

Abstract
Focal facial dermal dysplasia (FFDD) Type IV is a rare syndrome characterized by facial lesions resembling aplasia cutis in a preauricular distribution along the line of fusion of the maxillary and mandibular prominences. To identify the causative gene(s), exome sequencing was performed in a family with two affected siblings. Assuming autosomal recessive inheritance, two novel sequence variants were identified in both siblings in CYP26C1—a duplication of seven base pairs, which was maternally inherited, c.844_851dupCCATGCA, predicting p.Glu284fsX128 and a missense mutation, c.1433G>A, predicting p.Arg478His, that was paternally inherited. The duplication predicted a frameshift mutation that led to a premature stop codon and premature chain termination, whereas the missense mutation was not functional based on its in vitro expression in mammalian cells. The FFDD skin lesions arise along the sites of fusion of the maxillary and mandibular prominences early in facial development, and Cyp26c1 was expressed exactly along the fusion line for these facial prominences in the first branchial arch in mice. Sequencing of four additional, unrelated Type IV FFDD patients and eight Type II or III TWIST2-negative FFDD patients revealed that three of the Type IV patients were homozygous for the duplication, whereas none of the Type II or III patients had CYP26C1 mutations. The seven base pairs duplication was present in 0.3% of healthy controls and 0.3% of patients with other birth defects. These findings suggest that the phenotypic manifestations of FFDD Type IV can be non-penetrant or underascertained. Thus, FFDD Type IV results from the loss of function mutations in CYP26C1.
INTRODUCTION
The focal facial dermal dysplasias (FFDDs) are a group of related developmental defects characterized by bitemporal or preauricular skin lesions resembling aplasia cutis congenita (1). These skin defects occur at the sites of facial fusion during embryogenesis, with temporal lesions situated at the junction between the frontonasal and maxillary facial prominences and preauricular lesions at the meeting point of the maxillary and mandibular prominences (2). The ectodermal lesions show consistent histologic abnormalities: atrophy and flattening of the epidermis, replacement of the dermis by loose connective tissue, reduced levels of fragmented elastic tissue and absence of the subcutaneous tissues and adnexal structures (1–4).
Recently, a revised classification of the FFDDs divided these disorders into four subtypes (1). Individuals with FFDD Type I, termed Brauer syndrome (MIM 136500), have temporal skin depressions that resemble ‘forceps marks’ (5–7). Other facial anomalies, comprising sparse lateral eyebrows, distichiasis and a flattened nasal tip, are usually mild (1,8). FFDD Type II, also known as Brauer–Setleis syndrome, is characterized by bitemporal skin lesions with variable facial findings, including thin and puckered periorbital skin, distichiasis and/or absent eyelashes, upslanting palpebral fissures, a flat nasal bridge with a broad nasal tip, large lips and redundant facial skin (7,9,10). FFDD Types I and II are inherited as autosomal dominant traits (1,5,11). In FFDD Type III or Setleis syndrome (MIM 227260), the same facial features as seen with Brauer–Setleis syndrome are present, but inheritance is autosomal recessive (8,12–14). FFDD Type IV is a new category reserved for a disorder characterized by isolated, preauricular skin lesions with autosomal dominant or recessive inheritance (Table 1; 1,3,4,15).
Table 1.
Summary of physical findings in focal facial dermal dysplasia type IVa
| Reference | Number of families/number of affected | Skin lesions | Other findings |
|---|---|---|---|
| 15 | 1/3 | Preauricular | – |
| 2 | 7/9 | Preauricular | – |
| 46 | 1/1 | Preauricular | – |
| 47 | 1/1 | Preauricular | – |
| 3 | 3/3 | Preauricular | Learning disability (one child) |
| 4 | 1/2 | Preauricular | – |
| This family | 1/2 | Preauricular | Intra-oral polyps (two children) |
aAfter reference (1).
To date, the molecular defect causing FFDD Type III has been identified in several families as loss of function mutations in TWIST2 (1,16,17). However, the gene(s) for the other subtypes remain unknown and genetic heterogeneity is probable. Herein, we report two siblings with FFDD Type IV who had classical, atrophic skin lesions in a preauricular distribution bilaterally. They also had polyps on the buccal mucosa along the same embryologic fusion line between the maxillary and mandibular prominences, a feature that has not been described in FFDD previously. By using exome sequencing to study this family with two affected siblings, two mutations in CYP26C1 were identified, consistent with autosomal recessive inheritance. Subsequent analysis of three additional unrelated Type IV FFDD patients also identified homozygosity for the same CYP26C1 duplication mutation.
RESULTS
Exome sequencing
For the first sibling (patient 1), 49.39 × 106 reads were obtained with 96.6% aligning to the hg19 human reference sequence. Mean coverage of target sequence was 69×, with 77.7% of the 4.3 × 109 base pairs covered on or near Nimblegen's target capture region (data not shown). Of the 49 000 variants that were within the target region, 97.2% were present in dbSNP132 (data not shown). For the second sibling (patient 2), we obtained 26.60 × 106 reads with 97.5% aligning to hg19. Of the 2.4 × 109 base pairs that we covered, 79.6% of them were on or near Nimblegen's targeted capture region, with a mean coverage of 38× for the target region (data not shown). In this study, 97.6% of the 47 000 variants in the target were found in dbSNP132 (data not shown).
