

Abstract
Protein synthesis is highly regulated via both initiation and elongation. One mechanism that inhibits elongation is phosphorylation of eukaryotic elongation factor 2 (eEF2) on threonine 56 (T56) by eEF2 kinase (eEF2K). T56 phosphorylation inactivates eEF2 and is the only known normal eEF2 functional modification. In contrast, eEF2K undergoes extensive regulatory phosphorylations that allow diverse pathways to impact elongation. We describe a new mode of eEF2 regulation and show that its phosphorylation by cyclin A–cyclin-dependent kinase 2 (CDK2) on a novel site, serine 595 (S595), directly regulates T56 phosphorylation by eEF2K. S595 phosphorylation varies during the cell cycle and is required for efficient T56 phosphorylation in vivo. Importantly, S595 phosphorylation by cyclin A-CDK2 directly stimulates eEF2 T56 phosphorylation by eEF2K in vitro, and we suggest that S595 phosphorylation facilitates T56 phosphorylation by recruiting eEF2K to eEF2. S595 phosphorylation is thus the first known eEF2 modification that regulates its inhibition by eEF2K and provides a novel mechanism linking the cell cycle machinery to translational control. Because all known eEF2 regulation is exerted via eEF2K, S595 phosphorylation may globally couple the cell cycle machinery to regulatory pathways that impact eEF2K activity.
INTRODUCTION
Global protein synthesis is subject to complex regulation that allows cells to control this energy-intensive process in response to diverse physiologic cues. Translation is regulated at the initiation and elongation levels. For example, translation is repressed in the G2/M phase of the cell cycle (1, 2), in which inhibition of eukaryotic initiation factors represses mitotic translation (2–4). Translational control is also exerted at the level of elongation, and this often involves inhibition of eukaryotic elongation factor 2 (eEF2) (5–7).
eEF2 is a GTP-dependent translocase that is responsible for the movement of nascent peptidyl-tRNAs from the A-site to the P-site of the ribosome. The only known normal mechanism that regulates eEF2 is an inhibitory phosphorylation at threonine 56 (T56), which falls within the eEF2 GTP-binding domain and prevents eEF2 from binding to the ribosome (8, 9). A single, atypical calmodulin-dependent kinase, termed eEF2 kinase (eEF2K), phosphorylates eEF2 on T56 (10–12). Many signals cause eEF2K to become phosphorylated on residues that inhibit or augment its activity (5, 13, 14). For example, the mitogen-activated protein kinase (MAPK) and mTOR pathways inhibit eEF2K in response to mitogen and nutrient signals (15–17). In contrast, AMP kinase- and protein kinase A/Ca2+-dependent signaling activates eEF2K in response to starvation, hypoxia, and oxidative stress (18–21). Thus, while many signaling pathways control eEF2 activity, this regulation is exerted exclusively via modification of eEF2K rather than eEF2. T56 phosphorylation is thus the only known eEF2 functional modification, other than its inhibition by ADP ribosylation catalyzed by bacterial toxins (6).
Cyclin-dependent kinases (CDKs) coordinate cell division by phosphorylating numerous downstream substrates. We developed a mass spectrometry-based proteomic approach employing ATP analog-sensitive CDK2 and substrate thiophosphorylation to identify cyclin A-CDK2 substrates in fractionated cell lysates (22). This screen identified 180 putative CDK2 substrates that function in diverse cellular processes. Several candidates have critical roles in protein synthesis, including eEF2, which we validated to be phosphorylated by cyclin A-CDK2 in vitro (22). Here, we show that eEF2 phosphorylation on serine 595 (S595) by cyclin A-CDK2 directly promotes its inhibitory phosphorylation by eEF2K. eEF2 S595 phosphorylation is highest in mitosis, when it is also sensitive to pharmacologic CDK inhibition. Remarkably, S595 phosphorylation is required for efficient eEF2 T56 phosphorylation by eEF2K. Mutation of either S595 or a nearby residue (H599) greatly inhibits eEF2 T56 phosphorylation in vivo and in vitro. Moreover, we reconstituted this regulation with purified components and found that eEF2 phosphorylation by cyclin A-CDK2 directly stimulates its phosphorylation by eEF2K and that this requires CDK2 activity, S595, and T56. Finally, a peptide spanning the eEF2 S595 region functions as a phosphorylation-dependent inhibitor of T56 phosphorylation. We speculate that S595 phosphorylation recruits eEF2K to promote T56 phosphorylation. S595 phosphorylation is the first known eEF2 modification that regulates its phosphorylation by eEF2K. Because all known eEF2 regulation is exerted via eEF2K, S595 phosphorylation may globally modulate elongation responses to diverse physiologic inputs.
MATERIALS AND METHODS
Antibodies, plasmids, and enzymes.
The antibodies used in the study were as follows: FLAG-M2 (Sigma); phospho-T56 eEF2 (catalog no. 2331; Cell Signaling); CDK2 (D12; Santa Cruz); Myc tag (9E10; prepared in-house); and eEF2 (Cell Signaling). eEF2 was cloned from HeLa cDNA into pCS2-3X-FLAG and sequenced. Mutations were generated by the QuikChange method (Stratagene) and sequenced. Glutathione S-transferase (GST)–eEF2K was purified as described previously (23). Recombinant cyclin A-CDK2 and cyclin B-CDC2 were purchased from New England BioLabs. GST-cyclin E-CDK2 was prepared from baculovirus-infected SF9 cells using standard methods and purified by ion exchange and gel filtration chromatography. p42 mitogen-activated protein kinase (MAPK) and glycogen synthase kinase 3β (GSK3β) were purchased from New England BioLabs.
Tissue culture, transfection, and drug treatments.
293A, 293T, U2OS, HeLa, and SK-N-AS cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and penicillin-streptomycin (Pen/Strep). Unless otherwise noted, all transfections utilized calcium phosphate (24). Plates (10-cm-diameter) of 293A or 293T cells were transfected as follows. For CDK assays, 10 μg of pCS2-FLAG-eEF2 vector was used. For preparation of eluted FLAG-eEF2, 15 μg of pCS2-FLAG-eEF2 was used. For cyclin A-CDK2, 9 μg of pCS2-myc-tagged cyclin A and 3 μg of pCS2-hemagglutinin (HA)-tagged CDK2 were used. For cell cycle experiments (see Fig. 4), HeLa cells were transfected with 9 μg of FLAG-eEF2 using Mirus Trans-IT HeLa Monster reagent and treated with 2 mM hydroxyurea, 10 μM aphidicolin, or 40 ng/ml nocodazole (Sigma) for 15 h. Roscovitine (25 μM) was added for 1 to 2 h where indicated. Unless otherwise noted, cells were lysed in 0.5% NP-40 buffer with phosphatase and protease inhibitors.
