

Abstract
We provide the first comprehensive analysis of the extracellular matrix (ECM) composition of peri-islet capsules, composed of the peri-islet basement membrane (BM) and subjacent interstitial matrix (IM), in development of type 1 diabetes in NOD mice and in human type 1 diabetes. Our data demonstrate global loss of peri-islet BM and IM components only at sites of leukocyte infiltration into the islet. Stereological analyses reveal a correlation between incidence of insulitis and the number of islets showing loss of peri-islet BM versus islets with intact BMs, suggesting that leukocyte penetration of the peri-islet BM is a critical step. Protease- and protease inhibitor–specific microarray analyses (CLIP-CHIP) of laser-dissected leukocyte infiltrated and noninfiltrated pancreatic islets and confirmatory quantitative real time PCR and protein analyses identified cathepsin S, W, and C activity at sites of leukocyte penetration of the peri-islet BM in association with a macrophage subpopulation in NOD mice and human type 1 diabetic samples and, hence, potentially a novel therapeutic target specifically acting at the islet penetration stage. Interestingly, the peri-islet BM and underlying IM are reconstituted once inflammation subsides, indicating that the peri-islet BM-producing cells are not lost due to the inflammation, which has important ramifications to islet transplantation studies.
Type 1 diabetes is a chronic autoimmune disease that targets the insulin-producing β-cells of the islets of Langerhans in the pancreas. Although it is a T-cell (CD4 and CD8)–mediated disease, macrophages, dendritic cells (DCs), and natural killer cells are also essential for disease progression (1). NOD mice are one of the best-studied animal models of type 1 diabetes (2) due to similarities with the pathogenesis and genetics of human type 1 diabetes. The precise signal that leads to disease initiation is not known and is considered to be a combination of genetic and environmental factors (3). However, once autoaggressive T cells are activated, several steps are crucial for disease progression, including extravasation of the CD4+, CD8+ T lymphocytes from the postcapillary venules surrounding the islets of Langerhans and their subsequent infiltration of the islets, leading to β-cell destruction and onset of disease symptoms. Progression from the first steps in disease induction to the presentation of symptoms occurs over a period of months in NOD mice and several years in humans, with clinical symptoms appearing only when 70–90% of β-cells are destroyed, leaving a very narrow therapeutic window (4,5).
In many cases, extravasated leukocytes can reside around the islets of Langerhans without induction of disease symptoms. Indeed, abundant infiltration has been described in several experimental models that never or rarely progresses to disease (6,7), indicating that penetration of the pancreatic islet is a crucial step in disease progression. Of all the steps in the progression of type 1 diabetes, this final step from peri-islet infiltration to invasion of islets is the least well understood. However, to understand the factors that convert a peri-islet infiltration to an overt intra-islet infiltration that leads to diabetes would provide enormous potential for controlling disease progression and therapeutic development. The present work addresses this important step in the development of type 1 diabetes in NOD mice and in humans, in particular, the contribution of the peri-islet basement membrane (BM) to this step.
In all tissues, the extracellular matrix (ECM) acts to separate tissue compartments but also provides specific molecular signals that control processes such as cell migration, differentiation, and survival (8). Two main types of ECM exist—the BM, which acts as a barrier limiting the movement of both soluble molecules and cells, and the interstitial matrix (IM), which confers flexibility and elasticity to tissues—both of which are present in the peri-islet capsule. Laminins and collagen type IV are the major constituents of the BM, each of which self-assemble to form two protein networks that are interconnected by nidogens and heparan sulfate proteoglycans, such as perlecan (9). Collagen type IV is considered to be the structural basis of BMs, whereas the laminins impart specific signals to cells attached to or moving through BMs and are thereby responsible for biological activity. Laminins are composed of an α, β, and γ chain, and 5α, 4β, and 3γ laminin chains exist that combine to form at least 18 different isoforms. These isoforms are differentially expressed in different BM types where they perform different functions (10) mediated by direct binding to a variety of different integrin and nonintegrin receptors (11). The IM layer, which in most tissues underlies the BM, is composed of fibrillar collagens, such as collagen type I and III, nonfibrillar collagens, like collagen type VI, and noncollagenous glycoproteins, like fibronectin, tenascins, vitronectin, and chondroitin, or dermatan sulfate proteoglycans (12,13).
The IM and BM influence immune cell migration into or within inflamed tissues (14). The existing data, which are extremely limited, suggest that the biochemical nature of the ECM may determine the mechanism used by the immune cells for its penetration, with protease-independent migration dominating (15–17). However, selective cleavage mechanisms play a role in some cases (18,19).
Limited information exists on the nature of the ECM of the pancreas and, in particular, on the composition of peri-islet capsule (20–22). Recent studies have reported the existence of laminins, collagen type IV, and perlecan surrounding the pancreatic islets and the absence of staining for these antigens in pancreata of NOD mice, however, without correlation to disease progression in mice or with type 1 diabetes in humans (23,24).
Here, we provide the first comprehensive analysis of the ECM of the peri-islet capsule, and the 3-dimensional relationship among the peri-islet BM, peri-islet blood vessels, and peri-islet IM in NOD mice during the course of development of type 1 diabetes and in samples from humans recently diagnosed with type 1 diabetes. We demonstrate global loss of peri-islet BM and IM components only at sites of leukocyte penetration of the peri-islet capsule, which contrasts to leukocyte extravasation from blood vessels and demonstrates fundamental differences in these two processes. The number of islets showing loss of peri-islet BM versus islets with intact BMs correlated with the incidence of diabetes, indicating that this is a crucial step in disease progression. Protease- and protease inhibitor–specific microarray analyses (CLIP-CHIP) of laser-capture microdissection (LCM) material revealed upregulation of cathepsin S, W, and C in leukocyte-infiltrated islets compared with noninvaded pancreatic islets. Parallel protein analyses revealed cathepsin S, W, and C expression in association with a subpopulation of macrophages at the front of leukocyte penetration of the peri-islet BM in NOD mice and in samples from humans with type 1 diabetes and, hence, potentially a novel therapeutic target specifically acting at the islet penetration stage. Once inflammation had subsided, the peri-islet BM and underlying IM were shown to be reconstituted in the mouse and human, indicating that the cells producing the peri-islet BM are not lost due to the inflammation, which has important ramifications to islet transplantation studies. Our data suggest that the ECM milieu influences the mode used by immune cells to infiltrate into tissues and raises novel possibilities for tissue-specific immune modulatory therapies.