We assumed autosomal recessive inheritance and compiled a list of (1) homozygous and (2) compound heterozygous novel sequence variants that were not recorded in dbSNP132 or 1000 Genomes that were shared by both siblings (Supplementary Material, Table S1). We selected CYP26C1 (NM_183374.2) and KRT24 (NM_019016.2) as candidates for Sanger sequencing to determine segregation in the family based on the expression patterns of both of these genes. Cyp26c1 is expressed at the site of the fusion of the maxillary and mandibular prominences during facial development (18), whereas Krt24 is expressed in keratinocytes (19). For KRT24, both sequence variants c.95G>A, predicting p.Arg32Lys and c.1465C>T, predicting p.Gln489X were verified in the affected siblings, but testing of other family members showed that these two sequence alterations were also present in an unaffected sibling (data not shown), thus prompting us to examine CYP26C1.
We verified the two CYP26C1 mutations: c.844_851dupCCATGCA, a 7 bp tandem duplication predicting p.Glu284fsX128 that was maternally inherited and c.1433G>A, predicting p.Arg478His, that was paternally inherited. Neither unaffected sibling had inherited both CYP26C1 mutations (Table 2). No other alterations were present in the remaining CYP26C1 exons in the proposita or her brother (data not shown). The duplication results in a frameshift mutation that predicts the incorporation of 128 different amino acids from wild-type residue 284 and then premature truncation before residue 412. As exons 5 and 6 of CYP26C1 contain three conserved cytochrome P450 domains—ETLR, PERF and the heme-binding domain, the duplication is highly likely to compromise protein function and was predicted to be disease causing (Mutation taster; P = 1.0). Exon 4 of CYP26C1 is 156 bp in length, and it is possible that splicing out of this exon to produce a catalytically intact allele could occur. However, an in silico mutation prediction program (Mutation taster) showed that although the 7 bp duplication increased the strength of the donor splice site for exon 4, it was not predicted to disrupt the normal CYP26C1 gene splicing. For the missense substitution, p.Arg478 His, Polyphen-2 predicted that the substitution was probably damaging (P = 0.959, sensitivity 0.63 and specificity 0.92).
Table 2.
CYP26C1 Sanger sequencing results in this family and other FFDD type IV patients
| Family memberd | Affected | CYP26C1 exon 4 | CYP26C1 exon 6 |
|---|---|---|---|
| I-2 | No | +a/c.844_851dupCCATGCAb | +/+ |
| I-3 | No | +/+ | +/c.1433A>Gc |
| II-1 | No | +/+ | +/+ |
| II-2 | No | +/c.844_851dupCCATGCA | +/+ |
| II-3 | Yes | +/c.844_851dupCCATGCA | +/ c.1433A>G |
| II-4 | Yes | +/c.844_851dupCCATGCA | +/ c.1433A>G |
| II-5 | No | +/c.844_851dupCCATGCA | +/+ |
| Patient 3, this paper (3) | Yes | c.844_851dupCCATGCA/c.844_851dupCCATGCA | +/+ |
| Patient 4, this paper (3) | Yes | c.844_851dupCCATGCA/ c.844_851dupCCATGCA | +/+ |
| Patient 5, this paper | Yes | c.844_851dupCCATGCA/ c.844_851dupCCATGCA | +/+ |
a+Wild type; c.844_851dupCCATGCA.
bc.844_851dupCCATGCA, predicting p.Glu284fsX128; c.1433G>A.
cc.1433A>G, predicting p.Arg478His.
dFor family pedigree information, please see Materials and Methods, exome sequencing.
We sequenced CYP26C1 in 12 additional patients diagnosed with FFDD, of whom 4 had Type IV FFDD and found that two previously reported patients (3) were homozygous for the same c.844_851dupCCATGCA. Parental samples from only one of these patients were available, and both parents were heterozygous for the duplication (data not shown). A further unpublished female with FFDD Type IV, patient 5, was also found to be homozygous for the duplication. No patient with FFDD Type II or III was found to have a CYP26C1 mutation. Although the c.844_851dupCCATGCA duplication was not present in public databases such as dbSNP and 1000 Genomes, sequencing of controls found the duplication in 1/318 control chromosomes from individuals of Caucasian ethnicity (0.4%), 0/200 control chromosomes from individuals of Hispanic ethnicity and 1/236 chromosomes from individuals with eye or diaphragmatic birth defects (0.4%), for an overall frequency of 2/754 chromosomes (0.3%). With this estimated frequency, the probability of three out of the four unrelated FFDD Type IV patients who we studied being homozygous for the same duplication was 7.2810−16. The sequence alteration p.Arg478His was not present in the Exome Variant Server that contains data from more than 10 000 individuals. As animal model studies had suggested functional redundancy between Cyp26a1 and Cyp26c1 (20,21), we verified wild-type CYP26A1 sequence by Sanger sequencing in the proposita and her brother (data not shown). Sequencing of CYP26A1 in the nine FFDD patients without the CYP26C1 duplication was also negative (data not shown).
Studies to assess retinoic acid (RA) metabolic activity of mutations
Transfection studies in an expression system with wild-type CYP26C1, CYP26C1 with c.1433G>A and CYP26C1 with c.844_851dupCCATGCA showed that the mutant constructs had activities similar to untransfected cells or cells transfected with empty vector, implying that both mutations resulted in loss of function (Table 3).
Table 3.
