

Abstract
The key cellular regulator p53 is a common target of viral oncoproteins. However, the mechanism by which p53 transcription regulation is modulated by hepatitis B virus X protein (HBx), a transcription cofactor implicated in hepatitis B virus-associated hepatocellular carcinoma (HCC), is poorly understood. By integrating p53 chromatin immunoprecipitation (ChIP)-on-chip and expression profiling of an HBx-expressing cell culture system, we report that HBx alters p53 binding site selectivity in the regulatory regions of genes, and this is associated with their aberrant expression. Using an HBx-deregulated gene, p53AIP1, as a model, we show that HBx aberrantly increases p53AIP1 expression by conferring p53 selectivity for a more conserved binding site in its regulatory region. We further demonstrate that HBx-deregulated increased p53AIP1 expression is relevant in HCC livers and define a functional role for p53AIP1 in mediating HBx-induced apoptosis in vitro. Significantly, we provide evidence that specific p53-associated transcription cofactors and coregulators are differentially recruited in the presence of HBx, effecting a PCAF-mediated “p53 Lys320 acetylation switch” that results in altered binding site selection of distinct p53 transcription cassettes. The findings here clarify the role of HBx in modulating p53 transcription regulation and provide a novel mechanistic insight into this deregulation.
INTRODUCTION
p53 is a critical regulator of important cellular processes, such as cell cycle arrest, DNA repair, and apoptosis (1). Predominantly functioning as a transcription factor (TF), tetrameric p53 transactivates or transrepresses its target genes by binding to their regulatory regions in a sequence-specific manner (2, 3). The p53 protein itself can be regulated by a myriad of posttranslational modifications. For instance, low cellular p53 protein levels are maintained by mdm2-mediated polyubiquitination and proteasomal degradation of the protein, but stress-induced phosphorylation in its N-terminal transactivation domain rapidly stabilizes p53 (4–6). Additionally, p53 site-specific phosphorylation, acetylation, and methylation have been shown to alter its selectivity for promoters of distinct classes of target genes and consequently to influence cell fate (7–10). Since proper regulation of p53 is central to maintaining normal homeostasis, any aberrations in its regulation would conceivably facilitate neoplastic transformation.
Viral oncoproteins are known to disrupt p53-dependent transcription regulation. For instance, human papillomavirus (HPV) E6, adenovirus E1B 55 kDa, and Epstein-Barr virus nuclear antigen 3C (EBNA3C) have been reported to inactivate p53 by promoting its ubiquitination and proteasomal degradation (11–15). The viral proteins also inhibited p53 DNA-binding and transcription activity (16–19), although the underlying molecular mechanisms remain less well defined. In contrast, much less is known about how the tumor suppressor p53 protein is modulated by hepatitis B virus X protein (HBx), a viral factor that is strongly implicated in the carcinogenesis process of hepatitis B virus-associated hepatocellular carcinoma (HCC) (20–23).
HBx is a small 17-kDa protein that deregulates cellular processes through various modes depending on its subcellular localization in the cytoplasm (24–26), nucleus (24–26), and mitochondria of liver cells (27, 28). In this study, we focus on the role of nuclear HBx as a transcription cofactor in deregulating cellular gene expression. Lacking a DNA-binding domain, nuclear HBx does not bind DNA directly but interacts with and modulates the DNA-binding ability and/or activity of transcription factors, such as p53 (29–31), ATF/CREB (32, 33), E2F1 (34, 35), YY1 (35), and CREB-binding protein/p300 (36). Of these, the HBx-p53 interaction is the most studied. Several independent groups have demonstrated that HBx interacts with p53 in vitro (29, 30, 37), but they report different effects of HBx on p53 sequence-specific binding to DNA. On one hand, HBx was found to inhibit p53 DNA binding (31, 38), while a subsequent study by Truant et al. described enhanced p53 oligomerization on DNA oligonucleotide in the presence of HBx (30). Importantly, Chung et al. demonstrated the biological significance of HBx-altered p53 recruitment to DNA that resulted in inhibition of the tumor suppressor PTEN gene (39). Recognizing its importance and the increasing accessibility to global profiling technologies, a more comprehensive understanding of p53 DNA-binding modulation and consequent gene deregulation by the viral X protein would clarify virus-host interactions and further advance our understanding of cellular p53-mediated transcription regulation.
In this study, we examined p53 modulation by HBx and report for the first time that HBx alters global p53 binding site selection that is associated with aberrant gene expression. By detailed characterization of an HBx-deregulated candidate p53-regulated apoptosis-inducing protein 1 gene (p53AIP1), we provide the first evidence of a novel shift in p53 binding in the regulatory region of p53AIP1 by HBx that directly results in its deregulated increased expression. Mechanistically, our findings further reveal that HBx enhances a PCAF-mediated p53 Lys320 “acetylation switch” that modulates binding site selection of p53, together with distinct transcription cofactors and coregulators termed p53 transcription cassettes, providing new insights into host transcription deregulation by the viral oncoprotein.
MATERIALS AND METHODS
Cell culture, viral transduction, and small interfering RNA (siRNA) transfection.
The human HCC cell lines HepG2 (p53 wild type) and Hep3B (p53 deficient) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Nontransformed THLE-3 (ATCC CRL-11233) normal human liver cells were cultured in bronchial epithelial basal medium (Clonetics; Lonza) without addition of gentamicin-amphotericin and epinephrine and supplemented with 10 ng/ml epidermal growth factor (EGF), 100 ng/ml phosphoethanolamine, and 10% FBS.
For viral transduction, recombinant HBx and control adenoviruses were prepared as described previously (40). Cells were transduced at multiplicities of infection (MOI) of 10 and 6, respectively, to achieve physiological levels of HBx expression, as well as high transduction efficiency, minimal cytotoxicity, and equivalent viral transduction. HepG2 cells were treated with UVC (254 nm) irradiation, as described previously, 48 h postransduction and harvested 24 h after UV irradiation (40). Hep3B and THLE-3 cells that were not exposed to UV irradiation were harvested 24 h postransduction.
For transfection of siRNA, chemical transfection using siPORT Amine Transfection Agent (Ambion) was used for Hep3B cells while electroporation was used for HepG2 and THLE-3 cells. siRNA (100 μM) specific for TP53 (s605) or TP53AIP1 (241781), negative-control siRNA (AM4611) (Ambion), and PCAF (sc-36198) (Santa Cruz Biotechnology) was used. Cells were harvested 24 h posttransfection unless otherwise stated.
Reverse-transcription real-time PCR.
Total RNA was prepared using an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions and reverse transcribed using Superscript II (Invitrogen). p53AIP1 transcript abundance was determined by quantitative real-time PCR (qPCR) using QuantiTect SYBR Green Master PCR mix (Qiagen) and primers described in Table 1. Transcript abundance was normalized against that of the β-actin housekeeping gene.
Table 1.
