


Abstract
High-mobility group B (HMGB) proteins bind duplex DNA without sequence specificity, facilitating the formation of compact nucleoprotein structures by increasing the apparent flexibility of DNA through the introduction of DNA kinks. It has remained unclear whether HMGB binding and DNA kinking are simultaneous and whether the induced kink is rigid (static) or flexible. The detailed molecular mechanism of HMGB-induced DNA ‘softening’ is explored here by single-molecule fluorescence resonance energy transfer studies of single yeast Nhp6A (yNhp6A) proteins binding to short DNA duplexes. We show that the local effect of yNhp6A protein binding to DNA is consistent with formation of a single static kink that is short lived (lifetimes of a few seconds) under physiological buffer conditions. Within the time resolution of our experiments, this static kink occurs at the instant the protein binds to the DNA, and the DNA straightens at the instant the protein dissociates from the DNA. Our observations support a model in which HMGB proteins soften DNA through random dynamic binding and dissociation, accompanied by DNA kinking and straightening, respectively.
INTRODUCTION
High-mobility group B (HMGB) proteins are small chromatin-associated eukaryotic proteins that alter the physical properties of DNA in vitro and in vivo (1,2). HMGB proteins have one or two highly conserved motifs (‘box A’ and ‘box B’) that bend DNA, and each homologous box motif contains amino acids that form three alpha helices to bind DNA as an ‘L’-shaped structure (1,2). Saccharomyces cerevisiae Nhp6A (yNhp6A) is an abundant (50 000–70 000 molecules per haploid cell), small (11 kD) and sequence-non-specific DNA-binding protein that is a member of the HMGB family of proteins (1,2). This single-box HMGB protein conforms to the ‘box B’ group of HMGB proteins (1,2). yNhp6A binds to DNA as a monomer through both intercalative and electrostatic interactions (3), bending DNA sharply (4,5). yNhp6A–DNA interactions are believed to enhance DNA flexibility so as to facilitate looping required for processes such as DNA replication, DNA repair, recombination, gene regulation and translation (1,2). HMGB proteins have also been proposed to be involved in nucleosome remodeling (6).
Although the sequence-non-specific HMGB family of proteins enhances DNA flexibility by binding DNA, the detailed physical mechanism of DNA flexibility enhancement by these proteins remains unknown. There has been uncertainty about association and dissociation binding rates for individual HMGB proteins, whether DNA binding and kinking are simultaneous, and the flexibility of DNA in the kinked complex (1,7–13). Biophysical studies of HMGB proteins have led to the proposal of two models by which these proteins enhance apparent DNA flexibility. In the first model, termed the ‘static kink model’, HMGB proteins bind transiently to DNA, creating fixed angle bends in the DNA at sites of protein binding (12–14). In the second ‘flexible hinge’ model, HMGB protein binding creates a protein–DNA complex that is anisotropic but highly dynamic, so there is little bending angle preference at the binding site (8–10).
In the present study, single-molecule fluorescence resonance energy transfer (smFRET) is used to detect the local conformational effects of yNhp6A binding to isolated DNA duplexes designed to be short enough so that single protein–DNA complexes can be monitored without protein–protein interactions on the DNA. smFRET provides a direct measure of the conformational state(s) of DNA molecules induced by yNhp6A binding and allows the observation of any long-lived kinetic intermediates in the kinking/un-kinking of DNA by yNhp6A. The smFRET results presented here demonstrate that yNhp6A simultaneously binds and kinks DNA without sequence specificity, producing a static kink that is short lived. It is also shown that yNhp6A binding affinity and induced DNA bend angle can be tuned by the presence of a pre-existing DNA deformation. These results support the static kink model for apparent DNA flexibility enhancement by HMGB proteins.
MATERIALS AND METHODS
Protein preparation
Recombinant S. cerevisiae Nhp6A (yNhp6A) protein was prepared as described previously (15).
Single-molecule experiments
Sample chambers (∼5-mm long, ∼0.1-mm wide and ∼0.1-mm deep) were formed by assembling polyethylene glycol-coated quartz slides and glass coverslips with double-sided tape and epoxy cement. Two holes in the quartz slides—one on each end of the sample chamber—allowed sample chamber fluid exchange. Because ∼0.1% of the polyethylene glycol surface in the sample chamber was biotinylated, biotinylated DNA could be attached to the surface by flowing 0.25 mg/ml streptavidin solution into the sample chamber, washing away any unbound streptavidin, and adding 10–50 pM biotinylated DNA to the streptavidin-coated surface. Samples were imaged at room temperature (∼22°C) in a single-molecule imaging buffer containing 10 mM HEPES, pH 7.5; 100 mM NaCl; 1 mM MgCl2; 5% glycerol; 0.1 mg/ml bovine serum albumin; 0.4% glucose; 0.1 mg/ml glucose oxidase; 0.02 mg/ml catalase and saturated Trolox® (16).
smFRET experiments were performed on a prism-type total internal reflection microscope built on an Olympus IX71 inverted microscope. This wide-field microscope system permits the simultaneous observation of ∼200 DNA molecules immobilized on the surface of a slide, allowing thousands of molecules to be observed within minutes for each reaction condition. Surface-immobilized donor (Cy3) fluorophores were excited with a 532-nm laser, and the fluorescence emission of the donor and acceptor dyes was collected with a ×60 water objective (1.2 NA, Olympus). The donor (Cy3) and acceptor (Cy5) emission intensities were separated using a dichroic mirror and recorded with a CCD camera (iXon, Andor Technology) at 32-ms time resolution. The FRET efficiency for each individual pair was calculated as IA/(IA + ID) (where IA is the acceptor emission intensity, and ID is the donor emission intensity) and was corrected for cross talk.