In vitro expression of human wild-type or mutant CYP26C1 constructs in Cos-1 cellsa
| Mean aqueous radioactivity (3H-retinoic acid; counts per minute) | Mean aqueous radioactivity—pCMV6-XL5 (empty vector) (3H-retinoic acid; counts per minute) | Radioactivity as % of CYP26C1/wild type | |
|---|---|---|---|
| pCMV6-XL5 (empty vector) | 3130.28 ± 41.67 | 0 | – |
| CYP26C1 wild type | 10 375.46 ± 8.13 | 7245.18 | 100% |
| CYP26C1/ c.844_851dupCCATGCA | 3421.78 ± 172.96 | 291.5 | 4% |
| CYP26C1/c.1433A>G | 3167.24 ± 122.0 | 36.96 | 0.01% |
aEmpty pCMV6-XL5 vector, CYP26C1 wild type, CYP26C1/ c.844_851dupCCATGCA and CYP26C1/c.1433A>G were transfected into Cos-1 cells. CYP26C1 enzyme activity was determined by the conversion of radiolabeled, released 3H-retinoic acid (counts per minute) as described in the Materials and Methods section. The data in the second column represent the mean ± standard error of the mean of triplicate samples of a representative experiment. The values in the third column have been corrected for background, based on subtraction of the background counts per minute obtained from the transfection of the empty pCMV6-XL5 vector.
Microsatellite marker studies
We examined microsatellite markers to determine if the duplication was present on a founder allele with the same haplotype shared between the FFDD Type IV patients with the duplication. For marker D10S1680, located at chr10:95 501 357–95 701 690 and within 1 Mb of CYP26C1 at chr10:94 821 021-94 828 454 (hg19), the allele sizes did not support inheritance of a common haplotype, as both siblings had allele sizes of 204/214, and the other three unrelated patients homozygous for the CYP26C1 duplication had allele sizes of 206/210 (patient 3), 206/214 (patient 4) and 210/210 (patient 5) (data not shown).
DISCUSSION
Homozygosity or compound heterozygosity for two CYP26C1 mutations were identified in five individuals diagnosed with FFDD Type IV—the proposita and her brother from the family reported here, two published patients (3) and an additional, unpublished affected child from Belgium. Both mutations were shown to cause CYP26C1 loss of function by in vitro expression studies (Table 3). The duplication found in homozygous form in affected patients was present in heterozygous form in apparently normal control chromosomes at a frequency of 0.3%. As FFDD type IV is exceedingly rare, it is possible that this relatively mild facial phenotype is frequently non-penetrant or underascertained. As patients who were homozygous for the duplication all had European ancestry, it was plausible that they shared a common haplotype or that the mutation arose due to a founder effect, but this was not supported by microsatellite marker studies. Genetic heterogeneity is likely for FFDD Type IV, as at least one patient with FFDD Type IV was not found to have a CYP26C1 mutation in this study. We were unable to find any relationship between CYP26C1 and TWIST2, the gene responsible for FFDD Type III.
In Type IV FFDD, developmental disabilities and extracutaneous findings are rare (1), and we do not consider that the mild speech delay exhibited by the proposita's brother was likely to be related to his CYP26C1 mutations. The intraoral polyps in the proposita and her brother have not previously been described, and it is interesting to note that Cyp26c1 expression has been observed in the murine buccopharyngeal membrane at embryonic day (E) 8.5 (18).
Retinoic acid (RA) is the major active derivative of vitamin A (retinol) and is critical for developmental regulation of the hindbrain, spinal cord and eye (22,23). Endogenous RA levels in humans are determined by the interaction of RA-synthesizing enzymes (retinol dehydrogenases and retinaldehyde dehydrogenases) and the trio of RA-degrading enzymes CYP26A1, CYP26B1 and CYP26C1. The Cyp26 enzymes are part of the P450 superfamily and catalyze hydroxylation at the C14 and C18 positions of the β-ionone ring in RA to convert it to less active, more polar metabolites such as 4-OH-RA, 18-OH-RA and 5,8-epoxy-RA (24–26). Cells expressing a Cyp26 enzyme have minimal to complete absence of RA (26). We could not find any information on other potential drug or metabolic targets for Cyp26c1.
The human and murine CYP26C1 genes both contain the canonical heme thiolate-binding motif (FxxGxxxCxG) found in P450 proteins (18). CYP26C1 and CYP26A1 are present in the endoplasmic reticulum (27) and share 43% amino acid identity in humans, whereas CYP26C1 and CYP26B1 share 51% amino acid identity (27). However, Cyp26c1 differs from Cyp26a1 and Cyp26b1 in that it is less sensitive to the inductive effects of RA and relatively resistant to the inhibitory effects of ketoconazole (25,28). The duplication mutation occurs in a position 5′ to the heme thiolate domain and eliminates this domain and its catalytic activity. This loss-of-function was confirmed by in vitro expression of the mutant protein and assay of its catalytic activity. The p.Arg478His mutation occurs within the L-helix domain that is largely conserved across the cytochrome p450 family. Both CYP26A1 and CYP26C1 have an arginine residue at position 478, whereas CYP26B1 has a serine residue. Presumably, the histidine substitution at this position may disrupt the folding or integrity of the adjacent catalytic domain (20).
