

Abstract
The kinetic and thermodynamic ligand exchange dynamics are important considerations in the rational design of metal-based therapeutics and therefore, require detailed investigation. Co(III) Schiff base complex derivatives of bis(acetylacetone) ethylenediimine [acacen] have been found to be potent enzyme and transcription factor inhibitors. These complexes undergo solution exchange of labile axial ligands. Upon dissociation, Co(III) irreversibly interacts with specific histidine residues of a protein, that alters structure and causes inhibition. In order to guide the rational design of next generation agents, understanding the mechanism and dynamics of the ligand exchange process is essential. To investigate the lability, pH stability, and axial ligand exchange of these complexes in the absence of proteins, the pD- and temperature-dependent axial ligand substitution dynamics of a series of N-heterocyclic [Co(acacen)(X)2]+ complexes [where X = 2-methylimidazole (2MeIm), 4-methylimidazole (4MeIm), ammine (NH3), N-methylimidazole (NMeIm), and pyridine (Py)] were prepared and characterized by NMR spectroscopy. The pD stability was shown to be closely related to the nature of the axial ligand with the following trend toward aquation: 2MeIm > NH3 >> 4MeIm > Py > Im > NMeIm. Reaction of each [Co(III)(acacen)(X)2]+ derivative with 4MeIm showed formation of a mixed ligand Co(III) intermediate via a dissociative ligand exchange mechanism. The stability of the mixed ligand adduct was directly correlated to the pD-dependent stability of the starting Co(III) Schiff base with respect to [Co(acacen)(4MeIm)2]+. Crystal structure analysis of the [Co(acacen)(X)2]+ derivatives confirmed the trends in stability observed by NMR spectroscopy. Bond distances between the Co(III) and the axial nitrogen atoms were longest in the 2MeIm derivative as a result of distortion in the planar tetradentate ligand, and this was directly correlated to axial ligand lability and propensity toward exchange.
INTRODUCTION
The introduction of cisplatin into the clinic was a milestone for metal-based therapeutics.1-3 In response to its clinical success, the mechanism and dynamics of platinum coordination to biological molecules has been extensively studied, resulting in more potent generations of platinum anti-cancer agents.2,4-7 The success of this approach highlights the need for a thorough mechanistic understanding of new metal-based therapeutics for the rational design of next generation agents.8-17 Specifically, the in vivo activity of biologically active coordination complexes ultimately depends upon the kinetic and thermodynamic relationship between the metal center and its ligands.1 The structure and reactivity of the coordination complexes must be well characterized under physiological conditions for further development in biological applications.
Cobalt(III) Schiff base complexes of the tetradentate ligand bis(acetylacetone)ethylenediimine [acacen] have been found to be potent inhibitors of chymotrypsin, carbonic anhydrase, thermolysin, matrix metalloproteinase-2, and α-thrombin enzymatic activity.18-21 Additionally, incubation with [Co(acacen)(NH3)2]Cl was shown to compromise protein structure and function in metmyoglobin and zinc finger transcription factors Sp1, Gli, Snail, SIP1, and Slug.21-25 Previous investigations of Co(III)-exposed peptide systems suggest that the disruption of protein structure and function is related to the coordination of active site histidine residues to the Co(III) center.5 This interaction is proposed to occur through a dissociative axial ligand exchange of the weak donor NH3 ligands for the incoming electron-rich imidazole nitrogen of a histidine side chain.18,26 The coordination of axial ligands that increase steric bulk near the acacen backbone (such as 2-methylimidazole (2MeIm)) have been shown to increase the rate of enzyme inhibition, presumably by reducing the activation energy for ligand dissociation.18 In contrast, [Co(acacen)(Im)2]+ (Im = imidazole) have shown no significant effect on enzymatic turnover in α-thrombin or thermolysin.18
In order to understand, and ultimately tune, the axial ligand exchange properties of Co(III) Schiff base complexes a comprehensive study of ligand lability under physiological conditions is essential. Therefore, several Co(III) complexes were investigated under varying temperature and pD to understand the axial ligand exchange mechanisms for each derivative of [Co(acacen)(X)2]Y [X = 4-methylimidazole (4MeIm), ammine (NH3), 2-methylimidazole (2MeIm), N-methylimidazole (NMeIm), imidazole (Im), pyridine (Py); Y = Br, Cl] (Figure 1). The pD-dependent stability as a function of axial ligand identity was investigated by titration with DCl using NMR spectroscopy (Scheme 1). Axial ligand hydrolysis was found to be affected by a combination of factors, including pKa, steric bulk of the axial ligands, and mechanism of ligand exchange.
Figure 1.

[Co(acacen)(X)2]+ derivatives (cation only).
Scheme 1.
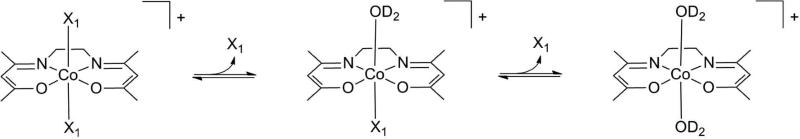
Substitution of the axial ligands of [Co(acacen)(X)2]+ with D2O. The pD-dependent stability of each complex was characterized by the exchange of the axial ligands for D2O with increasing DCl using NMR spectroscopy. Axial ligand hydrolysis was found to be affected by a combination of factors, including pKa, steric bulk of the axial ligands, and mechanism of ligand exchange.
