

Abstract
Cleavage of human cytomegalovirus (HCMV) genomes as well as their packaging into capsids is an enzymatic process mediated by viral proteins and therefore a promising target for antiviral therapy. The HCMV proteins pUL56 and pUL89 form the terminase and play a central role in cleavage-packaging, but several additional viral proteins, including pUL51, had been suggested to contribute to this process, although they remain largely uncharacterized. To study the function of pUL51 in infected cells, we constructed HCMV mutants encoding epitope-tagged versions of pUL51 and used a conditionally replicating virus (HCMV-UL51-ddFKBP), in which pUL51 levels could be regulated by a synthetic ligand. In cells infected with HCMV-UL51-ddFKBP, viral DNA replication was not affected when pUL51 was knocked down. However, no unit-length genomes and no DNA-filled C capsids were found, indicating that cleavage of concatemeric HCMV DNA and genome packaging into capsids did not occur in the absence of pUL51. pUL51 was expressed mainly with late kinetics and was targeted to nuclear replication compartments, where it colocalized with pUL56 and pUL89. Upon pUL51 knockdown, pUL56 and pUL89 were no longer detectable in replication compartments, suggesting that pUL51 is needed for their correct subnuclear localization. Moreover, pUL51 was found in a complex with the terminase subunits pUL56 and pUL89. Our data provide evidence that pUL51 is crucial for HCMV genome cleavage-packaging and may represent a third component of the viral terminase complex. Interference with the interactions between the terminase subunits by antiviral drugs could be a strategy to disrupt the HCMV replication cycle.
INTRODUCTION
Infection with human cytomegalovirus (HCMV) is relatively frequent in the human population, with a seroprevalence ranging from 40 to 100%, depending on the population's socio-economic status (1, 2). In healthy individuals, primary infection is often asymptomatic or is associated with mild symptoms only. Acute infection can, however, last for several weeks or even months (especially in children) before it is resolved by the immune system. Inevitably, a latent infection is established that lasts life-long and bears the risk of reactivation. Recurrent infections resulting from reactivation events are a major risk in immunocompromised patients, such as transplant recipients, leading to severe and sometimes life-threatening disease and loss of function of the transplanted organ (3–5). Primary infection of pregnant women is another major concern, because in the absence of protective immunity, high-level viremia can develop, which facilitates vertical transmission of the virus to the fetus. Accordingly, HCMV is the leading viral cause of birth defects, which manifest as disabilities such as mental retardation and deafness (6, 7).
The commonly used medication against HCMV targets the viral DNA polymerase and aims to disrupt viral DNA replication. However, use of drugs like the nucleoside analog valganciclovir or the pyrophosphate analog forscarnet is impacted by adverse effects, such as myelosuppression or nephrotoxicity, and their long-term application can lead to the emergence of drug-resistant HCMV strains. Thus, there is a strong need for the development of new antivirals that target other processes of the HCMV infection cycle (8–12). One promising target is the cleavage of the viral genome and its packing into capsids, which is mediated by the viral terminase. This virus-specific enzymatic process appears to be especially vulnerable to antiviral inhibitors, and consequently, several substances have been described that interfere with the function of the terminase (13–16), with the testing of the drug AIC246 in a phase 2b clinical trial being the most advanced (17, 18).
Genome packaging in herpesviruses is reminiscent of that in tailed bacteriophages, which utilize a protein complex referred to as a terminase that interacts both with the DNA and the portal protein to initiate genome encapsidation (19, 20). The terminase recognizes specific sequences termed packaging signals (or pac sites) on the viral genomes, docks at the portal vertex of the capsid, and by ATP hydrolysis provides the energy necessary for genome insertion, followed by cutting of the DNA after exactly one genome length is packaged. Encapsidation of herpesvirus genomes may be more complex than that of bacteriophages and seems to involve more proteins than just a terminase. Based on data obtained mainly with alphaherpesvirus mutants, it has been suggested that besides the genuine terminase subunits pUL56 and pUL89, at least five additional HCMV proteins, namely, pUL51, pUL52, pUL77, pUL93, and pUL104, contribute to this process. Thus, there may be additional options for interference and for the development of inhibitors. However, these HCMV proteins have been only scarcely characterized, and thus their functions remain elusive.
The nuclear phase of the HCMV life cycle comprises replication of the viral DNA genome, its packaging into procapsids, and maturation of the DNA-filled capsids, followed by their egress into the cytoplasm by primary envelopment and deenvelopment at the nuclear membrane. The further steps of virion assembly occur in distinct compartments in the cytoplasm (1, 21), before the fully assembled particles are finally released from infected cells by the exocytotic pathway (for a review, see references 22 and 23). Replication of the 235-kbp HCMV genome produces “endless” DNA concatemers of head-to-tail-linked viral genomes that are believed to be branched molecules. The precise mechanism for how concatemeric HCMV DNA is resolved into unit-length genomes that undergo packaging is not completely understood, but it is clear that the viral terminase plays a major role. Several lines of evidence imply that the HCMV terminase is made of pUL56 and pUL89, although HCMV mutants with a deletion in UL56 or UL89 are not available yet to confirm their expected functions in genome cleavage-packaging. pUL56 has been demonstrated to bind to the viral packaging sequences and to have ATPase activity, probably providing the energy for spooling of the viral DNA into the capsids (24, 25). As would be expected of a terminase subunit, pUL56 interacts with the UL104 protein that forms the portal through which the viral genome enters the capsids (26–28). pUL89 was found to build a complex with pUL56 and to possess DNA-metabolizing activity (29–32). Finally, the fact that resistance to inhibitors of HCMV genome maturation maps to both pUL56 and pUL89 (as well as to pUL104) suggests that the putative pUL56/pUL89 terminase complex is involved in HCMV DNA encapsidation (18, 28, 33–36).
The role of the other HCMV proteins suggested to participate in genome packaging is much more speculative. In analogy to their herpes simplex virus type 1 (HSV-1) counterparts UL25 and UL17, the HCMV proteins pUL77 and pUL93 may have a function in the formation or stabilization of C capsids (37). One has to point out, however, that the similarities between the amino acid sequences of these HSV-1 and HCMV proteins are moderate, and one cannot exclude that there may be differences in their functions. Indeed, the UL25 protein of pseudorabies virus plays a role during nuclear egress of C capsids (38), and for UL25 of HSV-1 an additional function during uncoating of the genome and its release into the nucleus at an early stage of infection has been described (39–42).
Many of the putative HCMV encapsidation proteins are yet to be characterized, ideally in the context of viral infection, and to learn about their function, the phenotypic consequences of the knockout of the respective genes have to be analyzed with suitable mutants. Since these viral genes are essential, complementing systems have to be set up to produce HCMV mutants lacking these genes. By using such an approach, we have recently demonstrated that the HCMV pUL52 protein is required for cleavage and packaging of the viral genome, too (43). Further insight into the precise functions of the HCMV genome-packaging proteins is expected from the definition of their interaction partners. Yeast two-hybrid screens and transient coexpression and coimmunoprecipitation assays performed with proteins from several herpesviruses implied that the herpesvirus encapsidation proteins form a close interaction network (44–46). One interesting member of the encapsidation network is the HCMV UL51 protein (and its homologs in other herpesviruses), as it seems to be central in connecting many of the viral proteins that are supposed to be important for this process. The homologs of pUL51 have been therefore termed “hub proteins” (45, 46) and hence might be an ideal target for antiviral inhibitors. Still, the proposed interactions have to be confirmed during the course of HCMV infection.
In this work, we provide the first characterization of the HCMV UL51 protein. To analyze the step at which the viral life cycle is interrupted in the absence of pUL51, a conditionally replicating HCMV recombinant was used in which pUL51 levels could be regulated by a small synthetic ligand (47). We found that pUL51 is a late protein that is present in nuclear replication compartments in infected cells. Upon knockdown of pUL51, viral concatemeric DNA was neither cleaved into unit-length genomes nor packaged into capsids. Moreover, pUL51 did interact with the pUL56 and pUL89 terminase subunits in infected cells, and it is possibly required for the correct subnuclear localization of these terminase subunits.
MATERIALS AND METHODS
Viruses and cells.
Recombinant viruses used in this study are based on the bacterial artificial chromosome (BAC)-cloned HCMV strain AD169 (48), and construction of the HG and HD viruses was reported before (43, 49). In these genomes the UL1 to UL10 open reading frames (ORFs) were previously deleted, leaving a Flp recognition target (FRT) site. HG additionally expresses enhanced green fluorescent protein (EGFP) under the control of the HCMV major immediate-early promoter. Disruption of the UL51 ORF and generation of the conditional mutant HCMV-UL51-ddFKBP, in which pUL51 levels can be regulated by the shield-1 ligand, have been described elsewhere (47). Propagation of human foreskin fibroblasts (HFF), preparation of virus stocks, and analysis of viral growth by plaque assay were performed as reported previously (48).
Plasmids.