The embryologic expression of Cyp26c1 has been studied in mouse, chick and zebrafish. At E8.0, murine Cyp26c1 was predominantly expressed in the region of the prospective rhombomeres r2 and r4 and in the rostral portion of the first pharyngeal arch, where expression persisted until E9.5 (18). The first pharyngeal arch later divides into the maxillary and mandibular processes, and Cyp26c1 is expressed along the fusion of these prominences (18; schematized in Supplementary Material, Fig. S1), thus, the expression pattern of Cyp26c1 encompasses the site of the skin lesions in FFDD Type IV. At E9.5, Cyp26c1 expression was observed in the lateral epibranchial placodes and cervical mesenchyme caudal to the otic vesicle and at E10.5, staining was present in the cervical mesenchyme and in a portion of the maxillary component of the first pharyngeal arch (18). The orthologous chick gene was expressed in rhombomeres 1, 2, 3 and 5, the neural crest-derived cephalic mesenchyme in the pre and postotic regions, the pharyngeal endoderm and the ectoderm of the first pharyngeal pouch and the epidermis of the first pharyngeal groove (24). In Danio rerio, Cyp26c1 was initially named as Cyp26d1 and Cyp26b1 like (21) and both genes are CYP26C1 orthologues (29). Expression in zebrafish was initially noted in rhombomeres 2–6 and the first pharyngeal arch before expanding to the eyes, otic vesicles, midbrain, telencephalon, diencephalons and pharyngeal arches (21).
Cyp26c1 homozygous null mice did not manifest overt anatomic abnormalities (30). However, the removal of Cyp26a1 together with Cyp26c1 in double homozygous null mice mimicked the teratogenic defects seen with RA (30,31), suggesting functional redundancy between these genes. Cyp26a1−/− and Cyp26c1−/− mice had microcephaly, hypoplasia of the first and second pharyngeal arches and deficient migration of the cranial neural crest cells with lethality at E9.5 to E10.5 (30). Depleting Danio rerio embryos of Cyp26b1 or Cyp26c1 resulted in a normal phenotype on a wild-type background, but subtle shortening of the hindbrain was observed after targeting both genes on a Cyp26a1 depleted background (29). Thus, the phenotype caused by absence or reduction in Cyp26c1 is mild or unchanged from wild type and there is functional redundancy between Cyp26a1 and Cyp26c1.
The effects of disordered retinoid metabolism on skin development have previously been studied in Cyp26b1−/− mice (32). At E16.5, the periderm of the skin degenerates, and the cornified layers of the epidermis are formed by keratinocyte differentiation in association with specific proteins to replace periderm as the initial skin barrier (32). At E17.5 and E18.5, Cyp26b1−/− mice have reduced thickness of the cornified layer of skin, reduced filaggrin and arrest of hair follicle growth (32). Interestingly, experiments using RA-soaked beads in embryonic chicken skin explant cultures also showed that the beads can create a radial zone of inhibition surrounded by a rim of disoriented buds (33). Skin equivalents treated with RA in vitro show altered epidermal stratification (34), and keratinocyte cultures treated with RA show increased sensitivity to apoptosis with upregulated p53 and caspase-3, -6, -7 and -9 (35). However, precisely which genes are altered by the excess RA in the regions of fusion of the facial prominences remains unknown.
In five patients with FFDD type IV, homozygosity or compound heterozygosity for loss of function mutations (c.844_851dupCCATGCA predicting p.Glu284fsX128 and p.Arg478His) were identified in the CYP26C1 gene. Therefore, it is concluded that mutations in CYP26C1 are responsible for the FFDD Type IV phenotype.
MATERIALS AND METHODS
Subjects
The proposita (patient 1) was a 6-year-old female evaluated in the Dermatology Genetics Clinic because of congenital hypopigmented skin lesions on both cheeks. Following a normal pregnancy and delivery, several white macules extending from the ear down to the lateral commissures of her mouth were noted on each cheek (Fig. 1A and B). The lesions were described as ‘blister-like’ and were reported to fill with fluid daily during the first 3 years of her life without a history of discharge. At 2 years of age, two oral, polypoid lesions measuring 1–2 cm were noted on her left buccal mucosa (Fig. 1C). The remainder of the dermatologic examination was normal, including her eyebrows and eyelashes, with the exception of numerous scattered dark brown nevi. Her developmental milestones were consistent with chronologic age. The proposita's medical history included multiple dental extractions and root canal surgery for dental caries. Her incisor teeth had minor ridging (Fig. 1D). She had a history of diurnal and nocturnal enuresis for which no cause was identified and which resolved spontaneously.
Figure 1.