RESEARCH DESIGN AND METHODS
Animals.
NOD mice were from Bomholtgaard (Ry, Denmark) and were screened for diabetes by urine analyses of glucosuria (Combur3 Test; Roche). Insulin pellets (LinShin Canada, Inc.) were inserted subcutaneously into diabetic NOD mice for long-term studies. Animal experiments were conducted according to the Swedish and German animal welfare guidelines.
Human samples.
Human healthy pancreas and type 1 diabetic samples were provided by the Network for Pancreatic Organ Donors with Diabetes (nPOD) organ bank.
Generation of anti-human laminin monoclonal antibodies.
Mouse monoclonal antibodies were generated to human laminin α5, α4, α2, β1, β2, and γ1 chains. Antibodies were characterized by enzyme-linked immunosorbent assay, tissue distribution as determined by immunofluorescence microscopy, and by Western blot analyses (25).
Microscopy.
Immunofluorescence staining of cryosections was performed as described previously (26). For confocal microscopy of whole mounts, pancreata were fixed in 4% paraformaldehyde and blocked in 1% BSA/0.3% TritonX-100. The primary and secondary antibodies used are listed in Supplementary Table 1. Sections were examined using a Zeiss AxioImager or a Zeiss LSM510 and documented using a Hamamatsu ORCA ER camera and Volocity 5.4 software (Improvision). Samples for electron microscopy were prepared according to standard protocols and analyzed with an EM-410 (Philips).
Stereological analyses and quantification of the peri-islet BM integrity.
Frozen pancreata from 5-week-old (normoglycemic) and 14-week-old NOD mice were sectioned completely and stained with antibodies to pan-laminin to label BMs, CD45, a pan-leukocyte marker, and insulin. At least 10 healthy and 10 invaded pancreatic islets were analyzed per mouse in 5- and 14-week-old NOD mice (n = 3/group). Volocity 6.0.1 software was used to trace and quantify (fluorescence intensity) the pan-laminin staining surrounding individual pancreatic islets. The mean fluorescence intensity per micrometer of BM length was measured at sites showing continuous pan-laminin staining (Supplementary Fig. 1A and C). In pancreatic islets showing disrupted panlaminin staining at sites coincident with CD45+ cells and loss of insulin+ cells, the mean fluorescence value per micrometer was significantly reduced (P < 0.0007; Supplementary Fig. 1B and C). Mean fluorescence values of peri-islet BM staining for laminin were therefore used to define healthy versus invaded pancreatic islets.
For investigation of correlations between peri-islet BM integrity and disease progression, frozen pancreata from 5-, 14- (normoglycemic) and 21-week-old (diabetic) NOD mice were sectioned completely, and every 10th section was immunofluorescently stained with pan-laminin antibody to label BMs, CD45, a pan-leukocyte marker, and insulin. Healthy islets with intact BMs were recorded when no inflammatory cells were found around the islet, peri-insulitis was scored when inflammatory cells were found around the islet but the peri-islet BM was intact, and islets showing disrupted pan-laminin staining at sites coincident with CD45+ cells and loss of insulin+ cells were scored as invaded islets (300–400 islets were counted per mouse). Statistical analyses were performed using two-way ANOVA.
LCM.
Pancreata from 4- and 12-week-old NOD mice were snap frozen, 12–16 μm cryosections were cut and mounted on polyethylene naphthalate membrane-coated slides (PALM MicroLaser Systems, Bernried, Germany), immediately fixed, dehydrated, and hematoxylin stained using the HistoGene Frozen Section Staining Kit (Arcturus). Inflamed and healthy islets were excised using a LCM microscope (Leica AS LCM; Leica).
RNA extraction from LCM samples.
Total RNA was isolated using the Pico-Pure RNA isolation Kit (Applied Biosystems, Carlsbad, CA), and its integrity was assessed using the RNA 6000 Pico LabChipon in an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Amplification of total RNA and CLIP-CHIP microarray analysis.
Owing to the low amounts of RNA isolated (7–16 ng/μL), the cDNA library generated from RNA was amplified using the TransPlex Complete Whole Transcriptome Amplification kit (Sigma). The cDNA was purified by the GenElute PCR Clean-Up Kit (Sigma) and applied to the CLIP-CHIP microarray, which was performed as previously described (27).
Quantitative real-time PCR.
Quantitative real-time PCR was performed with 35 ng amplified cDNA per sample in an ABI PRISM 7300 cycler (Applied Biosystems, Darmstadt, Germany) using Brilliant SYBR Green QPCR Master Mix (Agilent Technologies). The results were interpreted using the comparative CT method, where the amount of the target, normalized to the endogenous hypoxanthine-guanine-phosphoribosyltransferase reference and relative to a calibrator, is given by 2−ΔΔCT. Expression analysis specific for mouse cathepsin C, S, and W was performed. The unpaired t test was applied for statistics, and P values < 0.05 were considered to be statistically significant.
In situ zymography.
Matrix metalloproteinase (MMP)-2 and -9 activity in pancreas sections was detected, as previously described (19).
RESULTS
ECM of the murine pancreatic islet.
A broad repertoire of antibodies specific for BM and IM proteins (Supplementary Table 1) were used to perform whole-mount immunofluorescence staining of the pancreas and confocal microscopy to generate a 3-dimensional view of the ECM associated with the healthy pancreatic islet (Fig. 1A and B; Supplementary Video 1). Optical sectioning throughout entire islets, defined by insulin+ staining, revealed a continuous BM encasing each islet that contained all major BM components (collagen IV, perlecan, nidogens, and laminins; Table 1) (23). Examples of this peri-islet BM staining are shown in Fig. 1A–D. A layer of IM occurred immediately subjacent to the peri-islet BM and was composed of the fibrillar collagens (types I and III; Fig. 1E), together with several other typical interstitial matrix molecules (Table 1). Electron microscopy confirmed that the pancreatic islet is ensheathed by a BM and an underlying IM composed of fibrillar collagens (Fig. 1F), which together comprise the peri-islet capsule. Electron microscopy also revealed that endocrine cells within the pancreatic islet are associated with one BM, that of intraislet blood vessels, as previously reported (22,28), whereas the endocrine cells at the islet border, which in the mouse tend to be the glucagon+ α-cells (Fig. 1A), are associated with the peri-islet BM (Fig. 1F).
