

Abstract
Long-term reduced hypothalamic estrogen signaling leads to increased food intake and decreased locomotor activity and energy expenditure, and ultimately results in obesity and insulin resistance. In the current study, we aimed to determine the acute obesity-independent effects of hypothalamic estrogen signaling on glucose metabolism. We studied endogenous glucose production (EGP) and insulin sensitivity during selective modulation of systemic or intrahypothalamic estradiol (E2) signaling in rats 1 week after ovariectomy (OVX). OVX caused a 17% decrease in plasma glucose, which was completely restored by systemic E2. Likewise, the administration of E2 by microdialysis, either in the hypothalamic paraventricular nucleus (PVN) or in the ventromedial nucleus (VMH), restored plasma glucose. The infusion of an E2 antagonist via reverse microdialysis into the PVN or VMH attenuated the effect of systemic E2 on plasma glucose. Furthermore, E2 administration in the VMH, but not in the PVN, increased EGP and induced hepatic insulin resistance. E2 administration in both the PVN and the VMH resulted in peripheral insulin resistance. Finally, sympathetic, but not parasympathetic, hepatic denervation blunted the effect of E2 in the VMH on both EGP and hepatic insulin sensitivity. In conclusion, intrahypothalamic estrogen regulates peripheral and hepatic insulin sensitivity via sympathetic signaling to the liver.
Estradiol (E2) plays a major role in the control of energy homeostasis (1,2), as is exemplified by increased body weight after ovariectomy (OVX) in female rats, and is reversible with E2 replacement (3–6). The effects of E2 on energy homeostasis are thought to be mediated primarily through the hypothalamus, as direct injections of E2 into the hypothalamic paraventricular nucleus (PVN), arcuate, or ventromedial nucleus (VMH) effectively reduce food intake and body weight after OVX in rodents (7–9). A link between hypothalamic E2 receptors and energy expenditure was elegantly shown by the obese phenotype induced by selective silencing of E2 receptor (ER)-α in VMH (10,11). Together with many more studies, these data convincingly have shown that reduced estrogen signaling in the hypothalamus increases body weight and is associated with impaired glucose tolerance and insulin resistance. At this stage it is less clear if estrogen affects glucose metabolism directly or if it works indirectly by inducing obesity. When rats that underwent OVX were studied before the onset of obesity, they exhibited higher glucose-to-insulin ratios (with decreased plasma insulin concentration) compared with intact rats, suggesting that OVX increases insulin sensitivity (12). This surprising finding could represent a more direct obesity-independent effect of estrogen on glucose metabolism, whose mechanisms thus far have been elucidated only partially.
The hypothalamus has emerged as a key player in the regulation of glucose production (13). The suppressive effects of peripheral hyperinsulinemia on endogenous glucose production (EGP) can be blocked by the central administration of neuropeptide Y (14) or insulin antibodies (13,15). Moreover, the suppressive effect of central insulin on EGP can be largely abolished by selective hepatic vagal denervation (16,17), whereas the intrahypothalamic administration of various neurotransmitters stimulates EGP via the sympathetic efferent nerves to the liver (18–20). These hypothalamic neurotransmitter systems probably act as targets for circulating hormones such as insulin (14), thyroid hormone (21), and glucocorticoids (22). ERs are abundantly expressed in the hypothalamic PVN and VMH (23), nuclei that are key players in the hypothalamic regulation of glucose metabolism via autonomic outflow toward the liver (24). Considering that the hypothalamus plays a key role in the regulation of body weight by estrogen and in controlling glucose metabolism, we hypothesized that the direct obesity-independent effects of estrogen on glucose metabolism are, at least in part, mediated via the hypothalamus and the autonomic nervous system.
To test our hypothesis, we performed a series of experiments that involved the application of reverse microdialysis, selective hepatic autonomic denervations, euglycemic hyperinsulinemic clamps, and stable isotope dilution. To prevent any effects of increased adiposity on glucose metabolism, all experiments were performed 1 week after OVX, i.e., before any increase in body weight or adiposity occurred.
RESEARCH DESIGN AND METHODS
Animals.
Female Wistar rats (Harlan, Horst) housed with a 12-h light–12-h dark schedule (lights on at 7:00 a.m.) were used for all experiments. Body weight was between 220 and 280 g. Food and drinking water were available ad libitum. All of the following experiments were conducted with the approval of the Animal Experimental Committee of the Academic Medical Center in Amsterdam.
Surgery.
Rats were anesthetized using a mixture of ketamine/xylazine (100:10 mg/kg intraperitoneally). All animals underwent a bilateral OVX (except for the control group in experiment 1). Silicon catheters were inserted into the right jugular vein and left carotid artery for intravenous (IV) infusions and blood sampling, respectively. With a standard Kopf stereotaxic apparatus, bilateral microdialysis probes were placed adjacent to PVN or VMH. The coordinates for the PVN were: anterior posterior, 1.6 mm; lateral,1.8 mm (angled at 10°); and 9.1 mm ventral from the surface of the bone. The coordinates for the VMH were: anterior posterior, 2.5 mm; lateral, 2.0 mm (angled at 10°); and 9.0 mm ventral from dura. We used dental cement to secure the microdialysis probes and the jugular and carotid outlets to four stainless steel screws inserted into the skull. Rats were allowed 1 week of postoperative recovery before the start of the actual experiment. The probe location was checked by thionin staining after sacrifice. Only the animals with correct probe placements were used for data analysis (Supplementary Data 1).