NMR spectroscopy was used to investigate the axial ligand exchange of a series of [Co(acacen)(X)2]+ derivatives with 4MeIm (Scheme 2). 4MeIm was chosen as a model for the imidazole side chain of a histidine residue preventing competitive coordination of the α-amine or α-carboxylate groups and/or bidentate chelation of the amino acid.26 X-ray structural determination of these Co(III) Schiff base derivatives was used to validate the axial ligand exchange dynamics that were observed in solution.
Scheme 2.

Reaction of [Co(acacen)(X)2] + with 4MeIm. NMR spectroscopy was used to investigate the axial ligand exchange [Co(acacen)(X)2]+ derivatives with 4MeIm. 4MeIm was chosen as a model for the imidazole side chain of a histidine residue preventing competitive coordination of the α-amine or α-carboxylate groups and/or bidentate chelation of the amino acid.
EXPERIMENTAL SECTION
Materials
Unless noted, materials and solvents were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA) and used without further purification. Methanol was purified using a Glass Contour Solvent System. Deionized water was obtained from a Millipore Q-Guard System equipped with a quantum Ex cartridge. Unless noted, all syntheses were performed under an inert nitrogen atmosphere. Complexes 1 – 6 were synthesized with modification to previously published procedures.27-29
Crystal Structure Analysis of Co(III) Schiff Bases
Crystals of 3 and 6 suitable for X-ray crystallography were grown by slow evaporation from the reaction mixture and vapor diffusion of ether into methanol, respectively. Complexes 2, 4, 5 were converted to their respective tetraphenylborate salts through the dropwise addition of a saturated solution of NaBPh4 in methanol to an aqueous solution of each Co(III) Schiff base complex. The tan colored precipitates were filtered and dried. Crystals of [Co(acacen)(Py)2]BPh4, [Co(acacen)(Im)2]BPh4 and [Co(acacen)(NMeIm)2]BPh4 suitable for X-ray crystallography were acquired by slow cooling of an ethanol solution. Cell dimensions and intensity data were measured on a Bruker APEX-II C diffractometer with graphite monochromated MoKα radiation. Data were collected using Bruker APEX2 detector, processed using SAINTPLUS (Bruker), and corrected for Lorentz and polarization effects (Table 1). A full summary of data collection and structure refinement can be found in the Supporting Information.
Table 1.
Crystallographic data for [Co(acacaen)(X)2]+ derivatives.
| X = | 2MeIm | NMeIm | Im | Py | NH3 |
|---|---|---|---|---|---|
| empirical formula | C41.20H63.10Cl2Co2N12O6.35 | C48H62BCoN6O4 | C44H51BCoN6O3 | C24H36BrCoN4O4 | C36H44BCoN4O2 |
| formula weight | 1016.90 | 856.78 | 781.65 | 583.41 | 634.49 |
| temperature, K | 100(2) | 100(2) | 100.09 | 100.14 | 99.97 |
| space group | P21/c | P21/c | P21/m | Pna21 | Cc |
| crystal system | monoclinic | monoclinic | monoclinic | orthorombic | monoclinic |
| a, Å | 24.8162(9) | 13.9254(5) | 9.6818(5) | 23.538(5) | 12.4323(5) |
| b, Å | 11.6873(4) | 18.0542(6) | 14.5560(8) | 14.372(3) | 17.3432 (7) |
| c, Å | 16.9470(6) | 17.8151(6) | 14.3710(8) | 15.616(3) | 15.4529(6) |
| α, deg | 90 | 90 | 90 | 90 | 90 |
| β, deg | 100.328(2) | 97.6470(18) | 98.273(2) | 90 | 93.308(2) |
| γ, deg | 90 | 90 | 90 | 90 | 90 |
| V, Å3 | 4835.6(3) | 4439.1(3) | 2004.20(19) | 5282.6(18) | 3326.3(2) |
| Z | 4 | 4 | 2 | 8 | 4 |
| Dcalc, g cm-3 | 1.397 | 1.282 | 1.295 | 1.467 | 1.267 |
| abs coeff, mm-1 | 8.55 | 0.437 | 0.476 | 2.198 | 0.553 |
| obsd relfns | 41559 | 76376 | 6066 | 6300 | 27717 |
| unique relfns | 10981 | 12872 | 6066 | 6300 | 6248 |
| Rint | 0.1431 | 0.0504 | 0.0000 | 0.0000 | 0.0513 |
| restraints/params | 0/645 | 75 / 581 | 0/339 | 1/629 | 128/403 |
| GOF on F2 | 0.914 | 1.302 | 1.013 | 1.156 | 0.809 |
| Ra [I >2σ(I)] | 0.0540 | 0.0479 | 0.0459 | 0.0362 | 0.0426 |
| Rwb [I >2σ(I)] | 0.1255 | 0.1297 | 0.1190 | 0.0867 | 0.1107 |
R1 = Σ| |Fo|-|Fc| |/Σ|Fo|.
Rw = [Σ[w(Fo2-Fc2)2]/Σ[w(Fo2)2]]1/2, w=1/[σ2(Fo2)+(0.0660P)2 + 0.0000P] where P=(Fo2+2Fc2)/3
NMR Spectroscopy
A 0.2 M deuterated sodium phosphate buffer was prepared by dissolution of the lyophilized salts from a volume of aqueous buffer into an equal volume of D2O. The initial pD of the buffer (8.00) was adjusted as needed with addition of DCl or NaOD. Unless otherwise noted, NMR spectra were obtained on a Bruker 600 MHz Avance III NMR spectrometer with a variable temperature unit. 1H and 13C NMR chemical shifts are reported in ppm with respect to the internal reference, 3-(trimethylsilyl)propionic acid-d4 sodium salt. The integral percentage of each species in solution is recorded with respect to time using the 1H ethylene peak and 1H methyl peak NMR chemical shifts of the resultant Co(III) species; integrations are calibrated with respect to the internal reference.