FIG. 1.

A 3-dimensional projection of a whole mount of a healthy mouse pancreas and visualization of the peri-islet capsule by immunofluorescence staining and electron microscopy are shown. A: Insulin+ and glucagon+ pancreatic islets are encased in the peri-islet BM. B: An optical section of a 3-dimensional projection of a whole-mount pancreatic islet shows in a simultaneous view of XY, XZ, and YZ planes, illustrating the peri-islet BM and endothelial BMs. Differential localization of laminin (LM) α4 and α5 chains in the pancreatic islet is shown by immunofluorescence staining of thin sections (C) and whole-mount staining (D). C: Meca32 staining marks the endothelial layer of blood vessels (insets show images at higher magnification). E: Collagen type III staining visualizes the IM of the peri-islet capsule that underlies the peri-capsular BM, marked by pan-laminin (PLM) staining. F: Electron micrography illustrates two BMs: the peri-islet BM (arrow) that envelops the β-cells and the endothelial BM of blood vessels (arrowhead; inset 1). Subjacent to peri-islet BM (arrow), fibrillar collagens are evident (*; inset 2). Scale bars are 100 μm for A–E,50 μm for insets in C, and 1 μm in F. (A high-quality digital representation of this figure is available in the online issue.)
TABLE 1.
Summary of ECM components of the healthy and leukocyte-infiltrated peri-islet capsule in NOD mice
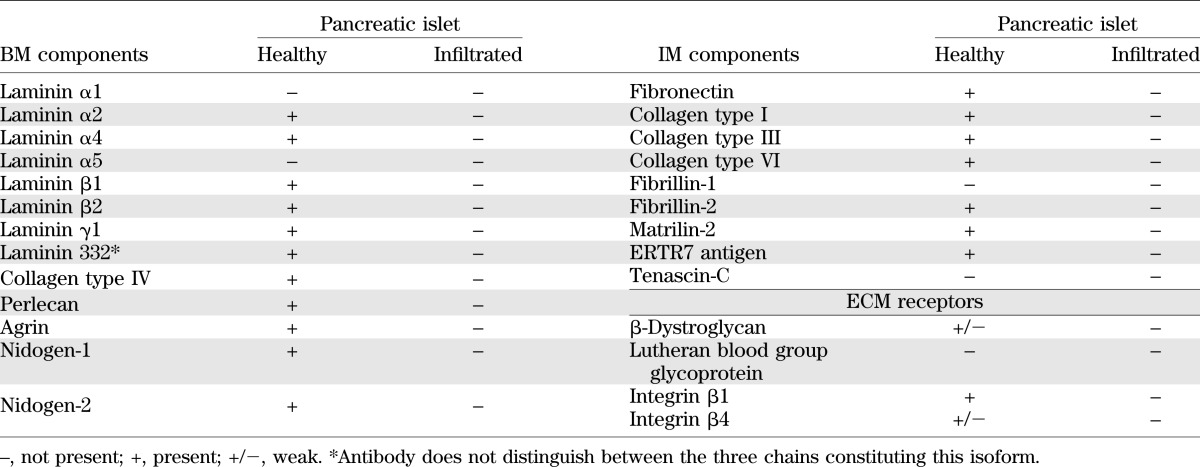
Analyses of the laminin family of BM proteins together with endothelial cell markers revealed that the peri-islet BM was biochemically distinct to the BMs surrounding blood vessels and contained laminin α2, α4, β1, β2, γ1, γ2, and γ3 chains (Fig. 1C and D) but not laminin α1, α3, and α5 chains (Fig. 1D). By contrast, blood vessel BMs within and surrounding islets contained laminin α4 and α5 chains (Fig. 1C and D) together with laminin γ1 and β1 chains. Laminin α5 expression was stronger in the arterioles where smooth muscle cells express this chain in addition to endothelial cells (Fig. 1D). The closeness of blood vessels to the peri-islet BM, surrounding the islets or entering them, resulted in the appearance of areas more intense BM staining (Fig. 1A, B, and D). Parallel analyses of the expression of the major ECM receptors revealed localization of Lutheran blood group glycoprotein and β-dystroglycan on endothelial cells and a weak immunosignal for β-dystroglycan on cells that were close to the peri-islet BM (Table 1). A summary of the BM and IM components of the peri-islet capsule and their receptors are listed in Table 1.
Loss of the peri-islet capsule in the inflamed NOD pancreas.
Optical sectioning of immunofluorescently stained whole-mount inflamed NOD pancreata suggested a striking loss of all BM and IM components of the peri-islet capsule specifically at sites of leukocyte infiltration into the pancreatic islets (Fig. 2A and B; Table 1; Supplementary Video 2) and loss of insulin staining within islets (Fig. 2B). However, the endothelial BM of blood vessels from the islet periphery and the acinar BMs remained intact (Fig. 2A and B).
FIG. 2.

Penetration of the peri-islet BM is a critical step in disease progression in NOD mice. Immunofluorescence staining of a whole mount (A) and thin sections (B; boxed areas show higher magnification) of leukocyte-invaded islets for pan-laminin (PLM) as a marker of the peri-islet BM, and the pan-leukocyte marker CD45 illustrates the correlation between the loss of the peri-islet BM and leukocyte infiltration into the pancreatic islet. The peri-islet BM remains detectable (arrows) at sites of leukocyte accumulation around the islet but not where leukocytes have invaded the islet (arrowhead). C: Quantification of peri-islet BM integrity was performed by measuring the mean fluorescence intensity per micrometer of the peri-islet BM (10 islets/mouse, n = 3/each group). The mean fluorescence intensity per micrometer of degraded peri-islet BMs is significantly lower than that of intact peri-islet BMs. ***P < 0.0007. D: Stereological analyses of pancreatic sections from 5- and 14-week-old normoglycemic and 21-week-old diabetic NOD mice (n = 3/each group) were used to quantify the number of islets with intact peri-islet BMs (healthy islets and islets in peri-insulitis stage) vs. invaded islets showing loss of peri-islet BM staining. W, weeks. Scale bars are 100 μm and 50 μm in insets (P < 0.0001). (A high-quality digital representation of this figure is available in the online issue.)
