

Abstract
Haploinsufficiency of Progranulin (PGRN), a gene encoding a secreted glycoprotein, is a major cause of frontotemporal lobar degeneration with ubiquitin (FTLD-U) positive inclusions. Single nucleotide polymorphisms in the TMEM106B gene were recently discovered as a risk factor for FTLD-U, especially in patients with PGRN mutations. TMEM106B is also associated with cognitive impairment in amyotrophic lateral sclerosis patients. Despite these studies, little is known about TMEM106B at molecular and cellular levels and how TMEM106B contributes to FTLD. Here, we show that TMEM106B is localized in the late endosome/lysosome compartments and TMEM106B levels are regulated by lysosomal activities. Ectopic expression of TMEM106B induces morphologic changes of lysosome compartments and delays the degradation of endocytic cargoes by the endolysosomal pathway. Furthermore, overexpression of TMEM106B correlates with elevated levels of PGRN, possibly by attenuating lysosomal degradation of PGRN. These results shed light on the cellular functions of TMEM106B and the roles of TMEM106B in the pathogenesis of FTLD-U with PGRN mutations.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) is one of the most prevalent forms of early onset dementia, second only to Alzheimer's disease (1,2). Mutations in the Progranulin (PGRN) gene were recently shown to be the major cause of FTLD with ubiquitin-positive inclusions of TDP-43 (FTLD-U) (3–5). Most PGRN mutations result in a decrease in the amount of PGRN expressed or secreted, rather than a gain of toxicity (3,4). Thus, PGRN haploinsufficiency is strongly associated with FTLD-U. PGRN encodes an evolutionarily conserved, secreted glycoprotein of 88 kDa involved in would healing, inflammation, tumorigenesis and neuronal survival (6). A member of the VPS10 family, sortilin, was recently identified as a binding partner for PGRN (7). Sortilin regulates PGRN trafficking by mediating PGRN endocytosis and targeting to lysosomes, thus controlling PGRN levels in the brain (7,8).
However, PGRN mutation carriers show a high variability in age of onset and pathologic presentation, even with identical mutations, suggesting that environmental influences or additional genetic factors modify the disease manifestation (9). Recent genome-wide association studies by several groups have pinpointed TMEM106B, a gene encoding a transmembrane protein of unknown function, as a bona fide risk factor for FTLD-U, especially in patients with PGRN mutations (10–13). TMEM106B is also associated with cognitive impairment in amyotrophic lateral sclerosis patients (14). However, studies with samples from FTLD patients have obtained conflicting results on the effect of TMEM106B polymorphisms on TMEM106B function and on the relationship between TMEM106B and PGRN. Firstly, it is not clear whether TMEM106B mRNA levels are affected by TMEM106B single-nucleotide polymorphisms (SNPs) (11,12). Secondly, it is not known whether the protein function of TMEM106B is altered by SNPs within the coding region of TMEM106B (10,11,13). Studies have identified a T185S coding variant in TMEM106B associated with the protective allele (10,11,13) and suggested T185S to be in perfect linkage disequilibrium with the strongest SNP associated with FTLD-U, rs1990622 (10,13). However, whether and how T185S affects TMEM106B function remains unknown. Finally, it is still under debate whether TMEM106B polymorphisms affect PGRN levels, although the T185S allele was reported to be protective and was correlated with higher plasma PGRN levels (10,11,13). Thus, despite a strong genetic linkage, the role of TMEM106B in FTLD-U and the relationship between TMEM106B and PGRN are not completely understood from these studies (10–17).
TMEM106B encodes a transmembrane protein of unknown function. To address the role of TMEM106B in FTLD-U with PGRN mutations, we investigated cellular functions of TMEM106B. Consistent with recent findings (18,19), we found that TMEM106B is a lysosomal protein and TMEM106B levels are modulated by lysosomal activities. We further showed that increased TMEM106B levels result in the accumulation of enlarged lysosomes and impair the degradation of endocytic cargoes. Exogenous expression of TMEM106B increases PGRN levels, possibly due to its regulation of the endolysosomal pathway.
RESULTS
TMEM106B is localized in late endosome/lysosome compartments
TMEM106B is a predicted type-II transmembrane protein of unknown function. To determine the expression of endogenous TMEM106B, we raised antibodies against the intracellular portion of TMEM106B (a.a. 1–96). The polyclonal antibody specifically detects a 43 kDa band on SDS-PAGE corresponding to endogenous TMEM106B detected in multiple cell lines (Fig. 1A). Because TMEM106B has 274 residues with a predicted molecular weight of 31 kDa, the discrepancy on migration indicates potential post translational modification, likely glycosylation of this protein (18). Consistent with studies by others (19), we found that TMEM106B protein exhibits as an 86 kDa band on SDS-PAGE when the samples are not boiled (data not shown), suggesting the presence of a SDS-resistant dimer. This is further confirmed by the co-immunoprecipitation of GFP-tagged TMEM106B with FLAG-tagged TMEM106B (Fig. 1B). The T185S variant of TMEM106B self-associates to a similar degree as wild-type TMEM106B.
Figure 1.

