independent infections previously described in Fig. 2, and then sequenced. (A) Distribution of mutation types in wild-type (top) and ΔVif (bottom) HIV-1 from all sequences. The total numbers of available nucleotides are indicated. (B) Percentage of G residues mutated over time for individual wild-type (at 4 h and 24 h) and ΔVif (at 4 h, 8 h, and 24 h) infections, with the mean values indicated. (C) Proportion of sequenced viral cDNAs from wild-type-infected (at 4 h and 24 h) and ΔVif-infected (at 4 h, 8 h, and 24 h) cells carrying at least one G-to-A mutation, with the mean values indicated. (D) Percentage of sequences with the indicated numbers of G-to-A mutations at various time points in ΔVif and wild-type virus infections.
The mean percentage of total G residues that were mutated in all sequenced cDNAs from each infection is presented in Fig. 5B (each point therefore represents multiple PCR amplicons). The accumulation of mutations was time dependent, rising from 3.5% of G residues at 4 h to about 10% at 24 h postinfection in the ΔVif virus infections. The temporal accumulation of mutations was also evident when the percentages of sequences carrying at least one mutation were depicted for each sample (Fig. 5C), as well as when all data for each time point were pooled and the sequences were stratified according to the number of G-to-A mutations found in each cDNA (Fig. 5D). This was to be expected and serves to confirm further that APOBEC3-mediated editing of reverse transcripts occurs postsynthetically (23, 60, 61, 65, 66): that is, the longer that a given cDNA substrate persists in a newly infected cell, the greater the opportunity for mutation. These data also demonstrate, as others have previously, that mutations were not evenly distributed among the cDNAs from a particular infection but that they tended to cluster in a subset of cDNAs (Fig. 5B). This was particularly obvious in cDNAs from wild-type virus infections at 24 h, where ∼1% of all G residues were mutated (Fig. 5B), but these mutations were limited to a small number of cDNAs, with ∼90% of the analyzed sequences escaping with zero mutations (Fig. 5D).
To investigate the site preferences for the observed G-to-A mutations, we pooled the sequencing data from all infections (wild type or ΔVif) (Fig. 6A). We initially looked solely at the +1 position, as this has previously been reported to have the greatest effect on APOBEC3 protein site selection (7–10, 23, 60–62). We carried out a chi-square test of independence of the +1 position, with expectation values calculated from the reference sequence +1 context of all G residues in the sequenced 500-bp region. As expected, the identity of the +1 nucleotide was strongly nonrandomly distributed (P < 0.0001), with a G residue present (i.e., 5′-GG) in 88% of cases and an A residue present (i.e., 5′-GA) in 11% of cases (Fig. 6A). We then went on to look at the influence of the +2 and −1 positions; the identity of the nucleotide at the +2 position was also strongly nonrandomly distributed (P < 0.0001), with the trinucleotide 5′-GGG present in 66% of cases, and there was a strong preference for a T in the −1 position (48% 5′-TGG; P < 0.0001). Considering next the 5′-GA dinucleotides, the nucleotide at the +2 position was again predominantly G (70% 5′-GAG; P < 0.0001), while that at the −1 position was less well defined, with a preference for 5′-CG or 5′-TG and a preference against 5′-GG (P < 0.0001).
Fig 6.

Dinucleotide sequence preferences for G-to-A mutations in HIV-1 cDNA from wild-type- and ΔVif-infected CD4+ T cells. Sequencing data described in Fig. 5 were analyzed for dinucleotide sequences at mutated sites. (A) Total number and percentage of mutated G residues in all four possible dinucleotide contexts for wild-type (top) and ΔVif (bottom) infections. (B) Correlation between the percentage of mutated G residues in the 5′-GG dinucleotide context and the total number of mutated G residues per viral sequence. Each data point represents the average of the percentage of mutations in the 5′-GG context for all cDNAs harboring the same number of G mutations. The Spearman rank correlation coefficient (rS) and the corresponding P value were determined for the two variables before averaging of the values.