To quantify differences between the peri-islet BM of healthy versus invaded islets, image analysis was used to measure the fluorescence intensity of peri-islet pan-laminin staining at sites were CD45 staining did and did not occur, revealing a statistically significant reduction in fluorescence intensity at sites of leukocyte invasion and loss of insulin+ cells within the islets (Fig. 2C; Supplementary Fig. 1). To investigate whether a correlation exists between leukocyte penetration of the peri-islet BM and progression of diabetes, a time-course study was undertaken. The number of islets showing disruption of the BM was compared with the number of islets with an intact BM, as defined by fluorescence intensity of pan-laminin staining, in NOD mice aged 5 (presymptomatic stages), 14 (early onset), and >21 weeks (diabetic stage). Stereological analyses of these immunofluorescence data revealed that the number of islets showing reduced insulin staining and presence of invading CD45+ cells and, therefore, defined as intrainsulitis, increased with age and revealed an inverse correlation between disease progression and the integrity of the peri-islet BM (Fig. 2B and D). In 5-week-old NOD pancreata, most islets were intact, few showed peri-insulitis, and islets with disrupted BMs were not present. This ratio changed in 14-week-old NOD pancreata, and <50% of the islets were invaded by leukocytes and showed degraded BMs. In 21-week-old NOD pancreata, 80% of the islets showed loss of peri-islet BM and IM staining (Fig. 2D). Taken together, these data suggest a correlation between degradation of the peri-islet capsule and disease progression and that penetration of the peri-islet BM is a critical step in disease progression.
Mechanism of loss of the peri-islet BM and IM in the NOD pancreas.
Owing to the different biochemical nature of BM and IM proteins, the complete loss of the pericapsular ECM at sites of leukocyte penetration into the islet suggests the involvement of several different proteases or of proteases with broad substrate specificity. To investigate whether proteases were involved, analyses were performed with the murine CLIP-CHIP microarray (29). LCM was used to isolate healthy islets from 4-week NOD pancreata, and islets showing early stages of immune cell penetration were isolated from >12-week NOD pancreata; mRNA was purified, and gene expression patterns were compared (Table 2; Supplementary Fig. 2). Data reveal upregulation of caspase 7 and 8 in infiltrated islets, which are members of the aspartic-specific cysteine protease family associated with apoptotic events (30). This is in accordance with enhanced β-cell apoptosis due to autoimmune attack, thereby validating our sample preparation. In addition, several members of the MMP family, including MMP-2, MMP-7, and MMP-14, were upregulated at the mRNA level in infiltrated islets. However, this could not be confirmed at the protein level because in situ gelatin zymography for the detection of MMP-2, MMP-9, and partially also for MMP-14 (because it is an upstream activator of MMP-2) (31) revealed gelatinase activity in association with the insulin-producing β-cells in the healthy part of the islet rather than at sites of leukocyte entry (Supplementary Fig. 3). Elevated MMP-7 activity, which can be detected by casein gel zymography, could also not be confirmed in islets showing leukocyte infiltration (data not shown).
TABLE 2.
Summary of murine CLIP-CHIP microarray analyses
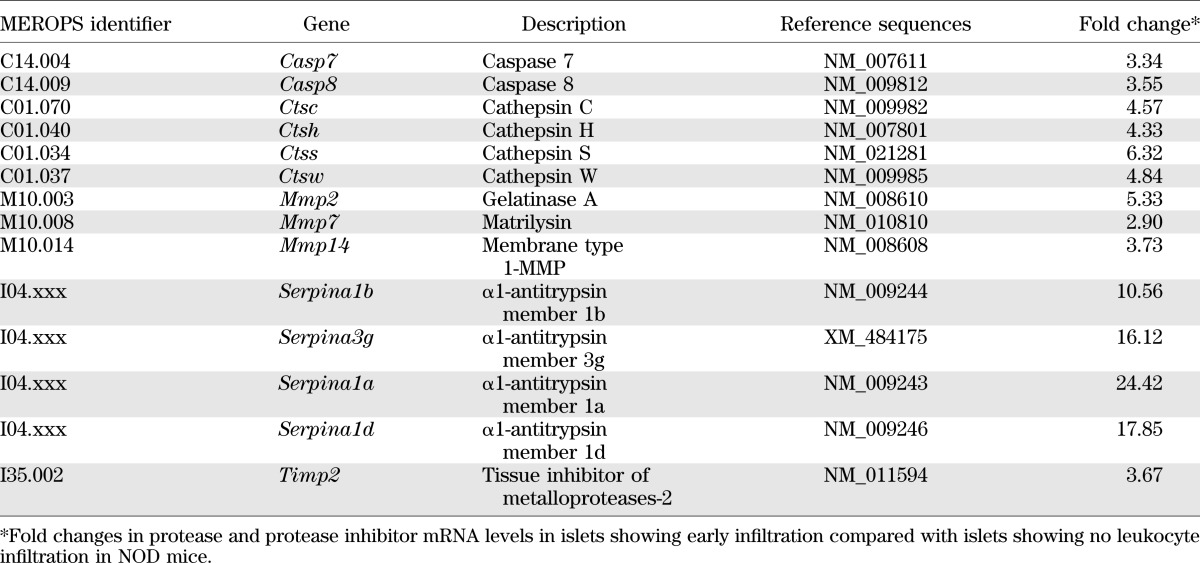
Several members of the cathepsin protease family, including cathepsin C, H, S, and W, showed a four- to sixfold increase in mRNA expression in infiltrated islets (Table 2), which was confirmed by quantitative real-time PCR (Supplementary Fig. 4). This was unusual, because cathepsins are best known as intracellular proteases; however, there is evidence that they can also act extracellularly (32). Consistent with this possibility was the upregulation of several members of α-1 antitrypsin family (Table 2), known to inhibit cysteine proteases as well as serine proteases (33). We therefore used immunofluorescence microscopy to investigate whether cathepsins C, S, and W colocalized with the islet-invading leukocytes. In situ localization of all three cathepsins correlated with sites of loss of peri-islet BM staining in NOD mice (Fig. 3A–C).
