

Abstract
Cell division control protein A7 (CDCA7) is a recently identified target of MYC-dependent transcriptional regulation. We have discovered that CDCA7 associates with MYC and that this association is modulated in a phosphorylation-dependent manner. The prosurvival kinase AKT phosphorylates CDCA7 at threonine 163, promoting binding to 14-3-3, dissociation from MYC, and sequestration to the cytoplasm. Upon serum withdrawal, induction of CDCA7 expression in the presence of MYC sensitized cells to apoptosis, whereas CDCA7 knockdown reduced MYC-dependent apoptosis. The transformation of fibroblasts by MYC was reduced by coexpression of CDCA7, while the non-MYC-interacting protein Δ(156–187)-CDCA7 largely inhibited MYC-induced transformation. These studies provide insight into a new mechanism by which AKT signaling to CDCA7 could alter MYC-dependent growth and transformation, contributing to tumorigenesis.
INTRODUCTION
The transcription factor MYC is a proto-oncogene that regulates the expression of hundreds of genes involved in cell cycle progression, adhesion, metabolism, and apoptosis (1–4). Overexpression of MYC is a hallmark of human cancer, contributing to the expression of numerous groups of genes involved in transformation, metastasis, and overall poor prognosis (5, 6). MYC has been estimated to be active in nearly 70% of human cancers; the mechanisms of activation include amplification, translocation, deregulated translation, and protein turnover (7, 8). As such, MYC has been the subject of extensive study in the search for treatment modalities (reviewed in references 3, 9, and 10).
Activation of MYC is induced by mitogenic stimuli to promote cell cycle progression (11–17). To prevent aberrant MYC expression from driving unsafe proliferation in the animal, a safeguard has evolved whereby MYC activation in the absence of mitogenic survival signals is opposed by cellular responses of apoptosis and/or cell cycle arrest, depending on the cellular context and p53 status (18–25). Despite these observations of almost 20 years ago and the realization that other growth-promoting transcription factors, such as E1A and E2F1, act similarly (26–29), the mechanism of MYC-induced apoptosis and cell cycle arrest is still poorly understood.
Expression of prosurvival oncogenes, the earliest example of which is Bcl-2, has been shown previously to counteract the death function of MYC (30–32). Additionally, activation of phosphoinositide-3-kinase (PI3K) and its downstream target AKT can protect against apoptosis induced by MYC (33). PI3K and AKT were shown in the mid-1990s to convey a strong prosurvival signal downstream of receptor tyrosine kinases (34–36) by impacting the apoptosis machinery directly (37–40) and by regulating FOXO transcription factors (41–45). Furthermore, loss of the tumor suppressor MMAC1/PTEN results in constitutive PI3K signals (46, 47) and can lead to tumors in humans (48, 49). Thus, aberrant MYC activation together with overactive AKT, a condition that is often achieved in tumor cells, can provide the cooperative growth and antiapoptotic signals necessary to promote tumorigenesis.
A recently identified protein, cell division control protein A7 (CDCA7; also called JPO1), is expressed from the MYC- and E2F-responsive gene cdca7 (50–52). MYC and E2F1 bind to the promoter of cdca7 to drive CDCA7 expression (50, 52), causing CDCA7 mRNA to be widely expressed, with high levels in the colon, thymus, and small intestine and lower levels in the testis, stomach, and bone marrow (52). High levels of CDCA7 mRNA have been found in patients with acute myeloid leukemia (AML) and blast crisis-stage chronic myeloid leukemia (CML) (51), while solid tumors displaying high levels of MYC have also been shown to be positive for CDCA7 (51). While CDCA7 has weak transformation properties when expressed alone, coexpression rescues the transformation of a transformation-defective MYC mutant with a MYC box II (MBII) deletion (52). Furthermore, recent work by Penn and colleagues has shown that JPO2, a protein with some homology with CDCA7, can associate directly with MYC, and this increases MYC-dependent transformation (53). Both CDCA7 and JPO2 contain a highly conserved cysteine-rich carboxyl-terminal region that might allow binding to DNA (50). However, it is not known whether CDCA7 also associates with MYC.
In the present study, we show that CDCA7 and MYC interact physically. We have mapped the domains of interaction and have discovered that AKT phosphorylates CDCA7 near this contact region, leading to loss of its association with MYC, binding to 14-3-3 proteins, and exclusion from the nucleus. Coexpression of CDCA7 with MYC sensitized cells to serum withdrawal-induced apoptosis, and this proapoptotic activity required the MYC-binding region. Short hairpin RNA interference (shRNAi)-mediated knockdown of CDCA7 rescued cells from MYC-dependent apoptosis following removal from serum. These findings point to a feed-forward loop whereby MYC activation upregulates CDCA7, with AKT activity controlling the accessibility of CDCA7 to nuclear MYC proportionately to growth factor signaling.
MATERIALS AND METHODS
Cell lines and cell culture.
HEK293 and Rat1 cells were obtained from the American Type Culture Collection and were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C under 5% CO2. AKT1/AKT2 (AKT1/2)-null mouse embryonic fibroblasts (MEFs) (54) were a gift from Veronique Nogueira of the University of Illinois at Chicago. Rat1-MYC cells were obtained by stable transfection of c-MYC and selection in 600 μg/ml G418 and 20% fetal bovine serum. Clones expressing both wild-type and mutant CDCA7 were established using the Tet-On advanced inducible gene expression system from Clontech. Specifically, stable clones expressing a tetracycline (Tet)-induced transactivator (rtTA) were obtained by stable transfection of Rat1 and Rat1-MYC cells with pIREShyg3-rtTA, followed by selection in 200 μg/ml hygromycin. Subsequently. the rtTA-Rat1 and rtTA-Rat1-MYC cells were cotransfected with plasmid pTRE-tight-3xFLAG-CDCA7 and a linearized puromycin plasmid at a 100:1 ratio, followed by selection in 2 μg/ml puromycin. All Rat1-MYC-derived inducible lines were maintained in 20% fetal bovine serum, while Rat1-CDCA7-derived inducible lines were maintained in 10% fetal bovine serum.
Antibodies and reagents.
Mouse monoclonal anti-Flag M2 (A2220) and rabbit polyclonal anti-CDCA7 (HPA005565) antibodies were purchased from Sigma-Aldrich. A rabbit polyclonal anti-CDCA7 antibody (Ab69609) was purchased from Abcam. Mouse monoclonal anti-14-3-3β (catalog no. SC-1657) and mouse monoclonal anti-MYC (9E10; catalog no. SC-40) antibodies were purchased from Santa Cruz. Rabbit polyclonal anti-c-MYC (5605), rabbit polyclonal anti-pan-AKT (9272), and mouse monoclonal anti-P-Ser473 AKT (4051) antibodies were purchased from Cell Signaling Technology. An anti-P-Thr163 CDCA7 rabbit polyclonal antibody was generated by GenScript Corporation (CA). AKT inhibitor VIII (A6730) and LY294002 (L9908) were purchased from Sigma-Aldrich.
Cloning.
The CDCA7 coding region was ligated into the mammalian expression vector p3XFLAG-CMV10 (Sigma-Aldrich) to introduce an amino-terminal FLAG epitope. Mutagenesis of p3XFLAG-CMV10-CDCA7 was performed using the QuikChange kit (Stratagene), and the various mutations were sequence verified. Amino-terminal and internal deletions were created by introducing silent mutations coding for unique restriction sites, followed by digestion and religation. Carboxyl-terminal deletions were created by introducing stop codons. All deletions were sequence verified. CDCA7 containing the 14-3-3-binding R18 peptide PHCVPRDLSWLDLEANMCLP or the control non-14-3-3-binding peptide PHCVPRDLSWLKLKANMCLP was created by ligating a double-stranded oligonucleotide to replace amino acids 139 to 164 of CDCA7 in the 3XFLAG-CMV10 vector. The C-terminally His tagged c-MYC plasmid was created by cloning the c-MYC coding region into pEcoli-C-terminal 6×HN (Clontech).
For transient and stable expression of cDNA in cells, HEK293, mouse NIH 3T3, or Rat1 cells were plated onto 100-mm-diameter dishes at 80% confluence and were transfected with 5 μg of plasmid by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The transfection medium was removed and was replaced with complete Dulbecco's modified Eagle's medium overnight.
For retroviral expression of CDCA7 in Rat1 cells, 3XFLAG-CDCA7 or CDCA7 with various mutations was cloned into the pQCXIH retroviral vector (Clontech) upstream of an internal ribosome entry site (IRES) sequence and the hygromycin resistance gene. Either an empty pQCXIH retroviral vector or pQCXIH-CDCA7-IRES-Hygro was transfected into the ecotropic-envelope protein packaging cell line EcoPack 2-293 (Clontech). Viral particles were collected 48 h later and were concentrated using a Retro-X concentrator (Clontech).