DNA preparation
DNA duplexes used in this work were assembled using oligonucleotides purchased from Integrated DNA Technologies. DNA duplexes were formed by annealing oligonucleotides in a ratio of 1:1.2 (strand B:strand A) in an annealing buffer containing 20 mM Tris, pH 8.0 and 500 mM NaCl by heating the strands to 80°C for 3 min followed by slow cooling to room temperature. All annealing reactions were performed in the dark, and the annealed DNA was stored at −20°C.
The oligonucleotide sequences used in the smFRET studies are as follows:
Linear_18a:
Strand A: 5′-/Cy3/TGG CGA CGG CAG CGA GGC-3′
-
Strand B: 5′-/Cy5/GCC TCG CTG CCG TCG CCA TTT TTT TTT TTT TTT/Biotin/-3′
Linear_18b:
Strand A: 5′-/Cy3/TGG TTA TAT CAT CTA GGC-3′
-
Strand B: 5′-/Cy5/GCC TAG ATG ATA TAA CCA TTT TTT TTT TTT TTT/Biotin/-3′
Bulge_18a:
Strand A: 5′-/Cy3/TGG CGA CGG AAC AGC GAG GC-3′
Strand B: 5′-/Cy5/GCC TCG CTG CCG TCG CCA TTT TTT TTT TTT TTT/Biotin/-3′
RESULTS
yNhp6A binds 18-bp linear DNA in a single kinked state
Preliminary bulk biochemistry experiments confirmed that yNhp6A binds a short DNA duplex with an equilibrium dissociation constant in the low nanomolar range (Supplementary Figure S1A) and with rapid exchange kinetics (Supplementary Figure S1B). To extend this result to the single-molecule regime to evaluate individual binding and bending events, we first used FRET to observe the effects on DNA of binding and dissociation of yNhp6A to 18-bp homoduplex (all Watson–Crick base pairs) DNA target linear_18a. As yNhp6A has a DNA footprint of ∼11 bp (17), this duplex should permit binding of only a single yNhp6A molecule. The linear_18a target was prepared by annealing two complementary oligonucleotides (Figure 1). Strand A was labeled on the 5′ terminus with a Cy3 (donor) fluorophore. Strand B was labeled on the 5′ terminus with a Cy5 (acceptor) fluorophore and on the 3′ terminus with a biotin residue. In addition, strand B contained a 15-nt oligo(dT) sequence separating the 3′ terminal biotin from the duplex region to prevent potential interactions of the fluorophores with the streptavidin-coated surface. The target design is such that the donor and acceptor fluorophores are on opposite termini of the duplex; thus, protein-induced duplex bending decreases the DNA end-to-end distance and results in an increase in the efficiency of energy transfer between the fluorophores of the FRET pair.
Figure 1.
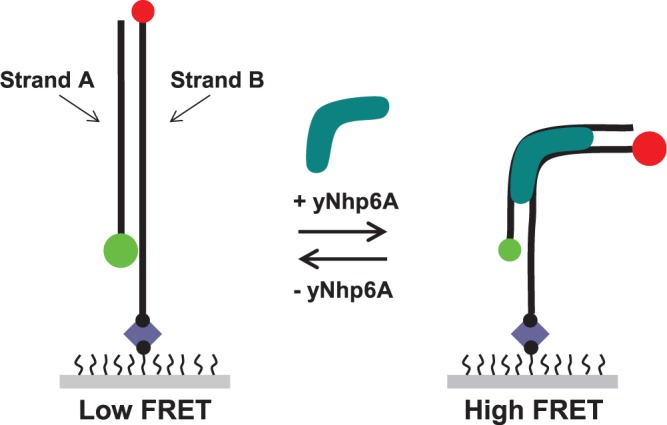
Schematic design of smFRET experiments. The DNA targets used in this study were designed so that the donor (Cy3, green circle) and acceptor (Cy5, red circle) are attached to opposite termini of the DNA duplex (black lines). Decreases in DNA end-to-end distance induced by yNhp6A (aqua) binding and kinking result in a measurable increase in the efficiency of energy transfer for each individual FRET pair. The DNA-only state is in a ‘low FRET’ state, and the yNhp6A-bound DNA is in a ‘high FRET’ state. Each DNA molecule is immobilized to the surface of a polymer-coated slide through a strong biotin (black circle)–streptavidin (blue diamond) interaction.
The distribution of FRET efficiency (EFRET) values and the dynamics of individual molecules were determined for hundreds of FRET pairs in the absence and presence of yNhp6A. In the absence of protein, the linear_18a target gives rise to a single narrow peak at EFRET ≈ 0.17 (the small peak at EFRET ≈ 0 represents targets with an inactive acceptor fluorophore and is not mentioned in EFRET histograms hereafter; Figure 2A, DNA only). Single-molecule time traces of the Cy3 (donor) and Cy5 (acceptor) emission intensities were plotted (Figure 2B, DNA only) along with the corresponding time traces of FRET efficiencies (Figure 2C, DNA only). No FRET transitions were observed for any of the linear_18a molecules in the absence of added protein.
Figure 2.

yNhp6A binds linear_18a DNA in a single kinked state. (A) FRET efficiency histograms for linear_18a DNA target in the absence and presence of yMhp6A indicating that yNhp6A binds to this DNA target in a single kinked state. (B) Time traces of donor and acceptor emission intensities and (C) corresponding FRET efficiency time traces showing no DNA target dynamics in the absence of protein but multiple transitions between the low (EFRET ∼0.17) and high (EFRET ∼0.30) FRET states in the presence of yNhp6A. (D) FRET efficiency time traces analyzed for a two-state system using a hidden Markov model to determine the average transition rates (v) from low FRET to high FRET states (v12) and from the high FRET to low FRET states (v21). (E) Dwell time distributions for the binding times (τ12) (low FRET states/DNA-only states) are shown at different yNhp6A concentrations. (F) Dwell time distributions for the binding times (τ21) (high FRET states/yNhp6A-bound states) are shown at different yNhp6A concentrations.