Table of primers and respective DNA sequences
| Primer use and name | Orientationa | DNA sequence (5′–3′)b |
|---|---|---|
| ChIP-qPCR | ||
| p53AIP1 promoter p53 RE | F | TCAGGGTGAGATGTCTTATC |
| R | CACAGGCAGAATTGTCATTT | |
| p53AIP 1 intron 1 p53 RE | F | CTCTTGCTAATGCCAGCCTG |
| R | GCATCAGGAAGTTCATCTCG | |
| RT-qPCR: p53AIP1 | F | CACCCAGTCACAGCAGCACA |
| R | CAGAGGAAGATCCCATCCAG | |
| Cloning and mutagenesis | ||
| Wild-type construct | F | AGGAACGATGGAATCAGAGTCAC |
| R | GCAGCAGCAAGGCACCATCATG | |
| Mutant promoter p53 RE | F | TAGaATtTCTGAAAGTTGGCAAgtgGTAAAAAGGC |
| R | TTACcacTTGCCAACTTTCAGAaATtCTATTCCG | |
| Mutant intron 1 p53 RE | F | CTCTaTTaCCCGGGtactTCGAGATGAAC |
| R | CATCTCGAagtaCCCGGGtAAtAGAGGAG | |
| Mutant YY1 RE | F | TACAATAAAAgacaGcCTAGGGAGAAATTACCCAGCAC |
| R | TTCTCCCTAGgCtgtcTTTTATTGTAGAGAATGGAAACCTG | |
| Mutant GATA-1 RE | F | GATGTCTTcTCCGGTTAACTGC |
| R | GCAGTTAACCGGAgAAGACATC | |
| Mutant Sp1 RE | F | CCTCATCttGCCCCCTGCAC |
| R | GTGCAGGGGGCaaGATGAGG | |
| p53 K320Q mutant | F | CAGCCAAAGcAGAAACCACTGGATGG |
| R | TGGTTTCTgCTTTGGCTGGGGAGAGG | |
| p53 K320R mutant | F | CAGCCAAAGAgGAAACCACTGGATGG |
| R | TGGTTTCcTCTTTGGCTGGGGAGAGG |
F, forward; R, reverse.
Lowercase letters denote introduced mutations.
Immunoblotting and antibodies.
Twenty micrograms of protein from each sample was subjected to gel electrophoresis on a 12% SDS-polyacrylamide gel. Western blotting was performed using standard techniques, and the following primary antibodies were used: mouse anti-p53 (DO-1; 1:10,000 dilution), mouse anti-PCAF (E-8; 1:5,000 dilution), mouse anti-YY1 (H-10; 1:1,000 dilution), rabbit anti-GATA-1 (H-200; 1:5,000 dilution), rabbit anti-histone deacetylase 1 (anti-HDAC1) (H-51; 1:10,000 dilution), mouse anti-Sp1 (1C6; 1:1,000 dilution), and goat antiactin (I-19; 1:10,000 dilution), purchased from Santa Cruz Biotechnology; rabbit anti-phosphorylated p53 Ser46 (1:1,000 dilution), purchased from Cell Signaling Technology; rabbit anti-acetylated p53 Lys320-, Lys373-, and Lys382-specific (1:5,000 dilution) and mouse anti-glyceraldehyde phosphate dehydrogenase (anti-GAPDH; 1:20,000 dilution) antibodies, purchased from Millipore; mouse anti-enhanced green fluorescent protein (anti-EGFP) antibody (1:20,000 dilution), purchased from Roche; and rabbit anti-HBx antibody (1:10,000 dilution), generated in our laboratory (40).
ChIP–real-time PCR.
Protein and DNA in THLE-3 and UV-treated HepG2 cells were cross-linked with 1% formaldehyde for 10 min at room temperature and quenched by addition of 125 mM glycine for 5 min. Cells (2 × 106) were used for each chromatin immunoprecipitation (ChIP) assay, performed according to the manufacturer's instructions (Upstate, Millipore). Essentially, cells were lysed using SDS lysis buffer supplemented with protease inhibitor cocktail (Roche Molecular Biochemicals). Chromatin was sheared to an average size of 300 bp using a Bioruptor sonicator (Diagenode) at medium setting for 12 cycles and 23 cycles of 30 s on followed by 30 s off for HepG2 and THLE-3 cells, respectively. Cell debris was removed by centrifugation at 10,000 × g, and cell lysate was precleared with bovine serum albumin (BSA)-blocked protein G beads (Upstate). A 100-μl aliquot of the cell lysate was saved as “input DNA.” The clarified lysates were immunoprecipitated with 1 μg of antibody specific for the protein of interest or 1 μg normal IgG antibody (nonspecific) at 4°C overnight and then incubated with BSA-blocked protein G beads. Following a series of washes, eluted protein-DNA complexes were reverse cross-linked using 5 M NaCl and incubated at 65°C overnight. ChIP DNA was recovered by phenol chloroform extraction and ethanol precipitation. Input DNA extracted from the total lysate that had not been immunoprecipitated but was similarly reverse cross-linked and recovered was used to normalize for differences in the starting amount of DNA in each sample. The enrichment of ChIP DNA at the promoter and intron 1 p53 response elements (REs) was determined by qPCR using QuantiTect SYBR Green Master PCR mix (Qiagen) and the following primer sets: 5′-TCAGGGTGAGATGTCTTATC-3′ (forward) and 5′-CACAGGCAGAATTGTCATTT-3′ (reverse) for the p53 RE-containing promoter region; 5′-CTCTTGCTAATGCCAGCCTG-3′ (forward) and 5′-GCATCAGGAAGTTCATCTCG-3′ (reverse) for the p53 RE-containing intron 1 region. The qPCR conditions were initial denaturation at 95°C for 10 min, followed by 45 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s.
ChIP-on-chip, expression profiling, and computational analyses.
ChIP-on-chip assays were performed on control and HBx UV-treated HepG2 cells with p53 DO-1 antibody (Santa Cruz) on NimbleGen 1.5-kb promoter arrays according to the manufacturer's recommendations. Differential p53 binding regions were identified using the Partek Genomics Suite with a minimum of five consecutive probes. Putative p53 REs in the binding regions were identified using the p53MH algorithm (41). Expression profiles of control and HBx UV-treated HepG2 cells were obtained using an Agilent whole genome expression array.
The transcription factor motif prediction tools TRANSFAC (Biobase) and MatInspector (Genomatix) were used to identify factors that bind in the vicinity of the p53 REs (±300 bp). GeneMANIA software (http://www.genemania.org) was employed to identify potential coregulators that associate with the transcription complexes but that may not bind DNA.
Generation of wild-type and mutant promoter constructs and beta-galactosidase (β-Gal) reporter assay.
To experimentally validate the predicted p53 REs of the p53AIP1 gene, a 3.8-kb fragment containing both the promoter and intron 1 p53 REs was PCR amplified from genomic DNA of human liver tissue using Expand High Fidelity Taq DNA polymerase (Roche) and primers 5′-AGGAACGATGGAATCAGAGTCAC-3′ (forward) and 5′-GCAGCAGCAAGGCACCATCATG-3′ (reverse) and cloned upstream of a β-Gal reporter gene. The promoter construct also contained the EGFP gene for visualization of transfection efficiency. In designing mutant promoter and intron 1 p53 REs, the transcription factor motif prediction tool MatInspector (Genomatix) was employed to identify mutations that abolish the respective p53 REs but that do not affect other proximal transcription factor binding sites. Mutant promoter constructs (M1, M2, and M3), as well as GATA-1, YY1, and Sp1 RE mutants, were generated by fusion PCR using primers containing the desired mutations (Table 1). All constructs were sequenced to verify the integrity of the DNA sequences and the successful introduction of only the appropriate mutations.