Expression of TMEM106B in different cell types. (A) Cell lysates from HEK293T, NSC-34 and BV-2 were loaded and blotted with anti-TMEM106B and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies. (B) TMEM106B forms homodimers. FLAG-tagged TMEM106B wild type (WT) and T185S variants were cotransfected with GFP-TMEM106B WT into HEK293T cells. Anti-FLAG antibodies were used to immunoprecipitate FLAG-TMEM106B. IP products were blotted for GFP and FLAG as indicated. (C) TMEM106B expression in rat cortical neurons. E17 rat cortical neurons were isolated and allowed to differentiate for indicated days. Cell lysates were prepared and blotted for sortilin, TMEM106B and GAPDH. Cortical neurons were treated with 2 µm Ara-C after DIV6 to inhibit the growth of glial cells. (D) N2A cells were treated with 5 mm 3-MA, 50 nm Baf1, 15 mm NH4Cl + 100 µm chloroquine or 10 µm MG-132 for 14 h as indicated. Cell lysates were prepared and blotted for TMEM106B and GAPDH. (E) Quantification of data shown in (D), n = 4, ±SEM, * P<0.05, ** P < 0.01 Student's t-test.
With our polyclonal antibodies, we found that TMEM106B is ubiquitously expressed in many different cell types, including neurons and microglia cells, with highest protein levels detected in neuronal cell lines (Fig. 1A). Analysis of TMEM106B expression levels in cortical neurons showed a positive correlation of TMEM106B protein levels with neuronal maturation in vitro (Fig. 1C). To investigate the cellular localization of endogenous TMEM106B, we stained neuroblastoma N2A cells with our polyclonal antibodies against TMEM106B. We found that a large pool of TMEM106B is localized in intracellular vesicles (Fig. 2A). The knockdown of TMEM106B expression by siRNA abolished this vesicular staining, confirming the specificity of our antibody (Fig. 2A). To determine the identities of these intracellular vesicles, we examined the colocalization of N-terminal FLAG-tagged TMEM106B with GFP-tagged Rab GTPases. TMEM106B shows strong colocalization with late endosome and lysosome markers Rab7 and Rab9, with little colocalization with the early endosome marker Rab5 and the recycling endosome marker Rab11, suggesting that TMEM106B mainly localizes to late endosomes and lysosomes (Supplementary Material, Fig. S1A). FLAG-TMEM106B also shows strong colocalization with LAMP1, a transmembrane protein localized mainly on the lysosomes (Supplementary Material, Fig. S1B). We further confirmed this with colocalization of endogenous TMEM106B with LAMP1 in N2A cells (Fig. 2A) and cortical neurons (Fig. 2B). Endogenous TMEM106B in N2A cells shows much better colocalization with LAMP1 than overexpressed TMEM106B (Figs 2; Supplementary Material, Fig. S1), suggesting that TMEM106B overexpression might cause mislocalization of the protein or disturbance of lysosomal compartments. In particular, endogenous TMEM106B usually appeared as a granular, but relatively evenly distributed coat on the lysosomal limiting membrane with one to several discrete TMEM106B puncta polarized on the lysosome surface occasionally. When TMEM106B was overexpressed, we saw a preponderance of this polarized/punctate localization pattern when compared with controls. Overexpressed TMEM106B also frequently appeared within the lumen of the LAMP1-positive vesicles. These data strongly indicate that TMEM106B localization is critically affected by its expression levels. A small pool of TMEM106B was also detected at the plasma membrane when overexpressed. Live cell staining with external antibodies against the myc epitope tag, which is inserted at the C-terminus of TMEM106B, confirms that TMEM106B is a type-II transmembrane protein with its C-terminus facing extracellularly or to topologically equivalent luminal spaces (Supplementary Material, Fig. S2).
Figure 2.
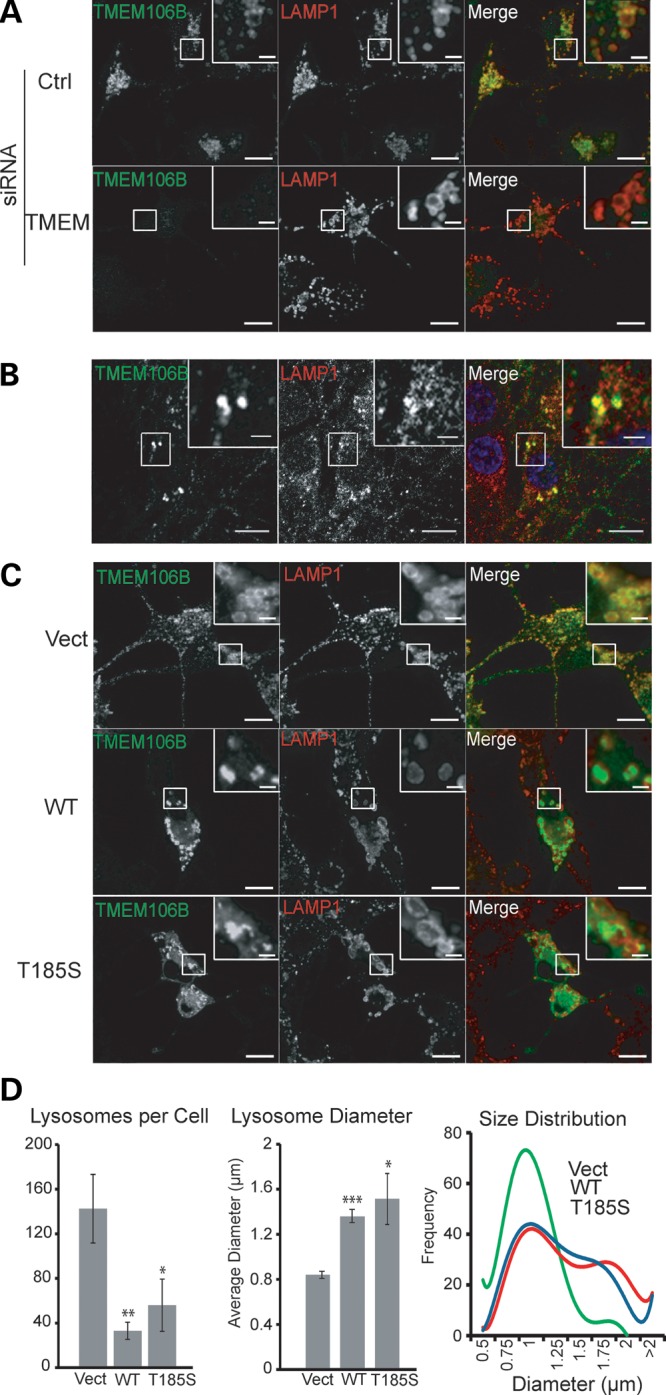