Hepatic sympathetic or parasympathetic branches were denervated according to our previously published methods (18). The effectiveness of the hepatic sympathetic denervation was checked by measuring the norepinephrine content in the liver (25). We previously have validated our method for selective hepatic parasympathectomy (parasympathetic denervation) by using retrograde viral tracing (18).
During the experiments, animals were connected to blood-sampling and microdialysis lines, which were attached to a metal collar and kept out of reach of the rats by means of a counterbalanced beam. This allowed all manipulations to be performed outside the cages without handling the animals. The metal collars were attached at least 24 h before the actual experiment. Animals were handled and sham blood was sampled (i.e., blood was withdrawn and immediately returned) regularly during the week before the first experiment began to familiarize them with all the experimental procedures.
One mg β-E2 (Sigma, St. Louis, MO) was dissolved in 1 mL pure dimethyl sulfoxide (DMSO) and diluted 100 times with Ringer solution. The ER antagonist ICI 182,780 (TOCRIS, Bristol, U.K.) was dissolved at a final concentration of 10 μg/mL in Ringer solution containing 1% DMSO. With a measured E2 recovery efficiency of 0.002% (in vitro), the drug concentrations used in the current study (i.e., 10 μg/mL) are expected to result in tissue concentrations of ∼1.0 nmol/L, which is close to the tissue concentration of E2, as measured in our previous study (26). The EC50 value of E2 is ∼0.15 nmol/L for both ERα and ERβ. Thus, the doses used for infusion are expected to activate both ERα and ERβ. We cannot measure the recovery efficiency of the antagonist. The IC50 value of antagonist is 0.29 nM. Thus, if the recovery is similar to that of E2, then it will inhibit both ERα and ERβ.
Plasma measurements.
Glucose enrichment was measured as described previously (27) (Supplementary Data 2). Plasma insulin and corticosterone were measured by a commercially available enzyme-linked immunosorbent assay. Plasma E2 concentration was determined by enzyme-linked immunosorbent assay kits (Biosource, Nivelles, Belgium).
Statistics.
Data were analyzed by ANOVA with repeated measures, with group (E2 or vehicle) as the between-animal factor and time as the within-animal factor. Post hoc tests (Tukey Honestly Significant Difference) were performed if ANOVA revealed a significant effect. Significance was defined at P ≤ 0.05.
Experiment 1 was designed to investigate the difference in plasma glucose concentrations between intact animals and animals that had undergone OVX, and the effect of IV infusion of E2 in OVX animals on plasma glucose concentrations. Blood samples from both intact and OVX animals were collected at 10:30 a.m. for the measurement of basal plasma glucose concentrations. In OVX animals, E2 (3.5 ng/min) or vehicle (saline containing 1% DMSO) was continuously infused via the jugular vein catheter for 165 min (start from t = 15 min). Blood samples were collected from the carotid artery at t = 0 (just before infusion) and at t = 30, 45, 60, 90, 120, 150, and 180 min after infusion. In the sham animals, blood samples were collected at the same time point.
Experiment 2 was designed to investigate the changes in plasma glucose induced by the reverse microdialysis of the ER antagonist ICI 182,780 into the PVN and VMH combined with the IV administration of E2 in OVX animals. Ringer dialysis (3 μL/min) in the PVN or VMH via the microdialysis probes was started at t = −60 min. E2 (3.5 ng/min) was IV-infused starting at t = 10 min, and E2 antagonist (10 μg/mL, 3 μL/min) was infused via the microdialysis probes into the PVN or VMH starting at t = 15 min. Blood samples were collected at t = 0, 30, 45, 60, 90, 120, 150, and 180 min.
Experiment 3 was designed to investigate the effects of reverse microdialysis of E2 into the PVN and VMH of OVX animals on glucose kinetics. To study glucose kinetics, [6,6–2H2] glucose (as a primed [8.0 μmol in 5 min] continuous [16.6 μmol/h] infusion) was used as tracer (>99% enriched; Cambridge Isotopes, Andover, MA). Blood samples were taken at t = −95 min for measuring background enrichment of [6,6–2H2] glucose at t = 0, 5, and 10 min for determining enrichment during the steady state, and at t = 30, 45, 60, 90, 120, 150, and 180 min for determining enrichment during the retrodialysis of E2 (nonsteady state). Ringer dialysis (3 μL/min) in the PVN or VMH via the microdialysis probes was started at t = −60 min. At t = 15 min, E2 (10 μg/mL, 3 μL/min) or vehicle (Ringer with 1% DMSO, 3 μL/min) was infused by retrodialysis into PVN or VMH.