Hep3B cells (p53 deficient) were chemically transfected with 1 μg TP53-expressing or control plasmid, 5 μg β-Gal reporter construct, and/or 100 μM TP53-specific or control siRNAs using siPORT Amine Transfection Agent (Ambion, TX) according to the manufacturer's protocol. For experiments that investigated the role of HBx, cells were transduced with either recombinant HBx or control vectors 24 h posttransfection. The crude lysates obtained were kinetically assayed for β-Gal reporter activity using chlorophenol red–β-d-galactopyranoside (CPRG) as the substrate and measured at 30-s intervals over 60 min at 570 nm using a microplate reader. The β-Gal activity of each construct was normalized against the protein concentration determined using a BCA protein assay kit (Pierce Thermo Scientific) according to the manufacturer's protocol, as well as the respective basal β-Gal activity to take into account the small inherent differences in β-Gal activity between promoter constructs in the absence of p53.
Generation of p53 Lys320 acetylation mutant constructs.
p53 Lys320 acetylation mutants K320Q and K320R were generated by fusion PCR using primers containing the desired mutations (Table 1).
Apoptosis assay.
HepG2 cells were electroporated with TP53AIP1 siRNA or negative-control siRNA (Ambion) and an HBx-expressing or control plasmid and subjected to UV treatment. The apoptosis profiles of the cells were analyzed by phycoerythrin (PE) annexin V and 7-aminoactinomycin D (7AAD) staining according to the manufacturer's protocol (BD Biosciences Pharmingen), followed by flow cytometry using the BD FACSCalibur (BD Biosciences) 24 h posttreatment. Cellular profiles were analyzed using FlowJo software (Tree Star).
p53AIP1 gene expression and HBx profiling of HCC patients.
Deidentified tumor (T) and paired nontumorous (NT) tissues from HCC patients were obtained from the National Cancer Centre Singapore (NCCS)/SingHealth Tissue Repository with prior approval from the SingHealth Centralised Institutional Review Board (2007/437/B). p53AIP1 gene expression profiles of 78 HCC patients were obtained using qPCR. The transcript abundance of p53AIP1 was normalized to the respective β-actin gene expression. HBx status was determined using immunoblot analysis with HBx-specific antibody.
Statistical analysis of experimental data.
Student's t test was performed to analyze the significance of differences between sample means obtained from at least three independent experiments.
RESULTS
HBx alters global p53 DNA binding with associated adjacent gene deregulation.
The role of HBx in p53 sequence-specific DNA binding and consequent transcription deregulation is poorly understood. Therefore, using p53 ChIP-on-chip, we first examined p53 occupancy profiles in a previously established HepG2 cell culture system expressing levels of HBx within the range that is observed in HCC patients (Fig. 1A and B) (40). Analysis of p53 ChIP-on-chip-identified binding regions containing p53MH-predicted p53 response elements revealed several patterns of altered p53 DNA binding in the regulatory regions of genes: HBx abolished p53-DNA binding at a subset of candidate binding sites but enhanced p53-DNA binding at other candidate sites (Table 2). Strikingly, global p53 ChIP analysis also revealed a novel shift in p53 DNA binding to a different candidate site within the same regulatory region of a target gene (Table 2). This is the first report to demonstrate that HBx can alleviate, as well as enhance, global p53 binding to its response elements, significantly clarifying the role of the viral protein in p53 sequence-specific DNA binding. To identify biologically significant p53 binding site selectivity alterations by HBx, gene expression profiles of HBx-expressing HepG2 cells were integrated with p53 ChIP-on-chip data. Table 2 shows a list of experimentally validated candidate genes with altered p53 DNA binding and their corresponding gene expression changes. Notably, only a subset of p53 DNA-binding alterations was associated with strong adjacent gene deregulation (>1.5-fold; P ≤ 0.05 [indicated in boldface in Table 2]). This observation was not surprising, since not all transcription factor-DNA-binding events affect gene expression (42). We also observed that each pattern of HBx-altered p53 DNA binding (abolishment, enhancement, or shift) did not associate with any gene deregulation pattern (up- or downregulation) (Table 2). A possible explanation is that the observed deregulated gene expression may be the result of an interplay of adjacent transcription cofactors and coregulators that influences p53-mediated transcription, typical of mammalian transcription regulation. To further understand the molecular mechanism underlying altered p53 binding site selection by HBx, a known p53-regulated candidate identified from the global study, p53AIP1, was selected for further characterization (Table 2).
Fig 1.
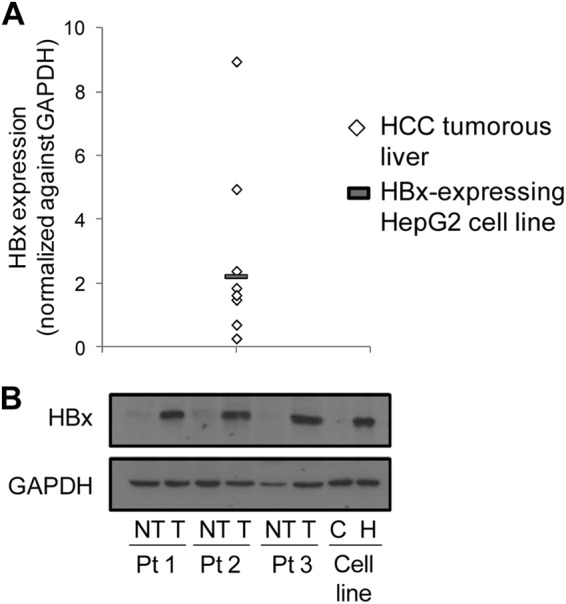
Physiological HBx expression levels in an in vitro cell culture system. (A) HBx protein expression levels in HCC tumorous livers and an in vitro HBx-expressing HepG2 cell culture system. (B) Immunoblots of HBx and GAPDH protein expression levels in 3 representative paired HCC T and NT livers, as well as in HBx-expressing UV-treated HepG2 cells. Pt, patient; C and H, control and HBx HepG2 cells, respectively.
Table 2.
Experimentally validated genes with HBx-altered p53 occupancy and corresponding gene expression from p53 ChIP-on-chip and expression profilinga
| Gene no. | Gene product name | p53 occupancy (ChIP-on-chip) |
Gene expressionb |
||
|---|---|---|---|---|---|
| Binding pattern | p53 binding (MAT scorec) | Fold change (HBx/control) | P value | ||
| 1 | p53AIP1 | Decrease | −15.17 | 1.66 | 5.0E−02 |
| 2 | SPINK6 | −9.12 | 3.10 | 1.0E−02 | |
| 3 | DUX4 | −4.98 | −1.50 | 1.0E−03 | |
| 4 | ERCC2 | −14.42 | 1.01 | 9.7E−01 | |
| 5 | C1D | −12.10 | 1.00 | 9.2E−01 | |
| 6 | FAS | Increase | 7.20 | 2.11 | 4.5E−08 |
| 7 | ABCD2 | 6.30 | 1.62 | 4.0E−02 | |
| 8 | AKT1S1 | 6.00 | −2.51 | 4.0E−03 | |
| 9 | GDNF | 4.00 | −1.44 | 2.0E−03 | |
| 10 | HNF4 | 10.60 | 1.18 | 9.1E−01 | |
| 11 | TNP1 | 4.40 | 1.11 | 8.8E−01 | |
| 12 | SLC7A13 | Shift | −13.80 | 1.61 | 4.0E−02 |
| 4.20 | |||||
| 13 | UNKL | −4.30 | −1.57 | 2.0E−03 | |
| 4.00 | |||||
| 14 | KALRN | −9.80 | −1.11 | 9.6E−01 | |
| 5.10 | |||||
Significantly deregulated candidate genes (fold change, >1.5; P ≤ 0.05) with altered p53 binding are indicated in boldface.