Fig 4.
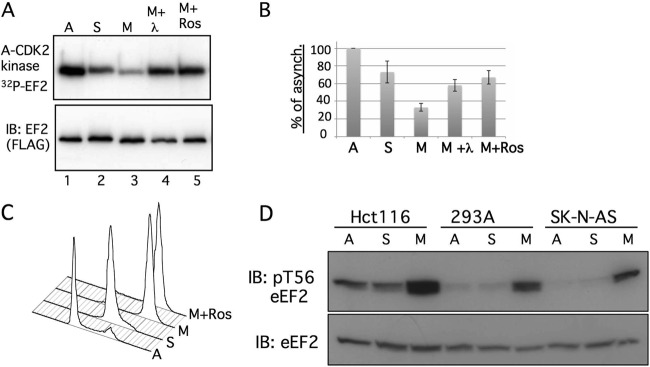
S595 phosphorylation is highest in mitotic cells. (A) HeLa cells were transfected with FLAG-eEF2 and synchronized in either early S phase (lane 2) or prometaphase (lanes 3 to 5). The immunoprecipitated eEF2 proteins were subjected to in vitro phosphorylation by cyclin A-CDK2. The top panel shows the amount of eEF2 phosphorylation, and the bottom panel indicates the amount of total eEF2 protein from a representative experiment. eEF2 from nocodazole-arrested mitotic cells was treated with lambda phosphatase prior to the cyclin A-CDK2 reaction represented in lane 4. Cells arrested in mitosis were treated with roscovitine to inhibit mitotic CDKs (lane 5). A, asynchronous; S, hydroxyurea; M, nocodazole; M+Ros, nocodazole plus roscovitine pulse. (B) Combined results of four independent experiments in which the amount of in vitro eEF2 phosphorylation by cyclin A-CDK2 was compared with the amount seen with asynchronous cells. The amount of eEF2 phosphorylation was first normalized to the total amount of eEF2 present in the immunoprecipitates. Error bars indicate the standard deviations of the means of the results of the four independent experiments. Three experiments used hydroxyurea to arrest S-phase cells, whereas the fourth used aphidicolin. (C) Flow cytometric cell cycle profiles of the various cell populations of a representative experiment shown in panel B. (D) Hct116 colon carcinoma cells, 293A cells, or SK-N-AS neuroblastoma cells were synchronized with aphidicolin (S) or nocodazole (M) or grown asynchronously (A). The amounts of total endogenous eEF2 and phospho-T56-eEF2 are shown.
Immunoprecipitation, immunoblotting, and CDK assays.
Immunoblotting and immunoprecipitation were performed using standard procedures (24). For the CDK assays, FLAG-eEF2 was immunoprecipitated from 200 to 300 μg of cell lysate and phosphorylated with recombinant cyclin A-CDK2 (NEB) (0.35 to 0.5 μl/reaction). Kinase reactions included 30 μM ATP and 50 to 75 nM [γ-32P]ATP (PerkinElmer) (3,000/mmol). The experiment represented in Fig. 1A utilized myc-tagged cyclin A-CDK2 immunoprecipitated from transfected 293A cells with 9E10 antibody and 500 ng eluted eEF2 (see below). The experiment represented in Fig. 1F to G used recombinant cyclin B-CDC2 (NEB) or GST-cyclin E-CDK2 and FLAG-eEF2 immunoprecipitated from 200 to 300 μg of cell lysate from transfected 293A cells. Histone H1 kinase assays were performed as described previously (24). Samples were treated with 200 U lambda phosphatase (NEB) for 15 min where indicated (see Fig. 4) and washed in kinase buffer. For the MAPK assays, FLAG-eEF2 or HA–c-Jun was immunoprecipitated from transfected 293A cells and phosphorylated with 100 U p42 MAPK in kinase buffer. For the GSK3β assays, FLAG-eEF2 or myc tag–cyclin E–dominant-negative CDK2 (dnCDK2) was immunoprecipitated from transfected 293A cells and phosphorylated with 500 U GSK3β in kinase buffer. Dominant-negative CDK2 was cotransfected with myc-tagged cyclin E to prevent cyclin E-CDK2 autophosphorylation during the GSK3β reaction.
Fig 1.

Cyclin A-CDK2 phosphorylates eEF2 on S595 in vitro. (A) Cyclin A-CDK phosphorylates eEF2. FLAG-eEF2 was eluted from immunoprecipitates (IP) and phosphorylated with myc tag-cyclin A-CDK2 isolated from transfected 293A cells. Top, autoradiograph of phosphorylated eEF2; bottom, eEF2 immunoblot (IB). (B) The six possible eEF2 phosphorylation sites. S435 and S595 contain CDK motifs (underlines). (C) Reduced phosphorylation of eEF2 595A by cyclin A-CDK2. The indicated eEF2 mutants were immunoprecipitated from transfected 293T cells and phosphorylated with recombinant cyclin A-CDK2. Top, autoradiograph of phosphorylated eEF2; bottom, Ponceau stain for input control. (D) FLAG-eEF2 was phosphorylated by recombinant cyclin A-CDK2 (AK2) or cyclin B-CDC2 (BK2). The amount of histone H1 phosphorylation by both enzymes is shown, as well as the FLAG-eEF2 input (IB: anti-FLAG). C, control. (E) eEF2 was phosphorylated with recombinant cyclin A-CDK2 or cyclin E-CDK2 as described for panel D. (F) FLAG-eEF2 was subject to kinase reactions with cyclin A-CDK2 (AK-2), MAPK, or GSK3β as indicated. GSK3β autophosphorylation is noted. (G) GSK3β phosphorylates myc-tagged cyclin E immunoprecipitated from transfected 293A cells. GSK3β autophosphorylation is noted. HC, heavy chain; LC, light chain. (H) MAPK phosphorylates HA–c-Jun immunoprecipitated from transfected 293A cells. (I) MAPK phosphorylates endogenous 4EBP1 immunoprecipitated from 293A cells. MT cyc E, myc-tagged cyclin E.
eEF2 purification and eEF2K assays.