The mutated UL51 versions were cloned into the shuttle plasmid pOri6K-MfeI (43). The shuttle contains a kanamycin resistance cassette, an FRT site, and the R6Kγ origin of replication, which is dependent on the presence of the phage lambda π protein (50). To construct pOri6K-UL51-CHA, in which the UL51 C terminus is fused to the influenza virus hemagglutinin (HA) epitope, the UL51 ORF, plus 255 nucleotides of upstream sequences providing putative promoter elements, were amplified from pHG with primers UL51-HA.for, providing sequences encoding the HA tag (5′-CGCGGATCCCTAAGCGTAGTCTGGGACGTCGTATGGGTATTTACCCGGCGCCGACTCGT-3′), and UL51-prom.rev (5′-GGCGATATCGCCATAGCAGCTCAGTTGTCAA-3′). The resulting PCR product was cut with BamHI and EcoRV and cloned into the BamHI and HpaI sites of pOri6K-MfeI. pOri6K-UL51-NHA encoding the HA epitope at the UL51 N terminus was generated by PCR, amplifying the UL51 sequences plus the putative promoter region using primers UL51.for (5′-CGCGGATCCCGCACCGACGCCACCGCCGAT-3′) and UL51-prom.rev, followed by cloning into the BamHI and HpaI sites of pOri6K-MfeI, resulting in pOri6K-Mfe-UL51-P. Subsequently, the UL51 ORF was subcloned into this plasmid from the BAC pHG by Red-mediated recombination (51). To this end, part of the plasmid was PCR amplified using primer pair UL51-NHA.for (5′-ATCGGCGGTGGCGTCGGTGCGATGGAGATGAACAAGGTTCTCCATCAGGATCTGGAGCTCTCCCGGGAATTCCA-3′) and UL51-NHA.rev (5′-CCAGAAACGGCACCCGCTGCTTAGCCCAGGAAGCGTAGTCTGGGACGTCGTATGGGTACATTCTTTTTTCCGCGTCCTC-3′), harboring regions homologous to the sequences upstream of the UL51 ATG start codon and downstream of the UL51 stop codon, with UL51-NHA.rev also comprising sequences encoding the HA tag. PIR1 bacteria (Invitrogen) containing pHG and pKD46, which encodes the phage lambda recombination enzymes Red-α, -β, and -γ (52), were transformed with the PCR product, which led to recombination of the amplicon with the desired UL51 sequences in pHG, yielding pOri6K-UL51-NHA. For construction of a shuttle plasmid expressing UL51 fused to the Strep-Flag (SF) tag, a BamHI restriction site was inserted at the 5′ end of the UL51 ORF of pOri6K-UL51-NHA by site-directed mutagenesis, giving rise to pOri6K-UL51-NHA-MB. The sequence enclosing the SF epitope was amplified from pDEST/N-SF-TAP (53, 54) with primers UL51-SF.for (5′-CGCGGATCCCCACCATGGATTATAAAGATG-3′) and UL51-SF.rev (5′-CGCGGATCCATCCTCTCCGCTAGCTCC-3′), and the resulting PCR product was cut with BamHI and ligated to pOri6K-UL51-NHA-MB cleaved with BamHI.
To express HCMV proteins or parts thereof as recombinant proteins in Escherichia coli, the respective DNA coding sequences were cloned into plasmid pQE-30 (Qiagen, Hilden, Germany). Primers for PCR amplification were UL51-His.for (5′-CGCAAGCTTTCATTTACCCGGCGCCGACTC-3′) and UL51-His.rev (5′-CGCGCATGCTCCTGGGCTAAGCAGCGGGT-3′) for the UL51 ORF, UL52-N-His.for (5′-CGCGCATGCAATCCGAGTACCCACGTGAGC-3′) and UL52-N-His.rev (5′-CGCAAGCTTTCAACAGCGCGTGTCGATGGC-3′) for the UL52 N terminus, UL52-C-His.for (5′-CGCGCATGCTACGATAACGTCGTCCTGCGC-3′) and UL52-C-His.rev (5′-CGCAAGCTTCTA GACATACTTGTCTATCAC-3′) for the UL52 C terminus, UL56-N.for (5′-CGCAAGCTTCCCGTACACGTAGTTCATT-3′) and UL56-N.rev (5′-CGCGGATCCGAGATGAATTTGTTACAGA-3′) for the UL56 N terminus, UL56-C.for (5′-CGCAAGCTTACGCAGACTACCAGGCACCA-3′) and UL56-C.rev (5′-CGCGGATCCGACCTAAACGTGCTGCAGAA-3′) for the UL56 C terminus, UL89-Ex1.for (5′-CGCAAGCTTATTCTTTTGAAACGGTTCG-3′) and UL89-Ex1.rev (5′-CGCGGATCCTTGCGCGGAGACTCGGCCGC-3′) for the UL89 N terminus, and UL89-Ex2.for (5′-CGCAAGCTTGCTGACCCTGAAACGGATGG-3′) and UL89-Ex2.rev (5′-CGCGGATCCGCCATCGCCGAGTGCGCGG-3′) for the UL89 C terminus. PCR products corresponding to UL51 and UL52 sequences were cut with HindIII and SphI and cloned into the respective sites of pQE-30, and PCR products representing parts of UL56 and UL89 were cloned via HindIII and BamHI sites into the vector. The integrity of all plasmids was verified by sequencing.
Recombinant protein expression and purification.
E. coli BL21(DE3) cells containing the pREP4 plasmid (encoding the lac repressor) were used for bacterial expression of 6×His-tagged recombinant proteins. Twenty milliliters of overnight culture of BL21(DE3) cells transformed with the expression vectors were inoculated into 1 liter of selective LB medium (100 μg/ml of ampicillin, 25 μg/ml of kanamycin) and grown at 37°C to an optical density at 600 nm of 0.6. Protein expression was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside and further shaking of the culture for 4 to 6 h at 37°C. Bacterial cells were harvested, resuspended in 40 ml of lysis buffer (50 mM Tris-HCl [pH 6.8], 0.3 M NaCl, 6 M guanidinium hydrochloride, 5 mM imidazole) and lysed by sonication. Soluble proteins were recovered by centrifugation and incubated with 1 ml of preequilibrated Ni-nitrilotriacetic acid–agarose beads (Qiagen, Hilden, Germany). Following incubation of the protein solution for 30 min, the slurry was allowed to drain by gravity and washed extensively with urea buffer, increasing the concentration of imidazole stepwise (0, 10, and 50 mM). Finally, the protein was eluted three times with 500 μl of elution buffer (50 mM Tris-HCl [pH 6.8], 0.3 M NaCl, 4 M urea, 450 mM imidazole). Eluted fractions and samples of solutions were analyzed by SDS-PAGE and Western blotting (WB; using a His-specific antibody).
Generation of MAbs.
BALB/c mice were injected subcutaneously with 50 μg of recombinant protein diluted in phosphate-buffered saline (PBS) and mixed with complete Freund's adjuvant in a 1:1 ratio. Two weeks later, mice were boosted with the same protein in incomplete Freund's adjuvant by injecting 2/3 of the volume subcutaneously and 1/3 intraperitoneally (i.p.). After an additional 2-week period, the sera of immunized mice were screened for the antibody titer against the immunogen by using an enzyme-linked immunosorbent assay (ELISA). The best responders were additionally boosted i.p. with the immunogen dissolved in PBS. Three days later, spleen cells were collected and, after lysis of red blood cells, fused with SP2/0 myeloma cells at a ratio of 1:1. The cells were seeded on 96-well tissue culture plates in RPMI 1640 medium containing 20% fetal calf serum, hypoxanthine, aminopterine, and thymidine for hybridoma selection. The cultures were screened for monoclonal antibodies (MAbs) reactive against the immunogens by ELISA. Positive mother wells were expanded and cloned and further on checked for reactivity in Western blot assays on purified recombinant proteins and lysates of HCMV-infected cells. Reactivities of the MAbs were also tested by immunofluorescence (IF) microscopy.
BAC mutagenesis, analysis of BAC DNA, and reconstitution of virus mutants.
HCMV BACs containing the UL51 ORF fused to different epitope tag sequences were constructed by inserting the respective sequences at an ectopic position of the viral genome (the formerly deleted UL1-10 locus, providing an FRT site) via Flp-mediated integration of a small shuttle plasmid containing one FRT site (49, 50). In brief, pOri6K-UL51-CHA was inserted into pHG-ΔUL51 to yield pHG-UL51-CHA and into pHG to obtain pHG-UL51-CHA-DN. In both BACs, the HA tag was fused to the C terminus of pUL51. Likewise, pOri6K-UL51-NHA was recombined with pHG-ΔUL51 and pHD-ΔUL51, which resulted in pHG-UL51-NHA and pHD-UL51-NHA, respectively. These BACs expressed the UL51 protein with an N-terminal fusion of the HA tag. To generate an HCMV mutant in which the N terminus of pUL51 was fused to the Strep-Flag tag, pOri6K-UL51-SF was integrated into pHD-ΔUL51, to produce pHD-UL51-SF. Here, pUL51 was tagged with both the Strep-Flag and the HA epitope. To isolate HCMV BAC DNA from E. coli cultures, an alkaline lysis procedure was used, followed by restriction analysis and gel electrophoresis, and large-scale BAC DNA preparations were performed as described previously (50). For reconstitution of virus mutants, HFF were transfected with the corresponding BACs by using an adenovirus-mediated gene delivery method developed by Baker and Cotten in 1997 (55) as reported previously (49).
