

Abstract
The glucagon-like peptides (GLP-1 and GLP-2) are processed from the proglucagon polypeptide and secreted in equimolar amounts but have opposite effects on chylomicron (CM) production, with GLP-1 significantly reducing and GLP-2 increasing postprandial chylomicronemia. In the current study, we evaluated the apparent paradoxical roles of GLP-1 and GLP-2 under physiological conditions in the Syrian golden hamster, a model with close similarity to humans in terms of lipoprotein metabolism. A short (30-min) intravenous infusion of GLP-2 resulted in a marked increase in postprandial apolipoprotein B48 (apoB48) and triglyceride (TG) levels in the TG-rich lipoprotein (TRL) fraction, whereas GLP-1 infusion decreased lipid absorption and levels of TRL-TG and apoB48. GLP-1 and GLP-2 coinfusion resulted in net increased lipid absorption and an increase in TRL-TG and apoB48. However, prolonged (120-min) coinfusion of GLP-1 and GLP-2 decreased postprandial lipemia. Blocking dipeptidyl peptidase-4 activity resulted in decreased postprandial lipemia. Interestingly, fructose-fed, insulin-resistant hamsters showed a more pronounced response, including possible hypersensitivity to GLP-2 or reduced sensitivity to GLP-1. In conclusion, under normal physiological conditions, the actions of GLP-2 predominate; however, when GLP-1 activity is sustained, the hypolipidemic action of GLP-1 predominates. Pharmacological inhibition of GLP-1 degradation tips the balance toward an inhibitory effect on intestinal production of atherogenic CM particles.
The prevalence of obesity and the metabolic syndrome has become a major health concern worldwide, and the health risks associated with dyslipidemia are well known (1). More recently, attention has been drawn to the role of postprandial hyperlipidemia as a risk factor for cardiovascular disease (2) and the metabolic syndrome (3). Apolipoprotein B48 (apoB48) is the primary structural component of the triglyceride (TG)-rich chylomicrons (CMs) secreted by the intestine, and excess apoB48 secretion has been associated with the formation of atherosclerotic plaques (2). Additionally, postprandial hyperlipidemia has been shown to be an important facet of the metabolic dyslipidemia associated with insulin resistance (3), principally due to intestinal apoB48 overproduction (4,5). Little is known of the hormonal and metabolic factors regulating intestinal lipid handling and CM production, although insulin action has been shown to inhibit the release of apoB48 (6). More recently we have demonstrated the ability of the gut-derived peptides, glucagon-like peptides 1 and 2 (GLP-1 and GLP-2), to regulate intestinally derived CM production (7,8). GLP-1 and GLP-2 are gut peptides secreted by ileal enteroendocrine L cells in response to dietary nutrients, particularly glucose and fatty acids (9). They are produced from the proglucagon gene and, as such, are cosecreted in equimolar quantities. Despite the similarities in their production and release, they have been shown to have opposing effects, particularly on intestinal lipid packaging (7,8).
GLP-1 promotes insulin secretion in a glucose-dependent manner and preserves pancreatic β-cell function (10). In addition to its proinsulinemic effects, GLP-1 has been shown to have extrapancreatic effects when administered systemically. GLP-1 slows gastric emptying and induces an anoretic effect (11). In addition, GLP-1 decreases postprandial intestinal CM production as shown by the reduction of apoB48 and TG in the TG-rich lipoprotein (TRL) fractions (8). Despite these beneficial effects, the presence of GLP-1 in the plasma is short lived as it is rapidly degraded by the enzyme dipeptidyl peptidase-4 (DPP-4). GLP-1 has a half-life of 1–2 min (12), with only 25% of newly secreted hormone leaving the gut intact (13,14). Due to the potential therapeutic nature of GLP-1, drug treatments have focused on preventing this rapid degradation. Inhibition of DPP-4 activity has been shown to lower fasting and postprandial glycemia and is a major drug target in patients with type 2 diabetes (15).