Figure 6A further provides the total numbers of potential editing sites in terms of each dinucleotide context, which enabled the percentages of mutagenesis at each context to be calculated. Mutations at 5′-GG were therefore ∼9 times more frequent than mutations at 5′-GA in ΔVif virus infections and 14 times more frequent in wild-type infections, with the preferred wider context at 5′-GG being 5′-TGGG. Because many previous analyses, both in cells and in vitro (8, 23, 60–62), have defined these motifs as the dinucleotide/tetranucleotide consensus sites for A3G-mediated mutagenesis, we have concluded that A3G is the predominant driver of viral cytidine deamination in cultured primary T cells. The wider context of mutations occurring at 5′-GA is 5′-YGAG, though it is not possible to assign the causative APOBEC3 protein with any degree of confidence. Both A3F and A3D prefer 5′-GA dinucleotides as the substrates; however, it is also important to note that ∼10% of A3G-driven mutations can occur at this dinucleotide, at least in transfected cell studies (8, 23). Since A3G appears to be so potent in these experiments, it seems that A3G, A3F, and A3D all contribute to the mutations seen at 5′-GA dinucleotides in CD4+ T cells.
The conclusion that A3G is the predominant anti-HIV-1 deaminase in T cells is further supported by the analysis shown in Fig. 6B. Here, the percentage of mutations found at 5′-GG is plotted against the number of mutations per sequenced reverse transcript. There is a significant trend for higher fractions of mutations to appear at non-5′-GG dinucleotides as the total number of mutations increases. In other words, the proportion of mutations at 5′-GG is very high for the initial deamination events (∼95%), implying that A3G is the mediator of the vast majority of initial APOBEC3 protein-mediated editing. We offer two, nonmutually exclusive explanations for why this percentage decreases as the numbers of mutations increase: (i) as A3G-mediated hypermutation progressively consumes potential 5′-GG substrate sites in an individual reverse transcript, the enzyme may start to utilize secondary 5′-GA targets with increasing efficiency, and/or (ii) when the enzymes that prefer 5′-GA sites (A3D and A3F) have engaged a given reverse transcript and are operative, the tendency could be for particularly excessive hypermutation, which would then favor multiple changes at 5′-GA targets within one cDNA. Our view is that the former is certainly plausible, particularly as A3G can deaminate 5′-GA sites, but that the latter, while theoretically possible, is not yet supported by empirical observation.
DISCUSSION
Despite the substantial effort toward understanding APOBEC3 protein-mediated suppression of HIV-1 infection and its antagonism by Vif that has been deployed, very few analyses have been conducted using primary humans CD4+ T cells, the principal site of virus replication in vivo. In particular, while it is irrefutable that APOBEC3 proteins cause hypermutation of nascent cDNA, there was a need to address whether the physiological expression levels of these proteins also impede reverse transcription, as had been observed in a number of transfection-based systems, as well as in vitro. We therefore undertook a detailed analysis of wild-type and ΔVif HIV-1 infections in cultured CD4+ T cells obtained from healthy volunteer donors. By using donors who lack anti-HIV-1 alleles of A3H, we presume that the antiviral effects observed in our analyses are imparted by A3G, A3F, and/or A3D. The data presented in this study document very clear effects of APOBEC3 proteins both on the accumulation of reverse transcripts and on the mutagenesis of those cDNAs. We propose that the employment of dual inhibitory mechanisms helps elaborate a more effective block to viral infection.
Consistent with earlier work, wild-type viruses are at least 17-fold (donor 1, 19-fold; donor 2, 17-fold; donor 3, 39-fold) more infectious than ΔVif/APOBEC3+ viruses in single-cell assays (Fig. 1B), and this has previously been demonstrated to correspond to ΔVif viruses being unable to replicate in primary T cells. Analyses of ERT using melittin-permeabilized virions established that ΔVif virions synthesize reverse transcripts less efficiently than wild-type viruses and that this deficiency is more pronounced as later-stage replication intermediates are measured (Fig. 1C and D). A polarity similar to the magnitude of inhibition was observed in multiple challenges of freshly isolated CD4+ T cells (Fig. 2), consistent with the general model that APOBEC3 proteins impede the processivity of reverse transcriptase.