Analysis of viral DNA.
For the viral DNA replication assay, HFF were seeded into 6-well plates, infected with HCMV-UL51-ddFKBP at a multiplicity of infection (MOI) of 1, and kept in the presence or absence of 1 μM shield-1. On days 1 to 6 postinfection (p.i.), cells were trypsinized and total DNA was extracted using the DNeasy blood and tissue kit (Qiagen). One half of each DNA sample was then applied to a slot blot apparatus, followed by hybridization to a 32P-labeled probe specific for the b-repeat of the HCMV genome, as described previously (43). Radioactive signals were quantified with a phosphorimager. To assess cleavage of viral concatemeric DNA by pulsed-field gel electrophoresis (PFGE), HFF (1.5 × 106 cells) infected with HCMV-UL51-ddFKBP (MOI, 1) and cultivated in the presence or absence of 1 μM shield-1 were harvested on day 5 p.i. and cast into agarose blocks. PFGE conditions were as reported elsewhere (43), and after gel electrophoresis the DNA was transferred to a nylon membrane which was hybridized to the b-repeat-specific probe.
Immunofluorescence microscopy and Western blotting.
HFF grown on coverslips were infected with the indicated viruses at an MOI of 0.5 and analyzed at the time points indicated in the figure legends. Immunofluorescence was done following a protocol described recently (43). Antibody dilutions were 1:500 for the rat anti-HA MAb (clone 3F10, catalog number 11 867 423 001; Roche, Mannheim, Germany), 1:10 for the UL51, UL56, and the UL89 hybridoma cell culture supernatants, 1:100 for the mouse anti-UL44 MAb (kindly provided by B. Plachter, University of Mainz, Mainz, Germany), and 1:100 for the anti-UL86 (major capsid protein) antibody (a kind gift of K. Radsak, University of Marburg, Marburg, Germany). Images were taken with a Zeiss Observer.Z1 microscope or a Zeiss LSM 510 Meta confocal laser scanning microscope and further processed with the Zeiss LSM image browser.
For immunoblotting, HFF were infected with the indicated viruses at an MOI of 1 and harvested on day 4 p.i. Inhibition of viral late gene expression by phosphonoacetic acid (PAA) was done as reported before (43). Cells were trypsinized, pelleted by centrifugation, and lysed by addition of the reducing buffer Roti-Load1 (Roth, Karlsruhe, Germany). The equivalent of 1 × 105 cells was loaded into each well of a 10% SDS-polyacrylamide gel, and after electrophoresis proteins were transferred onto nitrocellulose membranes. Antibody dilutions were 1:1,000 for the rat anti-HA antibody, 1:200 for the anti-UL44 antibody, 1:50 for the mouse MAbs anti-UL52, anti-UL56, and anti-UL89, 1:400 for the anti-IE1 antibody (catalog number NEA-9221; PerkinElmer, Boston, MA), and 1:2,000 for the anti-actin antibody (catalog number A2066; Sigma, Hamburg, Germany). Secondary horseradish peroxidase (HRP)-labeled anti-rat–HRP (P0450), anti-rabbit–HRP (P0448), and anti-mouse–HRP conjugated immunoglobulins (P0260) were from Dako (Hamburg, Germany) and used at a 1:2,000 dilution. Signals were visualized using the ECL Advance substrate (GE Healthcare, Freiburg, Germany) and a LAS-3000 imager. Pictures were further processed with Adobe Photoshop CS4.
Affinity purification of Strep-tagged proteins and coimmunoprecipitation.
HFF (4 × 106 cells) were infected with HD-UL51-SF or HD at an MOI of 1 and harvested on day 4 p.i. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM dithiothreitol, 0.5% NP-40) containing protease inhibitors (protease inhibitor cocktail set III; catalog number 535140; Merck, Darmstadt, Germany). Cell lysates were mixed with a Dounce homogenizer, and insoluble material was removed by centrifugation for 15 min, 16,000 × g, 4°C. Supernatants were then incubated with 50 μl of Strep-Tactin–Sepharose (catalog number 2-1201-002; IBA, Göttingen, Germany) for 1 h at 4°C with rotation. Beads were washed five times with lysis buffer before incubation with biotin elution buffer (100 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 2 mM biotin) for 30 min at 4°C with rotation. Proteins were denatured by adding reducing buffer Roti-Load1 and boiling at 99°C for 5 min. A 1/40 volume of the whole-cell lysate before incubation with Strep-Tactin–Sepharose and 1/9 volume of the eluted material, respectively, were analyzed by Western blotting. For coimmunoprecipitation, infected cells were lysed in 10 mM Tris-HCl (pH 7.5)–150 mM NaCl–0.5 mM EDTA–0.5% NP-40 in the presence of protease inhibitors, and insoluble material was pelleted as described above. The supernatants were precleared through incubation with protein A-Sepharose (protein A–Sepharose 4 fast flow; catalog number 17-5280-01; GE Healthcare) for 1 h at 4°C with rotation. Three micrograms of either UL56 or UL89 antibody was added to the precleared lysates, and immune complexes were collected by rotating for 2 h at 4°C in the presence of protein A–Sepharose. Samples were washed three times with lysis buffer, and proteins were recovered by boiling for 5 min at 99°C in Roti-Load1 buffer. A 1/20 volume of the whole-cell lysate of each sample before immunoprecipitation (IP) and 1/5 of the precipitated material, respectively, were then analyzed by SDS-PAGE and Western blotting. The secondary antibody was Clean-Blot IP HRP detection reagent (1:500 dilution; catalog number 21230; Pierce).
Electron microscopy.
HFF (4 × 105 cells) were infected with HCMV-UL51-ddFKBP at an MOI of 1 and harvested on day 5 p.i. Cells were fixed for 1 h at room temperature with 2% (wt/vol) glutaraldehyde in 130 mM cacodylate buffer at pH 7.4 containing 2 mM CaCl2 and 10 mM MgCl2. Then, the cells were washed and postfixed with 1% (wt/vol) OsO4 in 165 mM cacodylate buffer at pH 7.4 containing 1.5% (wt/vol) K3Fe(III)CN6 for 1 h, followed by incubation in 0.5% (wt/vol) uranyl acetate in 50% (vol/vol) ethanol overnight. The cells were flat embedded in Epon, and 50-nm sections were cut parallel to the substrate. Images were taken with an FEI Tecnai G2 T20 electron microscope. Nuclear A, B, and C capsids were counted, and the percentages of total nuclear capsids were calculated.
RESULTS
Disruption of pUL51 expression prevents cleavage of concatemeric genomes.
To investigate the role of the UL51 gene during HCMV infection, we made use of a conditional UL51 mutant, HCMV-UL51-ddFKBP, in which pUL51 is fused to a destabilizing ddFKBP domain (47). As reported by Banaszynski et al. in 2006, ddFKBP fusion proteins are rapidly degraded but can be stabilized by the addition of a small synthetic ligand termed shield-1 (56). We showed in a previous study (47) and confirmed here that cells infected with the mutant and kept in the presence of shield-1 express pUL51, whereas removal of the ligand results in efficient knockdown of pUL51 (data not shown, but see Figure 6A and C, below).
Fig 6.

Expression and localization of the terminase proteins pUL56 and pUL89 in the presence or absence of pUL51. (A) HFF were infected with HCMV-UL51-ddFKBP and cultivated with or without shield-1. On day 4 p.i., cells were examined by immunofluorescence microscopy using the indicated antibodies. EGFP expression marks the infected cells. (B) Cells from the same experiment were also investigated by confocal laser scanning microscopy. (C) Lysates from cells infected as described for panel A were prepared on day 4 p.i. and analyzed by immunoblotting using an anti-HA antibody (for pUL51) or antibodies directed against the indicated proteins. n.i., noninfected cells.
We first asked at which step the HCMV infection cycle is interrupted when pUL51 expression is knocked down, and we started with the analysis of viral DNA replication. HFF were infected with HCMV-UL51-ddFKBP and cultivated in the presence or absence of shield-1. On days 1 to 6 p.i., total DNA was extracted from the cells, and accumulation of HCMV DNA was analyzed by slot blot hybridization using an HCMV-specific probe, followed by quantification of the hybridization signals with a phosphorimager. As is shown in Fig. 1A, viral DNA replication was independent of the shield-1 ligand, demonstrating that pUL51 is not needed for this step of the HCMV infection cycle.
Fig 1.

pUL51 is required for cleavage of HCMV genome concatemers, but not for viral DNA replication. (A, left) DNA replication of the HCMV-UL51-ddFKBP mutant in the presence or absence of shield-1. HFF infected with the mutant were harvested on days 1 to 6 p.i. Total DNA was isolated and applied to slot blot hybridization with a probe specific for the b-repeat region. (Right) Signals were quantified using a phosphorimager. (B) Detection of unit-length genomes by pulsed-field gel electrophoresis. Cells infected with the HCMV-UL51-ddFKBP mutant were kept in the presence or absence of shield-1 for 5 days, and total DNA was subjected to pulsed-field gel electrophoresis followed by hybridization to the HCMV-specific probe. (C) Detection of free genomic ends. (Left) HCMV-UL51-ddFKBP-infected HFF cultivated with or without shield-1 were harvested on day 5 p.i. Total DNA was extracted, cut with HpaI, and analyzed by Southern blotting using the b-specific probe. (Right) Schematic drawing of the four isomeric forms of the HCMV genome, showing the orientation of the unique long (UL) and unique short (US) regions (arrows) and the fragments detected by the probe, with their sizes given below the diagrams.