The pD-dependent chemical shift assignments of Co(III) Schiff base complexes were performed using 2D NMR spectroscopy. Co(III) Schiff base complexes 1 – 6 were dissolved in phosphate buffer to create approximately 30 mM solutions at alkaline pD as indicated. A second set of samples of Co(III) Schiff base complexes 1 – 6 were adjusted with DCl to an acidic pD as indicated. 1H NMR, 13C NMR, COSY, HSQC spectra were obtained of each sample on a Bruker 500 MHz Avance III NMR Spectrometer. Solutions of 4MeIm, 2MeIm, NMeIm, Im, and Py in phosphate buffer were made for comparison of the free ligand chemical shifts (See Part 1 in Supporting Information).
Conditions for pD-Dependent Stability of [Co(acacen)(X)2]+ Derivatives
The pD-dependent stability of each Co(III) Schiff base complex was monitored by 1H NMR spectroscopy. Stock solutions of 1 – 6 (1.8 mL, 16.0 mM) were prepared in phosphate buffer. The pD of each sample was measured and 600 μL removed for 1H NMR analysis at 30 °C. The NMR sample was returned to the stock solution, an aliquot (4 μL) of DCl added, and the stock solution was incubated at room temperature for 10 minutes. The pD of each sample was measured and 600 μL used for 1H NMR analysis. Repeated aliquots were analyzed in this manner with decreasing pD until DCl addition limited buffer capacity. Identity and abundance of each unique [Co(acacen)(X)2]+ species were recorded as a function of pD.
Conditions for Ligand Exchange of [Co(acacen)(X)2]+ and 4MeIm
The pD- and temperature-dependent axial ligand exchange of Co(III) Schiff base complexes with 4MeIm was monitored by 1H NMR spectroscopy. A 2.0 M stock solution of 4MeIm was prepared in the NMR buffer. Complexes 2 – 6 were dissolved separately in the NMR buffer (to a final concentrations of 16 mM) and an aliquot of the 4MeIm stock equivalent to 2 molar equivalents of each [Co(III)(acacen)(X)2] derivative added. Additionally, incubation of 1 with 2MeIm, NMeIm, Im, and Py illustrated the reversibility of the 4MeIm coordination to the Co(III) complex. The resultant solutions were analyzed by 1H NMR spectroscopy with water suppression over 90 minutes with spectra collected every 72 seconds. NMR spectra of each species were obtained at pD 7.20 – 7.00 (pD 7.80-7.50 for 2), pD 6.30 – 6.10 (pD 6.40-6.20 for 2), and pD 5.60-5.40 and at 18 °C, 25 °C, 30 °C, and 37 °C. Identity, abundance, and equilibrium of each unique [Co(acacen)(X1X2)]+ species were recorded as a function of time.
Electrospray Ionization Mass Spectrometry (ESI-MS)
ESI-MS spectra were obtained using a Varian 1200 L single quadrupole spectrometer. Complexes 2 – 6 were each dissolved to a final concentration 16 mM in 0.2 M sodium phosphate buffer with 2 equivalents of 4MeIm (pH 8.00). Similarly, 1, 3, 4, and 6 (final concentration of 16 mM) were dissolved with two molar equivalents of Im. Control samples were prepared by dissolving 3 with two molar equivalents of 2MeIm, 4 with two molar equivalents of NMeIm, and 6 with two molar equivalents of Py. The samples were incubated overnight at 25 °C and diluted to 80 μM (200X) with methanol. Each sample was infused at a flow rate of 50 μL min-1. Mass spectra were obtained in positive mode in the range of 100-1500 m/z at a capillary temperature of 100 °C with spray voltage 1.1 kV and capillary voltage 10 V. The spectra were analyzed to determine the major species in each ligand exchange equilibrium.
RESULTS AND DISCUSSSION
Crystal Structure Analysis of [Co(acacen)(X)2]+
Crystals of 3 suitable for X-ray crystallography were grown by slow evaporation of the reaction mixture (Figure 2). Crystals of 4 and 5 suitable for X-ray crystallography were obtained by anion exchange to yield the tetraphenyl borate salt followed by slow cooling of an ethanol solution. Crystals of 2 and 6 were obtained by vapor diffusion of ether into methanol. The structure of 1 has been previously determined.27 The expected octahedral geometry is observed for all complexes with the acacen ligand in the equatorial plane. The N-bases occupy the two axial positions in all species.
Figure 2.

Molecular structure of 3 (plot with 30% probability ellipsoids, cation only) shown with crystal structure labeling scheme. Select bond distances (Å) for [Co(acacen)(X)2]+ derivatives show an increased Co-N(3) and Co-N(4) bond distance for 3. The axial Co-N bond distance of 3 is the longest (averaging 1.965 Å) due to unfavorable steric interactions between the methyl groups of the axial ligand with the planar acacen backbone. a Crystal structure of 1-Br reported in Inorg. Chem. 1997, 36, 2498-2504.