Short hairpin RNA (shRNA) cloning and quantitative reverse transcription-PCR (qRT-PCR).
Oligonucleotides 5′-GATCCCCAATGAAGTTCCCAGCACGATTCAAGAGATCGTGCTGGGAACTTCATTTTTTTA-3′, 5′-GATCCCCAGTTCCCAGCACGAAATACTTCAAGAGAGTATTTCGTGCTGGGAACTTTTTTA-3′, and 5′-GATCCCCCTGTGGACGGCTATATGAATTCAAGAGATTCATATAGCCGTCCACAGTTTTTA-3′, targeting rat CDCA7 at nucleotides 212, 220, and 658, respectively, were annealed, ligated into the pSuper vector, and transfected into Rat1-MYC cells. Several colonies of each targeting region were isolated following puromycin selection.
Total RNA was purified and cDNA synthesized using the ProtoScript Moloney murine leukemia virus (M-MuLV) first-strand cDNA synthesis kit from New England Biolabs (NEB). shRNA knockdown was quantified, and clones were screened, by using RT2 SYBR green qPCR master mix (SABiosciences) and the 7500 Fast RT-PCR system (Applied Biosystems).
Cell lysis, immunoprecipitation, and immunoblotting.
Cells were lysed in either radioimmunoprecipitation assay (RIPA) lysis buffer (10 mM NaPO4 [pH 7.6], 150 mM NaCl, 5 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 0.25% deoxycholic acid, 1% Triton X-100, plus protease and phosphatase inhibitors) or Triton X-100 lysis buffer (50 mM HEPES [pH 7.9], 250 mM NaCl, 0.1% Triton X-100, 10% glycerol, plus protease and phosphatase inhibitors). Ten microliters of anti-FLAG M2 agarose-conjugated beads (Sigma-Aldrich) was added to lysates, and the mixture was incubated overnight at 4°C. The beads were washed five times with lysis buffer, and proteins were first eluted with 200 μl of lithium dodecyl sulfate (LDS) sample buffer and then heated to 70°C for 10 min. Portions of the lysates were also reserved prior to immunoprecipitation and were boiled with a lithium dodecyl sulfate-containing sample buffer. Lysates and immunoprecipitates were fractionated by SDS-polyacrylamide gel electrophoresis (PAGE), transferred to a polyvinylidene difluoride (PVDF) membrane, blocked in 5% skim milk for 30 min, and probed with the appropriate antibody overnight at room temperature. Primary antibodies were decorated with an IR700 anti-mouse and/or an IR800 anti-rabbit secondary antibody (Li-Cor Biosciences) for 3 h at room temperature, and fluorescent images on the PVDF membranes were visualized using an infrared laser scanning system (Odyssey; Li-Cor Biosciences).
Microscopy.
Rat1, NIH 3T3, or HEK293cells were plated at 80% confluence on no. 1 glass coverslips. Following the desired treatment, the coverslips were washed twice in phosphate-buffered saline (PBS) and were then fixed and permeabilized in methanol-acetic acid (3:1) for 25 min. The coverslips were then washed three times in PBS, followed by blocking in 1% bovine serum albumin (BSA) in PBS. Primary antibodies at varying concentrations were applied for 1.5 h in a humidified chamber at room temperature. Coverslips were washed three times in PBS prior to the application of secondary antibodies for 2 h in a humidified chamber at room temperature in the dark. The coverslips were washed three times in PBS before mounting with ProLong Gold antifade reagent (Invitrogen) with 4′,6-diamidino-2-phenylindole (DAPI). The slides were viewed on an Olympus microscope, and images were taken via a QImaging 2000R camera and QCapture Pro software.
Live-cell imaging was performed on an inverted Olympus microscope equipped with an Ultraview ERS spinning-disc unit, a 37°C heated stage, and a 60× heated objective. NIH 3T3 cells were transfected in no. 1 coverslip chambers overnight, serum starved for 4 h, and stimulated with platelet-derived growth factor (PDGF). Confocal images were taken every 5 min for the duration of the time course and were analyzed with Perkin-Elmer Ultraview ERS software.
Peptide competition assay.
The Thr163 peptide (CDSKSPRRRTFPG) and phosphopeptide [CDSKSPRRR(p)TFPG] corresponding to the sequence surrounding Thr163 were synthesized by GenScript. Triton X-100 lysates containing FLAG-CDCA7 were immunoprecipitated with anti-FLAG M2 agarose-conjugated beads overnight at 4°C. Following washing, beads were incubated at 4°C for 1 h with various concentrations of the peptide or phosphopeptide. The beads were washed three times, and proteins were eluted and resolved by SDS-PAGE. Coimmunoprecipitated 14-3-3 was detected by using a rabbit polyclonal antibody against 14-3-3β.
In vitro pulldown assay.
HEK293 cells were transfected overnight with FLAG-CDCA7 and various mutants. Cells were lysed in RIPA lysis buffer and were immunoprecipitated with FLAG M2 agarose-conjugated beads overnight at 4°C. Agarose beads were washed 3 times with RIPA lysis buffer and then 3 times with Triton X-100 lysis buffer. Immunoprecipitates were then eluted by incubation at 4°C with 20 μM FLAG peptide in Triton X-100 lysis buffer.
For the generation of recombinant MYC, DH5α bacteria expressing 6×HN-MYC were cultured overnight in 100 ml Terrific broth (TB) at 37°C with shaking. The following day, 1 liter of TB from the overnight culture was inoculated. Cells were stimulated with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when the optical density at 600 nm (OD600) reached 1.0. The culture was then incubated at 16°C overnight. Cells were pelleted by centrifugation and were resuspended in 60 ml of binding buffer (50 mM Tris [pH 7.5], 500 mM NaCl, 5 mM imidazole). Cells were lysed by sonication at 30% amplitude, 2 s on, 3 s off, for a total of 2 min. Insoluble precipitates were removed by centrifugation, and the supernatant was bound to Ni-nitrilotriacetic acid (NTA) beads at 4°C for 20 min with gentle rocking. Ni-NTA beads were washed with 10 column volumes of wash buffer (50 mM Tris [pH 7.5], 500 mM NaCl, 30 mM imidazole). 6×HN-MYC was eluted in 5-ml fractions with elution buffer (50 mM Tris [pH 7.5], 500 mM NaCl, 250 mM imidazole). Purified 6×HN-MYC was dialyzed against three changes of HKG buffer (20 mM HEPES [pH 7.4], 125 mM KCl, 20% [vol/vol] glycerol).
For pulldown experiments, 500 ng of purified His-MYC protein was bound to Ni-NTA agarose beads and was incubated with purified FLAG-CDCA7 protein for 4 h at 4°C in Triton X-100 lysis buffer. The beads were washed 5 times, and the proteins were eluted with 200 μl of lithium dodecyl sulfate sample buffer heated to 70°C for 10 min. Portions of the input protein prior to binding were also boiled with lithium dodecyl sulfate-containing sample buffer. Input and bound proteins were fractionated by SDS-PAGE.
Apoptosis assays.
For annexin assays, cells were harvested with EDTA-free trypsin, washed twice at 4°C with ice cold PBS, and stained by resuspension in 200 μl annexin binding buffer (10 mM HEPES [pH 7.4], 140 mM NaCl, 2.5 mM CaCl2) containing 5 ml annexin V–Alexa Fluor 488 conjugate and 1 μg/ml propidium iodide. The proportion of apoptotic cells (annexin V positive, propidium iodide negative) was determined by using a FACScan flow cytometer and CellQuest software (Becton Dickinson). Caspase assays were performed by scraping approximately 106 cells in caspase assay buffer (25 mM HEPES [pH 7.4], 5 mM EDTA, 2 mM dithiothreitol [DTT], 137 mM NaCl, 10% glycerol). Cells were sonicated at low power, and insoluble materials were pelleted by centrifugation. One hundred fifty microliters of the lysate was incubated at 37°C with 10 μM DEVD substrate (N-acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin; Sigma). Samples were read every 15 min at an excitation wavelength of 380 nm and an emission wavelength of 450 nm, and caspase-3 activity was normalized to total protein as determined by a Bradford assay.
Metabolic labeling.
HEK293 cells were transfected overnight with FLAG-CDCA7, washed, and incubated in a phosphate-free medium containing 1 mCi/ml 32Pi for 4 h. Cells were lysed in 1 ml RIPA buffer, and FLAG-CDCA7 was immunoprecipitated overnight at 4°C with beads prebound to anti-FLAG M2 monoclonal antibody. Immunoprecipitates were fractionated by 8% SDS-PAGE and were Coomassie stained, and 32P-labeled CDCA7 was detected by autoradiography.
Tryptic mapping.
32P-labeled CDCA7 was isolated as described above, excised from the gel, and digested with 10 μg/ml tosylphenylalanylchloromethyl ketone-treated trypsin (Promega) in 50 mM (NH4)HCO3 (pH 7.8) overnight at 37°C. Peptides were isolated as described elsewhere (55).