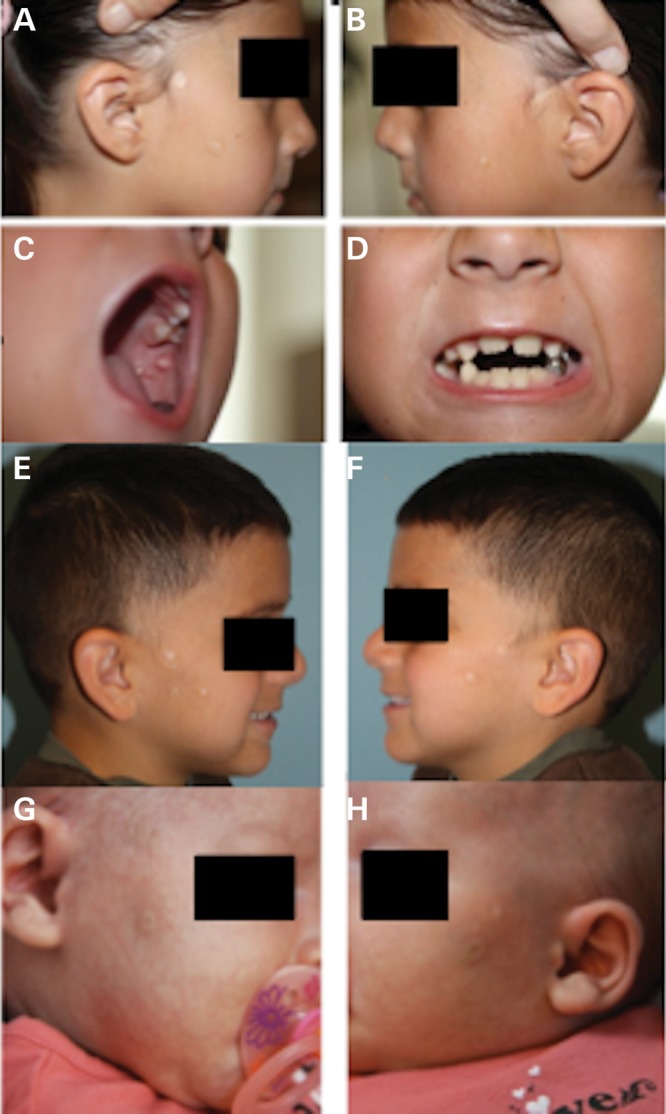
Focal facial dermal dysplasia in the proposita. (A and B). White, shiny, atrophic papules measuring 3–10 mm were present on the right cheek (A) and left cheek (B). (C). Intraoral polyps in the proposita that were seen as small, polypoid lesions measuring 2–3 mm protruding 5–8 mm from the left buccal mucosa. (D). Teeth of the proposita, showing minor dental ridges. (E and F). Focal facial dermal dysplasia in the brother of the proposita. White, shiny, atrophic papules similar to those in the proposita are present on the right cheek (E) and the left cheek (F). (G and H). Facial features of the fifth patient with FFDD Type IV. Atrophic skin lesion on the right (G and left H) cheeks.
The proposita's 4-year-old brother (patient 2) was delivered vaginally at 37 weeks of gestation with a weight of 4220 g (>97th centile) after an uncomplicated pregnancy. He had a history of similar facial lesions that were also reportedly filled with fluid daily for the first year of his life. He walked at 1 year of age and was described as having normal cognition, although he received speech therapy for a brief period because of speech delay and unclear pronunciation. On examination, he had two to four round skin lesions measuring up to 1.7 cm in diameter on both cheeks in a preauricular distribution, and the defects had a small center of cutis aplasia and were bordered by a hyperpigmented rim with long thin fine hairs (Fig. 1E and F). He had sparse hair growth in a small area in front of both ears. He had three small, intraoral polyps measuring less than 3 cm on the left buccal mucosa, a location similar to that seen for these lesions in his sister. The remainder of his physical examination was unremarkable. The siblings were diagnosed with FFDD type IV based on their clinical findings, and no dermal biopsies were performed.
The parents had normal skin and the family history was negative for significant skin findings. The parents had two unaffected daughters aged 8 years and 2 years and the mother had one unaffected daughter from a previous marriage. Maternal ethnicity was English, Nordic and Native American and paternal ethnicity was Mexican. There was no known history of parental consanguinity.
We sequenced CYP26C1 in four unrelated Type IV and eight TWIST2-negative Type II or Type III FFDD patients. Relevant clinical findings in those with Type IV FFDD who were homozygous for CYP26C1 sequence variants follow: the third patient (patient 1) had three, small, fluid-containing skin lesions on each cheek at birth, a right-sided cleft lip, a small scar implying late closure of the left lip and a cutaneous hemangioma of the right palm. Growth and development were unremarkable (3). The fourth patient (patient 2) was noted to have two, preauricular, vesicular lesions on both cheeks at birth. These congenital lesions deflated, but one new lesion erupted on each cheek at 2–3 weeks of life. At 18 months of age, he had moderate to severe developmental delays, right-sided hemiparesis, hydrocephalus with a shunt and focal epileptic seizures that were all attributed to a large, unexplained left-sided intracranial hemorrhage in the prenatal period (3). Microscopy of a biopsied skin lesion from this child revealed fragmentation of elastic fibers with striation and a diffuse increase in dermal mast cells, resulting in a histologic diagnosis of non-reactive deep and superficial elastolysis (3). The parents were not known to be consanguineous, but were from the same region in Norway. The fifth patient (Fig. 1G and H) was born at term after an uncomplicated pregnancy. After delivery, both cheeks were noted to have symmetrical, preauricular, ivory, round to oval patches with an atrophic aspect. There were no other abnormalities, and the skin lesions remained stable after birth. The child was investigated for frequent respiratory infections and was diagnosed with α-antitrypsin deficiency of the ZZ genotype.