293T cells transfected with FLAG-eEF2 were lysed in eEF2K buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM dithiothreitol [DTT]). FLAG-eEF2 was immunoprecipitated with M2-agarose (Sigma), washed in lysis and elution buffers, eluted with FLAG peptide (Sigma), and concentrated (Amicon centrifugal filter unit; 50-kDa cutoff). eEF2K assays were performed in eEF2K cocktail (150 nM [γ-32P]ATP, 0.2 μg eEF2K, eEF2K buffer, 50 U calmodulin), and reaction mixtures were incubated at 30°C for 10 min as described previously (23). For sequential cyclin A-CDK2–eEF2K reactions, 1 μg eluted FLAG-eEF2 (or FLAG-eEF2 S595A) was phosphorylated by 0.375 μl cyclin A-CDK2 (high efficiency; gift of T. Barshevsky, NEB) immobilized on anti-CDK2 antibody-bound Sepharose beads in kinase buffer containing 250 μM ATP. Reactions were terminated with roscovitine (25 μM), and the phosphorylated FLAG-eEF2 was transferred into fresh tubes and subsequently phosphorylated with eEF2K cocktail as described above.
In vivo labeling.
293T or U2OS cells transfected with the indicated vectors were labeled with [32P]orthophosphate (catalog no. 64014; MP Biomedicals) (1 mCi/ml) for 3 h and lysed in Tween buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 10% glycerol, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween). FLAG-eEF2 was immunoprecipitated and washed with radioimmunoprecipitation assay (RIPA) buffer five times and lysis buffer four times prior to electrophoresis.
Peptide competition.
A 30-fold molar excess of phosphorylated or unphosphorylated S595 peptide (biotin-TVSEESNVLCLSKS595PNKHNRLYMKARPFF) (purchased from Pi Proteomics) was preincubated with 0.2 μg eEF2K in the presence of 100 mM ATP and 150 nM [γ-32P]ATP at 4°C. These cocktails were mixed with 0.075 nmol of FLAG-eEF2 and incubated at 25°C.
Phosphopeptide mapping and phosphoamino acid analysis.
Phosphopeptide and phosphoamino acid analyses were performed as described previously (25). Briefly, FLAG eEF2 proteins phosphorylated by cyclin A-CDK2 in vitro or immunoprecipitated from orthophosphate-labeled cells were electrophoresed, blotted onto nitrocellulose, and exposed to film. Filter pieces containing labeled eEF2 were excised, digested with trypsin, oxidized, again subjected to digestion with trypsin, and subjected to phosphopeptide mapping in which peptides were separated on thin-layer chromatography (TLC) plates in two dimensions (2D) (electrophoresis followed by treatment with isobutyric buffer [pH 1.9]) and autoradiographed. These methods are illustrated in Fig. 2B. For phosphoamino acid analysis, phosphopeptides were eluted from the cellulose plates, acid hydrolyzed, separated by 2D electrophoresis, and analyzed as described previously (25). Marker phosphoamino acids were visualized with ninhydrin.
Fig 2.

Phosphopeptide and phosphoamino acid analyses of eEF2 phosphorylation by cyclin A-CDK2 in vitro. (A) Predicted eEF2 tryptic peptides. The S595 and T56 peptides are in grey, and the extended peptide caused by H599P is underlined. (B) Schematic of phosphopeptide mapping procedure. Trypsinized peptides were run on TLC plates in two dimensions. The first was electrophoretic separation based on charge, and the second was chromatographic separation based on hydrophobicity. The positions of the three major peptides discussed in the paper are shown. (C) Calculated charges and hydrophobicity of the three major phosphopeptides. (D) Two-dimensional phosphopeptide maps of WT eEF2 (map 1) and the indicated eEF2 mutants (maps 2 to 4) after in vitro phosphorylation by cyclin A-CDK2. The corner spots on each map are orientation markers. The two S595 peptides are indicated (spots A and B). Three minor phosphorylation sites (map 2, arrows) that are eliminated by the 279/435/502 alanine mutations (maps 3 and 4) are also indicated. (E) A S595-to-threonine (S595T) mutation is normally phosphorylated. The two S595A mutants were overloaded to reveal the residual phosphorylations shown in panel H. Note the reduced amount of phosphorylation of S595A and S595A/H599P despite their overexpression relative to the other eEF2 proteins. A-K2 Kinase, cyclin A-CDK2. (F) Phosphopeptide map of eEF2 H599P. Phospho-S595 shifts to a single phosphopeptide (spot B), and phosphoamino analysis of spot B reveals only phosphoserine. Circles indicate the positions of marker phosphoamino acids, and the asterisk denotes a minor phosphopeptide that comigrates near spot A. (G) Analyses similar to those shown in panel F show that the S595T mutant converts spot B from phosphoserine to phosphothreonine. (H) Mapping of residual phosphorylations of eEF2 S595A/H599P by cyclin A-CDK2. The dotted circle indicates loss of spot B.
In vitro translation assay.
Diphtheria toxin (DT) (gift of J. Lund, Fred Hutchinson Cancer Research Center [FHCRC]) was cleaved with 0.1 μg/ml trypsin for 30 min at 25°C to activate the ADP-ribosylating activity of the A subunit (26). Rabbit reticulocyte lysate (TNT SP6 coupled; Promega) was treated with 10 to 25 ng of the cleaved DT and 1 μM NAD+ for 30 min at 25°C. Lysates were supplemented with firefly luciferase plasmid supplied in the TNT kit, 1 μg eEF2 or eEF2 S595A, and the remainder of the in vitro translation kit components. Luciferase was assayed with britelite plus (PerkinElmer).
RESULTS
eEF2 is phosphorylated on serine 595 by cyclin A-CDK2 in vitro.
We initially identified eEF2 in a proteomic screen for CDK2 substrates in fractionated cell lysates (10). This approach employed recombinant cyclin A-CDK2 containing a CDK2 mutant that utilizes bulky ATP analogs and 2-phenylethyl-ATP-γ-S to thiophosphorylate substrates. This strategy allowed substrate enrichment based upon thiophosphate chemistry, and candidate substrates were subsequently identified by mass spectroscopy (27). eEF2 was among the initial group of substrates that we used to validated the mass spectrometry results (22).