To begin to address whether the suppression of reverse transcriptase is manifested as stalling, pausing, and/or termination at preferred sites, sequences, or structures within the viral genomic RNA (gRNA) template, we established a novel sequencing-based assay to define the 3′ termini of viral cDNAs synthesized in ERT reactions. Owing to the sensitivity of cDNA recovery, these experiments were carried out using virus stocks produced from 293T cells transfected with a ΔVif provirus and various doses of an A3G expression vector (Fig. 3 and 4). The effects of time and A3G dose were evident, with escalating levels of A3G causing the production of increasingly shorter cDNAs throughout the time course, supporting the notion that the elongation phases of reverse transcription are impaired by A3G (46, 48). However, clear hot spots for preferred 3′ termini could not be detected in this data set, with the ends being relatively well distributed along the strong stop sequence. Future efforts will strive to deepen the data sets from these types of experiments, as well as extend the analyses to virus-infected cells.
Because earlier work showed that tRNA primer placement and first nucleotide addition are not affected by A3G (46), we consider two, not necessarily mutually exclusive, molecular mechanisms to explain the inhibition of reverse transcription by APOBEC3 proteins. (i) Since APOBEC3 proteins are naturally RNA binding proteins (61, 65, 67–70), they can engage the viral gRNA and potentially block reverse transcriptase progression by steric hindrance. In light of the lack of obviously preferred sites of pausing or termination and the calculated approximately seven A3G molecules present per virion (71), this model seemingly requires that APOBEC3 proteins bind throughout the length of the gRNA, as well as translocate relatively freely from one binding site to another. Indeed, in support of this idea, this behavior has been documented for the binding of recombinant A3G to single-stranded DNA in vitro (65, 72, 73). Planned future studies that map APOBEC3 protein binding sites on viral RNA will help address the relationship between RNA interactions and effects on reverse transcription. (ii) APOBEC3 proteins could interact with reverse transcriptase itself (directly or indirectly) and perturb enzymatic function. This model is appealing, since relatively few p66-51 reverse transcriptase dimers would have to be bound and inhibited, and is further supported by recent coimmunoprecipitation studies indicating that A3G can bind to reverse transcriptase in an RNA-independent manner (74). Future efforts will focus on defining this interaction in greater detail and addressing possible mechanisms for enzymatic inhibition.
Our sequencing analyses of nascent reverse transcripts afforded the opportunity to examine APOBEC3 protein-induced mutational spectra in the context of natural levels of protein expression. As discussed above, our data indicate strongly that A3G is the deaminase responsible for the bulk of HIV-1 cDNA editing in CD4+ T cells and that A3F and A3D play minor roles. This is inferred from the target site preferences for all mutations (Fig. 6A) yet is even more apparent when examining mutations in cDNAs harboring lower numbers of G-to-A changes (Fig. 6B). Indeed, this conclusion is not unexpected, given that A3G is more highly expressed than A3F and A3D in T cells (55, 56) and exhibits more potent HIV-1-inhibitory effects in transfected cell experiments (6–8, 30, 75).
Sequencing from wild-type virus infections revealed that significant levels of G-to-A mutations are seen in the presence of Vif (Fig. 5A), consistent with the incomplete antagonism of the APOBEC3 proteins in cultured CD4+ T cells (Fig. 1A). It is important to note that most (∼90%) cDNAs recovered from wild-type infections were unmutated but that when mutations were present, there were frequently, though not always, several within a single cDNA (Fig. 5D). This is consistent with studies indicating that APOBEC3 proteins can translocate and reposition along single DNA templates (65, 72). It was also clear that some reverse transcripts contain just one or two mutations, arguing against the notion that APOBEC3 proteins can induce only hypermutation (76). Indeed, we and others have previously suggested that infrequent G-to-A mutations driven by APOBEC3 proteins may provide a source of beneficial viral sequence diversification upon which selective pressures can act, resulting in viral evolution (77–80).
In conclusion, this is the first detailed examination of the editing and nonediting effects of APOBEC3 proteins on HIV-1 in the context of primary human T cells. We find ample evidence for both activities and suggest that their combined effects contribute to the inhibition of infection. The analyses of mutation spectra indicate that A3G is the dominant anti-HIV-1 protein in T cells, implying that pharmacologic perturbation of Vif's regulation of A3G (rather than of A3F, A3D, or A3H) should be the primary focus of therapeutic initiatives targeting this area.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dan Stetson for advice on the recovery and sequencing of short cDNAs.