Conversely, GLP-2 activates the GLP-2 receptor (GLP-2R), a G protein–coupled receptor that is located on enteroendocrine cells (16), enteric neurons (17), subepithelial myofibroblasts (18), and neurons located in the central nervous system (19). GLP-2 activity has been shown to enhance hexose transport through upregulation of sodium-dependent glucose transporter-1 (20) and glucose transporter-2 (21) in the brush border membrane. GLP-2 has also recently been found to induce pronounced stimulatory effects on intestinal lipid uptake. Acute GLP-2 treatment enhances lipid uptake in healthy humans (22). An interesting observation considering that long-chain fatty acids stimulate the secretion of proglucagon-derived peptides from the gut (23), implying that there may be a feedback loop involving the proglucagon peptides and intestinal lipid uptake.
More recent studies from our laboratory have evaluated the specific effects of GLP-1 and GLP-2 on intestinal function, particularly focusing on fatty acid uptake and lipoprotein secretion. We have shown that acutely administered GLP-2 not only promoted triolein uptake but also increased secretion of TG-rich, apoB48-containing CM particles (7). This was found to be a CD36-dependent process (7). In contrast to these findings, we have shown that chronic DPP-4 inhibition reduced diet-induced dyslipidemia and postprandial production of TG-rich, apoB48-containing CM particles (8). These effects were likely due to the increased presence of GLP-1, as acute treatment with GLP-1 could induce similar effects (8). As such, we have shown that, although GLP-1 and GLP-2 are secreted from the same stimuli at the same time in equimolar amounts, they exert opposing effects on CM production with GLP-1 inhibiting postprandial lipemia and GLP-2 promoting lipid absorption and postprandial CM production. In the current study, we investigated the interactions between GLP-1 and GLP-2 under physiological conditions in the regulation of intestinal lipid absorption and lipoprotein metabolism.
RESEARCH DESIGN AND METHODS
Animals and diets.
Male Syrian golden hamsters (Mesocricetus auratus) weighing 110 g were purchased from Charles River (Montreal, QC, Canada) and maintained under controlled environmental conditions (temperature; humidity and airflow condition; 12-h light-dark cycle). After a 1-week acclimatization period, the hamsters were fed ad libitum with a standard chow diet or a fructose-enriched pelleted hamster diet containing 60% fructose and 20% casein (Dyets, Bethlehem, PA) for 10 days to induce insulin resistance (24). All animal protocols were approved by the Animal Ethics Committee of the Hospital for Sick Children, University of Toronto.
Determination of TRL apoB48 secretion in vivo.
Hamsters were anesthetized using isoflurane and were cannulated with a silastic catheter (VWR) inserted into the right jugular vein, exteriorized at the back of the neck, filled with heparinized saline (40 IU/mL), and sealed. Animals were allowed to recover for 24 h and then were fasted for 16 h. Baseline blood samples (400 µL) were collected into lithium heparin–coated tubes (BD, Franklin Lakes, NJ) from the jugular catheter. Hamsters were then given a 200-µL olive oil load via oral gavage, followed by a 30-min intravenous infusion of vehicle (VEH) (PBS), GLP-1 (20 pmol/kg body weight/min), GLP-2 (20 pmol/kg body weight/min), or both GLP-1 and GLP-2. The GLP-1 and GLP-2 dosing protocols were designed to ensure that GLP-1 and GLP-2 reached physiological concentrations after 30 min of infusion. Twenty minutes postgavage, Pluronic F-127 (20% in saline, 0.5 g/kg body weight) was injected intraperitoneally to inhibit lipoprotein catabolism and uptake. Blood (400 µL) was sampled at 30-min intervals until 120 min postgavage. The oral gavage, peptide administration, Pluronic injection, and blood collection were all performed on conscious animals.
Isolation of TRL.