Italics indicates significantly upregulated genes; underlining represents significantly downregulated genes.
MAT (model-based analysis of tiling arrays) score indicates ChIP-enriched regions (positive score, increased p53 binding; negative score, decreased p53 binding).
A novel HBx-induced shift in p53 binding in the regulatory region of p53AIP1 aberrantly increased its gene expression.
Global p53 ChIP profiling revealed that p53 bound to a previously unreported candidate p53 RE at the p53AIP1 promoter and that p53 DNA binding at this RE decreased in the presence of HBx (Fig. 2A, left). Located approximately 400 bp upstream of the p53AIP1 transcription start site, the novel promoter p53 RE consists of two half-sites separated by a 9-bp spacer. Notably, the promoter RE exhibits less sequence similarity (65%) to the p53 consensus sequence than the previously reported p53AIP1 intron 1 p53 RE (80%) (10) that was not captured by the 1.5-kb promoter chip (Fig. 2A, right). Importantly, both p53 REs were found to be functional and necessary for p53-mediated p53AIP1 transcription. Promoter assay of the p53AIP1 regulatory region containing both promoter and intron 1 REs (wild type [WT]) in p53-deficient Hep3B cells showed a 6-fold increase (P < 0.01) upon addition of p53, and this increase was abrogated by depleting p53 using p53-specific siRNA (P < 0.01) (Fig. 2B). Moreover, mutation of either promoter (M1) or intron 1 RE (M2) significantly reduced reporter activities compared to that of the WT promoter (P < 0.05), confirming that both REs are bona fide p53 REs essential for regulation of adjacent gene expression (Fig. 2C). We next examined the effect of HBx on p53 binding at both REs. Consistent with global p53 ChIP profiling, HBx decreased p53 binding at the p53AIP1 promoter RE (P < 0.01), as determined by p53 ChIP-qPCR on HBx and control HepG2 cells (Fig. 2D, left). Conversely, HBx increased bound p53 at the intron 1 RE (P < 0.01) (Fig. 2D, right), suggesting a novel shift in p53 binding from the promoter to intron 1 RE of p53AIP1. The shift in p53-DNA binding in the p53AIP1 regulatory region was also confirmed using another p53 antibody, sc-6243 (Santa Cruz) (data not shown). Importantly, this phenomenon is not cell line or treatment specific, since the HBx-induced shift in p53 DNA binding was consistently detected in nontransformed THLE-3 liver cells without DNA-damaging treatment (Fig. 2E). Next, since HBx binds DNA indirectly via transcription factors such as p53, we examined if HBx was also recruited to p53AIP1 intron 1 using HBx ChIP-qPCR. Indeed, HBx bound the intron 1 region in both cell lines (P < 0.05) (Fig. 2F), suggesting that an HBx-p53 transcription complex is recruited to the intron 1 p53 RE. Taken together, these findings demonstrate a novel shift in p53 selectivity for its binding sites in the regulatory region of p53AIP1 by the viral X protein.
Fig 2.

HBx shifts p53 binding at the p53AIP1 regulatory region. (A) (Left) Illustration of the ChIP-on-chip-identified p53 binding region at the p53AIP1 promoter, generated using SignalMap software (NimbleGen Systems). Decreased p53 occupancy at the novel p53AIP1 promoter RE by HBx is indicated by the absence of a peak in the HBx-p53 ChIP sample. (Right) Schematic of p53 RE positions relative to the p53AIP1 gene. Each p53 RE is depicted as two black boxes, each representing one half-site of the p53 consensus sequence. Nucleotides in capital letters represent identity of the genomic sequence to the consensus; nucleotides in lowercase letters represent disparity with the consensus. The transcription start site (+1) and translation start site (ATG) of the p53AIP1 gene are shown. p53 motifs and percent similarity to the consensus sequence of promoter and intron 1 REs are shown. (B) p53 stimulates p53AIP1 promoter activity. p53-deficient Hep3B cells were cotransfected with the wild-type promoter construct and the indicated plasmids and/or siRNA and assayed for β-Gal activity. (C) Both promoter and intron 1 p53 REs are functional and necessary for p53AIP1 regulation. Hep3B cells were cotransfected with the indicated wild-type or mutant promoter constructs and p53 or control plasmid. Basal β-Gal activity is denoted by the vertical black dashed line. Mutant p53 REs are indicated by xx. (D and E) p53 ChIP-qPCR validation of decreased p53 occupancy at the promoter RE (left) and increased p53 occupancy at the intron 1 RE (right) in HBx and control HepG2 (D) and THLE-3 (E) cells. Nonspecific ChIP control was performed using normal IgG. (F) ChIP-qPCR using HBx-specific antibody performed on HBx and control HepG2 (left) and THLE-3 (right) cells. All error bars show standard errors of the mean from triplicate experiments.
We next ascertained the biological significance of HBx-altered p53 DNA binding at p53AIP1. Expression profiling of HBx-deregulated genes revealed a 1.6-fold increase in p53AIP1 expression in the presence of HBx (Table 2). This was consistently validated in both cell lines by qPCR (Fig. 3A). Moreover, depletion of p53 using p53-specific siRNA negated this increase (Fig. 3A), confirming that p53 mediates HBx-increased p53AIP1 expression. Next, we tested whether the increased gene expression is a direct consequence of the shift in p53 binding in the p53AIP1 regulatory region by HBx. Indeed, only WT and promoter RE mutant M1 (which mimicked the shift in p53 occupancy) showed significantly enhanced reporter activities in the presence of HBx (P < 0.01) (Fig. 3B). In contrast, HBx did not significantly alter the reporter activities of mutants that prohibit the shift in p53 DNA binding (intron RE mutant M2 and double mutant M3) (Fig. 3B), providing strong evidence that the HBx-induced shift in p53 DNA binding directly results in the deregulated increase in p53AIP1 expression.
Fig 3.

A shift in p53 DNA binding directly results in a deregulated increase in p53AIP1 expression. (A) p53AIP1 expression in p53-specific or control siRNA-treated HBx and control HepG2 (left) and THLE-3 (right) cells measured by qPCR. (B) β-Gal activity of the indicated WT or mutant promoter constructs cotransfected with p53 in HBx or control Hep3B cells. The x axis shows a comparison of the normalized β-Gal activity of each construct in the presence or absence of HBx, and the difference is expressed as relative β-Gal activity in the presence of HBx versus β-Gal activity in the absence of HBx (i.e., control). (C) p53AIP1 gene expression and HBx protein status of T and paired adjacent NT samples from 78 HCC patients analyzed by qPCR and immunoblotting, respectively. The median ratios (horizontal lines) of p53AIP1 expression (T/NT) in patients with low (T/NT < 2) and high (T/NT > 2) HBx protein expression levels are shown.
HCC tumors with high HBx expression have significantly increased p53AIP1 expression.