Next, we tested if pUL51 is required for cleavage of concatemeric HCMV genomes. Total DNA isolated from HFF infected with HCMV-UL51-ddFKBP and kept with or without shield-1 was examined for the presence of unit-length genomes. Pulsed-field gel electrophoresis revealed that unit-length genomes (∼230 kbp) were detectable only when pUL51 was present (Fig. 1B, first lane). After knockdown of pUL51, only concatemeric viral DNA was found, which was too large to migrate into the gel and was thus retained in the wells (Fig. 1B, second lane). This result implied that genome cleavage does not occur when pUL51 is missing.
To substantiate this finding, we also checked for the presence of free genomic ends after knockdown of pUL51. The DNA from infected cells was cut with HpaI and investigated by Southern hybridization using a probe specific for the b-sequence (Fig. 1C, right panel). Note that in any case, i.e., independent of genome cleavage, this probe is expected to detect fragments of 12.5 and 18.3 kbp, originating from the repeat regions of either genome concatemers or unit-length genomes (with the sizes of the fragments resulting from the orientation of the UL and US regions of the HCMV genome). When cleavage occurs and unit-length genomes are generated, an additional 7.4-kbp fragment is expected. This fragment, corresponding to the left genomic terminus, was detected only when pUL51 expression was allowed (Fig. 1C, first lane), and not when shield-1 was omitted (Fig. 1C, second lane). The faint bands migrating with a slightly higher molecular weight than the 7.4-kbp fragment resulted from terminal fragments containing several copies of the a-sequence, as shown before (43), and are indicative of genome cleavage, too. These data confirmed that pUL51 is essential for viral genome cleavage.
Capsid types obtained in the absence of pUL51.
Cleavage and packaging of herpesvirus genomes into preformed capsids are tightly linked processes. The observation that viral genome concatemers were not cleaved after knockdown of pUL51 predicted that mature capsids are not generated in this setting. To investigate which capsid types are present, cells infected with HCMV-UL51-ddFKBP and with shield-1 either added to or omitted from cultures were analyzed by electron microscopy on day 5 p.i. A, B, and C capsids can be distinguished in herpesvirus-infected cells. Spherical procapsids that consist of capsid proteins arranged around an internal protein scaffold are the substrate for DNA packaging. However, procapsids are hard to detect, because they seem to be unstable and undergo spontaneous angularization, which leads to B capsids that still contain the scaffold but no DNA. C capsids represent DNA-filled capsids, and A capsids that contain neither scaffold nor DNA presumably result from unsuccessful genome packaging events. As is shown in Fig. 2A, C capsids characterized by their electron-dense inner material representing the viral DNA could only be detected when expression of pUL51 was allowed by addition of shield-1. In contrast, when cells were kept in the absence of the ligand, mainly B capsids along with a few A capsids were found, but not a single C capsid was detected. We examined a series of images and counted a substantial number of capsids (Fig. 2B), since the numbers of A and C capsids that we found were low, which was probably due to the slightly delayed production of infectious progeny of the conditional UL51 mutant as described before (47). We conclude from these experiments that pUL51 is required for the successful packaging of HCMV genomes and the generation of DNA-filled C capsids.
Fig 2.

Electron microscopy analysis of capsid types obtained in the presence or absence of pUL51. (A) HFF infected with HCMV-UL51-ddFKBP were kept with or without shield-1 and examined on day 5 p.i. for the presence of A capsids (white arrowhead), B capsids (open arrowhead), and C capsids (black arrowhead). Bar, 200 nm. (B) Quantification of capsid types observed with or without shield-1 (as the percentage of the total capsid number). The number of capsids analyzed was 1,567 (with shield-1) and 2,162 (without shield-1).
HCMV mutants for analysis of the UL51 protein and its interaction partners.
In order to characterize the UL51 protein and to identify potential interaction partners, we generated several HCMV mutants that express pUL51 variants with epitopes suitable for detection by antibodies or for affinity purification (Table 1 and Fig. 3A and B). Mutagenesis of the UL51 ORF at its original genomic position is hampered by its close proximity to the neighboring essential UL50 and UL52 genes, since sequence modifications in UL51 may also affect the promoter regions of those genes and thus their expression and function. We therefore disrupted the UL51 ORF at its original genomic position and inserted mutant versions of the UL51 gene at an ectopic position of the HCMV genome (Fig. 3A). After transfecting HFF with the BAC pHG-ΔUL51, which carries a small deletion and an in-frame stop codon within the original UL51 ORF (47), only single green cells but no plaques were obtained, confirming that UL51 is essential for growth of HCMV. Next, we inserted a version of the UL51 ORF with a 3′-terminal addition of the sequence for the HA epitope together with the putative UL51 promoter sequence at the ectopic position of BAC pHG-ΔUL51. However, the resulting HCMV genome, pHG-UL51-CHA (Fig. 3A, construct 2), did not give rise to infectious progeny upon transfection, suggesting that the C terminus of pUL51 is crucial for protein function. Since the HCMV genome of pHG-UL51-CHA-DN, which encodes the authentic as well as the C-terminally tagged pUL51 (Fig. 3A, construct 3) readily led to plaque formation, we concluded that the pUL51-CHA variant is nonfunctional but does not interfere with the function of the authentic pUL51 in a dominant-negative manner. When the HA tag was fused to the N terminus of pUL51, the resulting HCMV genome, pHG-UL51-NHA (Fig. 3A, construct 4), turned out to be infectious, and growth of the recombinant virus was similar to that of the parental HG virus (Fig. 3C). Tagging of the pUL51 N terminus with the HA epitope on the background of the pHD genome, which does not carry the EGFP gene (Fig. 3A, construct 6; pHD-UL51-NHA), led to an HCMV recombinant that was used for IF microscopy and colocalization studies. Finally, a virus mutant was generated in which the sequence encoding a Strep-Flag tag was inserted in front of the HA tag at the 5′ end of the UL51 ORF (Fig. 3A, construct 7; pHD-UL51-SF). This epitope tag has been successfully used before for identifying interaction partners of proteins by affinity purification (53, 54).
Table 1.
HCMV UL51 mutants generated in this study
| HCMV mutant | UL51 disrupted? | Ectopic UL51 taga | EGFP fluorescence | Growth |
|---|---|---|---|---|
| pHG-ΔUL51 | Yes | + | − | |
| pHG-UL51-CHA | Yes | HA tag (C) | + | − |
| pHG-UL51-CHA-DN | No | HA tag (C) | + | + |
| pHG-UL51-NHA | Yes | HA tag (N) | + | + |
| pHD-ΔUL51 | Yes | − | − | |
| pHD-UL51-NHA | Yes | HA tag (N) | − | + |
| pHD-UL51-SF | Yes | Strep-Flag–HA tag (N) | − | + |
Indicated in parentheses is the placement of the tag, i.e., at the C or N terminus.
Fig 3.