TMEM106B localizes to late endosomes/lysosomes. (A) Colocalization of endogenous TMEM106B with LAMP1-positive vesicles. N2A cells were fixed and stained with polyclonal anti-TMEM106B and monoclonal anti-LAMP1 antibodies. siRNA knockdown of TMEM106B confirms the specificity of our anti-TMEM106B antibody. (B) Colocalization of endogenous TMEM106B with LAMP1 in DIV15 cortical neurons. (C) Overexpression of TMEM106B induces enlarged LAMP1-positive vesicles. Vector control, pCMV-TMEM106B WT and T185S transfected N2A cells were stained with anti-TMEM106B and anti-LAMP1 antibodies. Green fluorescence exposure time was reduced for cells overexpressing TMEM106B to highlight differences in expression levels when compared with nearby non-transfected cells. Scale bars: 10 µm (2 µm in the inset). (D) Quantification of average number of lysosomes per cell and mean lysosome diameter ±SEM along with best fit curves of lysosome size histograms from cells imaged in (C). A minimum of 200 lysosomes were measured from six randomly selected cells in each condition. * P < 0.05, ** P < 0.01, ***P < 0.001 Student's t-test.
Because TMEM106B is mainly localized on lysosomes, we further investigated whether protein levels of TMEM106B are regulated by lysosomal activities. Treatment with inhibitors of lysosomal acidification, such as bafilomycin (Baf1), ammonium chloride or chloroquine, leads to a significant increase in TMEM106B levels in N2A cells, suggesting that TMEM106B levels are regulated by the lysosomal degradation pathway (Fig. 1D and E). Treatment with 3-methyladenine (3-MA), an inhibitor of VPS34, a PI3K involved in autophagy and formation of multivesicular bodies (20), also increases TMEM106B levels (Fig. 1D and E). This indicates that TMEM106B levels may be regulated by membrane-trafficking events. On the other hand, treatment with the proteasome inhibitor MG-132 had minimal effects on TMEM106B levels, suggesting that the ubiquitin-proteosomal pathway is not a major regulator of TMEM106B protein levels.
Increased TMEM106B levels induce enlarged lysosomes
Close examination of lysosome morphology in TMEM106B overexpressing N2A cells revealed an enlargement of lysosomes and a reduction in lysosome numbers (Fig. 2C and D). Examination of lysosomal morphology upon TMEM106B overexpression in other cell lines, including HEK293T, COS-7, T98G and motor neuron cell line NSC-34, indicated an enhanced sensitivity of lysosomes to increased TMEM106B levels in neurons, although lysosomal enlargement was also occasionally seen in non-neuronal cell lines (Supplementary Material, Fig. S3 and data not shown). Abnormal morphology was also seen in some of the enlarged lysosomes, some of which are reminiscent of intermediates of lysosomal fission or fusion (Fig. 2C). This phenotype is also seen with GFP-TMEM106B overexpression, which tends to give higher expression levels and results in large vacuoles positive for LAMP1, but negative for the early endosome marker EEA1 (Fig. 3). These vacuoles are prominent under regular phase microscope in GFP-TMEM106B overexpressing N2A cells and are present in N2A cells overexpressing untagged TMEM106B when treated with 3-MA (Supplementary Material, Fig. S4). We failed to detect any significant differences between wild type and the T185S variant of TMEM106B in inducing abnormalities in lysosomal morphology (Fig. 2 and Supplementary Material, Fig. S4).
Figure 3.

GFP-TMEM106B overexpression results in LAMP1-positive vacuoles in N2A cells. (A) GFP-TMEM106B expression in N2A cells results in enlarged vacuoles that are rimmed by TMEM106B and LAMP1. (B) GFP-TMEM106B-induced vacuoles are EEA1 negative. (C) Colocalization of PGRN with GFP-TMEM106B in some of the vacuoles. Scale bar: 10 µm (2 µm in the inset).
To further confirm the origin of these membranes, we preloaded lysosomes with dextran and then transfected the cells with TMEM106B constructs. It has been shown that preloaded dextran accumulates in lysosomes (21). We found that many of the enlarged vesicles induced by TMEM106B overexpression are positive for dextran (Fig. 4A), confirming that these are lysosome derived. Although TMEM106B overexpression induces lysosome enlargement, it does not induce apoptosis or TDP-43 mislocalization or cleavage (Supplementary Material, Fig. S5).