Bond distances and bond angles between the coordinated atoms and the Co(III) center are dependent upon the identity of the axial ligand (See Part 7A in Supporting Information). The axial Co-N bond distance of 3 is the longest (averaging 1.965 Å) due to unfavorable steric interactions between the methyl groups of the axial ligand with the planar acacen backbone. Equatorial bond distances are independent of the heteroatom and conserved across all species averaging 1.895 Å. Bond distances are similar to those observed for [Co(NH3)6]Cl.30 In all [Co(acacen)(X)2]+ derivatives, the equatorial O-Co-O and N-Co-N bond angles are less than the expected 90°. This deviation is compensated by an average increase in the equatorial O-Co-N bond angles (> 94°). The axial bond angles between the heteroatoms through the Co(III) center show only minor deviations from the expected 90°.
A comparison of these structures shows that the planarity is distorted with respect to the nature of the axial ligand. A N2O2 plane drawn through the coordinating equatorial nitrogen [N(1), N(2)] and oxygen atoms [O(1), O(2)] of the acacen ligand and the distance of the Co(III) and carbon atoms on the equatorial ligand from this plane gives a measure of the distortion in each [Co(acacen)(X)2]+ derivative (See Part 7B in Supporting Information). While the Co(III) atom is not significantly deviated from this plane in any species (< 0.015 Å), significant distortions from planarity are observed in the equatorial acacen carbons parallel to the methyl-substituted axial ligand in 3 [C(1), C(3), C(5)]. These carbons are shifted away from the N2O2 plane at distances greater than 0.390 Å due to increased strain from the 2MeIm axial ligand. The ethylenediimine backbone carbons [C(6), C(7)] are distorted above and below the plane in a similar manner to other transition metal acacen crystal structures.27,31,32
Dihedral angles measured from the N2O2 planes to the left [N(1), O(1), C(2), C(4)] and right [N(2), O(2), C(9), C(11)] planes of acacen show that 1 and 4 deviate the least from the idealized equatorial plane (See Part 7C in Supporting Information). Although minor deviations in the bond distances and bond angles were observed in 5 and 6, dihedral angles between the acacen planes are substantial (11.12°, 13.05°). Large dihedral angles are observed in 3, due to the steric influence of the 2MeIm on the equatorial ligand structure (18.57°, 13.32°, 31.87°).
pD-Dependent Stability of [Co(acacen)(X)2]+ Derivatives
The influence of the axial ligand on the pD stability of Co(III) Schiff base complexes was determined by 1H NMR spectroscopy. Alkaline solutions of each [Co(acacen)(X)2]+ derivative in deuterated phosphate buffer were titrated with DCl until acidic pD was observed. The quantification, identity, and assignment of all [Co(acacen)(X)2]+ derivatives at alkaline and acidic conditions was facilitated by 2D NMR analysis. The 1H and 13C NMR chemical shifts of the free ligands were determined at alkaline and acidic conditions for comparison (See Part 1 in Supporting Information).
The NMR assignment of 2, under alkaline conditions (pD 8.32), exhibits two distinct complexes; 2 and [Co(acacen)(NH3)(D2O)]+ (Figure 3). Singlets at 3.62, 2.09, and 2.26 ppm are assigned to 2; the integrals of these peaks correspond to 85% of the [Co(acacen)(X1X2)]+ species in solution. Ligand monosubstitution to give [Co(acacen)(NH3)(D2O)]+ corresponds to singlets at 3.80, 2.20, 2.37 ppm; the integrals of these peaks correspond to the remaining 15% of the [Co(acacen)(X1X2)]+ species in solution. Titration of DCl inverts the relative amounts of these two species with [Co(acacen)(NH3)(D2O)]+ reaching a maximum concentration (68% abundance) at pD ~5.5. Increasing aliquots of DCl gives a third species, [Co(acacen)(D2O)2]+, with singlets at 2.27, 0.90, and 0.74 ppm. The rapid increase in the abundance of [Co(acacen)(D2O)2]+ below pD 5.50 is coupled to a dramatic decrease in [Co(acacen)(NH3)(D2O)]+ and a near extinction of 2.
Figure 3.

DCl titration of 2. (A) 1H NMR spectra of 2 with successive additions of DCl; the alkyl region is highlighted showing the presence of three distinct [Co(acacen)(X1X2)]+ species at acidic conditions (arrows). (a) pD 8.32, (b) pD 7.42 , (c) pD 6.98, (d) pD 6.47, (e) pD 5.48 , (f) pD 4.63, and (g) pD 3.01. (B) Relative abundance of each [Co(acacen)(X1X2)]+ derivative in solution as a function of pD;  = 2,
= 2,  = [Co(acacen)(NH3)(D2O)]+,
= [Co(acacen)(NH3)(D2O)]+,  = [Co(acacen)(D2O)2]+.
= [Co(acacen)(D2O)2]+.
Similarly to 2, 3 indicates the presence of two different species at alkaline conditions (pD 8.34); 3 at 80% and [Co(acacen)(2MeIm)(D2O)]+ at 20%. Integrations of 3 correspond to the expected acacen:2MeIm NMR peak integration ratios (1 aliphatic proton per 2 aromatic protons). Upon the addition of DCl, the NMR spectra of [Co(acacen)(2MeIm)(D2O)]+ exhibits NMR peak integration ratios characteristic of the displacement of one axial ligand by D2O (1 alphatic proton per 1 aromatic proton). This displacement is confirmed by the presence of free 2MeIm peaks of equivalent abundance in the 1H NMR aromatic region (7.20 ppm, 2.57 ppm); that shift downfield with decreasing pD due to the pD sensitivity of imidazole.16 Aquation of the complex results in a loss of symmetry along the equtorial plane, splitting the 1H NMR peaks of the ethylenediimine backbone into a doublet of multiplets (3.87 – 3.51 ppm). The conformational inversion of these protons between axial and equatorial orientation becomes more pronounced upon loss of the vertical mirror plane and C2 rotational axis of 3.17 Below pD 6.00, complete dissociation of the 2MeIm ligand from [Co(acacen)(2MeIm)(D2O)]+ was observed to give [Co(acacen)(D2O)2]+ with no evidence of 3.