We first confirmed that myc-tagged cyclin A-CDK2 immunoprecipitated from transfected 293A cells robustly phosphorylates eEF2 in vitro (Fig. 1A). Our initial screen was not saturated with respect to phosphorylation site identifications, and we used site-directed mutants to determine which residue(s) is phosphorylated by cyclin A-CDK2. eEF2 contains 6 potential proline-directed phosphorylations, two of which, T435 and S595, are CDK consensus sites (S/T-P-X-K/R) (Fig. 1B). We mutated most of these sites to alanines, either individually or in combinations, and phosphorylated these mutants with cyclin A-CDK2 (Fig. 1C and not shown). The S595A mutation, either as a single mutation or when combined with other mutations, resulted in greatly reduced eEF2 phosphorylation by cyclin A-CDK2 in vitro (Fig. 1C, lanes 5 and 9). In contrast, the remaining mutations had minimal impact on bulk eEF2 phosphorylation (Fig. 1C and not shown).
Cyclin-CDKs often redundantly phosphorylate substrates, and we next examined the ability of other cyclin-CDKs to phosphorylate eEF2. Both cyclin B-CDC2 and cyclin E-CDK2 phosphorylated eEF2 to roughly the same extent as cyclin A-CDK2, when normalized to histone H1 kinase activity (Fig. 1D and E). Thus, like most CDK substrates, eEF2 is phosphorylated by multiple cyclin-CDKs. We also assessed the specificity of eEF2 phosphorylation by CDKs by testing two additional proline-directed kinases. Neither glycogen synthase kinase 3β (GSK3β) nor p42 mitogen-activated protein kinase (MAPK) phosphorylated eEF2 (Fig. 1F), whereas they phosphorylated known substrates (GSK3β-cyclin E and MAPK) (c-Jun and 4EBP1) (Fig. 1G to I).
Because site-directed mutations can impact phosphorylation of other sites, we used two-dimensional mapping of phosphopeptides generated by trypsin digestion to identify eEF2 phosphorylations. The immunoprecipitated FLAG-eEF2 proteins shown in Fig. 1C were excised from the membrane, digested with trypsin, and subjected to phosphopeptide mapping. The predicted eEF2 tryptic map is shown in Fig. 2A, whereas the mapping procedures used and the properties of the major peptides discussed below are represented in Fig. 2B and C. Phosphorylation of eEF2 by cyclin A-CDK2 produced two major phosphopeptides (Fig. 2D, map 1, spots A and B). Both spots were eliminated by the S595A mutation (Fig. 2D, map 2) but were unaffected by a triple S279A/T435A/S502A mutation (Fig. 2D, map 3), suggesting that they both contained S595. In addition to the lack of spots A and B, the S595A map revealed three minor spots (Fig. 2D, map 2, arrows) that were abrogated by a S279A/T435A/S502A mutation (Fig. 2, maps 3 and 4), indicating these were likely minor CDK2-phosphorylation sites. Although our initial proteomic screen identified eEF2 T435 phosphorylation by engineered cyclin A-CDK2 in cell lysates (22), the in vitro phosphorylation experiments and peptide maps revealed that S595 is the major phosphorylation site and that the contribution of additional residues, including T435, to overall eEF2 phosphorylation is minor. This discrepancy likely resulted from properties of the small S595 peptide (SPNK) that prevented its efficient identification by our proteomic methods, as well as from methodological differences between the screen and our current studies.
The presence of a single phosphorylation site in two spots often results from partial tryptic digestion. We hypothesized that the more positively charged spot B was derived from spot A by fusion with the next C-terminal peptide, since partial cleavage at the N terminus would result in the production of a neutral peptide (oxidized cysteine residues are negatively charged under pH 1.9 conditions). We thus mutated histidine 599 to proline (H599P), which prevents trypsin cleavage at lysine 598 and mimics the partial digestion product by extending the S595-containing peptide to arginine 601 (Fig. 2A and C). As predicted, the H599P mutant resulted in a single major S595 phosphopeptide corresponding to spot B (Fig. 2F) that was eliminated when S595 was also mutated (Fig. 2H). A minor spot that comigrates with spot A, or migrates nearby, was seen in the H599 maps (asterisks, Fig. 2F to H) that likely corresponds to a minor CDK site seen with eEF2 S595A (Fig. 2D, map 2). Of note, the partial trypsin digestion of the S595 peptide resulting in spot B differs somewhat between experiments, and in some cases, nearly complete digestion leads to reduced amounts of spot B (not shown).
In order to prove that S595 is phosphorylated by cyclin A-CDK2 in vitro, we mutated S595 to threonine (S595T) in both the wild-type (WT) and H599P backgrounds, which rescued normal amounts of eEF2 phosphorylation (Fig. 2E). We purposely overexpressed the S595A mutants relative to the WT and S595T eEF2 proteins to reveal residual phosphorylations (Fig. 2G) and to highlight the reduced eEF2 phosphorylation caused by S595A mutants even when they are relatively overexpressed (Fig. 2E). Importantly, phosphoamino acid analysis demonstrated that spot B was converted from phosphoserine to phosphothreonine when S595 was mutated to T595, thereby demonstrating that S595 is the phosphorylated residue in this phosphopeptide (Fig. 2F and G). In sum, these data indicate that S595 is the major site of eEF2 phosphorylation by cyclin A-CDK2 in vitro.
eEF2 is phosphorylated on serine 595 in vivo.
We also used phosphopeptide mapping to characterize eEF2 phosphorylation in vivo by incubating transfected cells with [32P]orthophosphate. These experiments utilized physiologic amounts of FLAG-eEF2 protein that were immunoprecipitated from transfected 293T cells (Fig. 3B), excised from membranes after electrophoresis and transfer, and subjected to phosphopeptide mapping as described above. In vivo eEF2 maps also contained spots A and B, and both were eliminated by the S595A mutation (Fig. 3A, maps 1 and 2). These maps also revealed a third major phosphopeptide, spot C, which represents the T56 phosphopeptide, as shown by its disappearance from the map of an eEF2 T56A mutant (Fig. 3C). Analogous to the in vitro maps, in vivo S595 phosphorylation shifted to spot B in the H599P mutant (Fig. 3A, map 3) and was eliminated by a concordant S595A/H599P mutation (Fig. 3A, map 4). Although it is difficult to quantitatively compare the amounts of phosphorylation of a specific peptide between independent maps, spot C appeared reduced in S595A and almost absent from H599P, suggesting a relationship between T56 phosphorylation and S595 (see below).
Fig 3.