Addition of yNhp6A to the linear_18a target led to the appearance of a new bound state with an EFRET peak centered at EFRET ∼0.30 (Figure 2A, +yNhp6A). The increase in EFRET on protein addition indicates that the protein binds and bends the DNA with a signal increase, suggesting DNA bending by ∼60° [with the simplifying assumption that changes in dye separation due to changes in DNA twist will be small relative to the effect of DNA bending (18,19); for details on angle calculations, see Supplementary Material]. This estimate is in agreement with previously reported values for yNhp6A-induced DNA bending [∼60° in an AFM study (13), ∼63° in a gel electrophoresis study (20) and ∼70° in a nuclear magnetic resonance (NMR) study (4)]. The population of DNA targets in the high EFRET state increased with protein concentration until all linear_18a DNA targets had conformations with EFRET ≈ 0.30 (Figure 2A, +yNhp6A). These results suggest that yNhp6A binds to the linear_18a DNA target to produce a single kinked state. Equilibrium dissociation constant (KD) values for yNhp6A/DNA interactions have been reported to be 1–10 nM (5,17,21). Consistent with this high affinity binding, the high FRET state saturated at a protein concentration in the low nanomolar range (Figure 2A, +yNhp6A).
With respect to kinetics, time trajectories for the emission intensities and corresponding EFRET values show that FRET dynamics for the linear_18a DNA target in the presence of yNhp6A are dependent on the protein concentration. In the yNhp6A concentration range where both binding and dissociation can be observed (0.5–5 nM), the frequency of binding events increases with protein concentration, while the duration of the binding events is independent of protein concentration (Figure 2B and C, +0.5–5 nM yNhp6A). This is the expected result for a reversible first-order binding reaction. At protein concentrations >5 nM, dwell times between binding (high FRET) events are shorter than the time resolution of the experiment. In this protein-saturated state, few DNA-only FRET states are detected in the individual time traces, and the transition rates cannot be accurately determined (Figure 2B and C, +50 nM yNhp6A). It was of great interest to deduce population average kinetic parameters from these experiments. To quantify binding and dissociation rates, a hidden Markov model (22) was applied to hundreds of time traces over three different yNhp6A concentrations. This quantitative analysis confirms that the association rate (v12) increases linearly with increasing protein concentration, while the dissociation rate (v21) is independent of protein concentration (Figure 2D). The values of the dissociation rate, v21, (and hence the dissociation rate constant, koff) are ∼1 s−1. The association rate (v12) at 1 nM yNhp6A concentration is ∼1 s−1, corresponding to an association rate constant (kon) of ∼1 × 109 M−1 s−1. Consistent with an equilibrium dissociation constant in the low nM range, the ratio of these rate constants is ∼1 nM. Again, these are the expected results for a reversible first-order binding reaction. Together, the observations that the EFRET value of the bound state is the same for low yNhp6A concentrations (0.5 nM) as for saturating concentrations (50 nM) and that the association rate increases linearly with increasing protein concentration demonstrate that the observed changes in EFRET reflect one yNhp6A binding to each DNA molecule (as expected for a yNhp6A DNA footprint of ∼11 bp on a DNA target that is 18 bp in length). Further supporting this simple binding model, the dwell time distributions for protein association (τon) and dissociation (τoff) all are characterized by single exponential decays (Figure 2E–F), consistent with the presence of a single kinetic step. The dwell time distribution results thus indicate that yNhp6A binding is simultaneous with the EFRET value increase from ∼0.17 (the DNA-only state) to ∼0.30 (the protein-bound state). Likewise, yNhp6A dissociation occurs at the moment the EFRET value returns from ∼0.30 to ∼0.17, suggesting no long-lived intermediates.
The evidence that DNA binding and bending by yNhp6A are simultaneous argues against a bound state sampling both bent and straight DNA conformations. Rapid yNhp6A binding is indicated by the observed trend in v12 (defined as the reciprocal of the average time in low FRET state); v12 increases linearly with yNhp6A concentration. If bound yNhp6A were bending/unbending the DNA without dissociation, v12 would be independent of concentration. Bulk fluorescence anisotropy experiments also demonstrate that yNhp6A pre-bound to dye-labeled double-stranded DNA (dsDNA) dissociates within seconds after competing excess unlabeled dsDNA is added to the reaction (Supplementary Figure S1B). Furthermore, regardless of protein concentration (i.e. 0.5 or 100 nM yNhp6A in the sample chamber), replacing the yNhp6A-containing sample chamber with imaging buffer (no protein) results in an immediate shift to the DNA-only FRET state, showing that yNhp6A binding is transient (lasting seconds) under these conditions. If yNhp6A were stably bound to the DNA, multiple washes would be required to remove the protein.
The interaction of yNhp6A with 18-bp linear DNA is sequence independent
Although HMGB proteins are believed to bind sequence non-specifically, it is possible that there may be a weak preference for certain AT regions because of intrinsic DNA curvature or local DNA flexibility (11,23,24). To test the effect of sequence on observed DNA bending by yNhp6A, we measured the binding and dissociation of yNhp6A to and from DNA duplex linear_18b, an 18-bp homoduplex DNA target with a sequence different from linear_18a. In contrast to linear_18a (G/C base content of 78%), linear_18b has a G/C base content of 39% (see ‘Materials and Methods’ section) and a correspondingly lower melting temperature than linear_18a. Apart from the difference in duplex G/C content, linear_18b target was prepared and analyzed identically to linear_18a. Note that duplexes linear_18a and linear_18b were designed so that the terminal base pairs were identical to ensure that local fluorophore environments were conserved between the two targets.
In the absence of protein, the distribution of EFRET values for the linear_18b DNA displayed a single narrow peak at EFRET ≈ 0.17 (Figure 3A, DNA only). Single-molecule fluorophore emission intensity and corresponding FRET efficiency time traces indicate no intrinsic FRET dynamics for linear_18b DNA. This result was expected and reflects behavior identical to that of the linear_18a target (Figure 3A–C, DNA only).