Figure 4.

The fluid-phase marker, dextran, accumulates in TMEM106B-positive vesicles. (A) N2A cells preloaded with dextran were transfected with TMEM106B and GFP-TMEM106B. Cells were fixed and stained for 24 h post transfection. Untagged and GFP-TMEM106B can be seen on the surface of dextran-containing vesicles. (B) Dextran-labeled endosomes are capable of fusion with TMEM106B enlarged lysosomes. N2A cells were transfected with vector control, TMEM106B and GFP-TMEM106B. After 28 h, cells were loaded with dextran for 16 h, washed and chased 4 h in growth medium. Cells were fixed and stained 48 h post transfection. Scale bars: 10 µm (2 µm in the inset).
TMEM106B overexpression impairs endolysosomal degradation
To investigate whether the abnormal morphology of enlarged late endosomes/lysosomes induced by TMEM106B impairs endolysosomal function, the cellular turnover rate of epidermal growth factor receptor (EGFR) was analyzed. T98G cells, which express high levels of endogenous EGFR, were treated with epidermal growth factor (EGF). EGF stimulation leads to the activation of EGFR and phosphorylation of ERK1/2. EGFR signaling continues after EGFR endocytosis, until the receptors are inwardly budded into intraluminal vesicles in multivesicular bodies (MVBs). EGFR is subsequently degraded through lysosomal fusion and degradation (22). We found that GFP-TMEM106B expression appears to attenuate the rate of EGFR degradation in T98G cells. However, EGF downstream signaling, as quantified by the levels of phospho-ERK1/2, is not affected (Fig. 5). These data suggest that TMEM106B may cause a defect in the later stages of late endosome/lysosome fusion or lysosomal degradation rather than early endocytic trafficking steps or MVB formation. Because the enlarged lysosome phenotype induced by TMEM106B overexpression is much more pronounced in neuronal cell lines than in T98G cell, a more severe endolysosomal dysfunction is expected in neuronal cell lines. Unfortunately, we were unable to detect appreciable amounts of endogenous EGFR in N2A cells, making them unsuitable for this assay.
Figure 5.

GFP-TMEM106B overexpression results in defects in EGFR degradation. (A) T98G cells transfected with vector control or GFP-TMEM106B were serum starved and stimulated with EGF in the presence of cycloheximide for indicated times. Levels of EGFR, phosphorylated ERK1/2 and GAPDH were quantified by western blots. A representative image of three experiments is shown. (B) Quantification of EGFR levels relative to loading control for experiment in (A). n = ±SEM.
To determine whether TMEM106B affects endosome/lysosome fusion, we incubated N2A cells overexpressing TMEM106B with the fluid-phase marker, dextran. Cells were then washed with PBS and incubated for additional 4 h in culture medium without dextran. Dextran signal is present in TMEM106B enlarged lysosomes to a similar extent as in control cells, suggesting that there are no major defects in fusion between TMEM106B-induced enlarged lysosomes and incoming endosomes (Fig. 4B).
TMEM106B modulates PGRN protein levels
Our previous published results have shown that sortilin mediates PGRN trafficking into lysosomes and plays a critical role in regulating PGRN levels (7). We hypothesized that TMEM106B may play a role in regulating PGRN levels by affecting lysosomal activities. To address this hypothesis, we measured intracellular and secreted PGRN levels in cells overexpressing the wild type or T185S allele of TMEM106B. Both wild-type and T185S alleles of TMEM106B increased endogenous PGRN levels in N2A cells (Fig. 6A–C). These changes in PGRN levels are not due to changes in PGRN mRNA levels as measured by qPCR (Fig. 6C), suggesting that TMEM106B regulates PGRN levels through post transcriptional mechanisms. Sortilin expression levels do not appear to be affected by TMEM106B (Fig. 6A), indicating it is unlikely that TMEM106B regulates PGRN levels through sortilin. Furthermore, in cells overexpressing GFP-TMEM106B, accumulation of endogenous PGRN, along with the prototypical lysosomal proteinase cathepsin D, can be detected in some of the GFP-TMEM106B-positive vacuoles (Fig. 3C). Together, these results suggest that TMEM106B may regulate PGRN levels through its function in the endolysosomal degradation pathway.
Figure 6.

Regulation of PGRN levels by TMEM106B. (A) Western blot analysis of N2A cells overexpressing TMEM106B WT and T185S. Transfected cells were changed to serum-free medium 24 h after transfection. After another 24 h, lysates and CM were collected. CM were further concentrated using TCA precipitation. (B) Western blot analysis of N2A cells transfected with control siRNA pool or siRNA pools against TMEM106B and sortilin. Transfected cells were changed to serum-free medium 48 h after siRNA transfection. After another 24 h, lysates and CM were collected. CM were further concentrated using TCA precipitation. (C) Overexpression of TMEM106B in N2A cells leads to increased intracellular and secreted PGRN levels as measured by western blot or ELISA, respectively (n = 5). PGRN levels were normalized to the mean of two vector transfected controls. No change in PGRN mRNA levels was detected via qPCR (n = 3). (D) Knockdown of TMEM106B in N2A cells has no effect on intracellular or secreted PGRN levels as measured by western blot or ELISA, respectively (n = 6). Sortilin knockdown leads to increased levels of PGRN in the media. * P < 0.05, **P < 0.01, Student's t-test.