To investigate if the observed HBx-deregulated increase in p53AIP1 expression is relevant in HCC tumors, p53AIP1 gene expression and HBx protein expression of 78 HCC T and paired adjacent NT tissues were profiled using qPCR and immunoblotting, respectively. Relative p53AIP1 expression levels (T/NT) were compared in patients with high HBx expression (>2-fold T/NT) versus patients with low HBx expression (<2-fold T/NT). Significantly higher p53AIP1 expression was observed in patients with high HBx expression levels (median, 1.46) than in patients with low HBx expression levels (median, −1.36) (P < 0.05) (Fig. 3C), highlighting the relevance of our findings in the HCC patient setting.
Increased p53AIP1 expression mediates HBx-induced apoptosis and is independent of p53 Ser46 phosphorylation.
Next, we examined the biological consequence of p53AIP1 deregulation by HBx. In our previous study, HBx was found to sensitize UV-treated HepG2 cells to apoptosis (40). Therefore, we hypothesized that the proapoptotic protein p53AIP1 may mediate HBx-induced apoptosis. To test this hypothesis, p53AIP1 was depleted in HBx- or control UV-treated HepG2 cells using p53AIP1-specific siRNA (Fig. 4A), and the apoptosis profiles were analyzed by annexin V/7AAD staining. As expected, the apoptotic-cell population increased from 12% to approximately 20% in the presence of HBx (P < 0.01) (Fig. 4B). Transient depletion of p53AIP1 abrogated this increase (Fig. 4B), suggesting a functional role for p53AIP1 in HBx-induced apoptosis.
Fig 4.

Increased p53AIP1 expression mediates HBx-induced apoptosis. (A) Transient p53AIP1 knockdown using specific siRNA compared to control siRNA measured by qPCR. (B) Apoptosis profiles of p53AIP1-specific or control siRNA-treated control and HBx UV-treated HepG2 cells from a representative set of experiments (top) and summarized from 3 independent experiments shown in percentages (bottom). (Top) The percentage of apoptotic cells in each sample is shown in the upper left quadrant. All error bars show standard errors of the mean from triplicate experiments. (C) Immunoblot of phosphorylated p53 Ser46 levels using a modification-specific antibody in HBx and control HepG2 (top) and THLE-3 (bottom) cells harvested at various time points over 72 h. Immunoblots of total p53, β-actin, and HBx expression are also shown.
As p53 serine 46 (Ser46) phosphorylation was reported to induce p53AIP1 expression and apoptosis (10), we examined if p53 phosphorylation at the site was altered by HBx. Immunoblot analysis using the modification-specific antibody showed comparable p53 Ser46 phosphorylation levels in HBx and control HepG2 (Fig. 4C, top) and THLE-3 (Fig. 4C, bottom) cells over 72 h despite increasing HBx expression, suggesting that HBx deregulates p53AIP1 expression independently of p53 Ser46 phosphorylation.
Differential recruitment of distinct p53 transcription coregulatory modules by HBx.
We further investigated the mechanism by which HBx alters p53 binding site selection at the p53AIP1 gene. Since integration of global p53 ChIP and expression profiling suggests a more complex mode of p53-mediated transcriptional deregulation by the viral X protein, we hypothesized that adjacent TFs and coregulators that modulate p53 transcription regulation may be involved. To this end, proximal (±300 bp) p53-interacting high-confidence TFs were identified using TRANSFAC and MatInspector TF binding site prediction tools and searched for reported interactions with p53. Notably, distinct sets of strong p53-interacting candidates were predicted in the two regions: transcription repressors YY1 and GATA-1 at the p53AIP1 promoter and transcription activator Sp1 in the intron 1 region (Fig. 5A). Strikingly, YY1/GATA1 and Sp1 recruitment to the respective regions assessed by ChIP-qPCR on HBx and control THLE-3 cells revealed binding patterns analogous to that of p53. YY1 and GATA-1 recruitment to the p53AIP1 promoter was abolished in the presence of HBx (P < 0.05 and P < 0.01, respectively) (Fig. 5B, left and middle, respectively), while Sp1 recruitment to intron 1 was enhanced in the presence of HBx (P < 0.01) (Fig. 5B, right). Furthermore, using RNA interference and promoter assays of each TF binding site mutant, we found that promoter-bound transcription repressors YY1 and GATA-1 negatively modulate p53AIP1 expression (Fig. 5C and D) while intron 1-bound transcription activator Sp1 positively modulates p53AIP1 expression (Fig. 5E and F). This suggests that the surrounding transcription cofactors recruited together with p53 at each RE also influence transcription of the target gene.
Fig 5.

Distinct transcription cofactors are differentially recruited by HBx and influence gene transcription. (A) MatInspector- and TRANSFAC-predicted promoter and intron 1 TFs and their positions relative to the respective p53 REs. p53-interacting TFs GATA-1, YY1, and SP1 are indicated in boldface. (B) ChIP-qPCR-validated YY1 (left), GATA-1 (middle), and Sp1 (right) occupancy in HBx and control THLE-3 cells using the respective specific antibodies. (C to F) Surrounding transcription cofactors modulate p53AIP1 expression. (C) Western blots showing efficient knockdown of YY1 (top left) and GATA-1 (top right) using specific siRNA. Depletion of YY1 or GATA-1 increases p53AIP1 expression in THLE-3 cells as measured by qPCR (bottom). (D) Mutation of YY1 or GATA-1 RE at the p53AIP1 promoter increases reporter activity in Hep3B cells. (E) (Top) Western blots showing efficient knockdown of Sp1 using specific siRNA. (Bottom) Depletion of Sp1 negates HBx-increased p53AIP1 expression in THLE-3 cells as measured by qPCR. (F) Mutation of the Sp1 RE at p53AIP1 intron 1 negates HBx-increased reporter activity in Hep3B cells. All error bars show standard errors of the mean from triplicate experiments.
To identify other potential transcription coregulators of the p53AIP1 promoter and intron 1 transcription complexes that do not exhibit sequence-specific DNA binding, the GeneMANIA association network prediction resource (http://www.genemania.org/) was used. Importantly, histone deacetylase 1 (HDAC1) was the only strongly associated factor predicted in the promoter region, associating with p53, YY1, GATA-1, and HSF1 (Fig. 6A). Similar to p53/YY1/GATA-1 recruitment, HDAC1 recruitment to the p53AIP1 promoter was markedly reduced in the presence of HBx (P < 0.01) (Fig. 6B). No strongly associated factors were predicted in the intron 1 region. Taken together, these findings suggest that HBx can perturb the recruitment of specific p53-associated transcription coregulatory modules that may modulate gene transcription.
Fig 6.

Transcription coregulator HDAC1 is recruited to the p53AIP1 promoter in the absence of HBx. (A) GeneMANIA-predicted coregulators at the p53AIP1 promoter transcription complex. The gray circles represent high-confidence TFs; the white circles represent GeneMANIA-predicted associated factors. The lines linking two circles indicate reported association between two factors. (B) ChIP-qPCR using HDAC1-specific antibody performed on HBx and control THLE-3 cells. All error bars show standard errors of the mean from triplicate experiments. (C) Immunoblots of YY1, GATA-1, HDAC1, and Sp1 expression levels in HBx and control THLE-3 cells using the specific respective antibodies. Immunoblots of GAPDH and HBx expression are shown, as well.