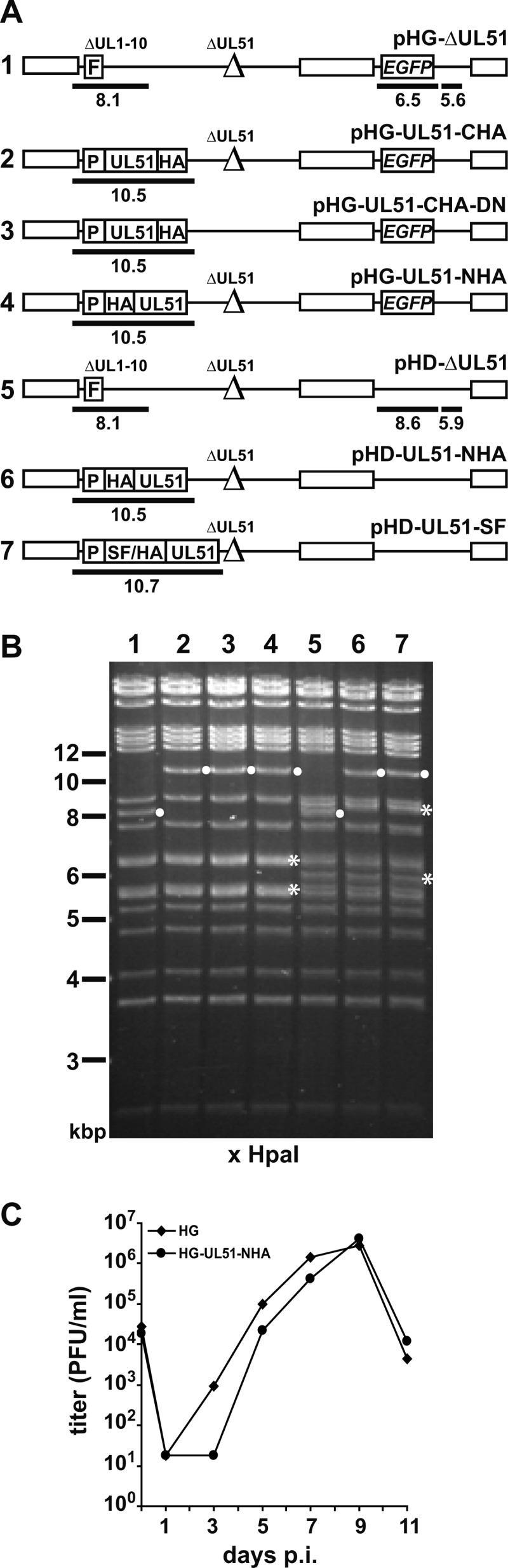
Construction of HCMV UL51 mutants used in this study. (A) Schematic drawing depicting the generated HCMV genomes with the following elements: open boxes, terminal and internal repeats; Δ, deleted or disrupted ORFs; F, an FRT site replacing the nonessential UL1 to UL10 ORFs; P, UL51 promoter region; HA and SF, sequences for the HA and Strep-Flag tags; EGFP, ORF encoding the enhanced green fluorescent protein. Fragments that resulted from successful insertion of the indicated elements are shown as black bars, with their sizes indicated below the construct. The illustration is not drawn to scale. (B) Restriction analysis results with the recombinant HCMV BAC genomes. BAC DNA was cut with HpaI and analyzed by agarose gel electrophoresis, followed by ethidium bromide staining. Lane numbers correspond to the genomes depicted in panel A. Fragments indicating successful mutagenesis are marked with white dots, and asterisks denote bands distinguishing pHG- and pHD-based genomes (with or without the EGFP ORF). (C) Growth kinetics of the HG-UL51-NHA mutant and the parental HG virus. HFF were infected at an MOI of 0.1 and viral titers in the supernatants were determined by plaque assay.
Expression of the UL51 protein in HCMV-infected cells.
To characterize the UL51 protein during infection, we first analyzed its expression kinetics by immunoblotting. HFF were infected with the recombinant virus HG-UL51-NHA, and cell lysates were prepared at different time points postinfection. pUL51 was detected using an antibody against the HA tag, and its appearance was compared to that of the early UL44 protein and the late UL52 protein (43). As is shown in Fig. 4A, pUL51 and pUL52 were observed 48 h after infection, whereas pUL44 was found already on day 1 p.i. This suggested that pUL51 is a late protein. When HFF cells infected with HG-UL51-NHA were treated with PAA, which inhibits viral DNA replication and arrests viral gene expression in the early phase, only a small amount of pUL51 was detected (Fig. 4B), indicating that the majority of pUL51 is produced at late times. This result was supported by immunofluorescence experiments performed with HFF infected with the parental virus HG and analyzed with a pUL51-specific monoclonal antibody (see below), which revealed that pUL51 was visible only in the absence of PAA (data not shown).
Fig 4.

Expression kinetics and subcellular localization of pUL51. (A) HFF were infected with HD-UL51-NHA, harvested at the time points indicated (hours postinfection), and analyzed by immunoblotting using antibodies against the HA tag (for pUL51), pUL44, pUL52, or actin. n.i., noninfected cells. (B) HFF infected with HD-UL51-NHA were kept with or without PAA and analyzed by immunoblotting on day 3 p.i. using the antibodies described in panel A. (C) HFF infected with HD-UL51-NHA were labeled with the anti-HA antibody on days 2 to 6 p.i. (d2 to d6) and analyzed by confocal laser scanning microscopy. Bar, 5 μm.
We next examined the subcellular distribution of pUL51 by infecting HFF with HD-UL51-NHA and staining for the HA-tagged pUL51 on days 1 to 6 p.i. (Fig. 4C). Whereas no signal was obtained on day 1 p.i. (data not shown), on day 2 and thereafter pUL51 was found in the nucleoplasm with an enrichment in structures that resemble viral replication compartments (57). The same localization was seen with the pUL51-specific antibody in cells infected with the parental virus (HD [data not shown]) or the HCMV-UL51-ddFKBP mutant (see Fig. 6A, below). This distribution of pUL51 did not change significantly over time, indicating that it is exclusively nuclear throughout the HCMV infection cycle.
pUL51 interacts with the terminase subunits pUL56 and pUL89.
The analysis of HCMV proteins implicated in cleavage and packaging of the viral genome has been hampered by a lack of suitable tools for their detection. We therefore considered it crucial to generate MAbs specific for these HCMV proteins. Recombinant proteins produced in E. coli and representing the full-length UL51 protein as well as N- or C-terminal domains of pUL52, pUL56, and UL89 (Table 2) were used to immunize mice. Hybridoma clones were chosen that produced antibodies exhibiting strong signals in immunofluorescence and/or Western blotting assays of infected fibroblasts and a minimal background with noninfected cells. Fluorescence microscopic analyses with these antibodies showed a distinct localization of pUL51, pUL56, and pUL89 to subnuclear compartments of HCMV-infected cells (see below). In immunoblotting assays, the antibodies recognized proteins of approximately 76 kDa (anti-UL52), 95 kDa (anti-UL56), and 78 kDa (anti-UL89) (data not shown), which is in agreement with their respective predicted molecular masses. The UL51 antibody was useful for immunofluorescence but displayed poor reactivity in Western blotting assays, as it was hardly distinguishable from background, prompting us to utilize the epitope-tagged pUL51 for immunoblotting experiments.
Table 2.
Monoclonal antibodies against HCMV encapsidation proteins
| HCMV protein | Immunogen used | Reactivitya |
Apparent molecular mass (kDa) | |
|---|---|---|---|---|
| IF | WB | |||
| pUL51 | Whole protein | +++ | (+) | 40 |
| pUL52 | N terminus | + | +++ | 76 |
| pUL56 | C terminus | +++ | +++ | 95 |
| pUL89 | N terminus | +++ | +++ | 78 |
+, good; +++, strong; (+), weak.
Since our experiments indicated that pUL51 is involved in genome cleavage and packaging, we hypothesized that pUL51 interacts with other viral proteins involved in this process. First, we checked whether pUL51 colocalizes with the known terminase subunits pUL56 and pUL89 in infected cells. As depicted in Fig. 5A, in cells infected with HD-UL51-NHA, pUL51 was enriched in the same nuclear compartments as pUL56 and pUL89. It has been shown that pUL56 localizes to viral DNA replication compartments (58) and, thus, we concluded that pUL51 is present in these subnuclear structures. In order to explore whether pUL51 does interact with pUL56 or pUL89, we applied affinity purification after infecting HFF with HD-UL51-SF, expressing pUL51 fused to the Strep-Flag tag. Following pulldown of the tagged pUL51 via StrepTactin–Sepharose, the proteins bound to pUL51 were analyzed by Western blotting. Figure 5B shows that pUL51 could be isolated from lysates of cells infected with HD-UL51-SF (first panel, second lane) by this approach and, remarkably, both pUL56 and pUL89 did copurify with pUL51 (Fig. 5B, second lane, second and third rows). These data provides evidence that pUL51, pUL56, and pUL89 form a complex in HCMV-infected cells. Conversely, pUL52, which is another nuclear viral protein required for genome cleavage and packaging (43), was not pulled down with pUL51 (Fig. 5B, second lane, row 4), implying that pUL52 is not part of the complex formed between pUL51, pUL56, and pUL89. To further substantiate our finding that pUL51 binds to the viral terminase subunits, coimmunoprecipitation studies with HD-UL51-SF-infected cells were carried out using the MAbs against pUL56 and pUL89. After immunoprecipitation with anti-UL56, pUL51 and pUL89 were indeed found in the precipitate (Fig. 5C, second lane), and when using the pUL89 antibody for immunoprecipitation, both pUL51 and pUL56 could be detected (Fig. 5C, fourth lane). Again, pUL52 did not coprecipitate with the terminase subunits. This result confirmed that pUL51 interacts with the terminase components pUL56 and pUL89 in infected cells.
Fig 5.

Colocalization and interaction of pUL51 with the terminase subunits pUL56 and pUL89. (A) Cells infected with HD-UL51-NHA were probed on day 3 p.i. with antibodies against the HA tag and pUL56 (upper panel) or pUL89 (lower panel) and then analyzed by confocal laser scanning microscopy. Bar, 5 μm. (B) HFF infected with HD-UL51-SF or the parental virus HD were harvested on day 4 p.i., and cell lysates were incubated with StrepTactin-Sepharose. Eluted proteins (bound) were analyzed by immunoblotting using antibodies against the HA tag, pUL56, pUL89, or pUL52. Input lanes contained whole-cell lysate before pulldown with StrepTactin-Sepharose. (C) HD-UL51-SF-infected fibroblasts were lysed on day 4 p.i., and immunoprecipitation was performed with either the pUL56- or the pUL89-specific antibody. Eluted proteins were subjected to immunoblotting (WB) with antibodies directed to the indicated proteins.
Influence of pUL51 on the pUL56 and pUL89 terminase subunits.