We also examined the effect of TMEM106B loss of function on PGRN levels by knocking down TMEM106B expression in N2A using siRNA. siRNA treatment resulted in ∼50–70% reduction of TMEM106B expression (Fig. 6B). To test the validity of our assay, we knocked down sortilin expression using siRNA as a positive control and observed an increase in secreted PGRN (Fig. 6B and D). However, TMEM106B knockdown did not appear to affect PGRN levels (Fig. 6D). Furthermore, reduced TMEM106B expression does not appear to affect lysosomal size or morphology (Fig. 2A). Thus, either residual TMEM106B expression is enough to maintain its function in the lysosomes or TMEM106B loss of function does not directly affect lysosomal morphology or function.
DISCUSSION
Endolysosomal function is essential for the health of neurons. Several genetic mutations found in FTLD, including those in CHMP2B and VCP/p97, result in the accumulation of enlarged vacuoles and a defect in endolysosomal trafficking or autophagosome maturation (23–26). Mutations in the PGRN gene are the main cause for FTLD-U (3–5). PGRN has also been implicated in regulating lysosome functions and is transcriptionally co-regulated with a number of essential lysosomal genes (27). PGRN knockout mice accumulate lysosomal byproducts, lipofuscin (28), and homozygous PGRN mutant human patients exhibit neuronal ceroid lipofuscinosis (29). In this study, we showed that the FTLD-U risk factor TMEM106B is highly expressed in neurons, mainly localized to late endosome/lysosome compartments and regulates lysosomal morphology (Fig. 2). Overexpression of TMEM106B results in the accumulation of enlarged lysosomes (Figs 2–4) and delays the degradation of endocytic cargoes such as EGFR (Fig. 5). TMEM106B does not affect the termination of EGFR signaling (Fig. 5), suggesting that TMEM106B does not cause defects in membrane invagination into multivesicular bodies. We speculate that TMEM106B may regulate the fusion of late endosomes with lysosomes or the fission of the hybrid organelle after endosome–lysosome fusion. Our examination of endosome–lysosome fusion events using dextran labeling suggested that TMEM106B-induced enlarged lysosomes are still capable of fusing with the incoming endosomes (Fig. 4). However, it is still possible that TMEM106B affects the kinetics of endosome–lysosome fusion, which requires detailed analysis of endosome–lysosome fusion using time lapse imaging.
It is still under debate whether TMEM106B polymorphisms result in changes in TMEM106B levels or changes in TMEM106B protein, as one variant, T185S, has been reported to be protective and to be in perfect linkage disequilibrium with the strongest SNP associated with FTLD-U (10–17). We find no differences between the wild type (WT) and T185S variant of TMEM106B in regards to regulating lysosomal morphology and number or PGRN levels. Our results are consistent with a model in which increased expression of TMEM106B perturbs the endolysosomal pathway. This is corroborated by findings of increased TMEM106B mRNA and protein levels in postmortem brain samples of FTLD-U patients (12,19). Surprisingly, reduced expression of TMEM106B by RNAi treatment does not have any obvious effect on lysosomal morphology. It is possible that residual TMEM106B is sufficient to maintain its function in the lysosomes or that TMEM106B is not essential for lysosomal function. Another possibility is that TMEM106B is functionally redundant and may be compensated by other genes such as TMEM106A and TMEM106C. A complete depletion of TMEM106B function using a mouse knockout model might be needed to determine TMEM106B function in vivo.
PGRN haploinsufficiency is strongly associated with FTLD-U. However, it is still under debate whether TMEM106B SNPs affect PGRN levels. Previous studies with FTLD patient samples resulted in contradictory conclusions on the regulation of PGRN by TMEM106B (10–13). Here, we show that exogenous expression of TMEM106B increases PGRN levels (Fig. 6) and results in the accumulation of PGRN in the lysosomes (Fig. 4C). Our results indicate that elevated TMEM106B levels could alter plasma PGRN levels in human patients, possibly due to its effect on endolysosomal trafficking. While the majority of the increased PGRN observed appears to be intracellular, likely as a result of abnormal lysosomal degradation, we do detect a modest increase in the levels of extracellular PGRN as measured by ELISA. One potential mechanism to explain this increase in PGRN levels is through an overflow of the increased intracellular PGRN into the extracellular space via lysosomal exocytosis, a well-described process in which lysosomes fuse with the plasma membrane and release their lumenal contents.
In summary, our molecular and cellular characterization of TMEM106B supports a role of TMEM106B in regulating lysosomal morphology and function. Together with a function of PGRN in lysosomes, as suggested by other studies, these results strongly argue for a critical role of lysosomal dysfunction in the progression of FTLD-U. Further studies to monitor lysosomal dynamics with live imaging, to examine the detailed morphology of TMEM106B-induced vacuoles with electron microscopy and to identify TMEM106B-binding partners will give us more insights into the function of TMEM106B in lysosomes. Examination of TMEM106B-induced lysosomal dysfunction in PGRN deficiency background will help illustrate the interaction of TMEM106B and PGRN in regulating lysosomal functions in FTLD-U.