eEF2 S595 phosphorylation in vivo. (A) Phosphopeptide maps of eEF2 (map 1) and the indicated mutants (maps 2 to 4) isolated from 293T cells labeled with [32P]orthophosphate. The peptides representing S595 (spots A and B) and T56 (spot C) are indicated. The S595A mutation abrogates the S595 spots in both the WT and H599P backgrounds (maps 2 and 4). Note that the amount of T56 phosphorylation is reduced in the S595A and H599P mutants (maps 2 to 4). Asterisks indicate a minor spot that comigrates with spot A. (B) Amount of overexpression of transfected FLAG-eEF2 relative to endogenous eEF2 (endog-eEF2). Vec, vector. (C) Identification of spot C as containing phosphorylated T56. Cells transfected with WT eEF2 or eEF2 T56A were labeled with orthophosphate and the immunoprecipitated eEF2 proteins analyzed by phosphopeptide mapping. (D) eEF2 phosphorylation is increased by cyclin A-CDK2 expression. U2OS cells were transfected with eEF2 and either cyclin A-CDK2 or empty vector (Vec), and eEF2 was immunoprecipitated after [32P]orthophosphate labeling (top two panels). The bottom panel shows total cyclin A-CDK histone H1 kinase activity. Phosphopeptide mapping reveals increased phosphorylation of S595 (spots A and B) and other sites (arrows). (E) Analysis similar to that described for panel D using eEF2 H599P.
To determine if cyclin A-CDK2 activity increases S595 phosphorylation in vivo, we labeled U2OS cells that were cotransfected with eEF2 and cyclin A-CDK2 with [-32P]orthophosphate. Ectopic cyclin A-CDK2 increased eEF2 phosphorylation in vivo (Fig. 3D, left), and this involved spots A and B as well as three additional spots (Fig. 3D, right, arrows) that correspond to the minor in vitro phosphorylation sites (Fig. 2D, map 2). Cyclin A-CDK2 similarly increased eEF2 H599P phosphorylation on S595 and the three minor spots (Fig. 3E). We speculate that the remaining phosphopeptides seen with eEF2 but not eEF2 H599P, which are not sensitive to cyclin A-CDK2, are likely derived from the T56 peptide. We conclude that eEF2 is phosphorylated on S595 in vivo and that eEF2 phosphorylation is augmented by ectopic cyclin A-CDK2 expression.
eEF2 S595 phosphorylation is increased in mitosis.
Cyclin A-CDK2 is active in the nucleus, whereas eEF2 is cytoplasmic. Thus, unlike the results seen with our initial lysate screen or in vitro experiments, subcellular localization may limit eEF2 access to cyclin A-CDK2 in vivo. Cyclin A-CDK2 shuttles from the cytoplasm to the nucleus and could phosphorylate eEF2 in interphase cells (28). However, we considered that cyclin A-CDK2 might have greatest access to eEF2 in mitosis, when nuclear compartmentalization is lost, and examined eEF2 S595 phosphorylation in different stages of the cell cycle. We do not have a phospho-S595-specific antibody, and we used the extent to which eEF2 isolated from cells can be further phosphorylated by cyclin A-CDK2 in vitro as a surrogate for preexisting phosphorylation, since in vitro phosphorylation occurs almost exclusively on S595. Figures 4A and B show that eEF2 immunoprecipitated from nocodazole-arrested prometaphase cells (M) was poorly phosphorylated by cyclin A-CDK2 in vitro compared with eEF2 isolated from asynchronous (A) or S-phase-arrested (S) cells. Figure 4A shows data from a single representative experiment, whereas Fig. 4B shows the combined data from four independent experiments. A representative cell cycle distribution of the various conditions is shown in Fig. 4C.
To determine if reduced in vitro phosphorylation of mitotic eEF2 resulted from its increased phosphorylation in vivo, we treated the immunoprecipitated mitotic eEF2 with phosphatase to remove preexisting in vivo phosphorylation. Phosphatase treatment restored the amount of mitotic eEF2 phosphorylation by cyclin A-CDK2 in vitro to nearly the amount seen with S-phase-derived eEF2, indicating that reduced in vitro phosphorylation did indeed reflect increased preexisting in vivo eEF2 phosphorylation in mitotic cells (Fig. 4A and B, M+λ). Finally, treating mitotic cells with roscovitine prior to lysis to inhibit endogenous CDKs also restored in vitro phosphorylation of mitotic eEF2, indicating that mitotic S595 phosphorylation is sensitive to roscovitine (Fig. 4A and C, M+Ros). These data demonstrate that eEF2 S595 is hyperphosphorylated in mitotic cells and that this is likely mediated by a CDK. In vitro phosphorylation of S-phase-derived eEF2 was modestly reduced compared with that seen with asynchronous cells, but this is not sensitive to the presence of phosphatase and is presently not well understood.
We also examined the extent to which endogenous eEF2 T56 phosphorylation differed under the cell cycle conditions described above in three cell types: Hct116, 293A, and SK-N-AS cells (Fig. 4D). Each of these cell lines exhibited large increases in T56 phosphorylation in prometaphase cells, consistent with the idea that eEF2 is inhibited in mitosis. Thus, both T56 and S595 are hyperphosphorylated eEF2 sites in mitotic cells.
S595 region mutants are poorly phosphorylated by eEF2K.
The in vivo maps suggested that eEF2 T56 phosphorylation is reduced by the S595A and H599P mutations (Fig. 2A), and we therefore used a phospho-T56-specific eEF2 antibody to assess their inhibitory phosphorylation in vivo. The S595A and H599 mutations each greatly reduced eEF2 T56 phosphorylation in vivo, which was normal in the S595T mutant (Fig. 5A). As expected, the pT56-specific antibody did not detect eEF2 when T56 was mutated. We also tested if glutamic acid could mimic S595 phosphorylation and restore T56 phosphorylation, but it did not (Fig. 5A, lane 6).
Fig 5.

An intact S595 region is required for efficient T56 phosphorylation. (A) eEF2 T56 phosphorylation in vivo is reduced by the S595A and H599P mutations. The indicated FLAG-eEF2 proteins were immunoprecipitated from transfected 293A cells and immunoblotted with anti-phospho-T56 eEF2 antibody and anti-FLAG. (B) eEF2 S595A is poorly phosphorylated by eEF2K in vitro. FLAG-eEF2 or FLAG-eEF2 S595A eluted from immunoprecipitates was subjected to phosphorylation by eEF2K in vitro, followed by autoradiography and immunoblotting with anti-pT56-eEF2. Input FLAG-eEF2 is shown. (C) S595A and H599P mutations prevent eEF2 phosphorylation by eEF2K in vitro. Immunoprecipitates of the indicated FLAG-eEF2 proteins were phosphorylated in vitro with either cyclin A-CDK2 (first panel) or recombinant eEF2K (fourth panel) or blotted for eEF2 (third panel) and pT56 eEF2 (second panel). eEF2K autophosphorylation is indicated (fourth panel). Panel 5 shows eEF2 and eEF2K abundance in the kinase assay shown in panel 4. (D) eEF2 S595A functions as a translocase. Diphtheria toxin and NAD+ were used to inhibit endogenous eEF2 in reticulocyte extracts (lane 2 to 4), and either WT eEF2 (lane 3) or 595A eEF2 (lane 4) was used to reconstitute translation of luciferase protein.