FIG. 3.

Validation of CLIP-CHIP data at the protein level by immunofluorescence in NOD samples. Immunofluorescence staining of NOD 21-week-old pancreas sections for cathepsin C (A), cathepsin S (B), and cathepsin W (C) shows localization at sites where peri-islet BM staining is lost (arrows), as shown here by laminin (LM) γ1 staining. The boxed areas are shown at higher magnification to the right. Scale bars are 100 μm. (A high-quality digital representation of this figure is available in the online issue.)
Macrophages and DCs are among the first cells recruited to pancreatic islets (34,35), and their localization was shown here to correlate with sites of loss of peri-islet BM (Fig. 4A). Immunofluorescence staining revealed some colocalization of F4/80+ macrophages with cathepsin C staining (Fig. 4B) and more extensive colocalization with cathepsin S and W (Fig. 4C and D), suggesting that a macrophage subpopulation may be one of the cellular sources that secrete these proteases at the site of leukocyte penetration of the peri-islet BM. A restricted colocalization of cathepsin S and W with some CD11c+ DCs was observed (data not shown).
FIG. 4.

Macrophages are a potential cellular source of cathepsins in leukocyte-invaded pancreatic islets in NOD mice. A: Immunofluorescence staining reveals F4/80+ macrophages close to the sites where peri-islet BM staining is lost (arrows), as shown here by pan-laminin (PLM) staining. The boxed area is shown at higher magnification to the right. Triple-staining for F4/80, as a marker for macrophages, the nuclei marker DAPI and cathepsin C (B), cathepsin S (C), or cathepsin W (D) show colocalization of all three cathepsins, with a subpopulation of macrophages. Scale bars are 100 μm. (A high-quality digital representation of this figure is available in the online issue.)
Peri-islet BM degradation in type 1 diabetic human pancreas.
To investigate whether leukocyte infiltration into the human pancreatic islets is also associated with loss of peri-islet ECM components, human type 1 diabetic samples were analyzed (Supplementary Tables 2 and 3). As in NOD samples, the healthy human pancreatic islet was surrounded by a capsule composed of a peri-islet BM and subjacent IM (Fig. 5A–C). Although most ECM proteins analyzed showed the same localization pattern as observed in NOD pancreata, only laminin α5 was found to mark the peri-islet BM, which was not the case in mouse samples, a result consistent with findings from previous studies (21,22). Sections from the pancreas head, body, and tail were analyzed by immunofluorescence staining. Data shown in Fig. 5D–H are from one sample (nPOD case 6052), a 12-year-old donor with type 1 diabetes who died 1 week after diagnosis, which still contained some insulin+ islets surrounded by leukocytes, according to the nPOD histological analysis (http://www.jdrfnpod.org/). Insulin+ islets occurred only in the pancreatic body and tail where, as in NOD samples, sites of leukocyte infiltration of the pancreatic islet were associated with loss of peri-islet BM staining (Fig. 5E and F; Supplementary Table 3); however, no changes were observed in the case of peri-insulitis alone. In the pancreatic head, only insulin− islets occurred, which were associated with little or no CD45+ cells, indicating that they represented islet remnants. As for the NOD mouse, triple-immunofluorescence staining in samples from humans with type 1 diabetes revealed that some of the CD45+ cells were also positive for cathepsin S and W staining at sites of peri-islet BM loss (Fig. 5I and J).
FIG. 5.

ECM of human healthy and type 1 diabetic pancreata. A: Immunofluorescence staining reveals the BM component laminin (LM) α5 surrounding the insulin and glucagon-containing islet. Glucagon+ cells are organized along the peri-islet BM, whereas the insulin+ β-cells fill the inner part of the islet. B: Double-staining for pan-laminin (PLM) and other BM components, such as perlecan, show some colocalization in the peri-islet BM (arrow) and in the endothelial BM (arrowhead). C: The peri-islet BM is further surrounded by a dense network of IM, represented here by collagen type III. (A–C, nPOD case 6024, healthy pancreas) D: Schematic representation of the areas analyzed. In a recently diagnosed individual with type 1 diabetes (nPOD case 6052), insulin+/CD45+ islets were detected only in the pancreatic tail (E and F) but not in the head (G) of the pancreas, where only insulin− (G) and glucagon+ (H) islets with intact BMs were present. The peri-islet BM is intact at sites where the leukocytes accumulate around the insulin+ islets (arrows in E) and is lost at sites of leukocyte invasion (arrowheads in E and F). H: Glucagon-producing α-cells become evenly distributed throughout the islet in type 1 diabetic CD45−/insulin− samples and are enclosed within an intact peri-islet BM, as shown by laminin α5 staining. I and J: Staining of samples from individuals with recently diagnosed human type 1 diabetes show loss of the peri-islet BM at the front of the CD45+ cell infiltration (arrows) and colocalization of immunoreactivity for cathepsin S (I) and for cathepsin W (J). The peri-islet BM is marked here by perlecan staining. The boxed areas are shown at higher magnifications to the right. Scale bars are 100 μm. (A high-quality digital representation of this figure is available in the online issue.)
Reconstitution of the peri-islet BM in long-term diabetic NOD mice and type 1 diabetic patients.