In vitro AKT phosphorylation assay.
FLAG-tagged CDCA7 was immunoprecipitated from cell lysates with 25 μl anti-FLAG M2 monoclonal antibody conjugated to agarose (Sigma-Aldrich). Immunoprecipitates were washed three times in RIPA buffer, followed by an additional two washes in a reaction buffer containing 40 mM morpholinepropanesulfonic acid (MOPS)-NaOH (pH 7.0) and 1 mM EDTA. The samples were then resuspended in reaction buffer plus 0.5 μg of active AKT (S472D) (Millipore). The kinase reaction was started by the addition of an ATP mixture containing 8 μM magnesium acetate, 10 μM ATP, and 1 μCi of [32P]ATP. Samples were incubated for 20 min at 30°C; the supernatant was then removed to fresh tubes; and CDCA7 immunoprecipitates were washed three times with reaction buffer and were finally resuspended in 1× LDS sample buffer. The supernatant containing AKT and the CDCA7 immunoprecipitates were resolved by SDS-PAGE. 32P-labeled CDCA7 was visualized with a phosphorimager (Bio-Rad Laboratories).
Soft-agarose colony formation assay.
Rat1 cells were infected with a retrovirus generated by the pQCXIH-CDCA7-IRES-Hygro vector. After 5 to 7 days of selection in hygromycin (200 μg/ml), resistant pools were counted and plated in complete medium containing 0.3% agarose and hygromycin over a bottom layer of complete medium containing 0.7% agarose and hygromycin. Fresh medium with serum was added to the plates every 2 days, and after 2 weeks, the plates were stained with crystal violet, destained, and visualized by infrared scanning on an Odyssey laser scanner. Macroscopic colonies were counted using ImageJ software.
RESULTS
CDCA7 binds to the carboxyl terminus of MYC.
Figure 1A depicts the various regions of CDCA7 and their postulated biological roles, including a C-terminal zinc finger domain that could interact with DNA, a leucine zipper (LZ) region that could promote hetero- and homodimerization, and a putative nuclear localization signal (NLS) between amino acids 170 and 190 of human CDCA7. Conservation within this region is very high between species. We performed coprecipitation experiments, and deletions of both N-terminal and C-terminal segments of CDCA7 revealed that a small region of CDCA7, amino acids 146 to 170, was essential for interaction with MYC (Fig. 1B and C). In reciprocal experiments we found that CDCA7 lost association with MYC harboring a deletion of the C-terminal region (Δ274–440) but not with MYC lacking the N-terminal MYC box I (MBI) and MBII domains (Δ5–149) or with MYC lacking the central region containing the MBIII domain (Δ151–274) (Fig. 1D). Finally, immunoprecipitation of endogenous MYC led to coprecipitation of endogenous CDCA7, indicating interaction of physiological levels of these proteins (Fig. 1E).
Fig 1.

Mapping of CDCA7 and MYC association. (A) Sequence alignment of amino acids 131 to 180 of human CDCA7 with the corresponding regions of CDCA7 from various other species. The asterisk indicates the position of a putative AKT phosphorylation/14-3-3 binding site. (B) (Top) Purified CDCA7 or various truncated versions of CDCA7 were incubated with His-MYC and were subjected to pulldown with Ni-agarose, followed by immunoblotting with an anti-FLAG (α-FLAG) antibody. (Bottom) A reserved portion of the input CDCA7 was also immunoblotted with anti-FLAG. WT, wild type. (C) Schematic diagram of the region of interaction. (D) Purified full-length His-MYC or various truncated forms of His-MYC were incubated with purified FLAG-CDCA7 or Δ156-187-CDCA7, subjected to pulldown with Ni-agarose, and immunoblotted with an anti-FLAG antibody. Input His-MYC was detected with an N-terminal anti-MYC antibody (D84C12) or a C-terminal antibody (9E10). (E) Endogenous MYC was immunoprecipitated (IP) from HEK293 cells, and endogenous CDCA7 was detected by immunoblotting with anti-CDCA7 (Sigma). An anti-FLAG antibody was used as a nonspecific immunoprecipitation control.
The region of CDCA7 that associates with MYC is remarkable for several reasons. First, there appears to be a putative bipartite NLS domain spanning amino acids 160 to 190 (Fig. 1A). Second, threonine 163 conforms to the consensus AKT phosphorylation site, with arginines at −5 and −3 and a bulky hydrophobic amino acid at +1. Furthermore, there is a proline at +2, and the same arrangement of residues is present in other AKT substrates that bind to the 14-3-3 phosphoadapter proteins, including FOXO3α (42, 43, 45). Based on these initial observations, we formulated the hypothesis that the association of MYC and CDCA7 could be regulated in a transient manner though phosphorylation at Thr163 by AKT and binding to 14-3-3.
To test whether Thr163 is a site of phosphorylation, we performed phosphotryptic mapping of CDCA7 isolated from cells labeled with 32Pi (Fig. 2A). One spot comigrated with a synthetic phosphotryptic peptide for this residue, which disappeared upon the mutation of Thr163 to alanine. Next, we generated a phosphospecific antibody to this site. The anti-phospho-Thr163 antibody reacted with wild-type CDCA7 but not with T163A-CDCA7 (Fig. 2B). Treatment of CDCA7 with calf intestinal phosphatase resulted in a significant reduction in immunoreactivity (Fig. 2B). These data establish that Thr163 is phosphorylated in cells.
Fig 2.
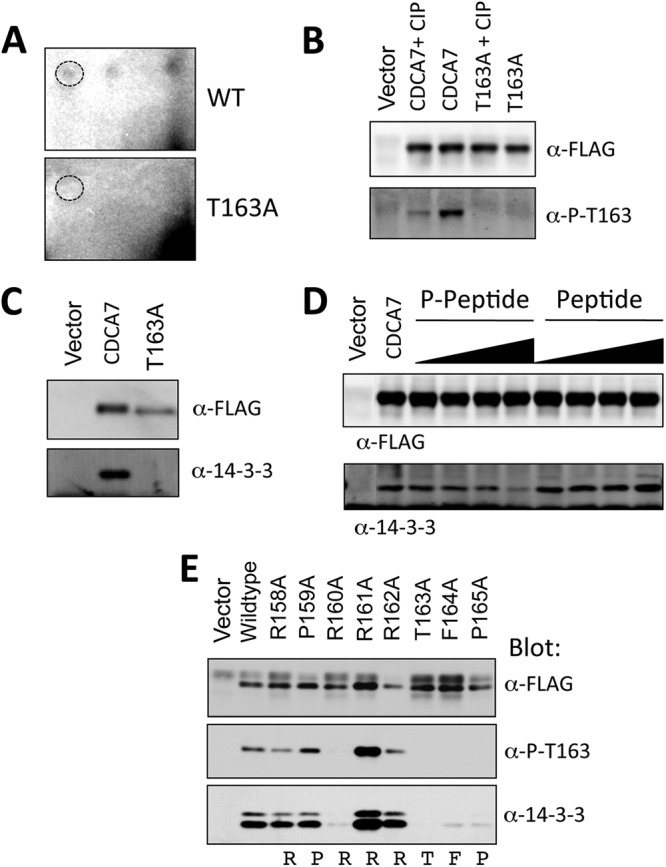
CDCA7 is phosphorylated at Thr163 and binds to 14-3-3. (A) Two-dimensional tryptic maps of wild-type CDCA7 and T163A-CDCA7. Dashed circles indicate the position of the cold synthetic Thr163 tryptic peptide that comigrated with 32P-labeled peptides. (B) A phospho-specific antibody raised against the phospho-Thr163 peptide was used to immunoblot CDCA7, CDCA7 pretreated with calf intestinal phosphatase (CIP), or T163A-CDCA7. (C) CDCA7 or T163A-CDCA7 was expressed in cells and was immunoprecipitated with anti-FLAG–agarose. Coimmunoprecipitated proteins were subjected to anti-FLAG or anti-14-3-3 immunoblotting to detect endogenous 14-3-3. (D) 14-3-3 bound to CDCA7 was competed with increasing concentrations of the synthetic phospho-Thr163 peptide or the unphosphorylated peptide. Following several washes, proteins were resolved by PAGE and were immunoblotted with an anti-FLAG or anti-14-3-3 antibody to detect coimmunoprecipitated endogenous 14-3-3. (E) CDCA7 containing the indicated mutations was immunoprecipitated, and proteins were immunoblotted with anti-FLAG, anti-phospho-Thr163, or anti-14-3-3.