Because reduced in vivo T56 phosphorylation of eEF2 S595A and eEF2 H599P could reflect many cellular processes, we determined if these mutations directly inhibit T56 phosphorylation in vitro. Figure 5B shows FLAG-wt-eEF2 and FLAG-eEF2 S595A proteins that were eluted from immunoprecipitates, subjected to in vitro phosphorylation by eEF2K, and analyzed either by autoradiography or by blotting with the T56 phospho-specific antibody. Remarkably, the S595A mutation prevented eEF2 phosphorylation by eEF2K.
We also immunoprecipitated a series of FLAG-eEF2 mutants from transfected 293T cells and subjected the bound eEF2 proteins to phosphorylation with recombinant cyclin A-CDK2 or eEF2K in vitro or to immunoblotting to reveal preexisting in vivo T56 phosphorylation and eEF2 abundance. As expected, S595A was poorly phosphorylated by cyclin A-CDK2 (Fig. 5C, top panel, lane 2). Moreover, T56 phosphorylation of the S595A, H599P, and T56A eEF2 mutants by recombinant eEF2K was greatly reduced compared with WT eEF2 (lane 1) or S595T eEF2 (lane 3) (Fig. 5C, fourth panel; eEF2K autophosphorylation is also indicated). In each case, the amount of in vitro T56 phosphorylation by eEF2K (fourth panel) matched the amount of in vivo T56 phosphorylation (second panel), demonstrating that the S595A and H599P mutations directly impede T56 phosphorylation by eEF2K. The ability of recombinant eEF2K to further phosphorylate eEF2 isolated from cells is consistent with the fact that T56 phosphorylation in vivo is substoichiometric. The residual in vitro phosphorylation of eEF2 T56A by eEF2K (lane 5, fourth panel) likely reflects low-level phosphorylation of T58, a known minor eE2FK site (8, 21). Overall, these data show that mutations of the S595 region greatly inhibit eEF2 T56 phosphorylation by eEF2K in vivo and in vitro.
Reduced T56 phosphorylation of the S595A and H599P mutants could simply reflect their deleterious structural consequences. To address this possibility, we examined eEF2 S595A function by measuring its ability to catalyze protein synthesis in reticulocyte extracts. Diphtheria toxin inactivates eEF2 by catalyzing its ADP ribosylation, and this requires NAD (NAD+). We used diphtheria toxin and NAD+ to inactivate endogenous reticulocyte eEF2 and then rescued the inhibited extracts with equal amounts of either eEF2 or eEF2 S595A (Fig. 5D). The amount of diphtheria toxin activity was titrated to inhibit endogenous eEF2 but not the exogenous eEF2 by limiting the amount of NAD+ (not shown). WT eEF2 and eEF2 S595A each restored translation of a luciferase plasmid in the inhibited extracts. eEF2 S595A is thus active as a translocase, strongly suggesting that its reduced T56 phosphorylation does not result from gross structural anomalies.
eEF2 S595 phosphorylation is required for efficient eEF2 T56 phosphorylation.
The previous experiments relied on mutants to demonstrate a role for S595 phosphorylation in regulating T56 phosphorylation. We next determined if S595 phosphorylation per se stimulates T56 phosphorylation. To accomplish this, WT eEF2 was prephosphorylated with cyclin A-CDK2 (or mock treated), the cyclin A-CDK2 was removed, and eEF2 was subjected to an eEF2K assay that included [32P]ATP and roscovitine (to inhibit any residual cyclin A-CDK2 activity). eEF2 phosphorylation by cyclin A-CDK2 greatly stimulated its phosphorylation by eEF2K, and this was not seen with eEF2 T56A, indicating that the enhanced phosphorylation occurred on T56 (Fig. 6A). We tested if cyclin A-CDK2 catalytic activity was required to stimulate T56 phosphorylation by inclusion of roscovitine in the prephosphorylation step to inhibit CDK2. Roscovitine completely prevented the stimulation of T56 phosphorylation by cyclin A-CDK2, indicating that CDK2 activity is required to stimulate eEF2 phosphorylation by eEF2K (Fig. 6B, compare lanes 2 and 3). Finally, we performed the sequential cyclin A-CDK2/eEF2K reaction using eEF2 S595A and found that the S595A mutation completely abrogated the impact of cyclin A-CDK2 (Fig. 6C). In sum, these data indicate that phosphorylation of eEF2 on S595 by cyclin A-CDK2 directly stimulates eEF2 phosphorylation by eEF2K on T56.
Fig 6.

Phosphorylation of S595 by cyclin A-CDK2 facilitates phosphorylation of eEF2 by eEF2K. (A) S595 phosphorylation stimulates T56 phosphorylation by eEF2K. eEF2 (lanes 1 to 3) or eEF2-T56A (lanes 4 to 6) was phosphorylated with cyclin A-CDK2 or mock treated and subsequently phosphorylated by eEF2K in the presence of [32P]ATP and roscovitine. Prephosphorylation by cyclin A-CDK2 greatly stimulated WT eEF2 phosphorylation by eEF2K but did not affect eEF2-T56A. Lane 7 does not contain eEF2. The amount of eEF2 is shown in the immunoblot. (B) Cyclin A-CDK2 kinase activity is required to stimulate eEF2 phosphorylation by eEF2K. Experimental conditions were similar to those described for panel A except that roscovitine was included in the prephosphorylation reaction to inhibit cyclin A-CDK2. Roscovitine prevented stimulation of eEF2K phosphorylation by cyclin A-CDK2 (compare lanes 2 and 3). The amount of eEF2 and eEF2K is shown in the stained gel. (C) The stimulation of T56 phosphorylation by cyclin A-CDK2 requires S595. Phosphorylation of S595A by eEF2K is not stimulated by cyclin A-CDK2 (compare lanes 2 and 4). Total eEF2 is shown in the immunoblot. (D) A 30-mer phosphopeptide representing the S595 region competitively inhibits eEF2 phosphorylation by eEF2K. The peptides were present in 30-fold molar excess compared with eEF2 (U, unphosphorylated; P, phosphorylated).