Figure 3.

yNhp6A binds linear_18b DNA in a single kinked state. (A) FRET efficiency histograms for linear_18b DNA target in the absence and presence of yMhp6A indicating that yNhp6A binds to this DNA target in a single kinked state. (B) Time traces of donor and acceptor emission intensities, and (C) corresponding FRET efficiency time traces showing no DNA target dynamics in the absence of protein but multiple transitions between the low (EFRET ∼0.17) and high (EFRET ∼0.30) FRET states in the presence of yNhp6A. (D) FRET efficiency time traces analyzed for a two-state system using a hidden Markov model to determine the average transition rates (v) from low FRET to high FRET states (v12) and from the high FRET to low FRET states (v21). (E) Dwell time distributions for the binding times (τ12) (low FRET states/DNA-only states) are shown at different yNhp6A concentrations. (F) Dwell time distributions for the binding times (τ21) (high FRET states/yNhp6A-bound states) are shown at different yNhp6A concentrations.
Addition of yNhp6A to the linear_18b target led to the appearance of a new EFRET peak, and as for the linear_18a target, the new population was centered at EFRET ∼0.30 (Figure 3A, +yNhp6A). Thus, the increase in EFRET on protein binding to the linear_18b target indicates that yNhp6A bends this DNA target indistinguishably from the more G/C-rich linear_18a target. As observed for the linear_18a target, the yNhp6A-bound population increased with protein concentration until all linear_18b DNA molecules had conformations with EFRET ∼0.30 (Figure 3A, +yNhp6A). Thus, yNhp6A binds linear_18b in a single kinked state, an interaction identical to that with the linear_18a DNA molecule. Time trajectories for the emission intensities (Figure 3B) and corresponding EFRET values (Figure 3C) show that the dynamics of the linear_18b DNA target in complex with protein are qualitatively similar to the dynamics observed for the linear_18a DNA target complex with yNhp6A: over the range of concentrations where both binding and dissociation can be observed (0.5–5 nM yNhp6A), the frequency of binding events increases with protein concentration, while the duration of binding events appears to be independent of protein concentration (Figure 3B and C, +0.5–5 nM yNhp6A). At protein concentrations >5 nM, intervals between individual binding events are shorter than the time resolution of the experiment, so transition rates cannot be accurately determined under these conditions (Figure 3B and C, +50 nM yNhp6A). The hidden Markov model (22) was again applied to estimate population average binding and dissociation rates. This analysis confirms again that the association rate (v12) increases linearly with increasing protein concentration, while the dissociation rate (v21) is independent of the protein concentration (Figure 3D). Thus, the behavior of the association and dissociation rates of yNhp6A to linear_18b is the same as for linear_18a. Association rates for yNhp6A binding to linear_18a and linear_18b are the same, within error (Figures 2D and 3D), whereas the dissociation rate of yNhp6A from linear_18a was approximately twice the rate of dissociation from linear_18b. This result implies that while yNhp6A binds to both DNA duplex targets similarly, yNhp6A persists on the more A/T-rich linear_18b target for about twice as long as it does to the more G/C-rich linear_18a, resulting in a ∼2-fold equilibrium binding preference for linear_18b. These kinetic differences are subtle; however, the general association and dissociation trends for the two targets are remarkably similar. As for the linear_18a target, the dwell time distributions for yNhp6A association (τon) and dissociation (τoff) to and from linear_18b DNA all have single exponential decays (Figure 3E and F), consistent with the presence of a single kinetic step. Taken together, these results emphasize the sequence independence of DNA binding by yNhp6A.
In summary, each single yNhp6A molecule binds to a canonical Watson–Crick DNA molecule in a relatively sequence-independent manner, kinking the DNA by ∼60°. There is no evidence for un-kinked intermediates. The yNhp6A–DNA complex is short lived, lasting only seconds. As yNhp6A has high affinity (Kd in the low nanomolar range) and rapid on/off kinetics for homoduplex DNA, the target DNA is quickly rebound by yNhp6A after dissociation, making DNA shape fluctuations highly dynamic in the presence of low nanomolar concentrations of yNhp6A.
yNhp6A binds 18-bp bulged DNA in an unique kinked state
Early results characterized HMGB protein binding as preferential for distorted DNA (25,26), with some studies showing HMGB protein binding more tightly to curved rather than linear DNA (3). To probe the conformation and kinetics of a yNhp6A complex with a non-canonical DNA structure, we designed a DNA target (bulge_18a) with the same sequence and fluorophore labeling as the linear_18a DNA target, but with a two-base (A–A) bulge in the center of strand A (see ‘Materials and Methods’ section for sequence information).
In the absence of protein, the distribution of EFRET values of the bulge_18a DNA has a single narrow peak at EFRET ≈ 0.17 (Figure 4A, DNA only). The time traces of fluorophore emission intensities and corresponding FRET efficiencies indicate no intrinsic conformational bending dynamics for this DNA target. This result demonstrates that the A–A bulge neither substantially bends the DNA backbone nor introduces a site of flexibility, as the conformations and dynamics of the bulge_18a and linear_18a targets in the absence of protein are indistinguishable.
Figure 4.

yNhp6A binds bulge_18a DNA in a single kinked state. (A) FRET efficiency histograms for bulge_18a DNA target in the absence and presence of yMhp6A indicating that yNhp6A binds to this DNA target in a single kinked state. (B) Time traces of donor and acceptor emission intensities, and (C) corresponding FRET efficiency time traces showing no DNA target dynamics in the absence of protein but multiple transitions between the low (EFRET ∼0.17) and high (EFRET ∼0.25) FRET states in the presence of yNhp6A. (D) FRET efficiency time traces analyzed for a two-state system using a hidden Markov model to determine the average transition rates (v) from low FRET to high FRET states (v12) and from the high FRET to low FRET states (v21). (E) Dwell time distributions for the binding times (τ12) (low FRET states/DNA-only states) are shown at different yNhp6A concentrations. (F) Dwell time distributions for the binding times (τ21) (high FRET states/yNhp6A-bound states) are shown at different yNhp6A concentrations.