MATERIALS AND METHODS
Cell culture procedures
T98G, N2A, HEK293T (from ATCC), NSC34 (30) and BV-2 (31) cells were grown in DMEM supplemented with 10% FBS, 1% Penicillin–Streptomycin at 37°C in a 5% CO2 atmosphere. Cells were transiently transfected with polyethyleneimine as described (32). E17 rat cortical neurons were isolated and cultured as described (7).
Plasmids and siRNAs
Human TMEM106B cDNA was obtained from Open Biosystems (the ORFome collection, T185; in the pCMV-Sport6, S185). FLAG-tagged TMEM106B constructs were generated by cloning TMEM106B into p3XFLAG-CMV7.1 vector (Sigma-Aldrich) using the enzymes HindIII and SalI. TMEM106B-myc was generated by cloning TMEM106B into pcDNA3.1myc his A vector (Invitrogen) using the enzymes HindIII and XhoI. GFP-TMEM106B was generated by cloning TMEM106B into pEGFP-C3 vector (Clontech) using the enzymes HindIII and XhoI/SalI. GFP -tagged Rab plasmids were generous gifts from Drs Bill Brown and Volker Vogt. PGRN and sortilin constructs were obtained as described (7). siRNA against Mouse TMEM106B (D-042561-01,-02,-03,-04), mouse SORT1 (D-041713-01,-02,-03,-04) and pooled control siRNAs (D-001206-13-05) were obtained from Dharmacon and transfected into N2A cells using DharmaFECT according to the manufacturer's instructions.
Antibodies and chemicals
The following antibodies were used in the study: sheep anti-mouse PGRN antibodies, goat anti-human PGRN antibodies and goat anti-mouse sortilin from R&D systems; mouse anti-myc (9E10) and anti-FLAG (M2) antibodies from Sigma-Aldrich; mouse anti-LAMP1 and EEA1 antibodies from BD biosciences; rabbit anti-EGFR, rabbit anti-phospho-Erk1/2 and rabbit anti-cleaved caspase 3 antibodies from Cell Signaling; rat anti-mouse LAMP1 (1D4B) antibodies from BioLegend; rabbit anti-TDP 43 antibodies from Proteintech; and mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies from Millipore. Rabbit anti-cathepsin D antibodies were a generous gift from Dr Bill Brown.
To generate anti-TMEM106B antibodies, the intracellular domain of TMEM106B (residues 1–96) was expressed and purified as glutathione S-transferase fusion proteins from E. coli. Recombinant proteins were sent to Covance laboratories to generate rabbit polyclonal antibodies. Serum from the final bleed was diluted 1:2000 for western blot analysis and 1:250 for immunostaining. Antibody specificity was confirmed via western blot and immunofluorescence by observing overlapping signals between FLAG and TMEM106B antibodies for overexpressed FLAG-TMEM106B and by decreased signals from cells treated with siRNAs against TMEM106B. The antibody does not cross-react with the TMEM106B family members TMEM106A and TMEM106C (data not shown).
The following reagents were used in this study: 3-MA, chloroquine, ammonium chloride, cycloheximide and staurosporine from Sigma-Aldrich; recombinant human EGF from Promega; puromycin from Calbiochem; trichloroacetic acid (TCA) from Acros Organics; MG-132 from Cayman chemical; and Baf1 from LC Laboratories.
Immunofluorescence microscopy
Cells were plated on coverslips the day prior to transfection. Cells were fixed with 3.7% formaldehyde in PBS 24 or 48 h post transfection and washed with PBS, followed by permeabilization and blocked with PBS containing 3% BSA, 0.1% Triton X-100 or 0.05% Saponin. Cells were stained with primary antibody in 1% BSA PBS blocking buffer overnight at 4°C. Cells were washed three times with PBS and incubated for 2 h at room temperature with a 1:500 mixture of Hoechst and secondary antibody (donkey-anti-rabbit/mouse IgG Alexa Fluor488 and 594, donkey-anti-rat AlexaFluor568 and donkey anti-sheep AlexaFluor680 Molecular Probes). For dextran-loading experiments, Texas Red-labeled dextran (70 000 MW, Lysine Fixable, Molecular probes) was used. Cells were either preloaded for 16 h with 0.5 µg/ml dextran, washed and chased 4 h in growth medium to label lysosomes, or cells were transfected and then loaded and chased. Cells were washed three times with PBS and mounted with Fluoromount G (Southern Biotech), then analyzed with a CSU-X spinning disc confocal microscope (Intelligent Imaging Innovations) with an HQ2 CCD camera (Photometrics) or a Zeiss LSM700 confocal microscope with a transmission photomultiplier detector. All confocal pictures were obtained through 63× or 100× objectives.
Lysosomal scoring and vacuolization assays
For lysosomal scoring, 0.56 µm confocal slices from N2A cells transfected with either vector control, WT or the T185S allele of TMEM106B were acquired. Slices were obtained through cells to yield the maximum number of lysosomes per field of view. Discrete LAMP1, positive vesicles between 0.3 and 4.0 µm were counted for each cell and measured through the widest part of the vesicle. Large swollen vacuoles greater than 4.0 µm were not included in these analyses. Histograms were generated using 200 randomly selected lysosomes from each of these datasets. Analysis was performed with Slidebook software (3I).
For the vacuolization assay, N2A cells were transfected with indicated constructs and treated 24 h later with any chemicals indicated. Cells were photographed with an ImagXpress Micro (Molecular Devices) at 20× and >200 cells counted per trial by an observer blind to the conditions. Cells were scored as positive by the presence of enlarged (>3 µm) phase-lucent vacuoles.