Experiment 4 was designed to investigate the effects of reverse microdialysis of E2 into the PVN and VMH of OVX animals on insulin sensitivity. Background blood samples and isotope tracer infusion were the same as for experiment 3. At t = 15 min, insulin was administered in a primed IV infusion (3.6 mU/kg · min in 5 min for the “low” clamp 1, and 7.2 mU/kg · min in 5 min for the “high” clamp 2), followed by a continuous IV infusion (1.5 mU/kg · min and 3 mU/kg · min, respectively). A variable infusion of a 25% glucose solution (containing 1% [6,6–2H2] glucose) was used to maintain euglycemia (5.5 ± 0.5 mmol/L) (Supplementary Data 3), as determined by carotid catheter blood sampling every 10 min. Thirty minutes after the start of the primary insulin infusion (t = 45 min), Ringer perfusion of the microdialysis probes was replaced by the E2 solution (10 μg/mL, 3 μL/min) or vehicle (Ringer containing 1% DMSO). At the end of the clamp, five blood samples were taken with a 5-min interval at t = 120, 125, 130, 135, and 140 min (Supplementary Data 4).
Experiment 5 was designed to investigate the effect of selective hepatic autonomic nerve denervations on plasma glucose changes induced by the reverse microdialysis of E2 into the PVN and VMH. The experimental design is similar to that for experiment 3.
Experiment 6 was designed to investigate the effects of a hepatic sympathetic nerve denervation combined with the reverse microdialysis of E2 into the VMH on hepatic insulin sensitivity. The experimental design was similar to that of experiment 4.
RESULTS
As expected, plasma E2 concentrations were lower in OVX than in intact animals (P = 0.004) (Fig. 1A). After 165 min of systemic E2 infusion, plasma E2 concentrations were higher in the E2 group (P = 0.002) and comparable with those of the intact animals (P = 0.357) (Fig. 1A). OVX animals showed a 17% decrease of basal plasma glucose concentrations as compared with intact animals (P = 0.035) (Fig. 1B). During systemic infusion of E2, plasma glucose concentrations significantly increased as compared with the vehicle-infused group (time effect: P < 0.001; group effect: P = 0.028; time × group effect: P = 0.001) (Fig. 1B).
FIG. 1.

A: OVX reduced plasma E2 concentrations compared with intact animals. IV E2 administration restored plasma E2 concentrations (different letters indicate a significant difference, P < 0.05). B: After OVX, plasma glucose concentrations were lower compared with those of intact animals (P = 0.035). IV E2 administration acutely increased plasma glucose to concentrations observed in intact animals. The increase in plasma glucose concentrations during IV E2 administration was attenuated by simultaneous administration of the ER antagonist ICI 182,780 (ICI) in (C) the PVN (P = 0.020 vs. vehicle) and (D) VMH (P = 0.009 vs. vehicle). The hatched bars indicate the continuous infusion of vehicle, E2, and/or ICI.
The effect of systemic E2 on plasma glucose was blunted by intrahypothalamic administration of the E2 antagonist ICI 182,780. Plasma glucose levels were significantly lower during systemic E2 infusion and simultaneous retrodialysis of ICI 182,780 in the PVN than during vehicle retrodialysis (time, P < 0.001; group, P = 0.02; time × group, P = 0.064) (Fig. 1C). A similar effect was found after systemic E2 infusion and simultaneous retrodialysis of ICI 182,780 in the VMH (time, P < 0.001; group, P = 0.009; time × group, P < 0.001) (Fig. 1D).
During retrodialysis of E2 in the PVN of OVX animals, plasma glucose concentrations increased compared with vehicle retrodialysis (group, P = 0.015; time × group, P < 0.001) (Fig. 2A). Similarly, E2 treatment in the VMH also resulted in higher plasma glucose levels (group, P = 0.006; time × group, P < 0.001) (Fig. 2D). During the retrodialysis of vehicle in the PVN and VMH, EGP showed a slow decline, probably because of the prolonged fasting (20). Retrodialysis of E2 in the PVN did not affect EGP (Fig. 2B), but the same treatment in the VMH increased EGP as compared with the vehicle treatment, i.e., EGP showed no steady decrease but showed significant effects of group (P < 0.001) and time × group (P < 0.001) (Fig. 2E). Importantly, plasma E2 concentrations were not affected by the intrahypothalamic infusions of E2 in either the PVN or the VMH (Fig. 2C and F).
FIG. 2.

A: Administration of E2 (n = 6) into the PVN increased plasma glucose concentrations (P = 0.015) (B), but not EGP as compared with vehicle (n = 7). D: E2 (n = 6) infusion into the VMH increased plasma glucose (P = 0.006). E: E2 infusion in the VMH increased EGP (overall group effect, P < 0.001). C and F: Plasma E2 concentrations were not different between vehicle and E2-infused animals, excluding leakage from the hypothalamus to the systemic circulation. The hatched bars indicate the continuous infusion of vehicle or E2.
We studied hepatic and peripheral insulin sensitivity using euglycemic hyperinsulinemic clamps at low and high insulin concentrations, respectively. Plasma insulin concentrations during the clamps were significantly higher than those during basal conditions in both the low-dose and high-dose clamp groups (time, P < 0.001). No significant differences between the different infusion groups (i.e., vehicle, PVN, and VMH) were detected (group, P > 0.27; time × group, P > 0.48) (Fig. 3C and F). The lower dose clamp experiment showed similar basal EGP levels between the vehicle and E2 infusion groups (P = 0.687), but the intrahypothalamic administration of E2 differentially affected the insulin-induced decrease of EGP (P = 0.003) (Fig. 3A). In the vehicle and PVN E2 infusion groups, EGP was suppressed by 30–40%, whereas in the VMH E2 infusion group the expected decrease in EGP induced by hyperinsulinemia was completely blunted (P = 0.017 versus vehicle) (Fig. 3B).