To isolate the TRL fraction of the plasma, blood samples were first centrifuged for 15 min at 4°C at 5,000 rpm to separate the plasma layer. The plasma was supplemented with a cocktail of protease inhibitors (Roche Diagnostics, Mannheim, Germany), and 150 µL were overlaid with 4 mL potassium bromide solution (density 1.006 g/mL) in an ultracentrifuge. This was centrifuged for 70 min at 35,000 rpm at 10°C using an SW55Ti rotor (Beckman Coulter, Mississauga, ON, Canada). The top 300 µL was collected as the TRL fraction (Svedberg floatation rate >400).
Chemiluminescent immunoblotting.
The apoB48 immunoblotting was performed on TRL fractions by SDS-PAGE analysis as previously described (7).
Determination of triolein absorption in vivo.
Hamsters were catheterized as described above and received an oral gavage of 3 µCi [9,10–3H(N)]triolein mixed with 200 µL olive oil. Hamsters were then infused (intravenous) with GLP-1, GLP-2, or both as described above. Four hundred microliters of blood was sampled from the jugular catheter into heparinized tubes at 30, 60, 90, and 120 min. The activity of tritium in 20 µL of plasma was determined by scintillation counting in triplicate.
Plasma measurements.
Plasma and TRL triacylglycerol and cholesterol levels were determined by an enzymatic-based colorimetric assay (Randox, Crumlin, U.K.). Plasma GLP-1 levels were determined using GLP-1 (Active 7–36) ELISA (Alpco, Salem, NH). To prevent degradation of GLP-1 in blood samples, blood was treated with the DPP-4 inhibitor sitagliptin (20 µmol) immediately after collection.
Statistical analysis.
Results were expressed as mean ± SEM. The statistical analyses were performed using two-way ANOVA with the Bonferroni post-test as indicated in the text and figure legends. All statistical analyses were performed using GraphPad Prism.
RESULTS
Acute coinfusion of GLP-1 and GLP-2 results in GLP-2–dominant effects on intestinal lipoprotein production.
To assess the relative contributions of GLP-1 and GLP-2 on the regulation of intestinal lipid metabolism and CM production in vivo, we intravenously infused VEH, GLP-1, GLP-2, or both GLP-1 and GLP-2 in chow-fed hamsters for 30 min to achieve prolonged peak levels of these peptides in all groups. All experiments were performed postprandially after a fat load and injection of poloxamer to block CM clearance (thus, plasma and TRL lipid levels reflect the rate of entry of apoB48 TG and cholesterol secretion into plasma). Physiological levels were determined based on plasma GLP-1 levels observed after oral fat load. Vehicle-treated hamsters had baseline GLP-1 levels of 0.25 pmol that peaked 30 min after fat load to 1.2 pmol. GLP-1–infused (20 pmol/kg body weight/min) hamsters reached a peak of 3.6 pmol at 30 min, but GLP-1 levels then decreased to the 2.9–0.92-pmol range for the remainder of the infusion. This infusion rate was selected because it maintained plasma GLP-2 levels at those seen postprandially in control animals. Intestinal fatty-acid absorption was measured in hamsters challenged with an oral fat load that contained 3 µCi of [9,10-3H(N)]triolein. By 90 min postgavage, there was a twofold increase in the entry of 3H radioactivity into the plasma of GLP-1/GLP-2– and GLP-2–treated hamsters (Fig. 1B), whereas GLP-1 infusion caused a significant decrease in triolein absorption by 120 min postgavage.
FIG. 1.