The identification of these interactions raised the question if pUL51 has an influence on the expression, stability, or subcellular localization of pUL56 or pUL89. To this end, HFF were infected with HCMV-UL51-ddFKBP, and shield-1 was either added to the cultures or not. On day 4 p.i., cells were analyzed by immunofluorescence microscopy using antibodies against pUL51, pUL56, pUL89, the early UL44 protein, and the late UL86 protein. As expected, pUL51 was efficiently knocked down in fibroblasts cultivated in the absence of shield-1 (Fig. 6A, compare left to right panel). pUL56 and pUL89 were predominantly found in nuclear replication compartments in the presence of pUL51 (Fig. 6A, left panel, and B, with shield-1), whereas the signals in the nucleus were strongly diminished, and the typical distributions of pUL56 and pUL89 were hardly visible when pUL51 was missing (Fig. 6A, right panel, and B, without shield-1). As controls, cells were probed with antibodies against pUL44, which is a marker for replication compartments, and the HCMV major capsid protein pUL86, which is also found in the nuclei of infected cells. Neither pUL44 nor pUL86 localization was influenced by the knockdown of pUL51, demonstrating that replication compartments are formed properly in the absence of pUL51 and that the lack of pUL51 does not interfere with expression of late structural proteins or their localization in general, as already shown by the electron microscopy studies (see above).
One explanation for the diminished immunoreactivities of pUL56 and pUL89 after knockdown of pUL51 could be that the terminase proteins become destabilized and are possibly degraded when pUL51 is missing. To evaluate this possibility, HFF were infected with HCMV-UL51-ddFKBP and cultivated in the presence or absence of shield-1. Cell lysates that were prepared on day 4 p.i. and analyzed for the levels of pUL56 and pUL89 as well as for several other proteins (immediate-early protein 1 [IE1], pUL44, and pUL52) contained comparable amounts of both pUL56 and pUL89, regardless of whether pUL51 was present or not, and there was also no effect of pUL51 on the levels of the control proteins (Fig. 6C). Taken together, these data suggest that pUL51 is necessary for either the correct localization to viral replication compartments or the appropriate folding and interaction of the known terminase subunits.
DISCUSSION
The aim of this work was to gain insights into the properties and function of the hitherto-uncharacterized HCMV UL51 protein. We focused on analyzing pUL51 in HCMV-infected cells, which in contrast to transient assays with isolated proteins guaranteed that pUL51 expression took place at physiological levels and with correct temporal kinetics. This approach also ensured the correct subcellular localization of the investigated viral protein, which may be dependent on the interaction with other virus proteins. To facilitate these studies, MAbs were raised against several viral proteins predicted to be involved in HCMV genome encapsidation. Our results showed that pUL51 is a late protein that was found to be enriched in viral replication compartments in the nucleus, where it colocalized with the known HCMV terminase subunits pUL56 and pUL89. Moreover, pUL51 did interact with the pUL56/pUL89 terminase complex. In the absence of pUL51, HCMV DNA replication was not disturbed, but cleavage of viral genomes and their packaging into capsids was abolished, and pUL56 and pUL89 were no longer detectable in DNA replication compartments with the specific MAbs. In summary, these data indicate that pUL51 plays a pivotal role in HCMV genome cleavage-packaging and may represent an additional component of the viral terminase.
According to in silico analysis, pUL51 is a 157-amino acid (aa) protein with a predicted molecular mass of 17 kDa, and alignment of its amino acid sequence revealed homology to the UL33 proteins of alphaherpesviruses. HSV-1 pUL33 is an essential protein required for cleavage-packaging of HSV-1 genomes and associates with the HSV-1 pUL15/pUL28 terminase complex, probably by direct interaction via pUL28 (59–63). Yet, the specific function of pUL33 within the HSV-1 terminase complex is still to be elucidated. We point out, however, that the similarity between HSV-1 pUL33 and HCMV pUL51 is limited and comprises only the C-terminal parts of the proteins (aa 73 to 149 of pUL51). The N-terminal half of pUL51 is not conserved among herpesviruses, and this part seems to be disordered, whereas the pUL51 C terminus is predicted to form α-helical structures. The in silico analysis results suggested that the conserved pUL51 C terminus might be essential, and this hypothesis was supported by the results of our attempts to tag the UL51 ORF in the BAC-cloned HCMV AD169 genome, namely, viable virus could only be obtained upon epitope tagging of the N terminus, but not of the C terminus. With respect to these findings, it is noteworthy that mutations in pUL33 that interfered with HSV-1 genome cleavage and also in part with interaction with the pUL28 terminase subunit were located exclusively within the conserved portion of the pUL33 C terminus (64). Our mutagenesis experiments together with the results of our previous study, in which we constructed a UL51 knockout genome (47), confirmed earlier reports that pointed to an essential role of the UL51 gene in the HCMV infection cycle (65, 66).
During the characterization of pUL51 in HCMV-infected cells, we observed that its migration in SDS-polyacrylamide gels was much slower than would be expected of a 17-kDa protein. Although using standard reducing and denaturing conditions, pUL51 exhibited an apparent molecular mass of ∼40 kDa. Similar results were obtained with pUL51 fused to the destabilizing ddFKBP domain (this study and reference 47). Likewise, HSV-1 pUL33 has been described to possess an apparent molecular mass of 19 kDa rather than the predicted 14 kDa (60), and it was hypothesized that this is due to the high proline content (5%) of pUL33. Similarly, pUL51 is also proline rich (10 aa out of 157 [6%]), and additionally it contains numerous acidic residues which result in a theoretical pI value of 4.05 (the predicted pI of HSV-1 pUL33 is 6.28). Both factors could account for the unusual electrophoretic mobility of pUL51.
When investigating the subcellular localization of pUL51, we found that it is a nuclear protein concentrated in replication compartments during the HCMV infection cycle. HSV-1 pUL33 was also present in replication compartments, but it was additionally detected in the cytoplasm adjacent to the nuclear membrane (60), a distribution we did not observe for HCMV pUL51. Remarkably, pUL51 colocalized with both pUL56 and pUL89 inside the replication compartments, which presumably also represent the sites of genome encapsidation. These findings are in line with a putative role of pUL51 in viral genome replication or cleavage-packaging.
Both proteins, HSV-1 pUL33 and HCMV pUL51, seem to be expressed mainly with late kinetics (reference 60 and this work), as only a small amount of pUL51 was detectable when late gene expression was prevented with PAA. The late kinetics was retained when pUL51 was expressed from the ectopically inserted UL51 ORF, indicating that the elements required for appropriate temporal expression were present in the transferred DNA fragment. Similarly, we previously showed that pUL52, another protein involved in genome encapsidation, is produced at late times of infection when encoded by the ectopically inserted UL52 gene (43). Using a newly generated MAb to pUL52, we now confirmed in this study the late expression kinetics of the UL52 gene located at its original genomic position.
In publications based on yeast two-hybrid and coimmunoprecipitation experiments that were performed with viral proteins expressed independently of viral infection, it was proposed that the homologs of pUL51 in other herpesviruses are central hub proteins that interact with various other viral proteins involved in the nuclear phase of the herpesvirus life cycle (44–46, 67, 68). Using different approaches and lysates from HCMV-infected cells, we found here that pUL51 interacts with the HCMV terminase subunits pUL56 and pUL89. Thus, as suggested for alphaherpesviruses (61–63), the terminase seems to comprise additional viral proteins in betaherpesviruses, too. If assembly of the pUL51/pUL56/pUL89 complex takes place in the nucleus or already in the cytoplasm will be the subject of further investigations. For the HSV-1 terminase, it has been postulated that assembly occurs in the cytoplasm followed by nuclear import of the complex via a nuclear localization signal (NLS) present within pUL15 (69). Notably, a functional NLS was identified in pUL56 of HCMV (70), which is the homolog of the other HSV-1 terminase subunit pUL28, and not in pUL89, the homolog of HSV-1 pUL15. Whether pUL51 binds to pUL56 or pUL89 or both and if a putative C-terminal dimerization domain in pUL51 is involved in these interactions remain to be analyzed. Other questions that have to be addressed in further studies is whether pUL51 stays associated with pUL56/pUL89 during genome cleavage-packaging and if it is present on capsids, as is the case for HSV-1 pUL33 (71, 72).
It is also conceivable that the HCMV terminase complex is even larger and contains additional viral or cellular proteins. However, for HSV-1 the testing of several viral proteins for terminase binding did not provide evidence for this hypothesis, and the formation of the pUL15/pUL28/pUL33 complex can apparently occur independently of other viral proteins (61). Since the nuclear steps in the herpesvirus life cycle (genome replication, cleavage-packaging, maturation, and nuclear egress of capsids) are thought to be tightly linked, it is possible that protein-protein interactions within such an encapsidation network are only weak or transient and therefore hard to detect (73). Interestingly, at least under the experimental conditions used here, we could not see an interaction of the pUL51/pUL56/pUL89 complex with pUL52, although this was predicted based on results obtained with infection-independent expression of the corresponding proteins of other herpesviruses (44–46). Short-lived interactions are probably easier to detect following overexpression, and since such interactions may occur during the nuclear phase of the herpesvirus infection cycle, cross-linking experiments that freeze such transient protein-protein interactions in infected cells may help to address this aspect in future studies.