FIG. 3.

A and B: Insulin decreased EGP by 30–40% during vehicle (VEH; n = 6) and E2 infusion in the PVN E2 (n = 6). Insulin-mediated suppression of EGP was completely blunted by simultaneous E2 infusion in the VMH (n = 6). D and E: Insulin infusion also stimulated glucose uptake (Rd), which was partly prevented by infusion of E2 in either the VMH or the PVN. C and F: Plasma insulin concentrations in the different groups during the “low” (1.5 mU/kg · min) (C) and “high” (3.0 mU/kg · min) (F) clamps. Different letters indicate a significant difference (P < 0.05).
During the higher-dose hyperinsulinemic clamp, the insulin-induced increase in glucose uptake was reduced by E2 treatment both in the PVN and VMH (both P = 0.012) (Fig. 3D). In the vehicle group, as expected, glucose uptake was increased by 130%, whereas in the PVN and VMH treatment group, the increase in glucose uptake by insulin was attenuated (Fig. 3E).
Liver noradrenalin levels were significantly lower (<10%) in all sympathectomy (sympathetic denervation) groups as compared with both the sham-denervated and parasympathectomy (parasympathetic denervation) groups (Supplementary Data 5). During E2 infusion in the PVN, the increase in plasma glucose did not differ between sham, hepatic sympathetic denervation, and parasympathetic denervation groups (group, P = 0.386; time × group, P = 0.163) (Fig. 4A). Likewise, there was no effect of hepatic denervations on EGP (group, P = 0.528; time × group, P = 0.939) either during steady-state or nonsteady-state conditions (Fig. 4B). During E2 infusion in the VMH, however, plasma glucose concentrations were lower in the sympathetic denervation group than in the parasympathetic denervation and sham groups (post hoc group effect: P = 0.002 and P = 0.001, sympathetic denervation versus parasympathetic denervation and sham, respectively) (Fig. 4C). The stimulatory effect of E2 infusion in the VMH on EGP (Fig. 2E) was abolished by sympathetic denervation, but not by parasympathetic denervation or sham denervation (group P = 0.006 and time × group P < 0.001 for sympathetic denervation versus sham, and group P = 0.604 and time × group P = 0.487 for parasympathetic denervation versus sham) (Fig. 4D). During E2 infusion in the VMH, insulin suppressed EGP by 35% in the sympathetic denervation group, but only by 10% in the group with intact sympathetic signaling to the liver (P = 0.014) (Fig. 5A and B), indicating the necessity of the sympathetic hepatic innervation for the modulation of hepatic insulin sensitivity by E2 in the VMH. Plasma insulin levels during the clamp were significantly increased as compared with basal conditions before the clamp, but no significant differences between the two groups (sham and sympathetic denervation) were found (P < 0.001 and P = 0.172, respectively) (Fig. 5C).
FIG. 4.

A and B: The effect of E2 administration in the PVN on plasma glucose concentration and EGP was not affected by sympathetic denervation (Sx) or parasympathetic (Px) denervation of the liver. C and D: In contrast, sympathetic denervation reduced plasma glucose (P = 0.002 Sx vs. sham) and EGP (P = 0.006 Sx vs. sham) during E2 infusion in the VMH compared with sham denervation. The hatched bars indicate the continuous infusion of E2.
FIG. 5.
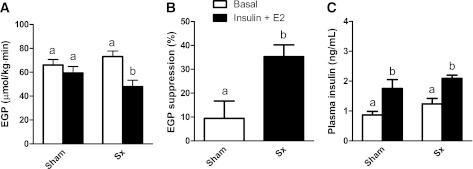
A and B: During E2 administration in the VMH, insulin-mediated EGP suppression was restored after sympathetic denervation (Sx) of the liver. C: Insulin concentrations under basal conditions and during IV insulin administration. n = 6 for all groups. Different letters indicate a significant difference (P < 0.05). Insulin administration rate: 1.5 mU/kg · min.
E2 effects on glucoregulatory hormones.