Exome sequencing
After obtaining written, informed consent, venous bloods or cheek swabs were sampled from the affected sibling pair with FFDD Type IV (II-3 and II-4), both parents (I-2 and I-3) and their two unaffected full siblings (II-1 and II-5) and unaffected half-sibling (II-1). DNA was extracted from all family members using standard methods. Mutations in TWIST2 were excluded using Sanger sequencing in the affected sibling pair (data not shown). One microgram of DNA from the affected sibling pair only was prepared for exome sequencing. Genomic DNA was sonicated (Covaris S2; Applied Biosystems, Foster City, CA, USA) and assembled into a library with TruSeq adaptors (Illumina, San Diego, CA, USA) containing barcodes that differentiate the libraries in a capture reaction and sequencing run. The two libraries (250 ng each) were pooled into a capture reaction that contained biotinylated DNA oligonucleotides (SeqCap EZ Human Exome Library v3.0; Roche Nimblegen, Madison, WI, USA) for 72 h. The DNA bait–DNA hybrids were then pulled out of the complex mixture by incubation with streptavidin-labeled magnetic beads, and the targeted DNA of interest was eluted and subjected to 18 cycles of DNA amplification prior to sequencing on a HiSeq2000 (Illumina, San Diego, CA, USA) for pair-end 100 cycles. Sequence reads collected from the HiSeq were processed with the Cloud BioLinux-based sequencing data analysis pipeline. Essentially, sequencing reads were aligned to the hg19 reference genome using the Burrows–Wheeler Alignment tool (BWA v0.5.9) using the default parameter that allows two mismatches. Indexing, realignment and duplicate removal were performed using Picard and Samtools. Variant quality score was recalibrated, and variants were subsequently called using Genome Analysis Toolkit v 1.3-21-gcb284ee (36,37). This procedure trained an adaptive error model using known variant sites from HapMap v3.3 and the Omni chip array from the 1000 Genomes Project [for single nucleotide polymorphisms (SNPs)] and the Natural indels (38) for indels to differentiate a true genetic variant from a machine artifact. Variants were filtered with the recommendations listed in the Best Practice Variant Detection with GATK v3 with ‘QD < 2.0’, ‘MQ < 40.0’, ‘FS > 60.0’, ‘HaplotypeScore > 13.0’, ‘MQRankSum <−12.5’, ‘ReadPosRankSum <−8.0’ for SNPs and ‘QD < 2.0’, ‘ReadPosRankSum <−20.0’, ‘InbreedingCoeff <−0.8’, ‘FS > 200.0’ for Indels. The filtered list was then annotated with both ANNOVAR 2012Mar08 version (39) and snpEff v2.0.5 in GRCh37.64 (40) with information from public databases to determine the significance of novel sequence alterations, including the National Heart Lung Blood Institute Exome Sequencing Server, 1000 Genomes and the Database of Single Nucleotide Polymorphisms. The potential deleteriousness of novel sequence variants was assessed using Polyphen-2, Sorting Intolerant from Tolerant and Mutation Taster. We utilized VarSifter (41) to explore the dataset and selected sequence variants were verified using Sanger sequencing (42). We then used Sanger sequencing to interrogate the CYP26C1 and CYP26A1 genes in 12 patients previously diagnosed with FFDD.
Studies to assess RA metabolic activity of mutations
We examined the effect of each CYP26C1 mutation on RA catabolic activity using an in vitro expression system (43). Cos-1 cells were grown in DMEM containing 10% fetal bovine serum and antibiotic-antimycotic (0.2 units/ml penicillin, 0.2 μg/ml streptomycin, 2.5 μg/ml fungizone). The day prior to transfection, cells were seeded in six well plates at 150 000 cells per well. Cells were transfected in triplicate with 1 µg of empty pCMV6-XL5, or vector containing wild-type CYP26C1, CYP26C1/c.1433 G>A or CYP26C1/Dup844_851CCATGCA. Cells were incubated for 48 h prior to analysis of CYP26 activity. To assess RA metabolic activity, cells were washed twice with PBS, trypsinized and reseeded into a 48-well plate in 200 µl of growth medium containing 0.2 µCi/ml 3H-RA. Cells were incubated for 3 h, and the reaction was stopped with the addition of 5 µl of 10% glacial acetic acid. Total lipids were extracted using a modified Bligh-Dyer procedure (44) and 500 µl of the aqueous layer was taken to measure radioactivity. Results were shown as mean counts per minute × standard error of the mean (Table 3).
Microsatellite marker studies
We selected polymorphic microsatellite markers in the region of CYP26C1 using the University of California, Santa Cruz Genome Browser and labeled the forward primer with 5′ HEX (Integrated DNA Technologies, San Diego, CA, USA). The markers were run according to previously published methods (45).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by The Eunice Kennedy Shriver National Institute of Child Health, National Institutes of Health (K08HD053476-01A1 to A.S.).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Andrew Annalora for discussion and thank the family for their help and participation. We are grateful to the Genome Core at the University of California, San Francisco for performing the microsatellite marker studies.
Conflict of Interest statement: The authors do not have any conflicts of interest to declare.