This work was supported by the United Kingdom Medical Research Council, the National Institutes of Health (AI070072), and the Department of Health via a National Institute for Health Research comprehensive Biomedical Research Centre award to Guy's and St. Thomas's NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust.
Footnotes
Published ahead of print 14 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02587-12.
REFERENCES
- 1. Albin JS, Harris RS. 2010. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 12:e4 doi:10.1017/S1462399409001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chiu YL, Greene WC. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317–353 [DOI] [PubMed] [Google Scholar]
- 3. Holmes RK, Malim MH, Bishop KN. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 32:118–128 [DOI] [PubMed] [Google Scholar]
- 4. Malim MH. 2009. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364:675–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650 [DOI] [PubMed] [Google Scholar]
- 6. Dang Y, Wang X, Esselman WJ, Zheng YH. 2006. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80:10522–10533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85:11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392–1396 [DOI] [PubMed] [Google Scholar]
- 9. Liddament MT, Brown WL, Schumacher AJ, Harris RS. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14:1385–1391 [DOI] [PubMed] [Google Scholar]
- 10. Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23:2451–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 78:6073–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. 2008. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ooms M, Majdak S, Seibert CW, Harari A, Simon V. 2010. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J. Virol. 84:7961–7969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. 2010. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J. Virol. 84:1902–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2012. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2012. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379 [DOI] [PubMed] [Google Scholar]
- 17. Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. 2012. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J. Virol. 86:2874–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu B, Sarkis PT, Luo K, Yu Y, Yu XF. 2005. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 79:9579–9587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marin M, Rose KM, Kozak SL, Kabat D. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398–1403 [DOI] [PubMed] [Google Scholar]
- 20. Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407 [DOI] [PubMed] [Google Scholar]
- 21. Stopak K, de Noronha C, Yonemoto W, Greene WC. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12:591–601 [DOI] [PubMed] [Google Scholar]
- 22. Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060 [DOI] [PubMed] [Google Scholar]
- 23. Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809 [DOI] [PubMed] [Google Scholar]
- 24. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103 [DOI] [PubMed] [Google Scholar]
- 25. Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eigen M. 2002. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. U. S. A. 99:13374–13376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anderson JL, Hope TJ. 2008. APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology 375:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bishop KN, Holmes RK, Malim MH. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 80:8450–8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. 2006. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 80:11710–11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282:2587–2595 [DOI] [PubMed] [Google Scholar]
- 31. Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 81:7099–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon JH, Malim MH. 1996. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J. Virol. 70:5297–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sova P, Volsky DJ. 1993. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type 1. J. Virol. 67:6322–6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. von Schwedler U, Song J, Aiken C, Trono D. 1993. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 67:4945–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harris RS, Sheehy AM, Craig HM, Malim MH, Neuberger MS. 2003. DNA deamination: not just a trigger for antibody diversification but also a mechanism for defense against retroviruses. Nat. Immunol. 4:641–643 [DOI] [PubMed] [Google Scholar]
- 36. Kaiser SM, Emerman M. 2006. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J. Virol. 80:875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langlois MA, Neuberger MS. 2008. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J. Virol. 82:4660–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166–170 [DOI] [PubMed] [Google Scholar]
- 39. Schumacher AJ, Hache G, Macduff DA, Brown WL, Harris RS. 2008. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 82:2652–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. 2006. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 103:8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16:480–485 [DOI] [PubMed] [Google Scholar]
- 42. Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C. 2006. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 281:22161–22172 [DOI] [PubMed] [Google Scholar]
- 43. Okeoma CM, Lovsin N, Peterlin BM, Ross SR. 2007. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 445:927–930 [DOI] [PubMed] [Google Scholar]
- 44. Stenglein MD, Harris RS. 2006. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 281:16837–16841 [DOI] [PubMed] [Google Scholar]