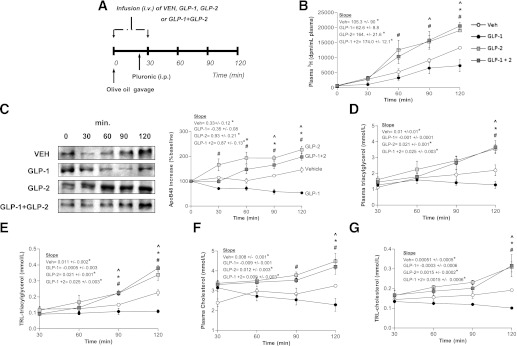
Short-term (30-min) coinfusion of GLP-1 and GLP-2 results in increased postprandial lipemia. A: Chow-fed hamsters received an oral fat load followed by a 30-min infusion (intravenous) of either VEH, GLP-1, GLP-2, or GLP-1 and GLP-2; poloxamer was given 20 min post–fat load (intraperitoneal) and blood was collected at 30, 60, 90, and 120 min after the fat load. B: Intestinal fatty-acid absorption was assessed using radiolabeled triolein; graph shows accumulation of radioactivity in plasma. C: A representative blot is shown of TRL-apoB48 along with a graph of apoB48 levels as quantified by densitometry. Plasma (D) and TRL-TG (E) levels were assessed as well as plasma (F) and TRL-cholesterol (G) levels. Each graph represents the mean ± SEM at each time point for the given parameter. Calculated slopes are shown within each graph (n = 4–5; *P < 0.05 vs. GLP-1, #P < 0.05 vs. GLP-2, ^P < 0.05 vs. GLP-1 + GLP-2).
As we have demonstrated previously (7), GLP-2 showed a stimulatory effect on CM production, whereas GLP-1 exerted a suppressive effect. The circulating TRL-apoB48 was increased twofold after the 30-min intravenous infusion of GLP-2 and decreased 1.5-fold 60 min post–fat load in GLP-1–treated hamsters. Coinfusion of GLP-1 and GLP-2 resulted in a 1.5-fold increase in TRL-apoB48 compared with VEH-treated hamsters (Fig. 1C). The amounts of TG and cholesterol in plasma and TRL fractions 120 min after the fat load were significantly increased (P < 0.05) in GLP-2– and GLP-1/GLP-2–treated hamsters. Conversely, TG and cholesterol were lower (P < 0.05) in GLP-1–treated hamsters versus control animals in both TRL and whole plasma. This was likely due to a significant lowering of the entry of apoB48 TG and cholesterol secretion into plasma, as CM clearance was blocked with poloxamer (Fig. 1D–G).
Prolonged coadministration of GLP-1 and GLP-2 results in GLP-1–dominant effects on intestinal lipoprotein production.
Since it is known that GLP-1 is more rapidly degraded than GLP-2, we evaluated whether the above observations were related to the more rapid degradation of circulating GLP-1. To overcome this confounding factor, we performed constant infusion of GLP-1, GLP-2, or GLP-1 and GLP-2 throughout the entire 120-min procedure. Infusion of GLP-2 resulted in a 1.5-fold increase in apoB48 levels in the TRL at 90 and 120 min after fat load (compared with VEH-treated animals). In contrast, GLP-1–treated animals exhibited a 1.5-fold decrease versus VEH-treated animals. Coinfusion of GLP-1/GLP-2 for 120 min had a drastically different effect on intestinal CM production compared with 30 min coinfusion. CM production was significantly decreased compared with VEH-treated animals after 90 and 120 min (P < 0.05) coinfusion of GLP-1/GLP-2, as shown by the decrease in TRL-apoB48 (Fig. 2B). In agreement with the TRL-apoB48 data, TRL-TG and TRL-cholesterol were higher in GLP-2–treated hamsters at 90 and 120 min postgavage (P < 0.05); conversely, the rate of entry of TRL-TG levels into plasma was decreased in GLP-1–infused hamsters (P < 0.05). TRL-TG and TRL-cholesterol accumulation declined when both peptides were infused at the same time compared with VEH-treated hamsters (Fig. 2D and E). Plasma TG accumulation was modestly but not significantly lower with GLP-1 infusion and GLP-1/GLP-2 coinfusion when compared with VEH-treated animals (Fig. 2C). This suggested that, when circulating GLP-1 levels are maintained, GLP-1 effects on intestinal CM and lipid production predominate.
FIG. 2.