DCl titrations of 1, 4, 5, and 6 show considerable conversion of each complex to a mono(aquo) species with decreasing pD, but not the complete aquation to [Co(acacen)(D2O)2]+ at acidic conditions (< pD 5). The pD required for formation of the mono(aquo) [Co(acacen)(X1X2)]+ species is dependent upon the identity of the axial ligand; 4 was found to have the greatest resistance to ligand exchange with the lowest pD of aquation (Figure 4).
Figure 4.

The pD-dependent stability of the [Co(acacen)(X)2]+ derivatives. The observed trends of pD stability shows deviation from the pKa of the free ligand. (A) the depletion of each derivative as a function of pD. Percentages of [Co(acacen)(X)2]+ were determined in relation to the aquated species with reference to an internal standard. (B) the pD at which fifty percent of the starting material remained was determined in order to compare the pD stability of the [Co(acacen)(X)2)]+ derivatives to resistance to aquation. The pD stability trend does not directly correlate to the pKa stability trends of the free ligands.  = 1,
= 1,  = 2,
= 2,  = 3,
= 3,  = 4,
= 4,  = 5,
= 5,  = 6.
= 6.
The pD at which 50% of each [Co(acacen)(X)2]+ derivative undergoes hydrolysis was used to compare the relative pH stability of the complexes (Figure 4). The pD stability was shown to be strongly related to the axial ligand; the following trend toward aquation was observed: 2MeIm > NH3 >> 4MeIm > Py > Im > NMeIm. The observed pD stability trends correspond to the known enzyme inhibitory activity of these complexes. Complexes 2 and 3 are potent enzyme and transcription factor inhibitors, and show a relatively high degree of hydrolysis. In contrast, 5 has been shown to have no affect on enzymatic activity and has greater pD stability.5
The observed pD stabilities suggest dissociative ligand exchange as a mechanism for the axial ligand exchange of the [Co(acacen)(X)2]+ imidazole-based derivatives. The trend in pD stability of the axial ligand coordination cannot be soley attributed to the inherent pKa of the free axial ligand; steric interactions contribute to the stability trend. Increases in steric bulk near the exchange site, such as in 3, are shown to promote ligand release under a dissociative exchange mechanism.21 The strong basic character of the imidazole derivatives without steric bulk near the exchange site reduce the likelihood of aquation due to electronic stablization of the metal.22 The aquation of 2 may be mediated by an interchange dissociative ligand exchange; the three protons of the NH3 have greater propensity to hydrogen bond with the incoming water molecule, weakly binding it to the Co(III) center, and ulimately facilitating ligand exchange.23
Ligand Exchange of [Co(acacen)(X)2]+ and 4MeIm
Enzyme and transcription factor inhibition through distortion of protein structure by active forms of [Co(acacen)(X)2]+ is thought to occur by the dissociative exchange of labile axial ligands for the tele imidazole nitrogen of a histidine residue. To determine the reaction dynamics of this exchange, each [Co(acacen)(X)2]+ derivative (with the exception of X = 4MeIm) was incubated with two equivalents of 4MeIm at a range of temperatures (18 °C, 25 °C, 30 °C, and 37 °C). The reaction was monitored by 1H NMR spectroscopy at adjusted pDs of 7.0 and 5.5 over 90 minutes; NMR spectra were acquired every 72 seconds. For each complex, the relative abundance of each species was found to be the same as the reverse reaction (Figure 5). The percentage of each [Co(acacen)(X1)(X2)]+ derivative was quantified at each time point with respect to an internal standard (See Part 3 in Supporting Information).
Figure 5.

The relative abundance of all [Co(acacen)(X)2]+ species over the time course of the reaction for 6 with 4MeIm (2 eq.). The reaction produces equal ratios of [Co(acacen)(4MeIm)2]+, [Co(acacen)(4MeIm)(Py)]+, and [Co(acacen)(Py)2]+ as in the reverse reaction of 1 with Py (2 eq.), validating an equilibrium.
The reaction of 2 with two equivalents of 4MeIm shows a complete conversion to 1, with a total depletion of 2. A doublet of multiplets splitting pattern is observed for the ethylenediimine backbone signifying a loss of symmetry along the equatorial plane (3.65 – 3.28 ppm). These peaks are attributed to the formation of the mixed intermediate species, [Co(acacen)(NH3)(4MeIm)]+ given the lack of symmetry in the axial plane. With increasing acidity, the formation of [Co(acacen)(4MeIm)(D2O)]+ is observed in greater quantity, consistent with the pD stability of 1 (Figure 6). Mass spectrometry competition studies of 2 and 4MeIm show 1 as the major product. Furthermore, the substitution reaction of 1 is temperature-dependent (Figure 7). At 18 °C, the reaction proceeds significantly slower than at higher temperatures. This corresponds to previously observed temperature-dependent protein inhibitory activity by targeted [Co(acacen)(NH3)2]+ complexes.10
Figure 6.