Interestingly, addition of yNhp6A to the bulge_18a target led to the appearance of a new EFRET peak that was centered at EFRET ≈ 0.25 (Figure 4A, +yNhp6A). This increase in EFRET on protein addition indicates that the protein binds the bulged DNA, kinking it by ∼45°. As observed for the linear homoduplex targets, the DNA population in the bound (high EFRET) state increases with protein concentration until all bulge_18a molecules displayed conformations with EFRET ∼0.25 (Figure 4A, +yNhp6A). Thus, yNhp6A binds the bulge_18a DNA target in a single kinked state, but the induced kink angle is ∼15° (i.e. 25%) less than the angle of the kink induced in the linear homoduplex target. Consistent with the observation that yNhp6A binds some non-canonical targets with higher affinity than linear homoduplex DNA (3), for each yNhp6A concentration, the proportion of the DNA population in the high FRET state was greater for the bulge_18a target than the linear_18a target. To determine whether the increased affinity of yNhp6A for the bulge_18a target versus linear_18a target originated through an increased association rate or a decreased dissociation rate (or both), time trajectories for the emission intensities and corresponding EFRET values were examined. The results indicate that (as for the linear_18a DNA target) dynamics of the bulge_18a DNA target FRET transitions in the presence yNhp6A were dependent on the protein concentration. In the range of concentrations where both binding and dissociation can be observed (0.5–5 nM yNhp6A), the frequency of the binding events again increased with protein concentration, while the duration of binding events again appeared to be independent of protein concentration (Figure 4B and C, +0.5–5 nM yNhp6A). At protein concentrations >5 nM, intervals between binding (high FRET) events were again shorter than the time resolution of the experiment, so transition rates could not be accurately determined in this protein-saturated state (Figure 4B and C, +50 nM yNhp6A). Hidden Markov model analysis (22) was applied again to estimate kinetic parameters, demonstrating that the association rate (v12) increases linearly with increasing protein concentration, while the dissociation rate (v21) is independent of protein concentration (Figure 4D). Association rates for yNhp6A binding to the bulge_18a DNA target were observed to be ∼2-fold higher than the association rates for yNhp6A binding to the linear_18a target, whereas dissociation rates were similar. Therefore, the increased binding affinity of yNhp6A for the bulge target relative to the homoduplex arises from the increased association rate for the bulged DNA, as the dwell times are comparable for both DNA targets. As for the linear_18a DNA target, the observations that the EFRET value for the bound state of the bulge_18a target DNA is the same at low yNhp6A concentrations (0.5 nM) as it is at saturating (50 nM) protein concentrations and that the association rate increases proportionately with increasing protein concentration demonstrate that the observed EFRET changes reflect one yNhp6A molecule binding to each DNA molecule. Again, this result was expected because the DNA footprint of yNhp6A is ∼11 bp, and the DNA target length is 18 bp. Also similar to results for the linear_18a DNA target, dwell time distributions for protein association (τon) and dissociation (τoff) all display single exponential decays (Figure 4E and F), consistent with the presence of a single kinetic step. This result implies that, as for the linear_18a and linear_18b targets, yNhp6A binding is simultaneous with the EFRET increase from ∼0.17 (DNA-only state) to ∼0.25 (protein-bound state), and that yNhp6A dissociation is simultaneous with the EFRET return from ∼0.25 to ∼0.17.
Similar results were obtained for yNhp6A binding to shorter (15 bp) homoduplex and bulge duplex DNA targets (Supplementary Figures S2 and S3). These experiments were undertaken because of the formal possibility that smFRET behavior might reflect the binding of more than one yNhp6A molecule to longer DNA targets. Demonstrating similar results with 15-bp DNA targets provides evidence that single yNhp6A molecules have this effect. Because the footprint of yNhp6A covers ∼11 bp, a second yNhp6A molecule should not be accommodated on a 15-bp target.
DISCUSSION
yNhp6A is a protein in the family of chromatin-associated eukaryotic HMGB proteins, and HMGB proteins are intriguing for their abilities to increase the apparent flexibility of DNA in vitro and in vivo (1). Here, we investigated the detailed physical basis for apparent DNA flexibility enhancement of HMGB proteins by studying the kinetics of yNhp6A association and dissociation to and from a series of DNA targets using smFRET. By design, the smFRET experiments allow the direct visualization of protein-induced bending by reporting on changes in the end-to-end distances of DNA targets sufficiently short that only single proteins can bind.
First, the results reported here demonstrate that single yNhp6A molecules bind homoduplex DNA with high affinity and kink the DNA with an angle of ∼60°, independent of the DNA sequence. The kink associated with the yNhp6A–homoduplex complex occurs simultaneously (within the ∼32-ms CCD camera time resolution of these experiments) with the yNhp6A–DNA binding event, and the kink remains static at ∼60° for the entire time the protein is bound to the DNA (seconds). As yNhp6A dissociates from the DNA, the DNA simultaneously straightens. This observation suggests a model for yNhp6A interaction in which transient binding creates a static bent state (kinking), and subsequent dissociation (un-kinking) straightens the DNA. According to this ‘static kink model’ (Figure 5A), it is the dynamic interchange of DNA conformations induced by random and transient static kinking events that leads to the appearance of enhanced DNA flexibility (softening) over multiple seconds.
Figure 5.