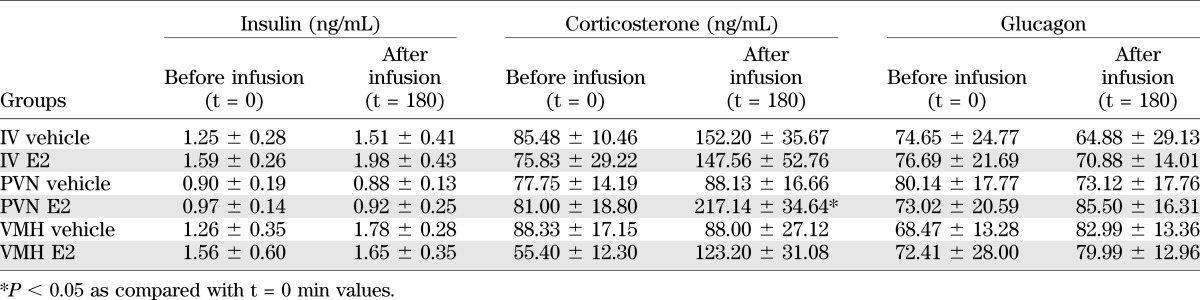
IV E2 infusions in experiment 1 did not affect plasma insulin, corticosterone, or glucagon levels when compared with the vehicle group (Table 1). Also, E2 administration in the PVN or VMH did not affect plasma insulin or glucagon concentrations (Table 1). Both PVN and VMH administration of E2 resulted in increased plasma corticosterone values at t = 180 min, but only the effect in the PVN reached significance (ANOVA basal values [t = 0 min], P = 0.637; t = 180 min, P = 0.009). The PVN–E2 corticosterone value differed significantly from both vehicle groups at t = 180 min (P < 0.005), whereas the VMH–E2 t = 180 value did not differ from the two vehicle groups or from the PVN–E2 group.
TABLE 1.
Plasma hormone measurements for experiments 1 and 3
DISCUSSION
The key finding of the current study is that local changes in hypothalamic E2 availability have site-specific effects on peripheral glucose metabolism and insulin sensitivity. More specifically, in rats that have undergone OVX, local supplementation of E2 in the PVN and VMH decreased peripheral insulin sensitivity and reduced glucose uptake, whereas local E2 supplementation in the VMH also reduced EGP and caused hepatic insulin resistance. Finally, we demonstrated that the effect of E2 in the VMH on hepatic insulin sensitivity is mediated by the sympathetic innervation to the liver.
The effects of OVX in animal models include increased food intake and decreased running activity, all of which are reversed on E2 replacement (28,29). A series of experiments by Clegg et al. (6) showed that local effects of E2 in the brain play an important role in these restorative effects of E2. Recent reports showed that a major part of the profound effects of E2 on energy metabolism are mediated via the hypothalamus. For instance, ICV infusion of E2 was sufficient to restore the normal pattern of body fat distribution in OVX females (6), and a local knockout of ERα in the VMH was sufficient for animals to become obese (10). Thus, E2 withdrawal and substitution have profound effects on adiposity and lipid metabolism (3,6,30). Direct effects of E2 on glucose metabolism via the brain, however, are not evident yet. However, previous studies by us and others revealed that the hypothalamus also plays a crucial role in the regulation of glucose production (19,21). To further examine the possible neural mechanisms behind the modulation of peripheral E2 on glucose metabolism, we used reverse microdialysis technology to be able to administer E2 locally into PVN or VMH. In our first set of experiments, strikingly lower plasma glucose levels in OVX animals were found, which could be reversed by a systemic E2 replacement. Interestingly, the increase in plasma glucose concentrations induced by systemic E2 was blocked by the hypothalamic administration of the E2 antagonist ICI 182,780, both in PVN and VMH. The data indicated that, like glucocorticoid and thyroid hormone, hypothalamic E2 also may play an important role in the regulation of peripheral glucose metabolism.
Next, we infused E2 directly into the PVN and VMH by reverse microdialysis. The increased plasma glucose levels were consistent with the results of the antagonist experiment. The E2-induced changes could not be explained by the changed corticosterone or insulin levels as during the VMH infusions, neither plasma corticosterone nor insulin concentrations were affected. Infusions in the PVN did not affect plasma insulin concentrations either. In accordance with the well-known effects of estrogens on the hypothalamo-pituitary-adrenal axis, corticosterone concentrations were changed by E2 administration in the PVN. However, the E2-induced corticosterone changes occurred after t = 150 min, which was much later than the E2-induced change in plasma glucose. Although both electrical stimulation of VMH neurons and direct insulin injections in the VMH have been reported to induce an increase of plasma glucagon concentrations (31–33), we did not find any change in plasma glucagon values after the different treatments in the current study. These data indicate that the increased EGP induced by VMH E2 is not mediated by an increased release of glucagon, and that different mechanisms may be activated via the VMH. Although our denervation experiments suggest that the E2-induced increased EGP is caused by an increased sympathetic activity, at present we cannot exclude the involvement of other hormonal regulators such as the catecholamines.
Recently Yonezawa et al. (34) reported that during exposure to a high-fat diet, both peripheral and central ERs are involved in the regulation of glucose metabolism. However, central and peripheral ERs seem to operate via different mechanisms. When treated with peripheral E2, fatty acid synthase was decreased in white adipose tissue, whereas treatment with central E2 changed both liver glucose production and peripheral tissue glucose uptake (34). Their results suggest that hypothalamic E2 may affect EGP and glucose uptake by, respectively, increasing and decreasing insulin sensitivity. Estrogenic effects on peripheral organs also were addressed by several other studies. In the liver, glucose homeostasis seems to be regulated mainly by estrogen acting via ERα, which was associated with a pronounced hepatic insulin resistance (35). Immunohistochemical analysis revealed that ERα and ERβ are coexpressed in the nuclei of most muscle cells. These studies also showed that ERα is a positive regulator of GLUT4 expression, whereas ERβ has a suppressive role (36).