Time course of the ligand substitution of [Co(acacen)(X)2]+ derivatives and 4MeIm (2 eq.) at pD 7.0 (A) and pD 5.5 (B) at 37 °C. 2 and 3 readily reacts with 4MeIm to form 1. Under acidic conditions, the formation of [Co(acacen)(4MeIm)(D2O)]+ is observed in greater quantity and reducing the abundance of 1; this is consistent with the pD stability of 1. Complex 1 is a minor product (< 20%) for 4 and 5 at pD 7.0, and 4, 5, and 6 at pD 5.5. For 6, the extend of 1 formation is pD dependent due to the relative basicity of 4MeIm (pKa 7.4) and Py (pKa 5.5).  = 2,
= 2,  = 3,
= 3,  = 4,
= 4,  = 5,
= 5,  = 6.
= 6.
Figure 7.

Ligand substitution of 2 and 4MeIm (2 eq.) at pD 7.0. The time course of the reaction toward 1 is temperature dependent.
The reaction of 3 with two equivalents of 4MeIm confirms a correlation between the pD instability of the 2MeIm axial ligand coordination and its susceptibility to ligand exchange with 4MeIm. An instantaneous conversion to 1 is observed (Figure 6). Insignificant amounts of [Co(acacen)(2MeIm)(4MeIm)]+ are observed with complete loss of 3. The lability and subsequent ligand exchange in favor of the 4MeIm is indicative of a dissociative mechanism. As with 2, the formation of [Co(acacen)(4MeIm)(D2O)]+ is observed in acidic conditions, consistent with the pD stability of 1. Due to the species having the same mass, the mass spectrometry results of this ligand exchange are ambiguous. Thus, the reaction of 3 with two equivalents of Im was used as an alternative strategy for mass spectrometry evaluation. The resulting data confirms the high reactivity of 3 by a near complete conversion to 5 with only a weak intensity for the mixed ligand in the mass spectrum. These studies indicate that the reaction of 2 and 3 with 4MeIm readily go to completion to 1.
The relative percentages of the [Co(acacen)(X1)(X2)] derivative following the reaction with 2 molar equivalents of 4MeIm with 4, 5, or 6, show a preference toward mono-substitution (40-50% in all conditions). Mass spectrometry studies showing a high intensity peak corresponding to [Co(acacen)(4MeIm)(Im)]+ confirms the mixed species as the dominant species (See Part 5 in Supporting Information). The complete di-substitution to 1 is a minor product (< 20%) for 4 and 5 at pH 7.0, and 4, 5, and 6 at pH 5.5 (Figure 6). For the 4MeIm substitution reactions with 6, the relative abundance of 6 and 1 are dependent upon the acidity of the solution. Although the pD stability of 6 and 1 are equivalent at pD 7.0, the formation of 1 is favored at neutral pD in ligand exchange reactions with 4MeIm. With increasing acidity the formation of 6 is favored in accordance with the relative stability of 6 and 1 at pD 5.50. The pD dependence of the relative species formation is not observed with the imidazole-based derivatives. This substitution behavior is attributed to the basicity of 4MeIm and Py; at acidic pD, 4MeIm (pKa 7.4) is more readily protonated than Py (pKa 5.5).
The reactions of [Co(acacen)(X)2]+ derivatives with 4MeIm provides perspective into the trends previously observed during enzymatic inhibition studies.18-20,33 Both 2 and 3 are potent inhibitors of thermolysin, α-thrombin and chymotrypsin. In contrast, 5 shows no inhibition of enzyme activity.18,19 Based on the observed NMR trends for the reaction of each derivative with 4MeIm, it appears that the exchange of both labile axial ligands for histidine residues is required for detectable enzyme inhibition. Complete conversion to 1 was only observed in reaction of 4MeIm with 2 and 3. Reaction of 4MeIm with 4, 5, and 6 showed formation of a stable mixed species preventing complete conversion to 1. Increasing the temperature of the reactions did not affect the relative abundance of each species, but did accelerate conversion to the equilibrium [Co(acacen)(X1X2)]+ species ratios suggesting an endothermic reaction. These studies demonstrate that the Co(III) Schiff base-4MeIm axial ligand exchange is dependent upon the pD stability of the starting material in comparison to the pD stability of 1.
The similarity of the trends observed for aquation and ligand substitution with 4MeIm of these [Co(III)(acacen)(X)2]+ derivatives suggest the occurrence of an aquo intermediate.34,35 Hydrolysis has dominated the mechanistic analysis for Co(III) octahedral complexes due to the pivotal role water plays in the ligand exchange reactions.34,36 Furthermore, previous experimental evidence suggests that hydrolysis occurs via a dissociative or interchange dissociative mechanism for [Co(acacen)(NH3)2]+ derivatives.37-39
Based on these observations, the conversion of [Co(acacen)(X)2]+ to 1 is proposed to occur through a sequential dissociative ligand exchange via an aquo intermediate. In this scheme, the release of the exchangeable axial ligand that allows for the coordination of the solvent molecule is the rate-determining step. The formation of the aquo intermediate is followed by rapid binding of the incoming N-donor ligand.