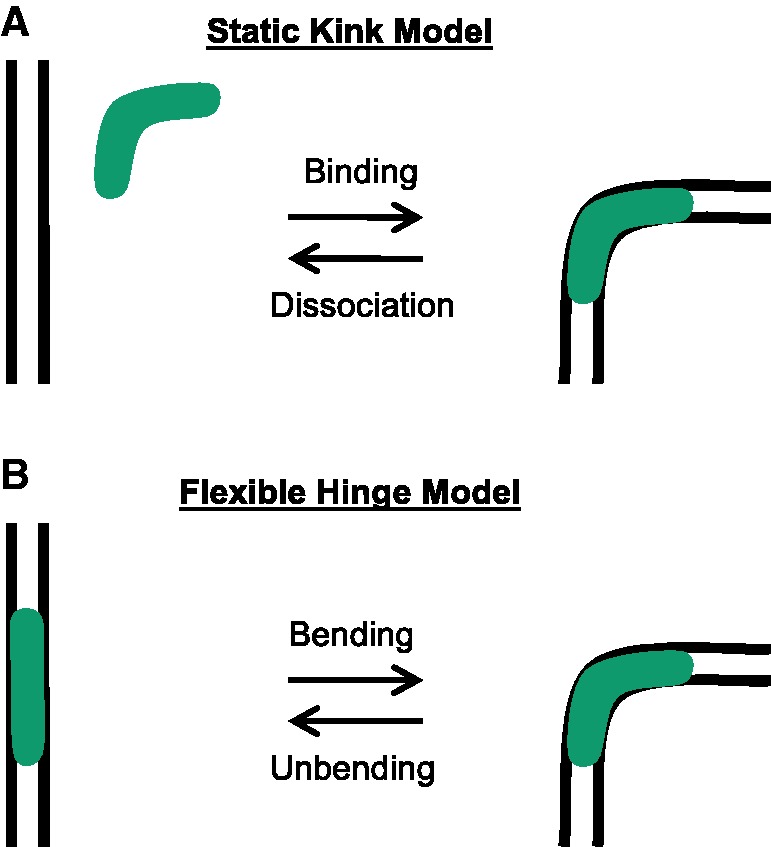
Models for DNA deformation by HMGB proteins. (A) Static kink model supported by the results for yNhp6A reported here. Protein binding and DNA kinking are simultaneous at the available time resolution of the present smFRET experiments, and the deformed DNA attains a static local structure that is relieved on protein dissociation. (B) Flexible hinge model that may apply to some HMGB proteins. The site of protein binding defines a locus of induced flexibility, which may be anisotropic.
If yNhp6A induced flexible hinge behavior in the DNA, the bound protein would dynamically induce multiple DNA conformational states. Rather than a steady induced FRET signal, the signal would likely vary and differ from complex to complex producing a wide FRET distribution. Dwell times in the various states would be independent of protein concentration because changes in FRET state would reflect single bound proteins bending and unbending the DNA in a concentration-independent manner. In contrast, we report that the dwell time of the low FRET state decreases with increasing protein concentration, a trend not expected in the flexible hinge model. Note that the time resolution of the present experiments (32 ms) means that DNA bending fluctuations faster than this rate cannot be observed. On the other hand, if an induced dynamic flexible hinge always holds the bent DNA at the same average bend angle, then the hinge is not evenly flexible. The lack of evidence for un-kinked intermediates reflects the observation of single exponential dwell time distributions (e.g. Figure 2E and F). Poisson statistics indicate an interpretation of single stochastic events. In contrast, detectable intermediates should produce non-exponential dwell time distributions.
Various studies probing the biophysical mechanism of HMGB protein-mediated DNA flexibility enhancement have produced differing results. Simulations based on the static kinking model supported by the present work were previously shown sufficient to explain the enhancement of ligase-mediated DNA cyclization rates by HMGB proteins (14). In these prior simulations, yNhp6A molecules are allowed to randomly decorate DNA in Monte Carlo simulations, introducing static kinks corresponding to the ∼70° bends observed by NMR (4). The stochastic formation of such structures was found to be sufficient to explain yNhp6A-induced enhancement of the J-factor (DNA effective end–end concentration) observed in cyclization kinetics experiments (12). Thus, although modeling yNhp6A binding events as sites of hinge flexibility in DNA might also predict enhanced DNA cyclization, such induced flexibility is not required to account for the experimental data.
Previous work using conventional ensemble experiments (cyclization kinetics and electrophoretic mobility shift assays) has shown that HMGB proteins (rat HMGB1 including both HMG box A and box B, and box A of human HMGB2) enhance ligase-mediated cyclization and rapidly exchange on and off DNA (12). That study (12) also supports the model in which DNA compaction and enhancement of ligase-catalyzed cyclization are promoted by HMGB proteins through random transient static DNA kinks, in agreement with our model, but the authors note that a flexible hinge model (Figure 5B) could not be excluded.
Single-molecule studies using DNA stretching with optical tweezers examined the effect of human HMGB2 box A (8) or both human HMGB2 box A and rat HMGB1 box A + box B (9) on dsDNA force-extension curves. Both studies showed that the dsDNA force-extension curves were strongly altered (indicating a dramatic decrease in apparent DNA persistence length) by the presence of HMGB proteins (8,9). The change in persistence length as a function of protein concentration was then used to determine protein equilibrium association constants and the average protein-induced kinking angle, with the model for determining the average kink angle based on the assumption that the bound protein induces a random flexible hinge (8,9). No direct measurement of the bend angle or the flexibility of the protein-bound site could be determined by this method.