EGFR degradation assay
T98G cells were transfected with indicated constructs. Cells were washed with PBS and media replaced with serum-free DMEM after 24 h. After 24 h in serum-free media, cells were stimulated with 100 ng/ml recombinant human EGF in the presence of 25 µg/ml cycloheximide to inhibit further protein synthesis and cells lyzed at indicated time points for western blot analysis.
Western blot analysis
Protein samples in SDS sample buffer containing β-mercaptoethanol were kept on ice or boiled for 2 min. Samples were run on 12% polyacrylamide gels and transferred to Immobilon-FL polyvinylidene fluoride membranes (Millipore). Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for 1 h, followed by incubation with primary antibodies overnight at 4°C. Membranes were washed for 5 min three times in Tris-Buffered Saline with 0.1% Tween-20 (TBS-T), incubated with secondary antibody for 2 h at room temperature and washed three more times with TBS-T. Blots were imaged and quantified using an Odyssey Infrared Imaging System (LI-COR Biosciences). For quantitation, all immunoreactive bands were normalized to a corresponding GAPDH reference band.
Immunoprecipitation
Cells were lyzed in 50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1%Triton, 0.1% sodium deoxycholate and proteinase and phosphatase inhibitors (Roche). Mouse anti-FLAG conjugated beads (Sigma) were allowed to immunoprecipitate FLAG-TMEM106B in the cell lysates. These beads were incubated with cell lysates for 4 h at 4°C and washed four times with the lysis buffer.
TCA precipitation of conditioned medium
One day after transfection, N2A cells were grown in serum-free medium. Conditioned media (CM) were collected 24 h later and centrifuged at 10 000 rcf for 10 min to remove the cell debris. The cleared media were incubated with 10% TCA at 4°C. Protein pellets were collected by centrifugation, washed with acetone and dissolved in SDS sample buffer prior to western blot to determine PGRN levels.
ELISA
PGRN levels were measured in N2A cells with TMEM106B overexpressed or knocked down with siRNAs. Cells were washed 48 h post transfection and incubated for 24 h in serum-free DMEM. CM were cleared at 10 000 rcf for 10 min and subjected to a Mouse PGRN ELISA (AdipoGen), according to the manufacturer's instructions. PGRN concentrations were converted to percentage of control to correct for interassay variability. Corresponding cell lysates from these samples were collected in RIPA buffer for western blot analysis.
RT-PCR
RNA was purified from cells 48 h after transfection using TRIzol Reagent (Invitrogen). Two micrograms of total RNA was reverse transcribed using a poly(T) primer and SuperScript III Reverse Transcriptase (Invitrogen). qPCR was performed on a LightCycler 480 (Roche Applied Science), and transcript levels were calculated using efficiency-adjusted ΔΔ-CT. All transcripts were normalized to the geometric mean of two reference genes, Tbp and Actb. The mouse Grn primer pair sequences were 5′AGTTCGAATGTCCTGACTCCGCCA3′ and 5′AAGCCACTGCCCTGTTGGTCCTTT3′. Mouse Tbp primers were 5′CCCCACAACTCTTCCATTCT3′ and 5′GCAGGAGTGATAGGGGTCAT3′ and mouse Actb primers were 5′ACGAGGCCCAGAGCAAGAG3′ and 5′TCTCCAAGTCGTCCCAGTTG3′.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by funding to F.H. from Weill Institute for Cell and Molecular Biology and from the Association of Frontotemporal Dementia (AFTD), Alzheimer's Association and NIH (R21 NS081357-01). O.A.B. was partially supported by the NIH training grant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Bill Brown, Volker Vogt, Haiyuan Yu for their generous gifts of plasmids and antibodies, Mrs Xiaochun Wu for technical assistance, Dr Scott Emr and Tony Bretscher for helpful discussions and Dr Yuxin Mao for critical reading of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Neary D., Snowden J., Mann D. Frontotemporal dementia. Lancet Neurol. 2005;4:771–780. doi: 10.1016/S1474-4422(05)70223-4. [DOI] [PubMed] [Google Scholar]
- 2.Ratnavalli E., Brayne C., Dawson K., Hodges J.R. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 3.Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S., et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 4.Cruts M., Gijselinck I., van der Zee J., Engelborghs S., Wils H., Pirici D., Rademakers R., Vandenberghe R., Dermaut B., Martin J.J., et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 5.Gass J., Cannon A., Mackenzie I.R., Boeve B., Baker M., Adamson J., Crook R., Melquist S., Kuntz K., Petersen R., et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 6.Bateman A., Bennett H.P. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- 7.Hu F., Padukkavidana T., Vaegter C.B., Brady O.A., Zheng Y., Mackenzie I.R., Feldman H.H., Nykjaer A., Strittmatter S.M. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrasquillo M.M., Nicholson A.M., Finch N., Gibbs J.R., Baker M., Rutherford N.J., Hunter T.A., DeJesus-Hernandez M., Bisceglio G.D., Mackenzie I.R., et al. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am. J. Hum. Genet. 2010;87:890–897. doi: 10.1016/j.ajhg.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Deerlin V.M., Wood E.M., Moore P., Yuan W., Forman M.S., Clark C.M., Neumann M., Kwong L.K., Trojanowski J.Q., Lee V.M., et al. Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch. Neurol. 2007;64:1148–1153. doi: 10.1001/archneur.64.8.1148. [DOI] [PubMed] [Google Scholar]