Next, we immunoprecipitated CDCA7 and found that it coprecipitated endogenous 14-3-3 in a phospho-Thr163-dependent manner (Fig. 2C). Addition of a molar excess of a synthetic Thr163-containing phosphopeptide, but not the corresponding unphosphorylated peptide, caused a loss of 14-3-3 binding (Fig. 2D). We mutated each residue surrounding Thr163 to alanine, starting at −7, and looked for 14-3-3 binding and reactivity to the phospho-specific antibody. Both the −5 and −3 arginines, Arg158 and Arg160, were absolutely required for 14-3-3 binding and Thr163 phosphorylation (Fig. 2E). Mutation of phenylalanine 164 or proline 165 resulted in loss of phospho-Thr163 reactivity and loss of 14-3-3 binding. Interestingly, mutation of Arg161 or Arg162 to alanine caused an increase in 14-3-3 binding and an apparent increase in Thr163 phosphorylation.
Binding of 14-3-3 is known to alter numerous biological functions of target proteins, including subcellular localization (56). In cells grown in 10% serum, overexpressed wild-type CDCA7 resided primarily in the nucleus and to a lesser extent in the cytoplasm (Fig. 3A). In contrast, T163A-CDCA7 was entirely nuclear, with no cytoplasmic localization. The CDCA7 mutation R161A caused both cytoplasmic and nuclear staining, correlating with the increased binding to 14-3-3 seen in Fig. 2E. The P165A mutation, which abolished 14-3-3 binding, resulted in entirely nuclear localization. We quantified these observations by calculating the ratio of the fluorescence intensity of the cytoplasm (Fc) to that of the nucleus (Fn) for each mutation. R161A-CDCA7, as expected, had the highest Fc/Fn ratio, which was similar to that for wild-type CDCA7 stimulated with fibroblast growth factor (FGF). The CDCA7 mutants with T163A, F164A, or P165A all had low Fc/Fn ratios, consistent with their predominantly nuclear staining (Fig. 3A) and their low level of 14-3-3 binding (Fig. 2E).
Fig 3.

Thr163 phosphorylation alters CDCA7 localization. (A) NIH 3T3 cells were transfected with FLAG-CDCA7 containing the indicated mutations on coverslips. After 24 h, cells were fixed in methanol, and immunofluorescence microscopy was performed to detect CDCA7. (B) NIH 3T3 cells were transfected with various FLAG-CDCA7 cDNAs for 24 h, followed by the stimulation of some samples with FGF for the indicated times. Cells were fixed, stained with anti-FLAG, and visualized by confocal microscopy. The fluorescence intensities of the cytoplasm (Fc) and the nucleus (Fn) were measured in a blind analysis for each sample, and the Fc/Fn ratio was calculated. Error bars represent the standard errors of the means for a minimum of 30 cells for each sample. (C) Cells expressing CDCA7, T163A-CDCA7, DE-CDCA7, or KK-CDCA7 were lysed, and CDCA7 was immunoprecipitated with anti-FLAG M2 agarose. Coimmunoprecipitated endogenous 14-3-3 was detected by anti-14-3-3 immunoblotting. (D) NIH 3T3 cells were plated on coverslips and were transfected with FLAG-DE-CDCA7 or FLAG-KK-CDCA7. After 24 h, cells were fixed, stained with an anti-FLAG antibody, and subjected to immunofluorescence microscopy.
Next, we asked if constitutive 14-3-3 binding independent of Thr163 phosphorylation could regulate localization. To test this, we replaced the 20 amino acids surrounding Thr163 with the R18 peptide (PHCVPRDLSWLDLEANMCLP). This peptide was isolated from a phage display screen for peptides that bind strongly to 14-3-3; the D and E residues underlined in the sequence above form contacts with 14-3-3 and substitute for phospho-amino acids (57). We found that DE-CDCA7 bound to 14-3-3 and resulted in cytoplasmic localization of CDCA7 (Fig. 3C and D). As a control, we generated a protein in which the D and E residues were replaced with lysine. KK-CDCA7 did not bind to 14-3-3 and was entirely nuclear (Fig. 3C and D). Thus, the binding of 14-3-3 at Thr163 of CDCA7 appeared to be responsible for its shift to the cytoplasm in cells.
The region comprising amino acids 160 to 176 is a bipartite nuclear localization signal.
To account for the shift in localization due to Thr163 phosphorylation and 14-3-3 binding, we searched CDCA7 for a proximal NLS domain that could be influenced by 14-3-3. The presence of basic arginine residues at positions 160, 161, and 162, and the presence of another cluster at positions 170, 171, 175, and 176, resembled the pattern of the bipartite NLS domains of RB and SWI5 (Fig. 4A). To test if these residues constituted an NLS domain, we asked whether the isolated region spanning amino acids 157 to 188 of CDCA7 could import a large cargo protein. We placed our test region between green fluorescent protein (GFP) and β-galactosidase (β-gal), creating a 140-kDa protein. A bona fide NLS isolated from simian virus 40 (SV40) caused the GFP–β-gal fusion protein to localize to the nucleus (Fig. 4B). The introduction of a point mutation within the SV40 NLS resulted in cytoplasmic localization, validating this model for testing NLS activity. The insertion of amino acids 157 to 188 of CDCA7 resulted in nuclear localization, as did the NLS of SV40 (Fig. 4B). In contrast, subdivision of the full 157-to-188 region into smaller regions, the region surrounding just the 14-3-3 binding site (amino acids 157 to 167) or the second cluster of arginines from amino acid 167 to 188, led to entirely cytoplasmic localization (Fig. 4B). This suggests that the entire region comprising amino acids 157 through 188 is needed for the nuclear import of the GFP–β-galactosidase fusion protein.
Fig 4.

Amino acids 160 to 176 of CDCA7 constitute a putative bipartite NLS. (A) Sequence alignment of CDCA7 with the bipartite NLS domains of human RB protein and SWI5 protein. (B) NIH 3T3 cells were transfected on glass coverslips with a vector expressing pFloc-GFP-SV40-β-gal, pFloc-GFP-SV40KE-β-gal, or pFloc-GFP-CDCA7peptide-β-gal, containing various regions of CDCA7, as indicated, for 24 h. The following day, GFP signals in live cells were visualized by fluorescence microscopy. (C) pFloc-GFP-CDCA7peptide-β-gal containing amino acids 156 to 188 of CDCA7 and the indicated mutations was transfected into NIH 3T3 cells plated on coverslips. The next day, cells were fixed with methanol and were visualized by confocal microscopy.
We next tested the importance of the specific arginine residues within the putative NLS. Mutation of Arg160, Arg161, or Arg162 shifted the localization of the fusion protein to the cytoplasm (Fig. 4C). Similarly, mutation of Arg171 or Arg176 individually to glutamic acid shifted the balance of localization from the nucleus to the cytoplasm, while the double mutation of R171E and R176E led to complete nuclear exclusion (Fig. 4C). Finally, mutation of Arg182 or Arg184 had no effect on localization (Fig. 4C). These results indicate that arginines 160, 161, 162, 171, and 176 compose the critical basic residues of a potential bipartite NLS within CDCA7.
AKT phosphorylates CDCA7 Thr163.
Next, we asked whether Thr163 phosphorylation changed in cells treated with mitogenic stimuli. We expressed CDCA7 in NIH 3T3 fibroblasts, followed by stimulation with PDGF, and found that Thr163 phosphorylation increased rapidly and remained sustained for several hours (Fig. 5A). Furthermore, we found that PDGF stimulation led to increased cytoplasmic localization, which lagged behind Thr163 phosphorylation (Fig. 5B). Finally, GFP-CDCA7 was visualized in live cells over a time course of PDGF stimulation (Fig. 5C and D). This experiment showed that the intensity of the cytoplasmic GFP-CDCA7 signal increased approximately 3-fold, which agreed well with the kinetics observed by immunoblotting (Fig. 5B).
Fig 5.

PDGF stimulates CDCA7 phosphorylation at Thr163 and cytoplasmic translocation. (A) NIH 3T3 cells were transfected with FLAG-CDCA7. On the following day, the cells were starved of serum for 6 h and were stimulated with PDGF for the indicated times. The cells were then solubilized in 0.25% NP-40 buffer, and nuclei were removed by centrifugation. Lysates were probed with anti-phospho-Thr163 (α-P-T163) and anti-FLAG (α-FLAG), and both signals were detected simultaneously on an Odyssey infrared scanner. The bottom panel shows the merge of the phospho-Thr163 (680 nm) and FLAG (800 nm) signals; the ratios of their intensities are given below. (B) Graphical representation of the data shown in panel A. (C) NIH 3T3 cells were transfected with GFP-CDCA7 in a no. 1 glass-bottom chamber. After 24 h, the cells were starved of serum for 6 h and were visualized on a spinning-disc confocal microscope. At 0 min (T = 0′), PDGF was added; the cells were then captured every 5 min for 1 h. (D) The intensity of fluorescence in the cytoplasm was determined using Ultraview software (Perkin-Elmer) and was plotted against the time of PDGF stimulation. Fluorescence intensity in the nuclei was also measured; it decreased by approximately 5% over the time course (not shown). (E) NIH 3T3 cells were transfected on glass coverslips with FLAG-CDCA7 or T163A-CDCA7 for 24 h. Following 6 h of serum starvation, cells were stimulated with PDGF for the indicated times, fixed with methanol, and visualized with anti-FLAG and anti-phospho-Thr163 immunofluorescence microscopy. WT, wild type. (F) NIH 3T3 cells were transfected with CDCA7 or T163A-CDCA7. On the following day, cells were incubated with 50 mM LY294002 for 1 h; then nuclear and cytoplasmic extracts were isolated. CDCA7, endogenous AKT, and P-Thr308 AKT were detected by immunoblotting with anti-FLAG, anti-AKT, and anti-P-Thr308 AKT antibodies, respectively.