Another study used magnetic tweezers to study mammalian HMGB1, yNhp6A and Escherichia coli HU (not an HMGB protein but often compared as an architectural protein that also binds and bends DNA, decreasing apparent DNA persistence length) (10). Three main observations were presented: First, for each protein (HMGB1, yNhp6A and HU), there is a well-defined protein concentration above which the proteins do not spontaneously dissociate from DNA. In this regime, the amount of protein bound to the DNA (as assayed by the degree to which DNA is compacted) is unperturbed either by replacing the surrounding protein solution with protein-free buffer or by straightening the molecule by applied force. Second, as observed elsewhere (27), HU is distinguished from the HMGB proteins in that it undergoes a switch to a DNA-stiffening function at the protein concentration where formation of highly stable complexes occurs. Third, introduction of competitor DNA into the surrounding solution disassembles within seconds the stable protein–DNA complexes for all the proteins. As spontaneous protein dissociation does not occur on the timescale of hours, the authors conclude that the observed rapid protein exchange in the presence of competitor DNA is driven by direct DNA–DNA contact. When comparing these results with those reported in our present study, it is important to note that smFRET allows the focus to be placed on kinetic and structural characterization of individual ‘isolated’ protein/DNA complexes. In contrast, micromanipulation experiments with long DNA molecules characterize global effects of protein binding (including cooperativity and other effects of protein–protein interactions).
An additional relevant biophysical study used AFM imaging on surfaces to measure local dsDNA bend distributions of rat HMGB1 box A + box B and human HMGB2 box A (11). The authors report that HMGB1 binding yielded a mean bend angle of 67° (standard deviation of 21°), and HMGB2 binding gave a mean bend angle of 78° (standard deviation of 23°). It was noted that the moderately broad induced bend angle distributions were not consistent with either the static kink model or a purely flexible hinge model. In a follow-up report (13), AFM is used to compare DNA bending by yNhp6A and variants of human HMGB2 box A. The authors report a model in which HMGB proteins, depending on N-terminal charge, create static kinks in DNA with various degrees of additional DNA flexibility induced at the protein binding site. The yNhp6A protein is reported to confer the greatest added flexibility.
Based on the present smFRET study and these previous results, we suggest a unifying model to understand this range of reported HMGB behaviors observed using different techniques. As suggested by X-ray, NMR and smFRET experiments (and consistent with Monte Carlo simulations), we propose that HMGB proteins induce static kinks of reproducible geometry when bound to short DNA segments in the absence of additional forces on the DNA. The DNA kink reflects the low-energy conformation for the isolated complex, and depends on the unique structure of each kind of HMGB protein. However, this complex is not absolutely rigid, as indicated dramatically by the results of force-extension experiments where HMGB proteins are not dissociated easily by DNA-straightening forces. In the presence of longer-range polymer strain (e.g. stretching of single molecules, or their deposition onto AFM surfaces), the preferred DNA static kink then reveals additional hinge flexibility that depends on characteristics of the HMGB protein. Thus, we envision HMGB proteins as endowing on DNA characteristics of ‘a hinged swinging door equipped with springs’: the door has a preferred low-energy conformation observed in the absence of external forces, but can be swung on its hinges by applied force.
Finally, we observed that yNhp6A kinks bulged DNA less severely than homoduplex DNA. This is an interesting and puzzling result, and no other biophysical studies have reported measurements for the interaction of yNhp6A with this kind of DNA target. The induced DNA kink angle apparently depends on the local structure of the DNA. Amino acid intercalation plays key wedging and unwinding roles in DNA kinking caused by HMGB protein binding in the DNA minor groove. It is likely that bulged DNA targets interact differently with the intercalating residues of HMGB proteins.
In conclusion, the present study demonstrates conclusively that the local effect of yNhp6A binding to DNA is induction of a single static DNA kink conformation that is homogenous from complex to complex. This deformation is short lived under physiological buffer conditions, and the kink angle is sequence independent for homoduplex targets. The presence of a bulge defect in the target DNA slightly increases yNhp6A affinity and slightly reduces the angle of induced DNA kinking, but the kink remains static and is homogenous across complexes. For both homoduplex and bulged targets, our observations are consistent with a model in which yNhp6A overcomes DNA stiffness by creating an ensemble of interconverting transient static kinks induced locally at the random sites of yNhp6A protein binding. In this static kink model, local DNA conformation is modulated between two discrete states (an unbent DNA-only state and a bent yNhp6A-bound state) by repeated binding and dissociation by yNhp6A. Future smFRET work will investigate the kinking dynamics of other HMGB proteins to determine whether this mechanism is conserved or to what extent flexible hinge characteristics are observed in DNA complexes with other members of the HMGB protein family.
It will also be appropriate to apply to HMGB proteins the fast-kinetic approaches that have previously been used to study DNA binding and bending by the E. coli IHF protein (28). These stopped-flow and temperature-jump (T-jump) methods allow analysis by FRET of binding and bending steps on the microsecond timescale, not accessible in the present smFRET study. Although the current work supports a concerted binding/bending model for yNhp6A with no evidence for spontaneous bending of naked DNA, T-jump studies (28) suggest that IHF binds straight DNA, and then bends the DNA in a separate step, perhaps limited by spontaneous DNA bending fluctuations that occur on the timescale of DNA base pair opening. It will be interesting to determine whether the mechanism of DNA bending by yNhp6A is amenable to similar kinetic dissection.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–5, Supplementary Materials and Methods and Supplementary References [4,11,19,20,29].