Surprisingly, in several human diabetic pancreas samples we observed many islets that were insulin−/glucagon+ but also lacked CD45+ leukocytes and were encased in an intact peri-islet BM (Fig. 5G and H). Such islets dominated the pancreata of donors who had been diagnosed with the disease for several weeks (nPOD cases 6062–6064; Supplementary Table 2) and type 1 diabetic medalists samples that were diagnosed with diabetes for more than 50 years (nPOD cases 6085 and 6086). These results suggest that the cells responsible for producing the peri-islet BM are not lost due to the inflammation and can reconstitute the peri-islet BM once the inflammation has subsided. We show here that this reconstituted peri-islet BM in human samples of type 1 diabetes contains laminin α5 (Fig. 5H), which has been shown to be a ligand for β-cells, interaction with which enhances insulin production (28). Hence, reconstitution of the peri-islet BM may have important ramifications to islet transplantation studies. We therefore used diabetic NOD mice (aged >21 weeks) supplemented with subcutaneous insulin pellets to investigate whether reconstitution of the peri-islet BM indeed occurs after inflammation has subsided and what is the potential cellular source or sources of these BM proteins. In both human and NOD samples, glucagon+ α-cell clusters were closely associated with the peri-islet BM, which were located mainly along the periphery of the islet (Fig. 5H, Fig. 6; Supplementary Video 3). During the course of inflammation in NOD mice, α-cells formed clusters that were encased by a BM of the same composition as the peri-islet BM and also surrounded by IM (Fig. 6A–C). This suggests that despite the massive infiltration of leukocytes, α-cell interaction with the BM can protect it from degradation. Whether this is due to direct secretion of ECM components by the α-cells or to their cross talk with other peri-islet cells that secrete ECM is not yet clear. Nevertheless, the data suggest that cellular interaction with the peri-islet BM is required for its integrity and barrier function.
FIG. 6.

Reconstitution of the peri-islet capsule after resolution of inflammation in NOD pancreata. A: Glucagon+ cell clusters are already detectable in heavily inflamed islets of nondiabetic mice, which are surrounded by a peri-islet BM shown here by immunofluorescence for laminin α2 (LMα2). The boxed area is shown at higher magnification to the right. B: In diabetic mice, glucagon+ clusters occur in close association with each other, and every cluster is covered by peri-islet BM and IM, shown here by perlecan, pan-laminin (PLM), and collagen type III staining. No or few leukocytes are present around such islets. The boxed areas are shown at higher magnification in the lower panel. C: A 3-dimensional projection of a whole-mount staining shows that glucagon+ cell clusters are individually ensheathed by a peri-islet BM, shown by perlecan staining. Scale bars are 100 μm in A–C and in B insets and are 50 μm in A insets. (A high-quality digital representation of this figure is available in the online issue.)
DISCUSSION
We show that the peri-capsular ECM is composed of a peri-islet BM embedded in an IM layer that interconnects with the surrounding blood vessels and exocrine tissue. The composition of the peri-islet BM is biochemically distinct to BMs of blood vessels surrounding or within the islet, with characteristic expression of laminin α4 in the mouse and of laminin α5 in human tissue (21,22). Quantitative analyses revealed significantly reduced mean fluorescence intensity of the peri-islet BM pan-laminin staining at sites of leukocyte invasion of islets and loss of insulin+ cells and that this correlates with disease progression, with a higher incidence of insulitis when more islets showed penetration of the peri-islet BM. This indicates that leukocyte penetration of the peri-islet BM is a critical step in disease progression and potentially represents a new therapeutic target.
The widespread disappearance of IM and BM components from the mouse and human islet capsule indicates that leukocyte infiltration into the pancreatic islet is fundamentally different from the mainly nonproteolytic mode of leukocyte extravasation from postcapillary venules (14). It also suggests the involvement of several proteases or of proteases with broad specificity. CLIP-CHIP microarray analyses support the latter possibility, with the identification of upregulation of several members of the cathepsin protease family and protease inhibitors in leukocyte-infiltrated islets. In vivo staining for cathepsin S, C, and W showed colocalization with some macrophages at the inflammatory front, suggesting their potential secretion from a macrophage subpopulation but not excluding other sources. This is consistent with the exclusive expression of cathepsins S and W by inflammatory cells (36,37) and broad cellular expression of cathepsin C (38), and may explain why only the BM and IM components of the peri-islet capsule are selectively lost and not BMs of peri-islet blood vessels or the acini of the exocrine pancreas, despite widespread leukocyte infiltration. Further, cathepsin S–null mice on a NOD background show significantly reduced incidence of diabetes (39). This has been attributed to cathepsin S effects on antigen presentation (40), but a role in penetration of the peri-islet BM cannot be excluded.
Although MMPs, in particular MMP-2 and MMP-14, were also upregulated at the transcriptional level, in situ zymography demonstrated the absence of correlation between gelatinase activity and sites of leukocyte infiltration. Rather, gelatinase activity was associated with healthy, insulin-producing portions of the islets. The upregulation of MMP-14 is consistent with its proposed role in T-cell extravasation into the pancreas via CD44 cleavage (41) but can be excluded as a candidate for degradation of the peri-islet capsule. The upregulation of several protease inhibitors, including tissue inhibitor of metalloproteinases-2 and the serpins (α1-antitrypsin members), broad specificity proteinase inhibitors, suggests a compensatory mechanism to counteract the destructive effect of proteases (33), the balance of which probably controls whether the peri-islet BM can be penetrated. The strong upregulation of the serpins shown here is also consistent with the reported anti-inflammatory effects in NOD mice (42) and its recent use in clinical trials for treatment of type 1 diabetes (http://clinicaltrials.gov/ct2/show/NCT01319331).
Precisely how the cathepsins function in leukocyte penetration of the peri-islet BM is not clear. The cathepsins are known for their terminal proteolytic activity in lysosomes but have been shown to be secreted in certain circumstances and can be active at neutral pH in an extracellular milieu (43). Human macrophages can also create a pericellular acidic microenvironment by producing vacuolar-type H+-ATPase components, thus creating an optimal micromilieu for cathepsin activity solely in their close vicinity (44). Cathepsins have also been reported to cleave ECM molecules; in particular, cathepsin S has been shown to cleave Matrigel, a mixture of BM components (45), as well as laminins γ2 and β1 (46) and collagen type IV (47), all of which are present in the peri-islet BM. Further, cathepsin S has been reported to cleave the native triple-helical region of fibrillar collagen type I (48), which is present in the peri-islet IM. Hence, the possibility exists for a direct involvement of cathepsins in degradation of the peri-islet ECM. However, an indirect involvement of cathepsins by initiating a proteolytic cascade resulting in activation of other proteases that degrade the ECM is also possible. In addition, cathepsins may be associated with degradation of endogenous inhibitors, resulting in increased activity of other proteases (49).