REFERENCES
- 1.Cervantes-Barragán D.E., Villarroel C.E., Medrano-Hernández A., Durán-McKinster C., Bosch-Canto V., Del-Castillo V., Nazarenko I., Yang A., Desnick R.J. Setleis syndrome in Mexican-Nahua sibs due to a homozygous TWIST2 frameshift mutation and partial expression in heterozygotes: review of the focal facial dermal dysplasias and subtype reclassification. J. Med. Genet. 2011;48:716–720. doi: 10.1136/jmedgenet-2011-100251. [DOI] [PubMed] [Google Scholar]
- 2.Drolet B.A., Baselga E., Gosain A.K., Levy M.L., Esterly N.B. Preauricular skin defects: a consequence of a persistent ectodermal groove. Arch. Dermatol. 1997;133:1551–1554. doi: 10.1001/archderm.133.12.1551. [DOI] [PubMed] [Google Scholar]
- 3.Prescott T., Devriendt K., Hamel B., Pasch M.C., Peeters H., Vander Poorten V., Tallerås O. Focal preauricular dermal dysplasia: distinctive congenital lesions with a bilateral and symmetric distribution. Eur. J. Med. Genet. 2006;49:135–139. doi: 10.1016/j.ejmg.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Krathen M.S., Rosenbach M., Yan A.C., Crawford G.H. Focal preauricular dermal dysplasia: report of two cases and a review of literature. Pediatr. Dermatol. 2008;25:344–348. doi: 10.1111/j.1525-1470.2008.00679.x. [DOI] [PubMed] [Google Scholar]
- 5.Brauer A. Hereditärer symmetrischer systematisierter naevus aplasticus bei 38 Personen. Derm. Wschr. 1929;89:1163–1168. [Google Scholar]
- 6.Jensen N.E. Congenital ectodermal dysplasia of the face. Br. J. Derm. 1971;84:410–416. doi: 10.1111/j.1365-2133.1971.tb02524.x. [DOI] [PubMed] [Google Scholar]
- 7.Graul-Neumann L.M., Stieler K.M., Blume-Peytavi U., Tzschach A. Autosomal dominant inheritance in a large family with focal facial dermal dysplasia (Brauer-Setleis syndrome) Am. J. Med. Genet. A. 2009;149A:746–750. doi: 10.1002/ajmg.a.32728. [DOI] [PubMed] [Google Scholar]
- 8.McGaughran J., Aftimos S. Setleis syndrome: three new cases and a review of the literature. Am. J. Med. Genet. 2002;111:376–380. doi: 10.1002/ajmg.10632. [DOI] [PubMed] [Google Scholar]
- 9.Di Lernia V., Neri I., Patrizi A. Focal facial dermal dysplasia: two familial cases. J. Am. Acad. Dermatol. 1991;25:389–391. doi: 10.1016/0190-9622(91)70211-j. [DOI] [PubMed] [Google Scholar]
- 10.Masuno M., Imaizumi K., Makita Y., Nakamura M., Kuroki Y. Autosomal dominant inheritance in Setleis syndrome. Am. J. Med. Genet. 1995;57:57–60. doi: 10.1002/ajmg.1320570113. [DOI] [PubMed] [Google Scholar]
- 11.McGeoch A.H., Reed W.B. Familial focal facial dermal dysplasia. Arch. Dermatol. 1973;107:591–595. [PubMed] [Google Scholar]
- 12.Setleis H., Kramer B., Valcarcel M., Einhorn A.H. Congenital ectodermal dysplasia of the face. Pediatrics. 1963;32:540–548. [PubMed] [Google Scholar]
- 13.al-Gazali L.I., al-Talabani J. Setleis syndrome: autosomal recessive or autosomal dominant inheritance? Clin. Dysmorphol. 1996;5:249–253. [PubMed] [Google Scholar]
- 14.Tay Y.K., Morelli J.G., Weston W.L. Focal facial dermal dysplasia: report of a case with associated cardiac defects. Br. J. Dermatol. 1996;135:607–608. [PubMed] [Google Scholar]
- 15.Kowalski D.C., Fenske N.A. The focal facial dermal dysplasias: report of kindred and a proposed new classification. J. Am. Acad. Dermatol. 1992;27:575–582. doi: 10.1016/0190-9622(92)70225-5. [DOI] [PubMed] [Google Scholar]
- 16.Tukel T., Šošić D., Al-Gazali L.I., Erazo M., Casasnovas J., Franco H.L., Richardson J.A., Olson E.N., Cadilla C.L., Desnick R.J. Homozygous nonsense mutations in TWIST2 cause Setleis syndrome. Am. J. Hum. Genet. 2010;87:289–296. doi: 10.1016/j.ajhg.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franco H.L., Casasnovas J.J., Leon R.G., Friesel R., Ge Y., Desnick R.J., Cadilla C.L. Nonsense mutations of the bHLH transcription factor TWIST2 found in Setleis syndrome patients cause dysregulation of periostin. Int. J. Biochem. Cell Biol. 2011;43:1523–1531. doi: 10.1016/j.biocel.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahayato A., Dollé P., Petkovich M. Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine development. Gene Expr. Patterns. 2003;3:449–454. doi: 10.1016/s1567-133x(03)00066-8. [DOI] [PubMed] [Google Scholar]
- 19.Sprecher E., Itin P., Whittock N.V., McGrath J.A., Meyer R., DiGiovanna J.J., Bale S.J., Uitto J., Richard G. Refined mapping of Naegeli-Franceschetti- Jadassohn syndrome to a 6 cM interval on chromosome 17q11.2-q21 and investigation of candidate genes. J. Invest. Dermatol. 2002;119:692–698. doi: 10.1046/j.1523-1747.2002.01855.x. [DOI] [PubMed] [Google Scholar]
- 20.Annalora A.J., Goodin D.B., Hong W.X., Zhang Q., Johnson E.F., Stout C.D. Crystal structure of CYP24A1, a mitochondrial cytochrome P450 involved in vitamin D metabolism. J. Mol. Biol. 2010;396:441–451. doi: 10.1016/j.jmb.2009.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu X., Xu F., Wang X., Gao X., Zhao Q. Molecular cloning and expression of a novel CYP26 gene (cyp26d1) during zebrafish early development. Gene Expr. Patterns. 2005;5:733–739. doi: 10.1016/j.modgep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Kumar S., Duester G. SnapShot: retinoic acid signaling. Cell. 2011;147:1422. doi: 10.1016/j.cell.2011.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kam R.K., Deng Y., Chen Y., Zhao H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012;2:11. doi: 10.1186/2045-3701-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reijntjes S., Gale E., Maden M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev. Dyn. 2004;230:509–517. doi: 10.1002/dvdy.20025. [DOI] [PubMed] [Google Scholar]