The phenotype of a viral null mutant can often provide valuable hints to the function of the mutated gene; in particular, one can learn which step of the viral replication cycle is affected. However, production of a mutant with a disruption of an essential gene, such as UL51, requires a complementing cell line that provides the corresponding protein. Since our attempts to generate a pUL51-expressing cell line were unsuccessful (data not shown), we utilized a conditionally replicating mutant—an approach that was recently established by us and others for the analysis of essential herpesvirus proteins (47, 74–76). Fusion of the sequences for a destabilizing domain (ddFKBP [56]) to the UL51 ORF led to a viral genome that in the presence of the stabilizing ligand shield-1 gave rise to a viable HCMV mutant. Importantly, pUL51 was efficiently knocked down in the absence of the ligand, thereby disrupting the infection cycle and preventing the production of virus progeny and plaque formation (47). Using the conditionally replicating UL51-ddFKBP mutant, we could examine the effect of loss of pUL51 without the need for a complementing cell line (77). In the absence of pUL51, viral DNA replication in infected cells was not affected, but cleavage of genome concatemers and DNA packaging into capsids were abrogated. Moreover, assembly of procapsids was not affected, but no DNA-filled C capsids were obtained. These data argue for an essential role of pUL51 in HCMV genome cleavage-packaging, as would be expected of a protein that binds to the terminase subunits or is possibly part of the viral terminase complex.
This is the first report about the phenotype of an HCMV mutant in which expression of one of the putative terminase proteins is disrupted, since no null mutants for UL56 or UL89 are available. Interestingly, when pUL51 was knocked down, pUL56 and pUL89 became almost undetectable in the nuclear replication compartments using the specific MAbs, yet the amounts of both the UL56 and the UL89 proteins remained unaffected, implying that they are not degraded when pUL51 is missing. One explanation for this observation is the mislocalization of pUL56 and pUL89 in the absence of pUL51, although a possible increase of the fluorescence signals in other subcellular compartments is difficult to quantify by the immunofluorescence technique. Alternatively, the interaction between pUL56 and pUL89 might be weakened in the absence of pUL51, making these proteins possibly more prone to extraction during the fixation and staining procedures. Moreover, one can imagine that folding of pUL56 and pUL89 is altered in the absence of pUL51, thus impairing recognition by the specific MAbs. Differentiation between these possibilities will have to be addressed in the future. When an HSV-1 UL33 deletion mutant was analyzed (78), the intranuclear localization of the two other HSV-1 terminase subunits (pUL15 and pUL28) remained unchanged. Conversely, in cells infected with a UL15 null mutant, neither pUL33 nor pUL28 was found in replication compartments, although their protein levels were unaltered (78). Based on these data, it was therefore proposed that the HSV-1 UL15 protein targets the other terminase subunits to viral replication compartments. Adding to that, the nuclear localization signal identified in pUL15 was found to be essential for the import of the HSV-1 terminase into the nucleus (69). Analogous to the scenario for HSV-1, we hypothesize that pUL51 is needed for the correct nuclear localization or interaction of the HCMV terminase subunits, which would be in agreement with a function of pUL51 in assembly or stabilization of the terminase complex or its transport to the nucleus. A stabilizing role was also proposed for the HSV-1 UL33 protein, which seems to enhance the interaction between pUL15 and pUL28, and furthermore may contribute to correct folding (63). It remains open to discussion whether pUL33 and pUL51 have active roles within the terminase complex, such as DNA binding or cleavage. As with HSV-1 pUL33 (62), HCMV pUL51 does not exhibit obvious DNA-, RNA-, or ATP-binding motifs. Also, the high content of negatively charged residues argues against a DNA-binding activity of pUL51, and, as with pUL33, HCMV pUL51 would represent the smallest component of the terminase, which may rather hint to a regulatory function. In agreement with this, the recombinant pUL56 and pUL89 proteins were found to be sufficient for cutting plasmid DNA carrying the HCMV packaging signal (31). In infection, however, additional viral proteins are obviously needed to complete the cleavage-packaging process. Other functions of terminase components or proteins associated with them may include the association with the procapsid, translocation of the viral DNA into the capsids (9), or sealing the capsids after genome insertion is accomplished.
In summary, this study for the first time provides functional and biochemical characterizations of pUL51 in the context of HCMV infection. Knowledge of the interplay between pUL51 and the known terminase subunits of HCMV can serve as a starting point for the development of novel antiviral inhibitors that target the interaction between these essential viral proteins.
ACKNOWLEDGMENTS
We thank Christian Johannes Glöckner for providing the plasmids encoding the Strep-Flag epitope.
This work was funded by grant ME 11-02/3-1 of the Deutsche Forschungsgemeinschaft (to M.M.). J.K.-A. and I.G. were supported by the DEWIN Program of the Center for Infection Biology and the Hannover Biomedical Research School.
Footnotes
Published ahead of print 21 November 2012
REFERENCES
- 1. Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2702–2772 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Cannon MJ, Schmid DS, Hyde TB. 2010. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 20:202–213 [DOI] [PubMed] [Google Scholar]
- 3. Ljungman P, Hakki M, Boeckh M. 2010. Cytomegalovirus in hematopoietic stem cell transplant recipients. Infect. Dis. Clin. North Am. 24:319–337 [DOI] [PubMed] [Google Scholar]
- 4. Kotton CN. 2010. Management of cytomegalovirus infection in solid organ transplantation. Nat. Rev. Nephrol. 6:711–721 [DOI] [PubMed] [Google Scholar]
- 5. Fisher RA. 2009. Cytomegalovirus infection and disease in the new era of immunosuppression following solid organ transplantation. Transpl. Infect. Dis. 11:195–202 [DOI] [PubMed] [Google Scholar]
- 6. Hyde TB, Schmid DS, Cannon MJ. 2010. Cytomegalovirus seroconversion rates and risk factors: implications for congenital CMV. Rev. Med. Virol. 20:311–326 [DOI] [PubMed] [Google Scholar]
- 7. Nigro G, Adler SP. 2011. Cytomegalovirus infections during pregnancy. Curr. Opin. Obstet. Gynecol. 23:123–128 [DOI] [PubMed] [Google Scholar]
- 8. Emery VC, Hassan-Walker AF. 2002. Focus on new drugs in development against human cytomegalovirus. Drugs 62:1853–1858 [DOI] [PubMed] [Google Scholar]
- 9. Yang K, Wills EG, Baines JD. 2011. A mutation in UL15 of herpes simplex virus 1 that reduces packaging of cleaved genomes. J. Virol. 85:11972–11980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andrei G, De Clercq E, Snoeck R. 2009. Drug targets in cytomegalovirus infection. Infect. Disord. Drug Targets 9:201–222 [DOI] [PubMed] [Google Scholar]
- 11. Bogner E. 2002. Human cytomegalovirus terminase as a target for antiviral chemotherapy. Rev. Med. Virol. 12:115–127 [DOI] [PubMed] [Google Scholar]
- 12. Price NB, Prichard MN. 2011. Progress in the development of new therapies for herpesvirus infections. Curr. Opin. Virol. 1:548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, Haerter M, Buerger I, Trappe J, Herrington JA, Haebich D, Ruebsamen-Waigmann H. 2001. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action. J. Antimicrob. Chemother. 48:757–767 [DOI] [PubMed] [Google Scholar]
- 14. Hwang JS, Kregler O, Schilf R, Bannert N, Drach JC, Townsend LB, Bogner E. 2007. Identification of acetylated, tetrahalogenated benzimidazole D-ribonucleosides with enhanced activity against human cytomegalovirus. J. Virol. 81:11604–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J. Virol. 72:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McVoy MA, Nixon DE. 2005. Impact of 2-bromo-5,6-dichloro-1-beta-d-ribofuranosyl benzimidazole riboside and inhibitors of DNA, RNA, and protein synthesis on human cytomegalovirus genome maturation. J. Virol. 79:11115–11127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, Ruebsamen-Schaeff H, Zimmermann H. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 54:1290–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 85:10884–10893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Casjens SR. 2011. The DNA-packaging nanomotor of tailed bacteriophages. Nat. Rev. Microbiol. 9:647–657 [DOI] [PubMed] [Google Scholar]
- 20. Rao VB, Feiss M. 2008. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42:647–681 [DOI] [PubMed] [Google Scholar]
- 21. Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gibson W. 2008. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 325:187–204 [DOI] [PubMed] [Google Scholar]
- 23. Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 20:392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bogner E, Radsak K, Stinski MF. 1998. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J. Virol. 72:2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scholz B, Rechter S, Drach JC, Townsend LB, Bogner E. 2003. Identification of the ATP-binding site in the terminase subunit pUL56 of human cytomegalovirus. Nucleic Acids Res. 31:1426–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dittmer A, Drach JC, Townsend LB, Fischer A, Bogner E. 2005. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-d-ribonucleosides. J. Virol. 79:14660–14667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dittmer A, Bogner E. 2005. Analysis of the quaternary structure of the putative HCMV portal protein pUL104. Biochemistry 44:759–765 [DOI] [PubMed] [Google Scholar]