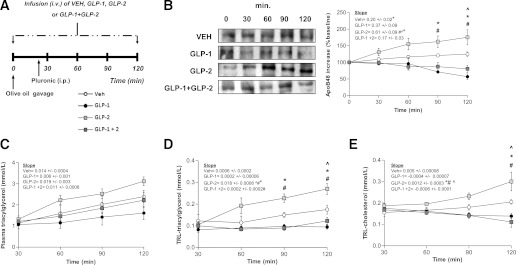
Long-term (120-min) coinfusion of GLP-1 and GLP-2 results in decreased postprandial lipemia. A: Chow-fed hamsters received an oral fat load followed by a 120-min infusion (intravenous) of either VEH, GLP-1, GLP-2, or GLP-1 and GLP-2; poloxamer was given 20 min post–fat load (intraperitoneal). Plasma was collected at 30, 60, 90, and 120 min post–fat load to assess TRL-apoB48. B: A representative blot of apoB48 is shown along with a graph of apoB48 levels quantified by densitometry. Plasma (C), TRL-TG (D), and TRL-cholesterol (E) levels were also quantified. Each graph represents the mean ± SEM at each time point for the given parameter. Calculated slopes are shown within each graph (n = 4–5; *P < 0.05 vs. GLP-1, #P < 0.05 vs. GLP-2, ^P < 0.05 vs. GLP-1 + GLP-2).
Sitagliptin treatment enhances the GLP-1 effects after 30 min of intravenous coinfusion.
Since it is known that the activity of DPP-4 is primarily responsible for the rapid degradation of GLP-1, using the drug sitagliptin we assessed how inhibition of DPP-4 activity affects the interaction between GLP-1 and GLP-2 and their respective effects on intestinal CM production. TRL-apoB48, TG, and cholesterol were significantly increased at 90 and 120 min postgavage in GLP-2–treated hamsters. In contrast, GLP-1 reduced these parameters at the same time points postgavage. Interestingly, after sitagliptin treatment, coinfusion of GLP-1 and GLP-2 for 30 min showed decreases in intestinal CM production similar to those seen with GLP-1 alone (Fig. 3B–F). Our data support the notion that increased GLP-1 activity achieved via DPP-4 inhibition is likely to be responsible for the reduced circulating TRL-TG, TRL-cholesterol, and intestinal CM production observed after sitagliptin administration. Moreover, the inhibition of DPP-4 activity may contribute to enhanced GLP-1 action, which is consistent with the lowered postprandial circulating levels of TG, cholesterol-rich TRL-apoB48, and CM-apoB48, as shown in GLP-1 + GLP-2–treated hamsters compared with control.
FIG. 3.
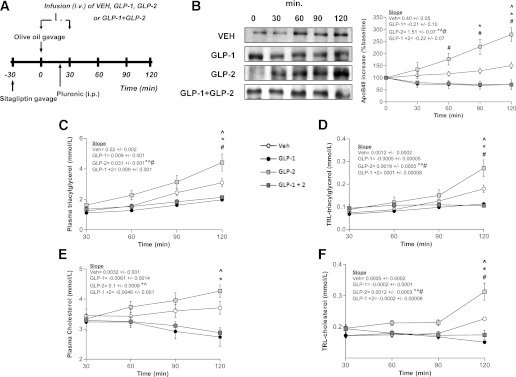
Inhibition of DPP-4 augments GLP-1 action, resulting in decreased postprandial lipemia in GLP-1/GLP-2–coinfused hamsters. A: Chow-fed hamsters received treatment with the DPP-4 inhibitor sitagliptin (intraperitoneal) 30 min prior to experiments, and then received an oral fat load followed by a 30-min intravenous infusion of either VEH, GLP-1, GLP-2, or GLP-1 and GLP-2; poloxamer was administered 20 min post–fat load (intraperitoneal). Blood was collected at 30, 60, 90, and 120 min to assess lipemia. B: A representative blot of TRL-apoB48 is shown along with a graph showing quantification of apoB48. Plasma (C) and TRL-TG (D) levels and plasma (E) and TRL-cholesterol (F) levels were quantified and represented in graphs (n = 4–5). Each graph represents the mean ± SEM at each time point for the given parameter. Calculated slopes are shown within each graph (n = 4–5; *P < 0.05 vs. GLP-1, #P < 0.05 vs. GLP-2, ^P < 0.05 vs. GLP-1 + GLP-2).