We then expressed CDCA7 or T163A-CDCA7 in NIH 3T3 cells and performed immunofluorescence with an anti-phospho-Thr163 antibody (Fig. 5E). This showed that there was a small amount of primarily nuclear phospho-Thr163 signal in unstimulated cells. Stimulation with PDGF for 15 and 60 min resulted in increases in both the nuclear and cytoplasmic phospho-Thr163 signals, corresponding to the appearance of cytoplasmic CDCA7. Finally, we isolated nuclear and cytoplasmic fractions of cells expressing CDCA7 following treatment with LY294002, a PI3K inhibitor (Fig. 5F). Cytoplasmic CDCA7 was present in cells maintained in serum but was abolished upon treatment with LY294002. The CDCA7 T163A mutation caused very little detectable cytoplasmic CDCA7.
We were able to detect endogenous CDCA7 in NIH 3T3 cells by immunofluorescence using an antibody raised against CDCA7 (Abcam). This experiment showed that under serum-starved conditions, endogenous CDCA7 was predominantly nuclear, with some cytoplasmic staining (Fig. 6A). Stimulation with PDGF for 30 min caused a pronounced shift to the cytoplasm. Pretreatment of cells with LY294002, wortmannin, or AKT inhibitor VIII, each of which is a potent inhibitor of PI3K and AKT, completely reversed the cytoplasmic localization of endogenous CDCA7 in response to PDGF, with pronounced staining of endogenous CDCA7 in the nucleus. Moreover, stimulation of cells with PDGF caused an increase in the amount of detectable endogenous Thr163 phosphorylation (Fig. 6B). The localization of endogenous CDCA7 phosphorylated at Thr163 was different from that of ectopically expressed CDCA7 or GFP-CDCA7: rather than being localized both to the nucleus and to the cytoplasm, the endogenous phospho-CDCA7 signal was detected primarily in the cytoplasm at 60 min. This difference might result from the smaller amount of endogenous CDCA7 protein than of overexpressed CDCA7, which could overwhelm the cell's resources for shuttling phospho-CDCA7 from the nucleus. Pretreatment with LY294002 abolished the elevated Thr163 phosphorylation induced by PDGF (Fig. 6B).
Fig 6.

Endogenous CDCA7 is phosphorylated at Thr163 and translocates to the cytoplasm upon PDGF stimulation. (A) NIH 3T3 cells were plated on glass coverslips and were starved of serum for 6 h. PDGF was added for 1 h together with either LY294002 (25 μM), wortmannin (100 nM), AKT inhibitor VIII (2.5 μM), or a vehicle only, as indicated. Cells were fixed in methanol, stained with anti-CDCA7 and DAPI, and visualized by florescence microscopy. (B) The experiment was similar to that described for panel A, except that cells were stained with anti-phospho-Thr163.
The observations that LY294002 could block the phosphorylation of endogenous Thr163 and reduce the cytoplasmic localization of CDCA7 suggested that a downstream effector of PI3K, such as AKT, might phosphorylate CDCA7 at this site. Treatment of cells with LY294002 reduced Thr163 phosphorylation (Fig. 7A). To determine whether this resulted from a loss of AKT activation, we coexpressed Gag-AKT, a membrane-bound, constitutively active form, with wild-type CDCA7 or T163A-CDCA7. Gag-AKT caused an elevation of Thr163 phosphorylation of CDCA7 and 14-3-3 binding (Fig. 7B) that was lost upon the mutation of Thr163 to alanine. Active recombinant AKT directly phosphorylated CDCA7, but not T163A-CDCA7, in vitro (Fig. 7C). Inhibition of AKT in cells by use of AKT inhibitor VIII blocked the phosphorylation of CDCA7 in cells (Fig. 7D). Coexpression of Gag-AKT with CDCA7 caused a redistribution of wild-type CDCA7, but not of T163A-CDCA7, from the nucleus to the cytoplasm (Fig. 7E). Finally, expression of CDCA7 in mouse embryonic fibroblasts in which AKT1 and AKT2 were deleted by homologous recombination resulted in diminished Thr163 phosphorylation (Fig. 7F). The level of PDGF-stimulated phosphorylation of Thr163 in AKT1/2−/− MEFs was lower than that in wild-type MEFs, a reduction similar to that in the level of total Ser473 phosphorylation detected for the remaining endogenous AKT3 (Fig. 7G). Together, these experiments provide strong evidence that AKT catalyzes Thr163 phosphorylation.
Fig 7.

AKT phosphorylates CDCA7 at Thr163. (A) Cells expressing CDCA7 were treated with the PI3K inhibitor LY294002 for 1 h and were then lysed. Immunoblotting was performed to detect Thr163 phosphorylation and total CDCA7. (B) 293 cells were transfected with CDCA7 together with an empty vector or Gag-AKT. Twenty-four hours later, CDCA7 was immunoprecipitated (IP), and phosphorylated Thr163 and coimmunoprecipitated endogenous 14-3-3 were visualized by immunoblotting. (C) Purified CDCA7 was phosphorylated in vitro with recombinant active S473D-AKT and [γ-32P]ATP. Proteins were resolved by PAGE and were visualized by autoradiography. Total CDCA7 and AKT were detected by immunoblotting. (D) Cells expressing CDCA7 were treated with AKT inhibitor VIII (0.5 and 2.5 μM) for 20 min and were then lysed. Phospho-Thr163 and total CDCA7 were detected by immunoblotting. (E) NIH 3T3 cells were transfected with WT CDCA7 or T163A-CDCA7 together with the vector or Gag-AKT. The next day, cells were fixed and were stained with anti-FLAG (α-FLAG) (for total CDCA7) and anti-phospho-S473 (α-P-T163) (for active AKT). (F) AKT1/2+/+ and AKT1/2−/− MEFs were transfected with CDCA7 on glass coverslips overnight in the presence of serum, fixed with methanol, stained with anti-FLAG and anti-P-T163, and visualized by immunofluorescence microscopy. (G) AKT1/2+/+ and AKT1/2−/− MEFs were transfected with CDCA7 for 24 h, starved of serum overnight, and then stimulated with PDGF for 1 h. (Top) CDCA7 was detected by immunoblotting with anti-FLAG and anti-P-T163 antibodies. The ratio of the P-T163 signal to the FLAG signal is given below each lane. The signals were obtained by direct immunofluorescence detection on an Odyssey infrared imager. (Bottom) Lysates were also probed with anti-pan-AKT, which detects AKT1, AKT2, and AKT3, and anti-P-S-473 AKT (both from Cell Signaling Technology). The arrowhead indicates the AKT band.
Binding of 14-3-3 regulates CDCA7-MYC interaction.
Since AKT phosphorylates CDCA7 and promotes 14-3-3 binding very near the residues required for MYC interaction, we thought that phosphorylation-dependent 14-3-3 binding could cause a change in association between CDCA7 and MYC. When we compared the abilities of wild-type CDCA7 and T163A-CDCA7 to coimmunoprecipitate MYC from cells, we found that more MYC bound to T163A-CDCA7 than to the equivalent amount of wild-type CDCA7 (Fig. 8A). Using purified proteins in pulldown experiments, we found that DE-CDCA7, which binds to 14-3-3 constitutively, immunoprecipitated less MYC; the loss of MYC interaction was similar to that seen with Δ(156–187)-CDCA7 (Fig. 8B). On the other hand, the non-14-3-3-interacting control protein, KK-CDCA7, did bind to MYC, about as well as wild-type CDCA7.
Fig 8.

14-3-3 competes with MYC for binding to CDCA7. (A) HEK293 cells were transfected with MYC together with an empty vector, FLAG-CDCA7, or FLAG-T163A-CDCA7. Detergent-solubilized cell extracts were prepared, and FLAG-CDCA7 was immunoprecipitated with the anti-FLAG M2 antibody. (B) Recombinant His-MYC was purified on Ni-agarose, and wild-type (WT) CDCA7, Δ(153–188)-CDCA7, DE-CDCA7, or KK-CDCA7 was added. The beads were rotated 1 h, washed, and boiled in sample buffer. Coprecipitated CDCA7 was detected by FLAG immunoblotting. (C) CDCA7 was immunoprecipitated from cells and was incubated with recombinant His-MYC with or without recombinant 14-3-3. CDCA7 bound to MYC was precipitated by Ni-agarose. 14-3-3 specifically reduced the amount of phospho-Thr163 CDCA7 bound to MYC. (D) Rat1 fibroblasts were cotransfected with FLAG-CDCA7 and CMV-MYC plasmids and were immunostained, and localization was visualized by confocal florescence microscopy. MYC colocalizes with CDCA7 in the nucleus when Thr163 is mutated to alanine, or with the non-14-3-3-binding mutant containing the R18 peptide (KK), but not with wild-type CDCA7 or the 14-3-3-binding CDCA7 mutant containing the R18 peptide (DE).