We next asked if the differential recruitment of the coregulatory factors to p53AIP1 promoter and intron 1 regions was due to possible alterations in their expression by HBx. It was postulated that HBx-induced degradation or stabilization of the factors could conceivably affect their availability to be recruited to the respective response elements. To this end, YY1, GATA-1, Sp1, and HDAC1 protein expression levels in HBx and control THLE-3 cells were determined by immunoblotting with their respective specific antibodies. Comparable protein expression levels of each factor were detected in the presence or absence of HBx (Fig. 6C). The data thus far suggest that HBx does not alter the expression of the transcription coregulatory factors and that another mechanism is responsible for their differential recruitment by viral protein.
A PCAF-mediated p53 Lys320 “acetylation switch” alters p53 binding site selection.
Since our findings indicate that HBx decreases HDAC1 recruitment at the p53AIP1 promoter and HDAC1 is known to deacetylate lysine residues (Lys or K) of both histone and nonhistone proteins, such as p53, we therefore examined if histone and p53 acetylation was altered in the presence of HBx. First, comparable levels of acetylated H3 and H4 were detected using ChIP-qPCR at the p53AIP1 promoter in HBx and control THLE-3 cells, suggesting that histone acetylation is unaffected by HBx-induced relief of bound HDAC1 (data not shown). Next, the acetylation status of known HDAC1-modifiable p53 Lys residues at positions 320, 373, and 382 was analyzed by immunoblotting of HBx and control THLE-3 cells. Strikingly, acetylation of p53 Lys320 was markedly enhanced in the presence of HBx, while the p53 Lys373 and Lys382 acetylation status remained unchanged (Fig. 7A). Since acetylation of p53 is reported to alter its sequence-specific DNA-binding property (9), we hypothesized that HBx may alter p53 RE selection through site-specific acetylation at Lys320. Strikingly, acetylated p53 Lys320 preferentially bound the more conserved p53AIP1 intron 1 RE in the presence of HBx, as demonstrated using acetyl p53 (Ac-p53) Lys320 ChIP on HBx and control THLE-3 cells (P < 0.01) (Fig. 7B). To further determine if acetylation of p53 Lys320 is essential for HBx-increased p53AIP1 expression, p53AIP1 expression was measured in HBx and control p53-deficient Hep3B cells with exogenously introduced wild-type p53, acetylation mimic K320Q, or nonacetylatable K320R. Notably, HBx increased p53AIP1 expression only in cells expressing either wild-type p53 (P < 0.05) or K320Q (P < 0.01), but not in cells expressing the acetylation-defective mutant K320R (Fig. 7C). Collectively, these data suggest that enhanced acetylation at p53 Lys320 by HBx alters p53 RE selection and is essential for the deregulated increase in p53AIP1 expression.
Fig 7.

HBx alters p53 occupancy by enhancing PCAF-mediated acetylation at p53 Lys320. (A) Immunoblots of Ac-p53 Lys320, -373, and -382 expression of control and HBx THLE-3 cells using site-specific modification antibodies. Immunoblots of total p53, GAPDH, and HBx expression are also shown. (B) ChIP-qPCR using Ac-p53 Lys320-specific antibody performed on HBx and control THLE-3 cells. (C) p53AIP1 expression measured by qPCR in HBx and control Hep3B cells transfected with wild-type p53, acetylation mimic K320Q, or nonacetylatable K320R. An immunoblot of p53 expression is shown. (D) Immunoblot of Ac-p53 Lys320 expression in HDAC1-specific and control siRNA-treated THLE-3 cells. Immunoblots of HDAC1 and GAPDH expression are shown. (E) ChIP-qPCR using PCAF-specific antibody performed on HBx and control THLE-3 cells. (F) p53AIP1 expression measured by qPCR in PCAF-specific or control siRNA-treated HBx and control THLE-3 cells. Immunoblots of Ac-p53 Lys320 and PCAF expression are shown. All error bars show standard errors of the mean from triplicate experiments.
To determine if the relief of HDAC1 activity by HBx is sufficient to enhance p53 Lys320 acetylation, Ac-p53 Lys320 levels were examined in HDAC1-depleted cells. Figure 7D shows that transient depletion of HDAC1 by RNA interference did not spontaneously increase Ac-p53 Lys320 levels, suggesting that the observed enhanced p53 Lys320 acetylation by HBx requires the action of a lysine acetyltransferase. Since the acetyltransferase p300/CBP-associated factor PCAF selectively acetylates p53 Lys320, we investigated if PCAF mediates HBx-induced p53 Lys320 acetylation and subsequent p53AIP1 expression. PCAF ChIP on HBx and control cells revealed significant PCAF recruitment to the p53AIP1 intron 1 region in the presence of HBx (P < 0.01) (Fig. 7E). Strikingly, transient depletion of PCAF by RNA interference abrogated increased p53AIP1 expression by HBx (P < 0.01), with concomitant blunting of p53 Lys320 acetylation (Fig. 7F). Taken together, the data suggest that PCAF plays a key role in mediating HBx-enhanced p53 Lys320 acetylation and consequent p53AIP1 deregulation.
DISCUSSION
Oncogenic viruses have devised various ways to deregulate the “guardian of the genome” and are invaluable tools for understanding the complexity of p53 regulation. A common mechanism by which viral oncoproteins, such as HPV E6 and EBNA3C, inactivate p53 is by interacting with and targeting the transcriptional regulator for ubiquitin-dependent proteasomal degradation. However, we and others had previously demonstrated that the hepatitis B virus X protein does not promote p53 degradation (31, 39, 40). Instead, nuclear HBx functions as a transcription cofactor and is thought to alter the sequence-specific DNA-binding property of p53, although this remains ill defined. Recognizing the importance of the HBx oncoprotein in viral hepatocarcinogenesis and the integral role of p53 in maintaining cellular homeostasis, this study addresses the mechanism of p53 modulation by the viral protein.
The findings of this study clarify the seemingly contradictory reports on the role of HBx in p53 sequence-specific DNA binding. We provide the first evidence that HBx globally changes p53 selectivity for its binding sites by potentiating, relieving, or shifting p53 binding at distinct sites of gene promoters with associated gene deregulation. Interestingly, HBx-altered p53 DNA binding either stimulated or repressed gene expression, suggesting a more complex mode of p53-mediated transcription deregulation by the viral X protein than previously thought. In an initial model, a repressive domain of HBx was proposed to be responsible for inhibiting the basal transcription machinery and repressing global gene expression (30). However, the findings of our study support the idea that p53 transcription deregulation by HBx involves an intricate interplay between transcription cofactors and coregulators, as well as p53 posttranslational modification(s) (collectively termed p53 transcription cassettes). Using the HBx-deregulated gene p53AIP1 as a model, we demonstrate that HBx modulates p53AIP1 expression by differentially recruiting distinct p53 transcription cassettes with altered p53 binding site selection. Specifically, our model postulates that a transcriptionally repressive p53 cassette comprising p53, YY1, GATA-1, and HDAC1 tightly regulates proapoptotic p53AIP1 under normal conditions (Fig. 8). Maintenance of low p53AIP1 levels is conceivable, as it would safeguard cells against aberrant apoptosis. Upon viral infection and HBx expression, HBx disrupts the bound repressive p53 cassette by tipping the HDAC1-PCAF balance in favor of the latter, enhancing PCAF-mediated acetylation of p53 Lys320. HDAC1-deacetylated p53 Lys320, which preferentially bound the weaker promoter RE, then becomes acetylated by PCAF, conferring p53 selectivity for the more conserved intron 1 RE. Together with the transcriptional coactivator Sp1, the activating acetylated p53 Lys320 cassette stimulates p53AIP1 transcription, which mediates HBx-induced apoptosis. Induction of apoptosis is thought to enhance compensatory proliferation (43) and regeneration of hepatocytes, creating a cellular environment that exerts selective pressure for premalignant hepatocytes that are resistant to apoptosis.