In the current studies, E2 administration in the VMH but not PVN increased EGP, indicating that the increasing plasma glucose concentrations observed after PVN infusion were mainly attributable to a decreased glucose uptake, whereas the increased plasma glucose concentrations after E2 administration in the VMH were mainly attributable to an increased EGP. The two classic ERs, ERα and ERβ, show a distinct hypothalamic distribution, with the VMH mainly containing ERα and the PVN mainly containing ERβ (23). At present it is not clear how this differential receptor distribution contributes to the different glucoregulatory effects of E2 in the PVN and VMH.
We used hyperinsulinemic-euglycemic clamps with two different insulin plasma levels. Consistent with the basal EGP results, PVN E2 treatment caused a peripheral insulin resistance. However, both peripheral and hepatic insulin resistance were found in the group treated with VMH E2. Previous studies in our group showed that the hypothalamus often increases hepatic glucose production by stimulating sympathetic efferent nerves (18,19,21). Also, in the current experiments the stimulatory effect of E2 via the VMH on EGP (and plasma glucose concentrations) was abolished by a sympathetic, but not a parasympathetic, denervation of the liver. However, autonomic denervation of the liver (either sympathetic or parasympathetic) had no effect on the stimulatory effect of E2 on plasma glucose concentrations via the PVN. There is no evidence for a direct neural connection between the VMH and autonomic nuclei in the brainstem or spinal cord, contrary to the PVN. However, the VMH has pronounced projections to the PVN, which functions as the hypothalamic integration center for autonomic and endocrine information and serves as the final neuroendocrine and autonomic output nucleus from the hypothalamus (18,37–40). ERα is expressed in the majority of glutamatergic neurons in VMH (41) and the PVN is known to receive a strong glutamatergic input from the VMH (42). Therefore, we propose that ERα-containing glutamatergic neurons in the VMH that project to the PVN are activated by local administration of E2, thereby exciting sympathetic preautonomic neurons in the PVN that, in turn, stimulate the hepatic sympathetic tone.
In line with our present findings, a number of previous experiments have provided evidence for hypothalamic effects on glucose uptake mediated via the autonomic nervous system (43,44). From a physiological viewpoint, the opposite effects of ERα stimulation in the VMH on hepatic glucose production and peripheral glucose uptake (stimulatory and inhibitory, respectively) are plausible, because in this way the two mechanisms will act in concert to increase plasma glucose concentration. To explain the opposite effect of the VMH on muscle-dedicated and liver-dedicated preautonomic neurons, we propose that either the glutamatergic projection of the VMH to the muscle-dedicated preautonomic neurons involves a GABAergic interneuron in the sub-PVN or the VMH contains GABAergic ERα-expressing neurons that contact and inhibit the muscle-dedicated preautonomic neurons directly. E2 treatment has been shown to increase GABAergic activity in the VMH (45). Finally, the preferential effect of E2 in the PVN on peripheral glucose uptake indicates that in all likelihood the muscle-dedicated preautonomic neurons, but not the liver-dedicated preautonomic neurons, in the PVN express the ERβ. Although the expression of ERβ mRNA in preautonomic PVN neurons has been reported (46), the peripheral targets of these neurons are not known.
Together, our results show differential effects of intrahypothalamic E2 on hepatic and peripheral glucose metabolism that are, at least partly, mediated by the sympathetic branch of the autonomic nervous system. However, the current results seem contradictory with earlier studies that indicated increased plasma glucose levels in OVX animals. We think this apparent difference is induced by the different models used. Most studies thus far used a “chronic” model in which animals were studied at least 1 month after the OVX (47–49). Therefore, the increased levels of plasma glucose observed most likely are the results of hyperphagia and obesity. The current results indicate that the first effect of reduced plasma E2 concentrations is a lowering of plasma glucose levels. During the second stage, the OVX animals have development of increased food intake and decreased energy expenditure and they become obese. At the end of the second stage, the impaired energy homeostasis will overrule the glucose-lowering effect of E2 removal. How the short-term effects of intrahypothalamic E2 on glucose metabolism as observed in the present set of experiments can be reconciled with the long-term effects of E2 deprivation on body weight and insulin sensitivity remains to be determined.
ACKNOWLEDGMENTS
This study was supported by a grant from the Chinese Academy of Sciences and the Royal Netherlands Academy of Arts and Sciences, within the framework of the China Exchange Program (P.H.B., E.Fl., J.-N.Z., Grant CEP-09CDP030), and an NWO-ZonMw (TOP 91207036) grant (A.K.).
No potential conflicts of interest relevant to this article were reported.
J.L. wrote the manuscript and contributed to the experiment. P.H.B. researched the data and reviewed and edited the manuscript. L.E. contributed to the experiment. E.Fl. researched the data. M.T.A. contributed to the EGP measurement. J.-N.Z. reviewed and edited the manuscript. E.Fo. contributed to the experiment. A.K. researched the data, reviewed and edited the manuscript and contributed to the discussion. J.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Wilma Verweij of the Netherlands Institute for Neuroscience for linguistic corrections.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0488/-/DC1.