Interestingly, in samples from humans with long-term type 1 diabetes, we found insulin− islets surrounded by an intact peri-islet BM and underlying IM. By implanting insulin capsules into diabetic NOD mice, we were able to observe the same phenomena and also identify the early formation of α-cell, glucagon+ clusters that were surrounded by a BM of the same composition as the peri-islet BM. This suggests that the cells responsible for formation of the peri-islet BM are not lost due to the inflammation and can regenerate the BM once the inflammation has subsided. Given that transplantation experiments have highlighted the importance of the regenerative ability of the peri-islet capsule to long-term graft survival, this is a significant finding. The ECM surrounding the islet is destroyed during islet isolation for transplantation experiments, leading to integrin-mediated cell death; however, preservation of an ECM mantel significantly increases the survival rate of transplanted islets (50).
More recently, developmental studies in the mouse have shown that β-cell adhesion to the BM components laminins α4 or α5 is essential for growth and strongly promotes insulin secretion via integrin-mediated processes (28). Taken together this suggests that inclusion of such peri-islet BM-specific laminins or the integrin-binding domains thereof, or cells capable of regenerating the BM, would enhance islet transplantation success. Although our data highlight the α-cells as a potential source of peri-islet BM components because of their tight association with the peri-islet BM in the reconstituted islets, other peri-islet cells, such as myofibroblasts or Schwann cells, which can synthesize BM and IM components, may also contribute to peri-islet BM restoration and represent appropriate support cells for islet transplantation.
In conclusion, our data demonstrate that leukocyte penetration of the peri-islet BM is a critical step in progression of type 1 diabetes, the mechanism of which differs fundamentally from that involved in leukocyte extravasation from blood vessels. This suggests that the ECM milieu influences the mode used by immune cells to infiltrate into tissues and raises novel possibilities for tissue-specific immune-modulatory therapies. We identify cathepsins at the front of leukocyte penetration of the peri-islet BM in NOD mice and in type 1 diabetic samples from humans and, hence, potentially a novel therapeutic target specifically acting at the islet-penetration stage. Demonstration that the peri-islet BM and underlying IM can be reconstituted, once inflammation has subsided in the mouse and human, opens new avenues for the use of peri-islet BM components or BM-producing cells in islet transplantation strategies.
ACKNOWLEDGMENTS
The study received support from the European Foundation for the Study of Diabetes/Juvenile Diabetes Research Foundation (JDRF)/Novo Nordisk A/S (BD21070), JDRF (1-2005-903). This research was performed with the support of nPOD, a collaborative type 1 diabetes research project sponsored by the JDRF. Organ Procurement Organizations partnering with nPOD to provide research resources are listed at http://www.jdrfnpod.org/our-partners.php. No other potential conflicts of interest relevant to this article were reported.
É.K. performed all experiments and was involved in writing the manuscript. N.K. provided advice on immunology, contributed to discussion, and reviewed the manuscript. R.K. and C.M.O. helped in the microarray analyses, with the CLIP-CHIP analysis, and with statistical analyses of the data. J.W. performed the quantitative real-time PCR experiments. E.W. provided antibodies and advice on the cathepsins. D.H. and S.C. were involved in project development and contributed significantly to compilation of the manuscript. L.S. supervised the work done in the laboratory and participated in writing the manuscript. L.S. is the guarantor of this work, and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Hartmut Halfter (Department of Neurology, University of Münster, Münster, Germany) for use of the LCM; Friedemann Kiefer, Stefan Volkery, and Ruben Böhmer (Max Planck Institute for Molecular Medicine, Münster, Germany) for assistance in the use of the LSM510 microscope; Karin Loser (Department of Dermatology, University of Münster, Münster, Germany) for help with quantitative real-time PCR; Horst Robenek and Karin Schlattmann (Arteriosclerosis Research, University of Münster, Münster, Germany) for help in the use of the electron microscope; and Frank Arfuso (Institute of Physiological Chemistry and Pathobiochemistry, University of Münster, Münster, Germany) for critical reading of the manuscript. The authors thank Petra Blumberg and Sigmund Bundy (Institute of Physiological Chemistry and Pathobiochemistry, University of Münster, Münster, Germany) for their excellent technical assistance.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0432/-/DC1.
REFERENCES
- 1.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 2010;10:501–513 [DOI] [PubMed] [Google Scholar]
- 2.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 1980;29:1–13 [DOI] [PubMed] [Google Scholar]
- 3.Eisenbarth GS. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol 2004;552:306–310 [PubMed] [Google Scholar]
- 4.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010;464:1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alanentalo T, Hörnblad A, Mayans S, et al. Quantification and three-dimensional imaging of the insulitis-induced destruction of beta-cells in murine type 1 diabetes. Diabetes 2010;59:1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duarte N, Stenström M, Campino S, et al. Prevention of diabetes in nonobese diabetic mice mediated by CD1d-restricted nonclassical NKT cells. J Immunol 2004;173:3112–3118 [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D. Genetic control of diabetes progression. Immunity 1997;7:873–883 [DOI] [PubMed] [Google Scholar]
- 8.Yurchenco PD, Patton BL. Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des 2009;15:1277–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Timpl R, Brown JC. Supramolecular assembly of basement membranes. Bioessays 1996;18:123–132 [DOI] [PubMed] [Google Scholar]
- 10.Durbeej M. Laminins. Cell Tissue Res 2010;339:259–268 [DOI] [PubMed] [Google Scholar]
- 11.Li S, Liquari P, McKee KK, et al. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol 2005;169:179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones PL, Jones FS. Tenascin-C in development and disease: gene regulation and cell function. Matrix Biol 2000;19:581–596 [DOI] [PubMed] [Google Scholar]