Fig 8.
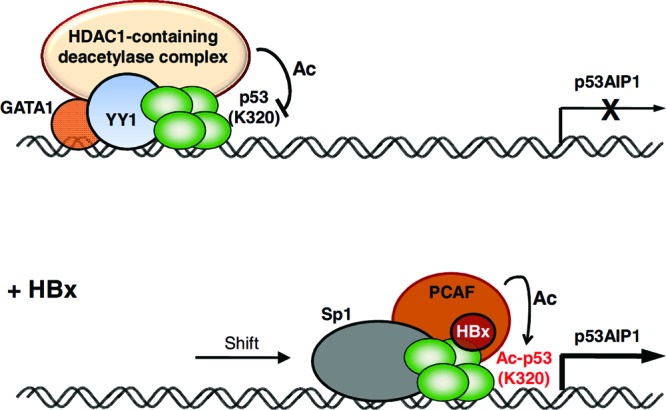
Model illustrating p53-mediated deregulation of p53AIP1 by HBx. In unstressed cells, a transcriptionally repressive p53–YY1–GATA-1–HDAC1 complex is bound to the p53AIP1 promoter, repressing p53AIP1 transcription. In the presence of HBx, PCAF-mediated p53 Lys320 acetylation is enhanced, endowing p53 with selectivity for the more conserved intron 1 RE. The transcriptionally activating HBx–PCAF–Ac-p53–Sp1 complex recruited to p53AIP1 intron 1 stimulates p53AIP1 transcription.
Importantly, our study provides a novel mechanistic insight into how the viral X protein deregulates p53 transcription. In contrast to the roles of other viral proteins, such as the adenovirus E1B-55 kDa and human immunodeficiency virus type 1 Tat proteins that deregulate p53-mediated transcription by interfering with PCAF-p53 interaction and inhibiting PCAF-mediated p53 Lys320 acetylation (44, 45), we have shown that, conversely, HBx hijacks and enhances the host PCAF acetylation pathway. How, then, does HBx enhance PCAF-mediated p53 Lys320 acetylation? Does HBx also enhance acetylation of other PCAF substrates, signaling a broader role for PCAF in HBx deregulation of cellular transcription? Future studies addressing this will undoubtedly provide greater insight into the function of the viral X protein. Furthermore, in support of our findings that HBx preferentially enhances the recruitment of distinct p53 transcription cassettes, global HBx ChIP revealed enrichment of known p53-interacting transcription cofactor motifs at the promoter's HBx direct target genes, including Sp1, E2F1, and YY1 genes (35). Identification of additional p53 transcription cassettes perturbed by HBx is in progress and will serve to advance our understanding of how HBx upsets cellular homeostasis in hepatocarcinogenesis.
Significantly, our work further refines the conceptual framework through which we can understand p53-mediated transcription regulation. A growing body of literature supports a p53 sensor system on which various stress signals converge in the form of a p53 posttranslational code, thereby determining the transcription of selective classes of p53 target genes to elicit the appropriate cellular response (46). In particular, Knights et al. proposed the concept of p53 acetylation cassettes, in which signal-dependent p53 acetylation clusters selectively transactivate/transrepress distinct functional classes of target genes to affect cellular outcomes (47). Using a lung carcinoma cell model, the authors show that acetylated p53 Lys320 is associated with stimulating prosurvival genes, while conversely, acetylated p53 Lys373 is associated with stimulation of proapoptotic genes. Through the study of the viral X protein in modulating p53, our findings support the broad concept of a p53 acetylation sensor-effector system that is important for fine-tuning p53 binding site selection and consequent transcription regulation. Moreover, our study highlights an additional layer of complexity in p53-mediated transcription regulation that was not examined by Knights et al. We show that sequence-specific transcription cofactors bound in the vicinity of p53 also influence transcription regulation of the adjacent gene. Noting that transcription factor expression profiles can vary in different cell types, this may explain in part the discrepancy in the prosurvival roles of acetylated p53 Lys320 in lung carcinoma cells (47) versus its proapoptotic role in liver cells (this study). Indeed, site-specific acetylation of p53 has been associated with different cellular outcomes in different cell types (47–49), supporting the idea that cell-type-specific repertoires of transcription factors and regulatory networks likely exert different effects on p53 transcription regulation. Our study therefore advocates dissecting p53-mediated transcription regulation in terms of transcription cassettes influenced by the unique proteome landscape of each cell type, rather than by analyzing specific posttranslational marks of p53 on its transcription regulation in isolation.
Furthermore, our study highlights the importance of identifying the p53 posttranslational mark(s) of each transcription cassette that is essential for transcription regulation. For example, although Knights et al. reported that acetylated p53 Lys373 preferentially associated with proapoptotic p53AIP1, they failed to show that acetylated Lys373 indeed enhances the transcription of p53AIP1 in their gene expression analysis of the gain-of-function mutant K373Q. In contrast, we provide strong evidence that acetylated p53 Lys320 not only binds to the p53AIP1 regulatory region but, importantly, also functions to stimulate its transcription. These findings suggest that although p53 can be marked with a variety of posttranslational modifications, it is imperative to identify the p53 posttranslational mark(s) of each transcription cassette that is functionally important for transcription of the target gene. Further, recognizing that these p53 posttranslational modifications can compensate and/or cross talk with each other, dissecting complete repertoires of p53 transcription cassettes specific to each cell type would be of paramount importance.
Finally, with the advent of ChIP coupled with high-throughput sequencing, we are discovering a rapidly increasing number of potential p53 binding sites in the genome. It would not be surprising, then, to find that more target genes may possess several p53 binding sites in their regulatory regions, adding to the complexity of p53-mediated transcription regulation. Using p53AIP1 transcription deregulation by the viral X protein as a model, we have shown here that transcription of a single gene can be coordinated by the selective recruitment of distinct p53 transcription cassettes to different functional p53 binding sites. In view of this, we believe it will be worthwhile to identify patterns of perturbed binding site selection of p53 transcription cassettes within genes, as well, to delineate the complexity of p53 transcription regulation. Through an appreciation of how p53 transcription is deregulated by the viral X protein, our study significantly provides a more comprehensive framework to gain a more precise understanding of p53 transcription regulation.