REFERENCES
- 1.Milewicz A, Bidzińska B, Mikulski E, Demissie M, Tworowska U. Influence of obesity and menopausal status on serum leptin, cholecystokinin, galanin and neuropeptide Y levels. Gynecol Endocrinol 2000;14:196–203 [DOI] [PubMed] [Google Scholar]
- 2.Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology 2001;142:4751–4757 [DOI] [PubMed] [Google Scholar]
- 3.Saengsirisuwan V, Pongseeda S, Prasannarong M, Vichaiwong K, Toskulkao C. Modulation of insulin resistance in ovariectomized rats by endurance exercise training and estrogen replacement. Metabolism 2009;58:38–47 [DOI] [PubMed] [Google Scholar]
- 4.Rachoń D, Vortherms T, Seidlová-Wuttke D, Wuttke W. Effects of dietary equol on body weight gain, intra-abdominal fat accumulation, plasma lipids, and glucose tolerance in ovariectomized Sprague-Dawley rats. Menopause 2007;14:925–932 [DOI] [PubMed] [Google Scholar]
- 5.Jones ME, Thorburn AW, Britt KL, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA 2000;97:12735–12740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clegg DJ, Brown LM, Woods SC, Benoit SC. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes 2006;55:978–987 [DOI] [PubMed] [Google Scholar]
- 7.Colvin GB, Sawyer CH. Induction of running activity by intracerebral implants of estrogen in overiectomized rats. Neuroendocrinology 1969;4:309–320 [DOI] [PubMed] [Google Scholar]
- 8.Ahdieh HB, Wade GN. Effects of hysterectomy on sexual receptivity, food intake, running wheel activity, and hypothalamic estrogen and progestin receptors in rats. J Comp Physiol Psychol 1982;96:886–892 [PubMed] [Google Scholar]
- 9.Butera PC, Czaja JA. Intracranial estradiol in ovariectomized guinea pigs: effects on ingestive behaviors and body weight. Brain Res 1984;322:41–48 [DOI] [PubMed] [Google Scholar]
- 10.Musatov S, Chen W, Pfaff DW, et al. Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA 2007;104:2501–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Y, Nedungadi TP, Zhu L, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab 2011;14:453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barros RP, Morani A, Moriscot A, Machado UF. Insulin resistance of pregnancy involves estrogen-induced repression of muscle GLUT4. Mol Cell Endocrinol 2008;295:24–31 [DOI] [PubMed] [Google Scholar]
- 13.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002;8:1376–1382 [DOI] [PubMed] [Google Scholar]
- 14.van den Hoek AM, van Heijningen C, Schröder-van der Elst JP, et al. Intracerebroventricular administration of neuropeptide Y induces hepatic insulin resistance via sympathetic innervation. Diabetes 2008;57:2304–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 2002;5:566–572 [DOI] [PubMed] [Google Scholar]
- 16.Pocai A, Lam TK, Gutierrez-Juarez R, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005;434:1026–1031 [DOI] [PubMed] [Google Scholar]
- 17.Acosta-Martínez M, Levine JE. Regulation of KATP channel subunit gene expression by hyperglycemia in the mediobasal hypothalamus of female rats. Am J Physiol Endocrinol Metab 2007;292:E1801–E1807 [DOI] [PubMed] [Google Scholar]
- 18.Kalsbeek A, La Fleur S, Van Heijningen C, Buijs RM. Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J Neurosci 2004;24:7604–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi CX, Serlie MJ, Ackermans MT, et al. A major role for perifornical orexin neurons in the control of glucose metabolism in rats. Diabetes 2009;58:1998–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi CX, Sun N, Ackermans MT, et al. Pituitary adenylate cyclase-activating polypeptide stimulates glucose production via the hepatic sympathetic innervation in rats. Diabetes 2010;59:1591–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klieverik LP, Janssen SF, van Riel A, et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc Natl Acad Sci USA 2009;106:5966–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi CX, Foppen E, Abplanalp W, et al. Glucocorticoid signaling in the arcuate nucleus modulates hepatic insulin sensitivity. Diabetes 2012;61:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol 1997;388:507–525 [DOI] [PubMed] [Google Scholar]
- 24.Kalsbeek A, Yi CX, La Fleur SE, Fliers E. The hypothalamic clock and its control of glucose homeostasis. Trends Endocrinol Metab 2010;21:402–410 [DOI] [PubMed] [Google Scholar]
- 25.Klieverik LP, Sauerwein HP, Ackermans MT, Boelen A, Kalsbeek A, Fliers E. Effects of thyrotoxicosis and selective hepatic autonomic denervation on hepatic glucose metabolism in rats. Am J Physiol Endocrinol Metab 2008;294:E513–E520 [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Hu P, Qi XR, Meng FT, Kalsbeek A, Zhou JN. Acute restraint stress increases intrahypothalamic oestradiol concentrations in conjunction with increased hypothalamic oestrogen receptor β and aromatase mRNA expression in female rats. J Neuroendocrinol 2011;23:435–443 [DOI] [PubMed] [Google Scholar]