- 13.Kresse H, Schönherr E. Proteoglycans of the extracellular matrix and growth control. J Cell Physiol 2001;189:266–274 [DOI] [PubMed] [Google Scholar]
- 14.Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol 2010;10:712–723 [DOI] [PubMed] [Google Scholar]
- 15.Wu C, Ivars F, Anderson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 2009;15:519–527 [DOI] [PubMed] [Google Scholar]
- 16.Lämmermann T, Bader BL, Monkley SJ, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 2008;453:51–55 [DOI] [PubMed] [Google Scholar]
- 17.Wolf K, Müller R, Borgmann S, Bröcker EB, Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood 2003;102:3262–3269 [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Voisin MB, Larbi KY, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med 2006;203:1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agrawal S, Anderson P, Durbeej M, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 2006;203:1007–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang FX, Naselli G, Harrison LC. Distinct distribution of laminin and its integrin receptors in the pancreas. J Histochem Cytochem 2002;50:1625–1632 [DOI] [PubMed] [Google Scholar]
- 21.Virtanen I, Banerjee M, Palgi J, et al. Blood vessels of human islets of Langerhans are surrounded by a double basement membrane. Diabetologia 2008;51:1181–1191 [DOI] [PubMed] [Google Scholar]
- 22.Otonkoski T, Banerjee M, Korsgren O, Thornell LE, Virtanen I. Unique basement membrane structure of human pancreatic islets: implications for beta-cell growth and differentiation. Diabetes Obes Metab 2008;10(Suppl. 4):119–127 [DOI] [PubMed] [Google Scholar]
- 23.Irving-Rodgers HF, Ziolkowski AF, Parish CR, et al. Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia 2008;51:1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavin EJ, Pinto GA, Zollner RL, Vassallo J. Immunohistochemical study of the pancreatic basement membrane in non obese diabetic mice (NOD) with spontaneous autoimmune insulitis. J Submicrosc Cytol Pathol 2003;35:25–27 [PubMed] [Google Scholar]
- 25.Sorokin LM, Conzelmann S, Ekblom P, Battaglia C, Aumailley M, Timpl R. Monoclonal antibodies against laminin A chain fragment E3 and their effects on binding to cells and proteoglycan and on kidney development. Exp Cell Res 1992;201:137–144 [DOI] [PubMed] [Google Scholar]
- 26.Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 2001;153:933–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kappelhoff R, Auf dem Keller U, Overall CM. Analysis of the degradome with the CLIP-CHIP microarray. Methods Mol Biol 2010;622:175–193 [DOI] [PubMed] [Google Scholar]
- 28.Nikolova G, Jabs N, Konstantinova I, et al. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev Cell 2006;10:397–405 [DOI] [PubMed] [Google Scholar]
- 29.Overall CM, Tam EM, Kappelhoff R, et al. Protease degradomics: mass spectrometry discovery of protease substrates and the CLIP-CHIP, a dedicated DNA microarray of all human proteases and inhibitors. Biol Chem 2004;385:493–504 [DOI] [PubMed] [Google Scholar]
- 30.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol 2003;15:725–731 [DOI] [PubMed] [Google Scholar]
- 31.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 1995;270:5331–5338 [DOI] [PubMed] [Google Scholar]
- 32.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 2006;6:764–775 [DOI] [PubMed] [Google Scholar]
- 33.Turk B, Turk D, Salvesen GS. Regulating cysteine protease activity: essential role of protease inhibitors as guardians and regulators. Curr Pharm Des 2002;8:1623–1637 [DOI] [PubMed] [Google Scholar]
- 34.Lund T, O’Reilly L, Hutchings P, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature 1990;345:727–729 [DOI] [PubMed] [Google Scholar]
- 35.Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes 2008;57:3025–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiederanders B, Brömme D, Kirschke H, von Figura K, Schmidt B, Peters C. Phylogenetic conservation of cysteine proteinases. Cloning and expression of a cDNA coding for human cathepsin S. J Biol Chem 1992;267:13708–13713 [PubMed] [Google Scholar]
- 37.Ondr JK, Pham CT. Characterization of murine cathepsin W and its role in cell-mediated cytotoxicity. J Biol Chem 2004;279:27525–27533 [DOI] [PubMed] [Google Scholar]
- 38.McGuire MJ, Lipsky PE, Thiele DL. Cloning and characterization of the cDNA encoding mouse dipeptidyl peptidase I (cathepsin C). Biochim Biophys Acta 1997;1351:267–273 [DOI] [PubMed] [Google Scholar]
- 39.Hsing LC, Kirk EA, McMillen TS, et al. Roles for cathepsins S, L, and B in insulitis and diabetes in the NOD mouse. J Autoimmun 2010;34:96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riese RJ, Wolf PR, Brömme D, et al. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity 1996;4:357–366 [DOI] [PubMed] [Google Scholar]
- 41.Savinov AY, Rozanov DV, Golubkov VS, Wong FS, Strongin AY. Inhibition of membrane type-1 matrix metalloproteinase by cancer drugs interferes with the homing of diabetogenic T cells into the pancreas. J Biol Chem 2005;280:27755–27758 [DOI] [PubMed] [Google Scholar]
- 42.Lu Y, Tang M, Wasserfall C, et al. Alpha1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum Gene Ther 2006;17:625–634 [DOI] [PubMed] [Google Scholar]
- 43.Jordans S, Jenko-Kokalj S, Kühl NM, et al. Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological pH and redox conditions. BMC Biochem 2009;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc Natl Acad Sci U S A 1995;92:3849–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gocheva V, Wang HW, Gadea BB, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 2010;24:241–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang B, Sun J, Kitamoto S, et al. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J Biol Chem 2006;281:6020–6029 [DOI] [PubMed] [Google Scholar]
- 47.Chang SH, Kanasaki K, Gocheva V, et al. VEGF-A induces angiogenesis by perturbing the cathepsin-cysteine protease inhibitor balance in venules, causing basement membrane degradation and mother vessel formation. Cancer Res 2009;69:4537–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdul-Hussien H, Soekhoe RG, Weber E, et al. Collagen degradation in the abdominal aneurysm: a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol 2007;170:809–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol 2011;21:228–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinkse GG, Bouwman WP, Jiawan-Lalai R, Terpstra OT, Bruijn JA, de Heer E. Integrin signaling via RGD peptides and anti-beta1 antibodies confers resistance to apoptosis in islets of Langerhans. Diabetes 2006;55:312–317 [DOI] [PubMed] [Google Scholar]