In addition to the S595A mutation, T56 phosphorylation is also inhibited by the H599P mutation but does not affect S595 phosphorylation but instead likely introduces a local structural alteration caused by the proline substitution. We thus speculated that the S595 phosphorylation serves to recruit eEF2K to eEF2 and that this is inhibited when phosphorylation is prevented by the S595A mutations or by a structural change imposed by the H599P mutation. Unfortunately, our efforts to coprecipitate eEF2 with eEF2K failed for technical reasons (not shown). We therefore synthesized 30-mer peptides spanning the eEF2 S595 region, with or without phosphorylated S595, and determined if they inhibited eEF2 phosphorylation by eEF2K. When present in 30-fold molar excess over eEF2, the phosphorylated peptide was a more effective inhibitor of T56 phosphorylation than the unphosphorylated peptide (Fig. 6D).
DISCUSSION
We describe a new mechanism of eEF2 regulation and show that S595 phosphorylation is required for efficient T56 inhibitory phosphorylation by eEF2K. Because all known regulatory pathways that control eEF2 converge upon eEF2K and T56, S595 phosphorylation can potentially impact global eEF2 regulation by augmenting T56 phosphorylation by eEF2K (Fig. 7A). Figure 7B shows the positions of T56, S595, and H599, superimposed on the structure of budding yeast eEF2, which is highly homologous to human eEF2 (29). Our previous work showed that eEF2K contains distinct C-terminal eEF2 targeting and N-terminal catalytic domains that are separated by a linker region (30). We thus suggest that the S595 region interacts with eEF2K via its eEF2-targeting domain to promote efficient T56 phosphorylation by its catalytic domain (Fig. 7B). However, this model remains speculative and other mechanisms, such as allosteric eEF2 regulation, could account for the impact of S595 phosphorylation on T56.
Fig 7.
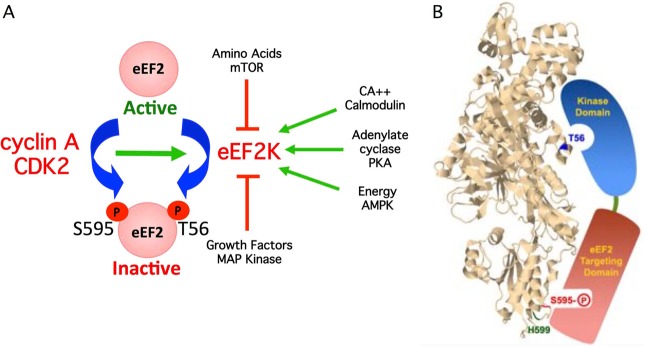
(A) Model depicting how S595 phosphorylation can globally regulate eEF2 activity by provoking T56 phosphorylation by eEF2K. Pathways that activate eEF2K are shown as green arrows, and pathways that inhibit eEF2K are shown in red. Increased S595 phosphorylation by cyclin A-CDK2 enhances T56 phosphorylation and eEF2 inhibition when eEF2K is activated. (B) Model depicting hypothesized interaction of the two functional eEF2K domains with eEF2 S595 and T56. The positions of T56, S595, and H599 were superimposed on the structure of budding yeast eEF2. The image is from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB; www.pdb.org) of PDB ID 2P8W (29). Residues were localized with Jmol, an open-source Java viewer for chemical structures in 3D (http://www.jmol.org/).
We focused on cyclin A-CDK2, but other kinases could also have roles in S595 phosphorylation. Multiple cyclin-CDKs phosphorylate eEF2 in vitro, and the impact of specific cyclin-CDKs on S595 phosphorylation may depend upon factors such as cell cycle position, relative binding affinities, and access to eEF2. Other proline-directed kinases also exhibit redundancy with cyclin-CDKs on some substrates. Although we found that GSK3β and MAPK, kinases that can exhibit redundancy with CDKs, do not phosphorylate eEF2 in vitro, other cellular kinases could have roles in S595 phosphorylation (25, 31). However, the fact that S595 phosphorylation is inhibited by roscovitine suggests that CDKs are the major S595 kinases in mitosis.
Our findings that both S595 and T56 are hyperphosphorylated in prometaphase cells support previous studies showing that mitotic cells exhibit high eEF2 T56 phosphorylation and suppressed translation (1–4, 6, 7, 32). Moreover, the concordant increase in both S595 and T56 phosphorylation in mitosis is consistent with the notion that S595 phosphorylation facilitates efficient T56 phosphorylation. In contrast, one group found that eEF2K is inhibited by phosphorylation on serine 359 by cyclin B-CDC2, which they suggest keeps eEF2 active in mitotic cells (33). The idea that CDC2 activity inhibits eEF2K seems opposed to our findings, as well as those of a recent study showing that phosphorylation of elongation factor 1 in mitosis, probably by cyclin B-CDK1, inhibits protein synthesis through hindered tRNA delivery to ribosomes (34). Although the basis for this discrepancy is unclear, it could reflect numerous experimental variables, including different cyclin-CDKs. Thus, the net impact of CDK activity on eEF2 may involve a balance between S595 phosphorylation and eEF2K inhibition in specific contexts.
In addition to intrinsic cell cycle oscillations, CDK activity is regulated by numerous processes, including checkpoints, apoptosis, DNA damage, and development. S595 phosphorylation may thus impact eEF2 activity in diverse physiologic contexts. However, because of the pleiotropic effects of general CDK inhibition, further physiologic functional studies of S595 phosphorylation will require dissociating S595 phosphorylation from overall CDK activity, such as with S595A knock-in models.
ACKNOWLEDGMENTS
We thank Jon Cooper for expert advice and the use of his phosphopeptide mapping apparatus and T. Barshevsky (NEB) for recombinant cyclin A-CDK2.
B.E.C. is supported by the NIH (CA102742 and CA084069).