ACKNOWLEDGMENTS
This work was supported by a grant from the BioMedical Research Council (BMRC06/1/21/19/449), the National Medical Research Council (NMRC/1238/2009), and the Singapore Millennium Foundation, as well as block funding from the National Cancer Center, Singapore, and Duke-NUS Graduate Medical School to Caroline G. Lee.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 12 November 2012
REFERENCES
- 1. Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408:307–310 [DOI] [PubMed] [Google Scholar]
- 2. Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C. 1991. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 65:1083–1091 [DOI] [PubMed] [Google Scholar]
- 3. Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. 1991. Identification of p53 as a sequence-specific DNA-binding protein. Science 252:1708–1711 [DOI] [PubMed] [Google Scholar]
- 4. Appella E, Anderson CW. 2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268:2764–2772 [DOI] [PubMed] [Google Scholar]
- 5. Shieh SY, Ikeda M, Taya Y, Prives C. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325–334 [DOI] [PubMed] [Google Scholar]
- 6. Unger T, Juven-Gershon T, Moallem E, Berger M, Vogt Sionov R, Lozano G, Oren M, Haupt Y. 1999. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 18:1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. 2004. Regulation of p53 activity through lysine methylation. Nature 432:353–360 [DOI] [PubMed] [Google Scholar]
- 8. Gu W, Roeder RG. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90:595–606 [DOI] [PubMed] [Google Scholar]
- 9. Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. 2004. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 101:2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. 2000. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102:849–862 [DOI] [PubMed] [Google Scholar]
- 11. Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. 1997. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 71:3788–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saha A, Murakami M, Kumar P, Bajaj B, Sims K, Robertson ES. 2009. Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J. Virol. 83:4652–4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505 [DOI] [PubMed] [Google Scholar]
- 14. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136 [DOI] [PubMed] [Google Scholar]
- 15. Steegenga WT, Riteco N, Jochemsen AG, Fallaux FJ, Bos JL. 1998. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene 16:349–357 [DOI] [PubMed] [Google Scholar]
- 16. Lechner MS, Laimins LA. 1994. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J. Virol. 68:4262–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lechner MS, Mack DH, Finicle AB, Crook T, Vousden KH, Laimins LA. 1992. Human papillomavirus E6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. EMBO J. 11:3045–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yi F, Saha A, Murakami M, Kumar P, Knight JS, Cai Q, Choudhuri T, Robertson ES. 2009. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 388:236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao LY, Santiago A, Liu J, Liao D. 2007. Repression of p53-mediated transcription by adenovirus E1B 55-kDa does not require corepressor mSin3A and histone deacetylases. J. Biol. Chem. 282:7001–7010 [DOI] [PubMed] [Google Scholar]
- 20. Kim CM, Koike K, Saito I, Miyamura T, Jay G. 1991. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351:317–320 [DOI] [PubMed] [Google Scholar]
- 21. Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, Kurokawa K. 1994. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 19:810–819 [PubMed] [Google Scholar]
- 22. Madden CR, Finegold MJ, Slagle BL. 2001. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J. Virol. 75:3851–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seifer M, Hohne M, Schaefer S, Gerlich WH. 1991. In vitro tumorigenicity of hepatitis B virus DNA and HBx protein. J. Hepatol 13(Suppl. 4):S61–S65 [DOI] [PubMed] [Google Scholar]
- 24. Doria M, Klein N, Lucito R, Schneider RJ. 1995. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 14:4747–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henkler F, Hoare J, Waseem N, Goldin RD, McGarvey MJ, Koshy R, King IA. 2001. Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 82:871–882 [DOI] [PubMed] [Google Scholar]
- 26. Hoare J, Henkler F, Dowling JJ, Errington W, Goldin RD, Fish D, McGarvey MJ. 2001. Subcellular localisation of the X protein in HBV infected hepatocytes. J. Med. Virol. 64:419–426 [DOI] [PubMed] [Google Scholar]
- 27. Rahmani Z, Huh KW, Lasher R, Siddiqui A. 2000. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 74:2840–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takada S, Shirakata Y, Kaneniwa N, Koike K. 1999. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 18:6965–6973 [DOI] [PubMed] [Google Scholar]
- 29. Feitelson MA, Zhu M, Duan LX, London WT. 1993. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene 8:1109–1117 [PubMed] [Google Scholar]
- 30. Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. 1995. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J. Virol. 69:1851–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. 1994. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc. Natl. Acad. Sci. U. S. A. 91:2230–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maguire HF, Hoeffler JP, Siddiqui A. 1991. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science 252:842–844 [DOI] [PubMed] [Google Scholar]
- 33. Williams JS, Andrisani OM. 1995. The hepatitis B virus X protein targets the basic region-leucine zipper domain of CREB. Proc. Natl. Acad. Sci. U. S. A. 92:3819–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choi BH, Choi M, Jeon HY, Rho HM. 2001. Hepatitis B viral X protein overcomes inhibition of E2F1 activity by pRb on the human Rb gene promoter. DNA Cell Biol. 20:75–80 [DOI] [PubMed] [Google Scholar]
- 35. Sung WK, Lu Y, Lee CW, Zhang D, Ronaghi M, Lee CG. 2009. Deregulated direct targets of the hepatitis B virus (HBV) protein, HBx, identified through chromatin immunoprecipitation and expression microarray profiling. J. Biol. Chem. 284:21941–21954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cougot D, Wu Y, Cairo S, Caramel J, Renard CA, Levy L, Buendia MA, Neuveut C. 2007. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 282:4277–4287 [DOI] [PubMed] [Google Scholar]
- 37. Lin Y, Nomura T, Yamashita T, Dorjsuren D, Tang H, Murakami S. 1997. The transactivation and p53-interacting functions of hepatitis B virus X protein are mutually interfering but distinct. Cancer Res. 57:5137–5142 [PubMed] [Google Scholar]
- 38. Ogden SK, Lee KC, Barton MC. 2000. Hepatitis B viral transactivator HBx alleviates p53-mediated repression of alpha-fetoprotein gene expression. J. Biol. Chem. 275:27806–27814 [DOI] [PubMed] [Google Scholar]
- 39. Chung TW, Lee YC, Ko JH, Kim CH. 2003. Hepatitis B virus X protein modulates the expression of PTEN by inhibiting the function of p53, a transcriptional activator in liver cells. Cancer Res. 63:3453–3458 [PubMed] [Google Scholar]
- 40. Lee AT, Ren J, Wong ET, Ban KH, Lee LA, Lee CG. 2005. The hepatitis B virus X protein sensitizes HepG2 cells to UV light-induced DNA damage. J. Biol. Chem. 280:33525–33535 [DOI] [PubMed] [Google Scholar]
- 41. Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. 2002. The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl. Acad. Sci. U. S. A. 99:8467–8472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, Liu J, Zhao XD, Chew JL, Lee YL, Kuznetsov VA, Sung WK, Miller LD, Lim B, Liu ET, Yu Q, Ng HH, Ruan Y. 2006. A global map of p53 transcription-factor binding sites in the human genome. Cell 124:207–219 [DOI] [PubMed] [Google Scholar]
- 43. Ryoo HD, Gorenc T, Steller H. 2004. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7:491–501 [DOI] [PubMed] [Google Scholar]
- 44. Harrod R, Nacsa J, Van Lint C, Hansen J, Karpova T, McNally J, Franchini G. 2003. Human immunodeficiency virus type-1 Tat/co-activator acetyltransferase interactions inhibit p53Lys-320 acetylation and p53-responsive transcription. J. Biol. Chem. 278:12310–12318 [DOI] [PubMed] [Google Scholar]
- 45. Liu Y, Colosimo AL, Yang XJ, Liao D. 2000. Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell. Biol. 20:5540–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olsson A, Manzl C, Strasser A, Villunger A. 2007. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 14:1561–1575 [DOI] [PubMed] [Google Scholar]
- 47. Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. 2006. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 173:533–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, Wang JY, Anderson CW, Appella E, Xu Y. 2006. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol. Cell. Biol. 26:6859–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Terui T, Murakami K, Takimoto R, Takahashi M, Takada K, Murakami T, Minami S, Matsunaga T, Takayama T, Kato J, Niitsu Y. 2003. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res. 63:8948–8954 [PubMed] [Google Scholar]