- 25.Taimi M., Helvig C., Wisniewski J., Ramshaw H., White J., Amad M., Korczak B., Petkovich M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J. Biol. Chem. 2004;279:77–85. doi: 10.1074/jbc.M308337200. [DOI] [PubMed] [Google Scholar]
- 26.Pennimpede T., Cameron D.A., MacLean G.A., Li H., Abu-Abed S., Petkovich M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 2010;88:883–894. doi: 10.1002/bdra.20709. [DOI] [PubMed] [Google Scholar]
- 27.Ross A.C., Zolfaghari R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 2011;31:65–87. doi: 10.1146/annurev-nutr-072610-145127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiboni G.M., Marotta F., Carletti E. Fluconazole alters CYP26 gene expression in mouse embryos. Reprod. Toxicol. 2009;2:199–202. doi: 10.1016/j.reprotox.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez R.E., Putzke A.P., Myers J.P., Margaretha L., Moens C.B. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development. 2007;134:177–187. doi: 10.1242/dev.02706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uehara M., Yashiro K., Mamiya S., Nishino J., Chambon P., Dolle P., Sakai Y. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev. Biol. 2007;302:399–411. doi: 10.1016/j.ydbio.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 31.Uehara M., Yashiro K., Takaoka K., Yamamoto M., Hamada H. Removal of maternal retinoic acid by embryonic CYP26 is required for correct nodal expression during early embryonic patterning. Genes Dev. 2009;23:1689–1698. doi: 10.1101/gad.1776209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okano J., Lichti U., Mamiya S., Aronova M., Zhang G., Yuspa S.H., Hamada H., Sakai Y., Morasso M.I. Increased retinoic acid levels through ablation of Cyp26b1 determine the processes of embryonic skin barrier formation and peridermal development. J. Cell Sci. 2012;125:1827–1836. doi: 10.1242/jcs.101550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuong C.M., Ting S.A., Widelitz R.B., Lee Y.S. Mechanism of skin morphogenesis. II. Retinoic acid modulates axis orientation and phenotypes of skin appendages. Development. 1992;115:839–852. doi: 10.1242/dev.115.3.839. [DOI] [PubMed] [Google Scholar]
- 34.Koenig U., Amatschek S., Mildner M., Eckhart L., Tschachler E. Aldehyde dehydrogenase 1A3 is transcriptionally activated by all-trans-retinoic acid in human epidermal keratinocytes. Biochem. Biophys. Res. Commun. 2010;400:207–211. doi: 10.1016/j.bbrc.2010.08.035. [DOI] [PubMed] [Google Scholar]
- 35.Mrass M., Rendl M., Mildner F., Gruber B., Lengauer C., Ballaun L., Eckhart E., Tschachler E. Retinoic acid increases the expression of p53 and proapoptotic caspases and sensitizes keratinocytes to apoptosis: a possible explanation for tumor preventive action of retinoids. Cancer Res. 2004;64:6542–6548. doi: 10.1158/0008-5472.CAN-04-1129. [DOI] [PubMed] [Google Scholar]
- 36.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Depristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mills R.E., Pittard W.S., Mullaney J.M., Farooq U., Creasy T.H., Mahurkar A.A., Kemeza D.M., Strassler D.S., Ponting C.P., Webber C., et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;21:830–839. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucl. Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cingolani P. 2012. SnpEff: variant effect prediction http://snpeff.sourceforge.net .
- 41.Teer J.K., Green E.D., Mullikin J.C., Biesecker L.G. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slavotinek A.M., Moshrefi A., Davis R., Leeth E., Schaeffer G.B., Burchard G.E., Shaw G.M., James B., Ptacek L., Pennacchio L.A. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1–15q26.2. Eur. J. Hum. Genet. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- 43.Helvig C., Taimi M., Cameron D., Jones G., Petkovich M. Functional properties and substrate characterization of human CYP26A1, CYP26B1, and CYP26C1 expressed by recombinant baculovirus in insect cells. J. Pharmacol. Toxicol. Methods. 2011;64:258–263. doi: 10.1016/j.vascn.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 44.White J.A., Ramshaw H., Taimi M., Stangle W., Zhang A., Everingham S., Creighton S., Tam S.P., Jones G., Petkovich M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc. Natl. Acad. Sci. USA. 2000;97:6403–6408. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slavotinek A., Lee S.S., Davis R., Shrit A., Leppig K.A., Rhim J., Jasnosz K., Albertson D., Pinkel D. Fryns syndrome phenotype caused by chromosome microdeletions at 15q26.2 and 8p23.1. J. Med. Genet. 2005;42:730–736. doi: 10.1136/jmg.2004.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stone N., Burge S. Focal facial dermal dysplasia with a hair collar. Br. J. Dermatol. 1998;139:1136–1137. doi: 10.1046/j.1365-2133.1998.2576u.x. [DOI] [PubMed] [Google Scholar]
- 47.Wells J.M., Weedon D. Focal facial dermal dysplasia or aplasia cutis congenita: a case with a hair collar. Australas. J. Dermatol. 2001;42:129–131. doi: 10.1046/j.1440-0960.2001.00496.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.