CONCLUSIONS
The ligand exchange dynamics of a series of [Co(acacen)(X)2]+ derivatives were examined by NMR spectroscopy. The pD-dependent trends were quantified to categorize the stability of the axial ligand coordination. The temperature- and pD-dependent susceptibility of the complexes to axial ligand exchange with 4MeIm showed features characteristic of a dissociative mechanism. However, an interchange dissociative is possible for the exchange of the labile NH3 (2), given the hydrogen bonding potential of this ligand with incoming water molecules and propensity toward formation of the corresponding conjugate base. The chemical equilibria for each system are reversible and dependent upon the molar equivalents of 4MeIm in the given reaction conditions.
In this study, we have shown that the axial ligand exchange dynamics correlate well with previously observed enzyme inhibitory activity of [Co(acacen)(X)2]+ derivatives.4,5 Axial ligands with increased lability, such as NH3, or unfavorable steric interactions, such as 2MeIm, have been shown to increase enzyme inhibition dramatically. Such complexes have significantly lower pD stability toward aquation (Figure 4) and undergo complete conversion to 1 when exposed to 4MeIm. In contrast, [Co(acacen)(X)2]+ derivatives with N-heterocyclic axial ligands show higher pD stability toward aquation and resistance to complete conversion to 1. Not surprisingly this observation is consistent with the absence of effect on enzymatic turnover of this complex.18 Thus there is a correlation between observed axial ligand lability of the [Co(acacen)(X)2]+ derivatives and their ability to inhibit histidine-rich proteins. The trends observed here will guide the rational design of more potent or activatable Co(III) Schiff base enzyme and transcription factor inhibitors.
Supplementary Material
Table 2.
Select bond distances (Å) and bond angles (deg) for [Co(acacen)(X)2]+ derivatives.
| Distances (Å) | ||||||
|---|---|---|---|---|---|---|
| 1a | 2 | 3 | 4 | 5 | 6 | |
| Co-N(1) | 1.899 | 1.898 | 1.888 | 1.902 | 1.885 | 1.906 |
| Co-N(2) | 1.895 | 1.895 | 1.913 | 1.898 | 1.885 | 1.902 |
| Co-O(1) | 1.897 | 1.888 | 1.903 | 1.898 | 1.880 | 1.903 |
| Co-O(2) | 1.901 | 1.889 | 1.905 | 1.899 | 1.880 | 1.905 |
| Co-N(3) | 1.953 | 1.942 | 1.967 | 1.941 | 1.936 | 1.971 |
| Co-N(4) | 1.941 | 1.939 | 1.962 | 1.940 | 1.926 | 1.946 |
| Angles (Deg) | ||||||
|---|---|---|---|---|---|---|
| 1a | 2 | 3 | 4 | 5 | 6 | |
| O(1)-Co-O(2) | 83.06 | 84.30 | 85.33 | 83.47 | 83.83 | 85.67 |
| N(1)-Co-O(1) | 94.58 | 95.00 | 93.94 | 95.20 | 95.29 | 94.68 |
| N(2)-Co-O(2) | 95.69 | 94.85 | 94.92 | 94.79 | 95.29 | 94.45 |
| N(1)-Co-N(2) | 86.75 | 85.85 | 85.81 | 86.58 | 85.60 | 85.19 |
| N(1)-Co-N(3) | 90.99 | 91.48 | 89.78 | 89.16 | 91.12 | 91.41 |
| N(2)-Co-N(3) | 91.21 | 90.47 | 90.61 | 91.47 | 91.12 | 91.20 |
| O(1)-Co-N(3) | 89.96 | 89.60 | 89.12 | 90.41 | 89.02 | 88.41 |
| O(2)-Co-N(3) | 85.27 | 90.02 | 90.10 | 89.51 | 89.02 | 86.91 |
| N(1)-Co-N(4) | 91.81 | 91.65 | 91.33 | 91.08 | 89.91 | 92.75 |
| N(2)-Co-N(4) | 89.16 | 92.87 | 89.37 | 87.88 | 89.91 | 92.03 |
| O(1)-Co-N(4) | 89.61 | 87.03 | 90.91 | 90.23 | 89.93 | 88.37 |
| O(2)-Co-N(4) | 91.93 | 86.81 | 88.79 | 90.27 | 89.93 | 88.94 |
Crystal structure of 1-Br reported in Inorg. Chem. 1997, 36, 2498-2504.