- 28. Komazin G, Townsend LB, Drach JC. 2004. Role of a mutation in human cytomegalovirus gene UL104 in resistance to benzimidazole ribonucleosides. J. Virol. 78:710–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hwang JS, Bogner E. 2002. ATPase activity of the terminase subunit pUL56 of human cytomegalovirus. J. Biol. Chem. 277:6943–6948 [DOI] [PubMed] [Google Scholar]
- 30. Thoma C, Borst E, Messerle M, Rieger M, Hwang JS, Bogner E. 2006. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry 45:8855–8863 [DOI] [PubMed] [Google Scholar]
- 31. Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. 2002. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res. 30:1695–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nadal M, Mas PJ, Blanco AG, Arnan C, Sola M, Hart DJ, Coll M. 2010. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. U. S. A. 107:16078–16083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 72:4721–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, Graeper S, Klenk HD, Ruebsamen-Waigmann H, Hallenberger S. 2001. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J. Virol. 75:9077–9086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Champier G, Couvreux A, Hantz S, Rametti A, Mazeron MC, Bouaziz S, Denis F, Alain S. 2008. Putative functional domains of human cytomegalovirus pUL56 involved in dimerization and benzimidazole D-ribonucleoside activity. Antivir. Ther. 13:643–654 [PubMed] [Google Scholar]
- 36. Thoma C, Bogner E. 2010. Short hairpin RNAs specific to human cytomegalovirus terminase subunit pUL89 prevent viral maturation. Antivir. Ther. 15:391–400 [DOI] [PubMed] [Google Scholar]
- 37. Meissner CS, Köppen-Rung P, Dittmer A, Lapp S, Bogner E. 2011. A “coiled-coil” motif is important for oligomerization and DNA binding properties of human cytomegalovirus protein UL77. PLoS One 6:e25115 doi:10.1371/journal.pone.0025115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klupp BG, Granzow H, Keil GM, Mettenleiter TC. 2006. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J. Virol. 80:6235–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cockrell SK, Sanchez ME, Erazo A, Homa FL. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J. Virol. 83:47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Hara M, Rixon FJ, Stow ND, Murray J, Murphy M, Preston VG. 2010. Mutational analysis of the herpes simplex virus type 1 UL25 DNA packaging protein reveals regions that are important after the viral DNA has been packaged. J. Virol. 84:4252–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rode K, Döhner K, Binz A, Glass M, Strive T, Bauerfeind R, Sodeik B. 2011. Uncoupling uncoating of herpes simplex virus genomes from their nuclear import and gene expression. J. Virol. 85:4271–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borst EM, Wagner K, Binz A, Sodeik B, Messerle M. 2008. The essential human cytomegalovirus gene UL52 is required for cleavage-packaging of the viral genome. J. Virol. 82:2065–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, Haas J. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239–242 [DOI] [PubMed] [Google Scholar]
- 45. Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, Schmidt T, Kraus T, Stellberger T, Rutenberg C, Suthram S, Bandyopadhyay S, Rose D, von Brunn A, Uhlmann M, Zeretzke C, Dong YA, Boulet H, Koegl M, Bailer SM, Koszinowski U, Ideker T, Uetz P, Zimmer R, Haas J. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5:e1000570 doi:10.1371/journal.ppat.1000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vizoso Pinto MG, Pothineni VR, Haase R, Woidy M, Lotz-Havla AS, Gersting SW, Muntau AC, Haas J, Sommer M, Arvin AM, Baiker A. 2011. Varicella zoster virus ORF25 gene product: an essential hub protein linking encapsidation proteins and the nuclear egress complex. J. Proteome. Res. 10:5374–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Glass M, Busche A, Wagner K, Messerle M, Borst EM. 2009. Conditional and reversible disruption of essential herpesvirus proteins. Nat. Methods 6:577–579 [DOI] [PubMed] [Google Scholar]
- 48. Borst EM, Hahn G, Koszinowski UH, Messerle M. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J. Virol. 73:8320–8329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Borst EM, Messerle M. 2005. Analysis of human cytomegalovirus oriLyt sequence requirements in the context of the viral genome. J. Virol. 79:3615–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Borst EM, Benkartek C, Messerle M. 2007. Use of bacterial artificial chromosomes in generating targeted mutations in human and mouse cytomegaloviruses, p 10.32.1–10.32.30 In Coligan JE, Bierer B, Margulies DH, Shevach EM, Strober W, Coico R. (ed), Current protocols in immunology. John Wiley & Sons, New York, NY: [DOI] [PubMed] [Google Scholar]
- 51. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 52. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gloeckner CJ, Boldt K, Schumacher A, Roepman R, Ueffing M. 2007. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7:4228–4234 [DOI] [PubMed] [Google Scholar]
- 54. Gloeckner CJ, Boldt K, Ueffing M. 2009. Strep/FLAG tandem affinity purification (SF-TAP) to study protein interactions. Curr. Protoc. Protein Sci. Chapter 19:19.20.1–19.20.19 [DOI] [PubMed] [Google Scholar]
- 55. Baker A, Cotten M. 1997. Delivery of bacterial artificial chromosomes into mammalian cells with psoralen-inactivated adenovirus carrier. Nucleic Acids Res. 25:1950–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. 2006. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126:995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Penfold ME, Mocarski ES. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46–61 [DOI] [PubMed] [Google Scholar]
- 58. Giesen K, Radsak K, Bogner E. 2000. Targeting of the gene product encoded by ORF UL56 of human cytomegalovirus into viral replication centers. FEBS Lett. 471:215–218 [DOI] [PubMed] [Google Scholar]
- 59. al-Kobaisi MF, Rixon FJ, McDougall I, Preston VG. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388 [DOI] [PubMed] [Google Scholar]
- 60. Reynolds AE, Fan Y, Baines JD. 2000. Characterization of the U(L)33 gene product of herpes simplex virus 1. Virology 266:310–318 [DOI] [PubMed] [Google Scholar]
- 61. Beard PM, Taus NS, Baines JD. 2002. DNA cleavage and packaging proteins encoded by genes U(L)28, U(L)15, and U(L)33 of herpes simplex virus type 1 form a complex in infected cells. J. Virol. 76:4785–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang K, Baines JD. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J. Virol. 80:5733–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang K, Poon AP, Roizman B, Baines JD. 2008. Temperature-sensitive mutations in the putative herpes simplex virus type 1 terminase subunits pUL15 and pUL33 preclude viral DNA cleavage/packaging and interaction with pUL28 at the nonpermissive temperature. J. Virol. 82:487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beilstein F, Higgs MR, Stow ND. 2009. Mutational analysis of the herpes simplex virus type 1 DNA packaging protein UL33. J. Virol. 83:8938–8945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. To A, Bai Y, Shen A, Gong H, Umamoto S, Lu S, Liu F. 2011. Yeast two hybrid analyses reveal novel binary interactions between human cytomegalovirus-encoded virion proteins. PLoS One 6:e17796 doi:10.1371/journal.pone.0017796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee S, Salwinski L, Zhang C, Chu D, Sampankanpanich C, Reyes NA, Vangeloff A, Xing F, Li X, Wu TT, Sahasrabudhe S, Deng H, Lacount DJ, Sun R. 2011. An integrated approach to elucidate the intra-viral and viral-cellular protein interaction networks of a gamma-herpesvirus. PLoS Pathog. 7:e1002297 doi:10.1371/journal.ppat.1002297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang K, Homa F, Baines JD. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 81:6419–6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Giesen K, Radsak K, Bogner E. 2000. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J. Gen. Virol. 81:2231–2244 [DOI] [PubMed] [Google Scholar]
- 71. Beard PM, Baines JD. 2004. The DNA cleavage and packaging protein encoded by the UL33 gene of herpes simplex virus 1 associates with capsids. Virology 324:475–482 [DOI] [PubMed] [Google Scholar]
- 72. Wills E, Scholtes L, Baines JD. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J. Virol. 80:10894–10899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J. Virol. 85:11675–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Perng YC, Qian Z, Fehr AR, Xuan B, Yu D. 2011. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J. Virol. 85:4841–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Qian Z, Leung-Pineda V, Xuan B, Piwnica-Worms H, Yu D. 2010. Human cytomegalovirus protein pUL117 targets the mini-chromosome maintenance complex and suppresses cellular DNA synthesis. PLoS Pathog. 6:e1000814 doi:10.1371/journal.ppat.1000814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tandon R, Mocarski ES. 2011. Cytomegalovirus pUL96 is critical for the stability of pp150-associated nucleocapsids. J. Virol. 85:7129–7141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ruzsics Z, Borst EM, Bosse JB, Brune W, Messerle M. 2013. Manipulating cytomegalovirus genomes by BAC mutagenesis: strategies and applications, p 37–57 In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1 Caister Academic Press, Hethersett, Norwich, United Kingdom [Google Scholar]
- 78. Higgs MR, Preston VG, Stow ND. 2008. The UL15 protein of herpes simplex virus type 1 is necessary for the localization of the UL28 and UL33 proteins to viral DNA replication centres. J. Gen. Virol. 89:1709–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]