FUNDING
Funding for open access charge: Mayo Foundation;National Institutes of Health [GM75965 to L.J.M.].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Maher LJ. Chapter 7. In: Williams MC, Maher LJ III, editors. Biophysics of DNA-Protein Interactions: From Single Molecules to Biological Systems. New York: Springer; 2011. pp. 143–162. [Google Scholar]
- 2.Stillman DJ. Nhp6: a small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2010;1799:175–180. doi: 10.1016/j.bbagrm.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yen YM, Wong B, Johnson RC. Determinants of DNA binding and bending by the Saccharomyces cerevisiae high mobility group protein NHP6A that are important for its biological activities—role of the unique N terminus and putative intercalating methionine. J. Biol. Chem. 1998;273:4424–4435. doi: 10.1074/jbc.273.8.4424. [DOI] [PubMed] [Google Scholar]
- 4.Masse JE, Wong B, Yen YM, Allain FHT, Johnson RC, Feigon J. The S. cerevisiae architectural HMGB protein NHP6A complexed with DNA: DNA and protein conformational changes upon binding. J. Mol. Biol. 2002;323:263–284. doi: 10.1016/s0022-2836(02)00938-5. [DOI] [PubMed] [Google Scholar]
- 5.Wong B, Masse JE, Yen YM, Giannikoupolous P, Feigon J, Johnson RC. Binding to cisplatin-modified DNA by the Saccharomyces cerevisiae HMGB protein Nhp6A. Biochemistry. 2002;41:5404–5414. doi: 10.1021/bi012077l. [DOI] [PubMed] [Google Scholar]
- 6.Travers AA. Priming the nucleosome: a role for HMGB proteins? EMBO Rep. 2003;4:131–136. doi: 10.1038/sj.embor.embor741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham JS, Johnson RC, Marko JF. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucleic Acids Res. 2011;39:2249–2259. doi: 10.1093/nar/gkq1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCauley M, Hardwidge PR, Maher LJ, Williams MC. Dual binding modes for an HMG domain from human HMGB2 on DNA. Biophys. J. 2005;89:353–364. doi: 10.1529/biophysj.104.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCauley MJ, Zimmerman J, Maher LJ, Williams MC. HMGB binding to DNA: single and double box motifs. J. Mol. Biol. 2007;374:993–1004. doi: 10.1016/j.jmb.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoko D, Wong B, Johnson RC, Marko JF. Micromechanical analysis of the binding of DNA-bending proteins HMGB1, NHP6A, and HU reveals their ability to form highly stable DNA-protein complexes. Biochemistry. 2004;43:13867–13874. doi: 10.1021/bi048428o. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JY, McCauley MJ, Maher LJ, Williams MC, Israeloff NE. Mechanism of DNA flexibility enhancement by HMGB proteins. Nucleic Acids Res. 2009;37:1107–1114. doi: 10.1093/nar/gkn1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmerman J, Maher J. Transient HMGB protein interactions with B-DNA duplexes and complexes. Biochem. Biophys. Res. Commun. 2008;371:79–84. doi: 10.1016/j.bbrc.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang JY, McCauley MJ, Maher LJ, Williams MC, Israeloff NE. Basic N-terminus of yeast Nhp6A regulates the mechanism of its DNA flexibility enhancement. J. Mol. Biol. 2012;416:10–20. doi: 10.1016/j.jmb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czapla L, Peters JP, Rueter EM, Olson WK, Maher LJ. Understanding apparent DNA flexibility enhancement by HU and HMGB architectural proteins. J. Mol. Biol. 2011;409:278–289. doi: 10.1016/j.jmb.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters JP, Becker NA, Rueter EM, Bajzer Z, Kahn JD, Maher LJ. In: Methods in Enzymology, Vol. 488: Biothermodynamics, Part C. Johnson ML, Holt JM, Ackers GK, editors. San Diego: Elsevier Academic Press Inc.; 2011. pp. 287–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasnik I, McKinney SA, Ha T. Nonblinking and longlasting single-molecule fluorescence imaging. Nat. Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 17.Allain FHT, Yen YM, Masse JE, Schultze P, Dieckmann T, Johnson RC, Feigon J. Solution structure of the HMG protein NHP6A and its interaction with DNA reveals the structural determinants for non-sequence-specific binding. EMBO J. 1999;18:2563–2579. doi: 10.1093/emboj/18.9.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang WH, Coats JE, Majka J, Hura GL, Lin YY, Rasnik I, McMurray CT. Conformational trapping of Mismatch Recognition Complex MSH2/MSH3 on repair-resistant DNA loops. Proc. Natl Acad. Sci. USA. 2011;108:E837–E844. doi: 10.1073/pnas.1105461108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iqbal A, Arslan S, Okumus B, Wilson TJ, Giraud G, Norman DG, Ha T, Lilley DMJ. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nuclelic acids. Proc. Natl Acad. Sci. USA. 2008;105:11176–11181. doi: 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang LJ, Li J, Katz DS, Feng JA. Determining the DNA bending angle induced by non-specific high mobility group-1 (HMG-1) proteins: a novel method. Biochemistry. 2000;39:3052–3060. doi: 10.1021/bi991819w. [DOI] [PubMed] [Google Scholar]
- 21.Ruone S, Rhoades AR, Formosa T. Multiple Nhp6 molecules are required to recruit Spt16-Pob3 to form yFACT complexes and to reorganize nucleosomes. J. Biol. Chem. 2003;278:45288–45295. doi: 10.1074/jbc.M307291200. [DOI] [PubMed] [Google Scholar]
- 22.McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog. Nucleic Acid Res. Mol. Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 24.Grosschedl R, Giese K, Pagel J. HMG domain proteins: architechural elements in the assembly of nucleoprotein structures. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 25.Churchill MEA, Changela A, Dow LK, Krieg AJ. Interactions of high mobility group box proteins with DNA and chromatin. Methods Enzymol. 1999;304:99–133. doi: 10.1016/s0076-6879(99)04009-4. [DOI] [PubMed] [Google Scholar]
- 26.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 27.van Noort J, Verbrugge S, Goosen N, Dekker C, Dame RT. Dual architectural roles of HU: formation of flexible hinges and rigid filaments. Proc. Natl Acad. Sci. USA. 2004;101:6969–6974. doi: 10.1073/pnas.0308230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ansari A, Kuznetsov SV. Chapter 6. In: Williams MC, Maher LJ III, editors. Biophysics of DNA-Protein Interactions: From Single Molecules to Biological Systems. New York: Springer; 2011. pp. 107–142. [Google Scholar]
- 29.Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha TJ. Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy. Biophys. J. 2004;86:2530–2537. doi: 10.1016/S0006-3495(04)74308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.