ACKNOWLEDGMENTS
The authors thank Dr. Josh Kurtz for experimental assistance and helpful discussions. We thank Dr. Amy Sargent and Ms. Charlotte Stern for crystal structure data collection and helpful discussions. We thank Lauren Matosziuk and Dr. Natsuho Yamamoto for helpful discussions. We acknowledge the Northwestern University Integrated Molecular Structure Education and Research Center (IMSERC); a description of the facility and full funding disclosure can be found at http://pyrite.chem.northwestern.edu/analyticalserviceslab/asl.htm. This work was supported by the National Institutes of Health's Centers of Cancer Nanotechnology Excellence initiative of the National Cancer Institute under award U54CA151880, the Rosenberg Cancer Foundation, and The Lurie Cancer Center. LMM acknowledges personal funding from the Northwestern University Biotechnology Training Program. MCH would like to acknowledge the NSF GRFP for personal funding.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supporting material includes NMR spectral characterization and time course plots of the pD stability and ligand exchange NMR experiments, in addition to the mass spectrometry measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Guo Z, Sadler PJ. Angewandte Chemie International Edition. 1999;38:1512. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1512::AID-ANIE1512>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Chemical Reviews. 1999;99:2467. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Thompson KH, Orvig C. Science. 2003;300:936. doi: 10.1126/science.1083004. [DOI] [PubMed] [Google Scholar]
- 4.Aris SM, Farrell NP. European Journal of Inorganic Chemistry. 2009;2009:1293. doi: 10.1002/ejic.200801118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung Y, Lippard SJ. Chemical Reviews. 2007;107:1387. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 6.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Nature. 1995;377:649. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 7.Mangrum JB, Farrell NP. Chemical Communications. 2010;46:6640. doi: 10.1039/c0cc01254h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruijnincx PCA, Sadler PJ. Current Opinion in Chemical Biology. 2008;12:197. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meggers E. Angewandte Chemie International Edition. 2011;50:2442. doi: 10.1002/anie.201005673. [DOI] [PubMed] [Google Scholar]
- 10.Orvig C, Abrams MJ. Chemical Reviews. 1999;99:2201. doi: 10.1021/cr980419w. [DOI] [PubMed] [Google Scholar]
- 11.Holm RH, Kennepohl P, Solomon EI. Chemical Reviews. 1996;96:2239. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 12.Storr T, Thompson KH, Orvig C. Chemical Society Reviews. 2006;35:534. doi: 10.1039/b514859f. [DOI] [PubMed] [Google Scholar]
- 13.Thompson KH, McNeill JH, Orvig C. Chemical Reviews. 1999;99:2561. doi: 10.1021/cr980427c. [DOI] [PubMed] [Google Scholar]
- 14.Valentine JS, Hart PJ. Proceedings of the National Academy of Sciences. 2003;100:3617. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang CX, Lippard SJ. Current Opinion in Chemical Biology. 2003;7:481. doi: 10.1016/s1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 16.Louie AY, Meade TJ. Chemical Reviews. 1999;99:2711. doi: 10.1021/cr9804285. [DOI] [PubMed] [Google Scholar]
- 17.Farrell N. Coordination Chemistry Reviews. 2002;232:1. [Google Scholar]
- 18.Böttcher A, Gray HB, Meade TJ, Simon MI, Takeuchi T. Journal of Inorganic Biochemistry. 1995;59:221. [Google Scholar]
- 19.Takeuchi T, Böttcher A, Quezada CM, Simon MI, Meade TJ, Gray HB. Journal of the American Chemical Society. 1998;120:8555. [Google Scholar]
- 20.Harney AS, Sole LB, Meade T. Journal of Biological Inorganic Chemistry. 2012 doi: 10.1007/s00775-012-0902-3. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Louie AY, Meade TJ. Proceedings of the National Academy of Sciences. 1998;95:6663. doi: 10.1073/pnas.95.12.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blum O, Haiek A, Cwikel D, Dori Z, Meade TJ, Gray HB. Proceedings of the National Academy of Sciences. 1998;95:6659. doi: 10.1073/pnas.95.12.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harney AS, Lee J, Manus LM, Wang P, Ballweg DM, LaBonne C, Meade TJ. Proceedings of the National Academy of Sciences. 2009;106:13667. doi: 10.1073/pnas.0906423106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harney AS, Meade TJ, LaBonne C. PLoS ONE. 2012;7:e32318. doi: 10.1371/journal.pone.0032318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurtado RR, Harney AS, Heffern MC, Holbrook RJ, Holmgren RA, Meade TJ. Molecular Pharmaceutics. 2012;9:325. doi: 10.1021/mp2005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujii Y. Bulletin of the Chemical Society of Japan. 1972;45:3084. [Google Scholar]
- 27.Böttcher A, Takeuchi T, Hardcastle KI, Meade TJ, Gray HB, Cwikel D, Kapon M, Dori Z. Inorganic Chemistry. 1997;36:2498. [Google Scholar]
- 28.Dori Z, Gershon D, Tivon K, States U. Sep, 1991.
- 29.Costa G, Mestroni G, Tauzher G, Stefani L. Journal of Organometallic Chemistry. 1966;6:181. [Google Scholar]
- 30.Kruger GJ, Reynhardt EC. Acta Crystallographica Section B. 1978;34:915. [Google Scholar]
- 31.Darensbourg DJ, Frantz EB. Inorganic Chemistry. 2007;46:5967. doi: 10.1021/ic7003968. [DOI] [PubMed] [Google Scholar]
- 32.Bukowska-Strzyżewska M, Maniukiewicz W, Bazylak G, Masłowska J. Journal of Chemical Crystallography. 1991;21:157. [Google Scholar]
- 33.Takeuchi T, Böttcher A, Quezada CM, Meade TJ, Gray HB. Bioorganic & Medicinal Chemistry. 1999;7:815. doi: 10.1016/s0968-0896(98)00272-7. [DOI] [PubMed] [Google Scholar]
- 34.Langford CH, Gray HB. Ligand Substitution Processes. W.A. Benjamin; Reading: 1966. [Google Scholar]
- 35.Basolo F. Chemical Reviews. 1953;52:459. [Google Scholar]
- 36.Basolo F, Pearson RG. Mechanisms of Inorganic Reactions. John Wiley & Sons, INC; New York: 1958. [Google Scholar]
- 37.Pearson RG, Boston CR, Basolo F. The Journal of Physical Chemistry. 1955;59:304. [Google Scholar]
- 38.Ni T-L, Garner CS. Inorganic Chemistry. 1967;6:1071. [Google Scholar]
- 39.Basolo F, Bergmann JG, Meeker RE, Pearson RG. Journal of the American Chemical Society. 1956;78:2676. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.