If 14-3-3 competes directly with MYC for sites of association on CDCA7, then recombinant 14-3-3 should compete with MYC for CDCA7 binding. Therefore, we incubated recombinant MYC with purified FLAG-CDCA7 that had been stripped of endogenous 14-3-3 by SDS treatment, in the presence or absence of recombinant 14-3-3 (Fig. 8C). We reasoned that a fraction of the purified CDCA7 binding to MYC would be phosphorylated at Thr163 and that if 14-3-3 competes with MYC, the addition of 14-3-3 should break this interaction. This was found to be the case: 14-3-3 dramatically reduced the amount of Thr163-phosphorylated CDCA7 coprecipitating with MYC (Fig. 8C). Together, these experiments indicate that 14-3-3 competes with MYC for binding to CDCA7. Furthermore, when coexpressed in Rat1 cells, MYC colocalizes with non-14-3-3-binding CDCA7 mutants T163A-CDCA7 and KK-CDCA7, while MYC colocalizes to a lesser degree with wild-type CDCA7 and the 14-3-3-binding DE mutant containing the R18 peptide substitution (Fig. 8D). Together, these results show that 14-3-3, by competing with MYC, alters the location of CDCA7 in the cell and, therefore, its degree of interaction with MYC.
CDCA7 expression sensitizes cells to serum withdrawal-induced apoptosis.
We next established Rat1 fibroblasts expressing MYC under the control of a constitutive cytomegalovirus (CMV) promoter (termed MYC-Rat1 cells). These cells underwent apoptosis following 4 to 24 h of serum withdrawal, reproducing the findings of Evan et al. (20). In order to investigate whether CDCA7 altered MYC-dependent apoptosis, we expressed CDCA7 under the control of a doxycycline-inducible promoter in either parental Rat1-TET-ON or MYC-Rat1-TET-ON fibroblasts. Several independent clones were isolated from each pool of cells, and all showed low levels of CDCA7 expression when grown in serum supplemented with 10 ng/ml doxycycline (Fig. 9A). When the concentration of doxycycline was increased to 1,000 ng/ml, the level of CDCA7 expression increased approximately 100-fold (Fig. 9A). Interestingly, we were not able to generate T163A-CDCA7-MYC-Rat1 cells. Colonies that were isolated were either negative for MYC expression or negative for T163A-CDCA7, indicating a possible prohibitive condition for coexpression.
Fig 9.

CDCA7 increases MYC-induced apoptosis. (A) Rat1 cells or Rat1 cells expressing MYC were established with CDCA7 expression under the control of a doxycycline-inducible promoter as described in Materials and Methods. After 24 h of treatment with doxycycline at various concentrations, cells were lysed, and CDCA7 was detected by immunoblotting. (B) Various cell lines from the experiment for which results are shown in panel A were immunoblotted for the presence of ectopically expressed MYC, indicated by an arrowhead. (C) CDCA7-Rat1 and CDCA7-MYC-Rat1 cells were treated with doxycycline at the indicated concentrations for 18 h, followed by serum starvation for 4 h. Apoptosis was measured by annexin V labeling and flow cytometry. Error bars show the ranges for duplicate samples and are representative of three independent experiments. (D) MYC-Rat1, CDCA7-Rat1, CDCA7-MYC-Rat1, and Δ(156–187)-CDCA7-MYC-Rat1 cells were cultured for 18 h in 10 ng/ml doxycycline, followed by serum starvation for 4 h. Apoptosis was determined by annexin V labeling and flow cytometry. Error bars show the ranges for duplicate samples and are representative of three experiments.
To see if CDCA7 upregulation had an effect on apoptosis, we pretreated CDCA7-Rat1 or CDCA7-MYC-Rat1 cells with doxycycline for 18 h, followed by 4 h of serum withdrawal. CDCA7-MYC-Rat1 cells were sensitized to apoptosis with increasing concentrations of doxycycline (Fig. 9C). Only a minor increase in apoptosis with CDCA7 upregulation was observed in the absence of ectopic MYC expression (Fig. 9C). Next, we asked if the non-MYC-binding CDCA7 mutant Δ(156-187)-CDCA7 would alter MYC-dependent apoptosis (Fig. 9D). Here we treated CDCA7-MYC-Rat1 or Δ(156-187)-CDCA7-MYC-Rat1 cells with doxycycline for 18 h and then starved them of serum for 4 h. This result showed that despite a high level of MYC expression (Fig. 9B), Δ(156-187)-CDCA7-MYC-Rat1 cells were no longer susceptible to serum withdrawal-induced apoptosis, suggesting that Δ(156-187)-CDCA7 was acting as a dominant negative mutant and repressing MYC-induced apoptosis.
Loss of CDCA7 expression reduces MYC-dependent apoptosis.
Endogenous CDCA7 mRNA levels in MYC-Rat1 cells were 2- to 5-fold higher than those in Rat1 cells not expressing ectopic MYC (data not shown), consistent with an earlier report that the cdca7 promoter is a target of MYC (52). Since we have shown that elevated levels of ectopic CDCA7 sensitized MYC-Rat1 cells to apoptosis upon serum withdrawal, we hypothesized that the elevated levels of both CDCA7 and MYC in the MYC-Rat1 cells may act together to cause the higher levels of apoptosis seen in these cells. In addition, the loss of apoptosis as a result of expression of Δ(156–187)-CDCA7 suggested that CDCA7 was necessary for MYC-sensitized apoptosis. To test this, we created MYC-Rat1 lines expressing shRNA sequences targeting endogenous CDCA7. This resulted in a knockdown of endogenous CDCA7 mRNA relative to the level in a control line expressing scrambled shRNA (Fig. 10A). These Rat1-MYC-sh lines maintained high levels of MYC as detected by immunoblotting (Fig. 10B). When cultured in 0.2% serum overnight, Rat1-MYC-sh cells exhibited less morphological evidence of apoptosis than their MYC-Rat1 parental lines (Fig. 10D) and experienced less cell death as measured by trypan blue exclusion (Fig. 10E) and less apoptosis as measured by an annexin V-binding assay (Fig. 10C) or by caspase-3 activity (Fig. 10F and G). Together, these results show that CDCA7 is necessary for MYC to promote apoptosis upon serum withdrawal.
Fig 10.

Knockdown of CDCA7 rescues Rat1-MYC cells from serum withdrawal-induced apoptosis. shRNA-expressing Rat1 cell lines in which endogenous CDCA7 was knocked down were established in the presence of ectopic MYC as described in Materials and Methods (Rat1-MYC-sh1, Rat1-MYC-sh2). Additionally, control lines were established using a scrambled version of the CDCA7 targeting sequence (Rat1-MYC-Scrmb). (A) Quantitative RT-PCR of ectopic MYC (black bars) and endogenous CDCA7 (gray bars) was performed on scrambled shRNA-expressing Rat1-MYC cells or on Rat1-MYC cells expressing shRNA-targeted CDCA7. Quantitative RT-PCR was performed in triplicate for each clone. (B) MYC levels of shRNAi Rat1-MYC lines were determined by immunoblotting. (C) Rat1-MYC, Rat1-MYC-sh1, and Rat1-MYC-sh2 cells were serum starved for 4 or 24 h, and apoptosis was measured by annexin V labeling and flow cytometry. Error bars represent the standard deviations for triplicate samples. (D) Rat1, Rat1-MYC, or Rat1-MYC-sh1 cells grown to 40% confluence were placed in 0.2% serum overnight. Differential interference contrast (DIC) images were captured using bright-field microscopy. (E) Rat1-MYC-Scrmb1 or Rat1-MYC-sh1 cells were plated as for the experiment for which results are shown in panel D. On the following day, trypan blue-positive cells were counted as a percentage of total cells by using a hemocytometer. (F) Rat1-MYC cells, as well as two scrambled lines and two lines expressing shRNA-targeted CDCA7, were plated in triplicate, starved in 0.2% serum for 4 h, and assayed for caspase-3 activity. (G) Rat1-MYC-Scrmb-1 cells and Rat1-MYC-sh1 cells were plated in triplicate in various concentrations of serum for 4 h and were then assayed for caspase-3 activity.
CDCA7 expression alters MYC-dependent transformation.