- 45. Turelli P, Mangeat B, Jost S, Vianin S, Trono D. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 303:1829. [DOI] [PubMed] [Google Scholar]
- 46. Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4:e1000231 doi:10.1371/journal.ppat.1000231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goncalves J, Korin Y, Zack J, Gabuzda D. 1996. Role of Vif in human immunodeficiency virus type 1 reverse transcription. J. Virol. 70:8701–8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35:7096–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Browne EP, Allers C, Landau NR. 2009. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 387:313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miyagi E, Opi S, Takeuchi H, Khan M, Goila-Gaur R, Kao S, Strebel K. 2007. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J. Virol. 81:13346–13353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fouchier RA, Meyer BE, Simon JH, Fischer U, Malim MH. 1997. HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J. 16:4531–4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swanson CM, Puffer BA, Ahmad KM, Doms RW, Malim MH. 2004. Retroviral mRNA nuclear export elements regulate protein function and virion assembly. EMBO J. 23:2632–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goujon C, Malim MH. 2010. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 84:9254–9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koning FA, Goujon C, Bauby H, Malim MH. 2011. Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J. Virol. 85:13448–13452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83:9474–9485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38:4274–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31 [DOI] [PubMed] [Google Scholar]
- 58. Simon JH, Miller DL, Fouchier RA, Soares MA, Peden KW, Malim MH. 1998. The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. EMBO J. 17:1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lecossier D, Bouchonnet F, Clavel F, Hance AJ. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112. [DOI] [PubMed] [Google Scholar]
- 60. Suspene R, Sommer P, Henry M, Ferris S, Guetard D, Pochet S, Chester A, Navaratnam N, Wain-Hobson S, Vartanian JP. 2004. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 32:2421–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11:435–442 [DOI] [PubMed] [Google Scholar]
- 62. Armitage AE, Katzourakis A, de Oliveira T, Welch JJ, Belshaw R, Bishop KN, Kramer B, McMichael AJ, Rambaut A, Iversen AK. 2008. Conserved footprints of APOBEC3G on hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J. Virol. 82:8743–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bishop KN, Holmes RK, Sheehy AM, Malim MH. 2004. APOBEC-mediated editing of viral RNA. Science 305:645. [DOI] [PubMed] [Google Scholar]
- 64. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. 2010. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84:9864–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chelico L, Pham P, Calabrese P, Goodman MF. 2006. APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13:392–399 [DOI] [PubMed] [Google Scholar]
- 66. Suspene R, Rusniok C, Vartanian JP, Wain-Hobson S. 2006. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 34:4677–4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, Heidmann T, Greene WC. 2006. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 103:15588–15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH. 2007. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 81:2165–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. 2009. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 5:e1000330 doi:10.1371/journal.ppat.1000330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Iwatani Y, Takeuchi H, Strebel K, Levin JG. 2006. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80:5992–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xu H, Chertova E, Chen J, Ott DE, Roser JD, Hu WS, Pathak VK. 2007. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology 360:247–256 [DOI] [PubMed] [Google Scholar]
- 72. Nowarski R, Britan-Rosich E, Shiloach T, Kotler M. 2008. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 15:1059–1066 [DOI] [PubMed] [Google Scholar]
- 73. Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, Rueda D. 2012. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J. Biol. Chem. 287:15826–15835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang X, Ao Z, Chen L, Kobinger G, Peng J, Yao X. 2012. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 86:3777–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zennou V, Bieniasz PD. 2006. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology 349:31–40 [DOI] [PubMed] [Google Scholar]
- 76. Armitage AE, Deforche K, Chang CH, Wee E, Kramer B, Welch JJ, Gerstoft J, Fugger L, McMichael A, Rambaut A, Iversen AK. 2012. APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete “all or nothing” phenomenon. PLoS Genet. 8:e1002550 doi:10.1371/journal.pgen.1002550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jern P, Russell RA, Pathak VK, Coffin JM. 2009. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 5:e1000367 doi:10.1371/journal.ppat.1000367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim EY, Bhattacharya T, Kunstman K, Swantek P, Koning FA, Malim MH, Wolinsky SM. 2010. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 84:10402–10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sadler HA, Stenglein MD, Harris RS, Mansky LM. 2010. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 84:7396–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, Shaw GM, Hahn BH, Korber B, Seoighe C. 2009. HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 5:e1000414 doi:10.1371/journal.ppat.1000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.