- 10.Cruchaga C., Graff C., Chiang H.H., Wang J., Hinrichs A.L., Spiegel N., Bertelsen S., Mayo K., Norton J.B., Morris J.C., et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 2011;68:581–586. doi: 10.1001/archneurol.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finch N., Carrasquillo M.M., Baker M., Rutherford N.J., Coppola G., Dejesus-Hernandez M., Crook R., Hunter T., Ghidoni R., Benussi L., et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Deerlin V.M., Sleiman P.M., Martinez-Lage M., Chen-Plotkin A., Wang L.S., Graff-Radford N.R., Dickson D.W., Rademakers R., Boeve B.F., Grossman M., et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Zee J., Van Langenhove T., Kleinberger G., Sleegers K., Engelborghs S., Vandenberghe R., Santens P., Van den Broeck M., Joris G., Brys J., et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134:808–815. doi: 10.1093/brain/awr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vass R., Ashbridge E., Geser F., Hu W.T., Grossman M., Clay-Falcone D., Elman L., McCluskey L., Lee V.M., Van Deerlin V.M., et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta. Neuropathol. 2011;121:373–380. doi: 10.1007/s00401-010-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rollinson S., Mead S., Snowden J., Richardson A., Rohrer J., Halliwell N., Usher S., Neary D., Mann D., Hardy J., et al. Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol. Aging. 2011;32:e751–e757. doi: 10.1016/j.neurobiolaging.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 16.van der Zee J., Van Broeckhoven C. TMEM106B a novel risk factor for frontotemporal lobar degeneration. J. Mol. Neurosci. 2011;45:516–521. doi: 10.1007/s12031-011-9555-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood H.B. TMEM106B is a susceptibility locus for Ftld. Nat. Rev. Neurol. 2010;6:184. doi: 10.1038/nrneurol.2010.22. [DOI] [PubMed] [Google Scholar]
- 18.Lang C.M., Fellerer K., Schwenk B.M., Kuhn P.H., Kremmer E., Edbauer D., Capell A., Haass C. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 2012;287:19355–19365. doi: 10.1074/jbc.M112.365098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen-Plotkin A.S., Unger T.L., Gallagher M.D., Bill E., Kwong L.K., Volpicelli-Daley L., Busch J.I., Akle S., Grossman M., Van Deerlin V., et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 2012;32:11213–11227. doi: 10.1523/JNEUROSCI.0521-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backer J.M. The regulation and function of class III PI3Ks: novel roles for Vps34. Biochem. J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 21.Ohkuma S. Use of fluorescein isothiocyanate-dextran to measure proton pumping in lysosomes and related organelles. Methods Enzymol. 1989;174:131–154. doi: 10.1016/0076-6879(89)74015-5. [DOI] [PubMed] [Google Scholar]
- 22.Eden E.R., White I.J., Futter C.E. Down-regulation of epidermal growth factor receptor signalling within multivesicular bodies. Biochem. Soc. Trans. 2009;37:173–177. doi: 10.1042/BST0370173. [DOI] [PubMed] [Google Scholar]
- 23.Urwin H., Authier A., Nielsen J.E., Metcalf D., Powell C., Froud K., Malcolm D.S., Holm I., Johannsen P., Brown J., et al. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 2010;19:2228–2238. doi: 10.1093/hmg/ddq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J.H., Ryu H.H., Jun M.H., Jang D.J., Lee J.A. The functional analysis of the CHMP2B missense mutation associated with neurodegenerative diseases in the endo-lysosomal pathway. Biochem. Biophys. Res. Commun. 2012;421:544–549. doi: 10.1016/j.bbrc.2012.04.041. [DOI] [PubMed] [Google Scholar]
- 25.Hirabayashi M., Inoue K., Tanaka K., Nakadate K., Ohsawa Y., Kamei Y., Popiel A.H., Sinohara A., Iwamatsu A., Kimura Y., et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–984. doi: 10.1038/sj.cdd.4400907. [DOI] [PubMed] [Google Scholar]
- 26.Lee J.A., Beigneux A., Ahmad S.T., Young S.G., Gao F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Belcastro V., Siciliano V., Gregoretti F., Mithbaokar P., Dharmalingam G., Berlingieri S., Iorio F., Oliva G., Polishchuck R., Brunetti-Pierri N., et al. Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res. 2011;39:8677–8688. doi: 10.1093/nar/gkr593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed Z., Sheng H., Xu Y.F., Lin W.L., Innes A.E., Gass J., Yu X., Wuertzer C.A., Hou H., Chiba S., et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith K.R., Damiano J., Franceschetti S., Carpenter S., Canafoglia L., Morbin M., Rossi G., Pareyson D., Mole S.E., Staropoli J.F., et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cashman N.R., Durham H.D., Blusztajn J.K., Oda K., Tabira T., Shaw I.T., Dahrouge S., Antel J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 31.Blasi E., Barluzzi R., Bocchini V., Mazzolla R., Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- 32.Vancha A.R., Govindaraju S., Parsa K.V., Jasti M., Gonzalez-Garcia M., Ballestero R.P. Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol. 2004;4:23. doi: 10.1186/1472-6750-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.