We next asked whether CDCA7 alters MYC-induced transformation. We assessed this by expressing CDCA7 in MYC-Rat1 cells and evaluating soft-agarose colony formation. To overcome the negative selection of prolonged expression of T163A-CDCA7 and MYC, we introduced CDCA7 and mutants into MYC-Rat1 cells by retroviral infection, followed by a short selection period and analysis of the polyclonal pools. Both wild-type and T163A mutant CDCA7 exhibited reduced numbers of macroscopic colonies resulting from MYC expression, indicating that increased expression of CDCA7 interferes with MYC-induced transformation (Fig. 11A and B). Surprisingly, Δ(156–187)-CDCA7 expression largely inhibited MYC-dependent colony formation, an effect similar to its effects on MYC-dependent apoptosis, suggesting that this protein was acting as a dominant negative mutant. Parental Rat1 cells did not form colonies (Fig. 11A), and we found no effect of CDCA7 expression alone in Rat1 cells (data not shown), in agreement with the previous results of Prescott and coworkers (52). Finally, stable knockdown of CDCA7 also reduced the number of macroscopic colonies formed from that for Rat1-Myc cells expressing scrambled shRNA (Fig. 11C and D). Together, these results suggest that CDCA7 is a necessary cofactor for Myc-induced transformation and apoptosis.
Fig 11.

CDCA7 influences MYC-induced transformation. (A) Rat1 cells or Rat1 cells stably expressing MYC were infected with a retrovirus containing either an empty vector, wild-type (WT) CDCA7, Δ(156–187)-CDCA7, or T163A-CDCA7. Hygromycin-resistant pools were isolated after 5 days of selection, and 5 × 103 cells were plated in complete medium containing 0.3% agarose over a 0.7% agarose bottom layer, as described in Materials and Methods. Two weeks later, colonies were stained with crystal violet, destained with water, and imaged on an Odyssey infrared scanner using a 4-mm offset and a 41-μm resolution setting. (B) Colonies were counted using ImageJ software. Error bars represent standard deviations for 6 individual wells. Asterisks indicate significant differences from the results for WT CDCA7 (*, P = 0.0005) or for the vector alone (**, P = 0.0003; ***, P = 0.004). Lysates of the hygromycin-resistant pools were immunoblotted with anti-FLAG. (C) Rat1-MYC-Scrmb or Rat1-MYC-sh cells were plated as described for panel A, and colonies were stained with crystal violet after 2 weeks. (D) Macroscopic colonies from 4 individual wells were counted using ImageJ. Error bars represent standard deviations. The asterisk indicates a significant difference from the results for Rat1-MYC-Scrmb cells (P = 0.001).
DISCUSSION
Aberrant proliferation induced by MYC is opposed by apoptosis and/or cell cycle arrest, providing cells with a safety mechanism against the onset of MYC-induced hyperproliferation and tumorigenesis (58). During normal development, proliferative signals result in the activation of pathways that instruct the cell to cooperate with MYC and divide. Phosphoinositide 3-OH kinase is at the apex of one important pathway activated by mitogenic stimuli, and one of its main effectors, AKT, cooperates with MYC to suppress proapoptotic gene expression while simultaneously instructing the cell to divide (33). Given the important roles of both AKT and MYC in cancer pathology, there is an urgent need to understand the mechanism of cooperative signaling between these two regulators.
In this study, we found that the transcriptional regulator CDCA7 was phosphorylated by AKT and that this caused its disengagement from MYC and its sequestration to the cytoplasm by binding to 14-3-3. 14-3-3 appeared to compete with MYC for binding to CDCA7, since the addition of recombinant 14-3-3 led to reduced coprecipitation of phospho-Thr163 CDCA7 and MYC. In addition, we found that the region surrounding Thr163 harbored nuclear localization activity. Mutation of predicted consensus sites derived from the bipartite NLS domains of RB (59) and SWI5 (60) led to the loss of nuclear localization of a large cargo protein. Mutation of two of these sites, Arg161 and Arg162, led to enhanced cytoplasmic localization of CDCA7, which was accompanied by increased Thr163 phosphorylation and 14-3-3 binding.
When CDCA7 was ectopically expressed with MYC, cells were sensitized to serum withdrawal-induced apoptosis. Surprisingly, the Δ(156-187)-CDCA7 mutation reduced serum withdrawal-induced apoptosis, suggesting that this protein was acting as a dominant negative mutant and interfering with the proapoptotic function of MYC. Conversely, knockdown of endogenous CDCA7 rescued MYC-expressing cells from apoptosis following serum withdrawal. These results argue that CDCA7 is an important cofactor in the ability of MYC to regulate target gene expression and that, under physiological conditions, CDCA7 may be important for the normal regulation of MYC target genes. On the other hand, dysregulation of this pathway becomes proapoptotic under pathophysiological conditions.
Consistent with this, transformation of fibroblasts by MYC was also influenced by CDCA7. Coexpression of CDCA7 reduced the level of anchorage-independent colony growth, indicating that overexpression of CDCA7 interferes with the process of MYC transformation. Importantly, Δ(156-187)-CDCA7 appeared to act as a dominant negative mutant under these conditions, since this protein greatly reduced the level of colony formation. These results suggest that CDCA7 influences MYC-induced transformation, with high-level expression causing interference, possibly by increasing apoptosis within the forming colonies, while expression of Δ(156-187)-CDCA7 interferes by disrupting the association of MYC with either endogenous CDCA7 or other factors that require full-length CDCA7 for interaction. In support of this, knockdown of endogenous CDCA7 also reduced the transformation potential of MYC, again arguing that CDCA7 is required for MYC transforming activity. These observations are, in general, consistent with the earlier findings of Prescott and coworkers (52), who found that CDCA7 rescued transformation by MYC containing a transformation-defective MYC box II deletion, supporting the hypothesis of a genetic link between CDCA7 and MYC. While Prescott and coworkers found that CDCA7 coexpression did not significantly increase or decrease MYC-induced transformation (52), the difference between our experiments and those of Prescott and coworkers could be due to the level of expression of MYC or CDCA7, or a combination of both. Future studies will investigate whether ectopic CDCA7 expression reduces transformation as a direct result of increased apoptosis.
Interestingly, the cdca7 gene itself has been reported to be a target of MYC (50, 52). In our own experiments, overexpression of MYC in Rat1 cells caused a 2- to 5-fold increase in the amount of cdca7 mRNA, suggesting that a feed-forward loop exists whereby MYC activation leads to elevated CDCA7 levels. We hypothesize that growth factor signals use AKT activity to finely tune the activation of MYC through CDCA7, with loss of AKT activating the prodeath effects of CDCA7/MYC, tipping the cell fate toward apoptosis.
An important question remaining is the alteration of global gene expression caused by the interaction between CDCA7 and MYC. The related protein JPO2 has been shown to bind to SP1 sites of the MAOA gene via its C-terminal zinc finger domain (61). CDCA7 and JPO2 are 77% identical within this region, suggesting that CDCA7 may also bind to similar SP1 elements. MYC-binding E-box sites are preferentially enriched with SP1 elements across the genome (62), suggesting that CDCA7 and MYC might cooperate to regulate these genes. What role CDCA7 plays in the context of cell fate determination during early development, where MYC expression is tightly regulated, remains to be determined, as does the potential relationship of CDCA7 with other transcription factors in non-MYC-expressing cells.
In summary, we have identified a new target of AKT that is required for the proapoptotic and transformation properties of MYC. AKT phosphorylates CDCA7 within a 14-3-3 binding site, disrupting its interaction with MYC and resulting in CDCA7 transit from the nucleus to the cytoplasm. The fine-tuning of CDCA7-MYC interaction by phosphorylation at Thr163 could represent an important physiological role for AKT, and one that becomes pathophysiological under conditions of an overactive AKT axis.
ACKNOWLEDGMENTS
This work was supported by funds from the Canadian Institute of Health Research, an Early Investigator award (to M.P.S.), and a Canadian Foundation of Innovation award (to M.P.S.).
We thank Veronique Nogueira of the University of Illinois at Chicago for the kind gift of Akt1/2-null MEFs.