- 27.Ackermans MT, Pereira Arias AM, Bisschop PH, Endert E, Sauerwein HP, Romijn JA. The quantification of gluconeogenesis in healthy men by (2)H2O and [2-(13)C]glycerol yields different results: rates of gluconeogenesis in healthy men measured with (2)H2O are higher than those measured with [2-(13)C]glycerol. J Clin Endocrinol Metab 2001;86:2220–2226 [DOI] [PubMed] [Google Scholar]
- 28.Asarian L, Geary N. Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats. Horm Behav 2002;42:461–471 [DOI] [PubMed] [Google Scholar]
- 29.Shimomura Y, Shimizu H, Takahashi M, et al. The significance of decreased ambulatory activity during the generation by long-term observation of obesity in ovariectomized rats. Physiol Behav 1990;47:155–159 [DOI] [PubMed] [Google Scholar]
- 30.Kannel WB, Wilson PW. Risk factors that attenuate the female coronary disease advantage. Arch Intern Med 1995;155:57–61 [PubMed] [Google Scholar]
- 31.Paranjape SA, Chan O, Zhu W, et al. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 2010;59:1521–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimazu T. Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia 1981;20(Suppl.):343–356 [PubMed] [Google Scholar]
- 33.Shimazu T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes Metab Rev 1987;3:185–206 [DOI] [PubMed] [Google Scholar]
- 34.Yonezawa R, Wada T, Matsumoto N, et al. Central versus peripheral impact of estradiol on the impaired glucose metabolism in ovariectomized mice on a high-fat diet. Am J Physiol Endocrinol Metab 2012;303:E445–E456 [DOI] [PubMed] [Google Scholar]
- 35.Bryzgalova G, Lundholm L, Portwood N, et al. Mechanisms of antidiabetogenic and body weight-lowering effects of estrogen in high-fat diet-fed mice. Am J Physiol Endocrinol Metab 2008;295:E904–E912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barros RP, Machado UF, Warner M, Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci USA 2006;103:1605–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams G, Bing C, Cai XJ, Harrold JA, King PJ, Liu XH. The hypothalamus and the control of energy homeostasis: different circuits, different purposes. Physiol Behav 2001;74:683–701 [DOI] [PubMed] [Google Scholar]
- 38.Saphier D. Electrophysiology and neuropharmacology of noradrenergic projections to rat PVN magnocellular neurons. Am J Physiol 1993;264:R891–R902 [DOI] [PubMed] [Google Scholar]
- 39.Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology 1980;31:410–417 [DOI] [PubMed] [Google Scholar]
- 40.Swanson LW, Kuypers HG. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J Comp Neurol 1980;194:555–570 [DOI] [PubMed] [Google Scholar]
- 41.Pompolo S, Pereira A, Scott CJ, Fujiyma F, Clarke IJ. Evidence for estrogenic regulation of gonadotropin-releasing hormone neurons by glutamatergic neurons in the ewe brain: An immunohistochemical study using an antibody against vesicular glutamate transporter-2. J Comp Neurol 2003;465:136–144 [DOI] [PubMed] [Google Scholar]
- 42.Csáki A, Kocsis K, Halász B, Kiss J. Localization of glutamatergic/aspartatergic neurons projecting to the hypothalamic paraventricular nucleus studied by retrograde transport of [3H]D-aspartate autoradiography. Neuroscience 2000;101:637–655 [DOI] [PubMed] [Google Scholar]
- 43.Flechtner-Mors M, Jenkinson CP, Alt A, Biesalski HK, Adler G, Ditschuneit HH. Sympathetic regulation of glucose uptake by the alpha1-adrenoceptor in human obesity. Obes Res 2004;12:612–620 [DOI] [PubMed] [Google Scholar]
- 44.Minokoshi Y, Okano Y, Shimazu T. Regulatory mechanism of the ventromedial hypothalamus in enhancing glucose uptake in skeletal muscles. Brain Res 1994;649:343–347 [DOI] [PubMed] [Google Scholar]
- 45.Luine VN, Grattan DR, Selmanoff M. Gonadal hormones alter hypothalamic GABA and glutamate levels. Brain Res 1997;747:165–168 [DOI] [PubMed] [Google Scholar]
- 46.Bingham B, Williamson M, Viau V. Androgen and estrogen receptor-beta distribution within spinal-projecting and neurosecretory neurons in the paraventricular nucleus of the male rat. J Comp Neurol 2006;499:911–923 [DOI] [PubMed] [Google Scholar]
- 47.Lemieux C, Picard F, Labrie F, Richard D, Deshaies Y. The estrogen antagonist EM-652 and dehydroepiandrosterone prevent diet- and ovariectomy-induced obesity. Obes Res 2003;11:477–490 [DOI] [PubMed] [Google Scholar]
- 48.Ulas M, Cay M. The effects of 17beta-estradiol and vitamin E treatments on oxidative stress and antioxidant levels in brain cortex of diabetic ovariectomized rats. Acta Physiol Hung 2010;97:208–215 [DOI] [PubMed] [Google Scholar]
- 49.Liu ML, Xu X, Rang WQ, Li YJ, Song HP. Influence of ovariectomy and 17beta-estradiol treatment on insulin sensitivity, lipid metabolism and post-ischemic cardiac function. Int J Cardiol 2004;97:485–493 [DOI] [PubMed] [Google Scholar]