Footnotes
Published ahead of print 26 November 2012
REFERENCES
- 1. Celis JE, Madsen P, Ryazanov AG. 1990. Increased phosphorylation of elongation factor 2 during mitosis in transformed human amnion cells correlates with a decreased rate of protein synthesis. Proc. Natl. Acad. Sci. U. S. A. 87: 4231– 4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pyronnet S, Sonenberg N. 2001. Cell-cycle-dependent translational control. Curr. Opin. Genet. Dev. 11: 13– 18 [DOI] [PubMed] [Google Scholar]
- 3. Bonneau AM, Sonenberg N. 1987. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J. Biol. Chem. 262: 11134– 11139 [PubMed] [Google Scholar]
- 4. Fan H, Penman S. 1970. Regulation of protein synthesis in mammalian cells. II. Inhibition of protein synthesis at the level of initiation during mitosis. J. Mol. Biol. 50: 655– 670 [DOI] [PubMed] [Google Scholar]
- 5. Browne GJ, Proud CG. 2002. Regulation of peptide-chain elongation in mammalian cells. Eur. J. Biochem. 269: 5360– 5368 [DOI] [PubMed] [Google Scholar]
- 6. Jørgensen R, Merrill AR, Andersen GR. 2006. The life and death of translation elongation factor 2. Biochem. Soc. Trans. 34: 1– 6 [DOI] [PubMed] [Google Scholar]
- 7. Sivan G, Kedersha N, Elroy-Stein O. 2007. Ribosomal slowdown mediates translational arrest during cellular division. Mol. Cell. Biol. 27: 6639– 6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ovchinnikov LP, Motuz LP, Natapov PG, Averbuch LJ, Wettenhall RE, Szyszka R, Kramer G, Hardesty B. 1990. Three phosphorylation sites in elongation factor 2. FEBS Lett. 275: 209– 212 [DOI] [PubMed] [Google Scholar]
- 9. Price NT, Redpath NT, Severinov KV, Campbell DG, Russell JM, Proud CG. 1991. Identification of the phosphorylation sites in elongation factor-2 from rabbit reticulocytes. FEBS Lett. 282: 253– 258 [DOI] [PubMed] [Google Scholar]
- 10. Nairn AC, Palfrey HC. 1987. Identification of the major Mr 100,000 substrate for calmodulin-dependent protein kinase III in mammalian cells as elongation factor-2. J. Biol. Chem. 262: 17299– 17303 [PubMed] [Google Scholar]
- 11. Ryazanov AG. 1987. Ca2+/calmodulin-dependent phosphorylation of elongation factor 2. FEBS Lett. 214: 331– 334 [DOI] [PubMed] [Google Scholar]
- 12. Ryazanov AG, Davydova EK. 1989. Mechanism of elongation factor 2 (EF-2) inactivation upon phosphorylation. Phosphorylated EF-2 is unable to catalyze translocation. FEBS Lett. 251: 187– 190 [DOI] [PubMed] [Google Scholar]
- 13. Proud CG. 2007. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem. J. 403: 217– 234 [DOI] [PubMed] [Google Scholar]
- 14. White-Gilbertson S, Kurtz DT, Voelkel-Johnson C. 2009. The role of protein synthesis in cell cycling and cancer. Mol. Oncol. 3: 402– 408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knebel A, Morrice N, Cohen P. 2001. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 20: 4360– 4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Redpath NT, Foulstone EJ, Proud CG. 1996. Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J. 15: 2291– 2297 [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. 2001. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20: 4370– 4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Althausen S, Mengesdorf T, Mies G, Olah L, Nairn AC, Proud CG, Paschen W. 2001. Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J. Neurochem. 78: 779– 787 [DOI] [PubMed] [Google Scholar]
- 19. Browne GJ, Finn SG, Proud CG. 2004. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J. Biol. Chem. 279: 12220– 12231 [DOI] [PubMed] [Google Scholar]
- 20. Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. 2002. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr. Biol. 12: 1419– 1423 [DOI] [PubMed] [Google Scholar]
- 21. Redpath NT, Proud CG. 1993. Cyclic AMP-dependent protein kinase phosphorylates rabbit reticulocyte elongation factor-2 kinase and induces calcium-independent activity. Biochem. J. 293(Pt 1):31–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chi Y, Welcker M, Hizli AA, Posakony JJ, Aebersold R, Clurman BE. 2008. Identification of CDK2 substrates in human cell lysates. Genome Biol. 9: R149 doi:10.1186/gb-2008-9-10-r149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dorovkov MV, Pavur KS, Petrov AN, Ryazanov AG. 2002. Regulation of elongation factor-2 kinase by pH. Biochemistry 41:13444– 13450 [DOI] [PubMed] [Google Scholar]
- 24. Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. 1996. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 10: 1979– 1990 [DOI] [PubMed] [Google Scholar]
- 25. Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM. 2003. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 12: 381– 392 [DOI] [PubMed] [Google Scholar]
- 26. Drazin R, Kandel J, Collier RJ. 1971. Structure and activity of diphtheria toxin. II. Attack by trypsin at a specific site within the intact toxin molecule. J. Biol. Chem. 246: 1504– 1510 [PubMed] [Google Scholar]
- 27. Chi Y, Clurman BE. 2010. Mass spectrometry-based identification of protein kinase substrates utilizing engineered kinases and thiophosphate labeling. Curr. Protoc. Chem. Biol. 2: ch100151 doi:10.1002/9780470559277.ch100151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jackman M, Kubota Y, den Elzen N, Hagting A, Pines J. 2002. Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol. Biol. Cell 13:1030– 1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor DJ, Nilsson J, Merrill AR, Andersen GR, Nissen P, Frank J. 2007. Structures of modified eEF2 80S ribosome complexes reveal the role of GTP hydrolysis in translocation. EMBO J. 26: 2421– 2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pavur KS, Petrov AN, Ryazanov AG. 2000. Mapping the functional domains of elongation factor-2 kinase. Biochemistry 39: 12216– 12224 [DOI] [PubMed] [Google Scholar]
- 31. Hydbring P, Larsson LG. 2010. Tipping the balance: Cdk2 enables Myc to suppress senescence. Cancer Res. 70: 6687– 6691 [DOI] [PubMed] [Google Scholar]
- 32. Sivan G, Elroy-Stein O. 2008. Regulation of mRNA translation during cellular division. Cell Cycle 7: 741– 744 [DOI] [PubMed] [Google Scholar]
- 33. Smith EM, Proud CG. 2008. cdc2-cyclin B regulates eEF2 kinase activity in a cell cycle- and amino acid-dependent manner. EMBO J. 27: 1005– 1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sivan G, Aviner R, Elroy-Stein O. 2011. Mitotic modulation of translation elongation factor 1 leads to hindered tRNA delivery to ribosomes. J. Biol. Chem. 286: 27927– 27935 [DOI] [PMC free article] [PubMed] [Google Scholar]