Footnotes
Published ahead of print 19 November 2012
REFERENCES
- 1. Hann SR, Eisenman RN. 1984. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol. Cell. Biol. 4:2486–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meyer N, Kim SS, Penn LZ. 2006. The Oscar-worthy role of Myc in apoptosis. Semin. Cancer Biol. 16:275–287 [DOI] [PubMed] [Google Scholar]
- 3. Meyer N, Penn LZ. 2008. Reflecting on 25 years with MYC. Nat. Rev. Cancer 8:976–990 [DOI] [PubMed] [Google Scholar]
- 4. Oster SK, Ho CSW, Soucie EL, Penn LZ. 2002. The myc oncogene: MarvelouslY Complex. Adv. Cancer Res. 84:81–154 [DOI] [PubMed] [Google Scholar]
- 5. Wolfer A, Ramaswamy S. 2011. MYC and metastasis. Cancer Res. 71:2034–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wolfer A, Wittner BS, Irimia D, Flavin RJ, Lupien M, Gunawardane RN, Meyer CA, Lightcap ES, Tamayo P, Mesirov JP, Liu XS, Shioda T, Toner M, Loda M, Brown M, Brugge JS, Ramaswamy S. 2010. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. U. S. A. 107:3698–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Albihn A, Johnsen JI, Henriksson MA. 2010. MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 107:163–224 [DOI] [PubMed] [Google Scholar]
- 8. Nilsson JA, Cleveland JL. 2003. Myc pathways provoking cell suicide and cancer. Oncogene 22:9007–9021 [DOI] [PubMed] [Google Scholar]
- 9. Grandori C, Cowley SM, James LP, Eisenman RN. 2000. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16:653–699 [DOI] [PubMed] [Google Scholar]
- 10. Soucek L, Evan GI. 2010. The ups and downs of Myc biology. Curr. Opin. Genet. Dev. 20:91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amati B, Littlewood TD, Evan GI, Land H. 1993. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 12:5083–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Born TL, Frost JA, Schonthal A, Prendergast GC, Feramisco JR. 1994. c-Myc cooperates with activated Ras to induce the cdc2 promoter. Mol. Cell. Biol. 14:5710–5718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. 2000. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. U. S. A. 97:3260–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. 2001. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 15:2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanson KD, Shichiri M, Follansbee MR, Sedivy JM. 1994. Effects of c-myc expression on cell cycle progression. Mol. Cell. Biol. 14:5748–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. 1997. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 8:1039–1048 [PubMed] [Google Scholar]
- 17. Rudolph B, Saffrich R, Zwicker J, Henglein B, Muller R, Ansorge W, Eilers M. 1996. Activation of cyclin-dependent kinases by Myc mediates induction of cyclin A, but not apoptosis. EMBO J. 15:3065–3076 [PMC free article] [PubMed] [Google Scholar]
- 18. Askew DS, Ashmun RA, Simmons BC, Cleveland JL. 1991. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6:1915–1922 [PubMed] [Google Scholar]
- 19. Eischen CM, Roussel MF, Korsmeyer SJ, Cleveland JL. 2001. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol. Cell. Biol. 21:7653–7662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. 1992. Induction of apoptosis in fibroblasts by c-myc protein. Cell 69:119–128 [DOI] [PubMed] [Google Scholar]
- 21. Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP, Lowe SW. 2005. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436:807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hermeking H, Eick D. 1994. Mediation of c-Myc-induced apoptosis by p53. Science 265:2091–2093 [DOI] [PubMed] [Google Scholar]
- 23. Pusapati RV, Rounbehler RJ, Hong S, Powers JT, Yan M, Kiguchi K, McArthur MJ, Wong PK, Johnson DG. 2006. ATM promotes apoptosis and suppresses tumorigenesis in response to Myc. Proc. Natl. Acad. Sci. U. S. A. 103:1446–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. 2004. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 431:712–717 [DOI] [PubMed] [Google Scholar]
- 25. Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12:2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mymryk JS, Shire K, Bayley ST. 1994. Induction of apoptosis by adenovirus type 5 E1A in rat cells requires a proliferation block. Oncogene 9:1187–1193 [PubMed] [Google Scholar]
- 27. Qin XQ, Livingston DM, Kaelin WG, Jr, Adams PD. 1994. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. U. S. A. 91:10918–10922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. 1992. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. U. S. A. 89:7742–7746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu X, Levine AJ. 1994. p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. U. S. A. 91:3602–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bissonnette RP, Echeverri F, Mahboubi A, Green DR. 1992. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature 359:552–554 [DOI] [PubMed] [Google Scholar]
- 31. Fanidi A, Harrington EA, Evan GI. 1992. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature 359:554–556 [DOI] [PubMed] [Google Scholar]
- 32. Vaux DL, Cory S, Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335:440–442 [DOI] [PubMed] [Google Scholar]
- 33. Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. 1997. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 385:544–548 [DOI] [PubMed] [Google Scholar]
- 34. Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. 1997. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275:661–665 [DOI] [PubMed] [Google Scholar]
- 35. Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. 1997. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11:701–713 [DOI] [PubMed] [Google Scholar]
- 36. Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. 1997. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16:2783–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241 [DOI] [PubMed] [Google Scholar]
- 38. del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. 1997. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687–689 [DOI] [PubMed] [Google Scholar]
- 39. She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J, Rosen N. 2005. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 8:287–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zundel W, Giaccia A. 1998. Inhibition of the anti-apoptotic PI(3)K/Akt/Bad pathway by stress. Genes Dev. 12:1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M. 2004. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 23:2830–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868 [DOI] [PubMed] [Google Scholar]
- 43. Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB. 2002. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 156:817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delpuech O, Griffiths B, East P, Essafi A, Lam EW, Burgering B, Downward J, Schulze A. 2007. Induction of Mxi1-SRα by FOXO3a contributes to repression of Myc-dependent gene expression. Mol. Cell. Biol. 27:4917–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh A, Ye M, Bucur O, Zhu S, Tanya Santos M, Rabinovitz I, Wei W, Gao D, Hahn WC, Khosravi-Far R. 2010. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Mol. Biol. Cell 21:1140–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. 1998. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 58:5667–5672 [PubMed] [Google Scholar]
- 47. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39 [DOI] [PubMed] [Google Scholar]
- 48. Dahia PL, Aguiar RC, Alberta J, Kum JB, Caron S, Sill H, Marsh DJ, Ritz J, Freedman A, Stiles C, Eng C. 1999. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in haematological malignancies. Hum. Mol. Genet. 8:185–193 [DOI] [PubMed] [Google Scholar]
- 49. Di Cristofano A, Pandolfi PP. 2000. The multiple roles of PTEN in tumor suppression. Cell 100:387–390 [DOI] [PubMed] [Google Scholar]
- 50. Goto Y, Hayashi R, Muramatsu T, Ogawa H, Eguchi I, Oshida Y, Ohtani K, Yoshida K. 2006. JPO1/CDCA7, a novel transcription factor E2F1-induced protein, possesses intrinsic transcriptional regulator activity. Biochim. Biophys. Acta 1759:60–68 [DOI] [PubMed] [Google Scholar]
- 51. Osthus RC, Karim B, Prescott JE, Smith BD, McDevitt M, Huso DL, Dang CV. 2005. The Myc target gene JPO1/CDCA7 is frequently overexpressed in human tumors and has limited transforming activity in vivo. Cancer Res. 65:5620–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prescott JE, Osthus RC, Lee LA, Lewis BC, Shim H, Barrett JF, Guo Q, Hawkins AL, Griffin CA, Dang CV. 2001. A novel c-Myc-responsive gene, JPO1, participates in neoplastic transformation. J. Biol. Chem. 276:48276–48284 [DOI] [PubMed] [Google Scholar]
- 53. Huang A, Ho CS, Ponzielli R, Barsyte-Lovejoy D, Bouffet E, Picard D, Hawkins CE, Penn LZ. 2005. Identification of a novel c-Myc protein interactor, JPO2, with transforming activity in medulloblastoma cells. Cancer Res. 65:5607–5619 [DOI] [PubMed] [Google Scholar]
- 54. Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N. 2006. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell 10:269–280 [DOI] [PubMed] [Google Scholar]
- 55. Matitau AE, Scheid MP. 2008. Phosphorylation of MEKK3 at threonine 294 promotes 14-3-3 association to inhibit nuclear factor κB activation. J. Biol. Chem. 283:13261–13268 [DOI] [PubMed] [Google Scholar]
- 56. Freeman AK, Morrison DK. 2011. 14-3-3 proteins: diverse functions in cell proliferation and cancer progression. Semin. Cell Dev. Biol. 22:681–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang B, Yang H, Liu YC, Jelinek T, Zhang L, Ruoslahti E, Fu H. 1999. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry 38:12499–12504 [DOI] [PubMed] [Google Scholar]
- 58. Wasylishen AR, Penn LZ. 2010. Myc: the beauty and the beast. Genes Cancer 1:532–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Efthymiadis A, Shao H, Hubner S, Jans DA. 1997. Kinetic characterization of the human retinoblastoma protein bipartite nuclear localization sequence (NLS) in vivo and in vitro. A comparison with the SV40 large T-antigen NLS. J. Biol. Chem. 272:22134–22139 [DOI] [PubMed] [Google Scholar]
- 60. Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. 1991. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell 66:743–758 [DOI] [PubMed] [Google Scholar]
- 61. Ou XM, Chen K, Shih JC. 2006. Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 103:10923–10928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Parisi F, Wirapati P, Naef F. 2007. Identifying synergistic regulation involving c-Myc and sp1 in human tissues. Nucleic Acids Res. 35:1098–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]