GLP-1 and GLP-2 modulation of intestinal TRL-TG excursion during a 6-h fat feeding period.
As has been shown by Yoder et al. (25), although GLP-1 and GLP-2 are primarily secreted from L cells in the ileum, the secretion of endogenous GLP-1 resulting from fat load occurs within 30 min of lipid ingestion and reaches its peak by 1 h (with the exception of very high doses of around 3–4 kcal, which peak at 2 h post–lipid load). Additionally, they found that lymph flow and lymphatic TG levels for lower levels of lipid dosing reach a peak within 1 h, decrease by 2 h, and at 3 h start to stabilize in accordance with the release of endogenous GLP-1. This indicates that the 2-h time frame used in the current study is appropriate to mimic the early stages of absorption seen with oral fat load. Nevertheless, we also performed experiments using an extended (6-h) fat absorption protocol to evaluate how GLPs can affect postprandial lipemia in a more physiologically relevant timeline. After a 30-min infusion of GLP-1, GLP-2, or both, hamsters were given a fat load and TRL-TG accumulation was monitored at 2, 4, and 6 h. Post–fat load TRL-TG excursions (Supplementary Fig. 1A) showed strikingly similar trends from 0 to 6 h when compared with the previous 2-h data. GLP-2 caused a significant increase in TRL-TG accumulation over the 6-h fat feeding period, whereas GLP-1 caused a decreasing trend; however, this did not reach statistical significance. The combination of both GLPs led to an effect similar to GLP-2, as previously observed over 2 h of fat feeding. We suspected that the lack of a prolonged GLP-1 effect was due to peptide degradation as we use native GLP-1 in these studies. To confirm whether GLP-1 degradation was a factor, we repeated the experiment in animals treated with sitagliptin, a DPP-4 inhibitor. As can be observed in Supplementary Fig. 1B, the stabilization of GLP-1 activity with sitagliptin led to a more pronounced inhibition of TRL-TG accumulation in plasma. These results further support the notion that when GLP-1 degradation is blocked, GLP-1’s effect predominates and leads to significant inhibition of CM production even up to 6 h post–fat load.
GLP-1 and GLP-2 coinfusion in a hamster model of diet-induced insulin resistance.
We next examined the effects of GLP-1 and GLP-2 coinfusion in an animal model of insulin resistance and postprandial dyslipidemia, the fructose-fed (FF) hamster (24). As we have shown previously, fructose feeding resulted in increased TRL-TG and apoB48 production compared with chow-fed controls starting as early as 30 min post–fat load (P < 0.05) (data not shown). TRL-apoB48 and TG levels were elevated in FF hamsters after 30 min intravenous infusion of VEH and GLP-2 and coinfusion of GLP-1/GLP-2. All three treatments showed similar increases in these parameters (Fig. 4B–D). Interestingly, GLP-1 alone was still able to significantly decrease the above parameters compared with VEH-treated hamsters (Fig. 4B–D). Total plasma and TRL-cholesterol levels were the same in all groups of treated animals during the course of the experiment (Fig. 4E and F). Similar to chow-fed hamsters, coinfusion of GLP-1/GLP-2 in FF hamsters resulted in an increase in apoB48 and TG levels (both TRL and plasma); however, unlike in chow-fed hamsters, this effect is seen as early as 30 min following treatment in FF hamsters, indicating that this model induces a more pronounced response with coinfusion and possible hypersensitivity to GLP-2 stimulation or reduced sensitivity to GLP-1 stimulation. This observation is based on the decline in apoB48 levels with GLP-1 infusion in chow-fed hamsters (Fig. 1) and the absence of this decline during GLP-1 infusion in FF hamsters (Fig. 4).
FIG. 4.
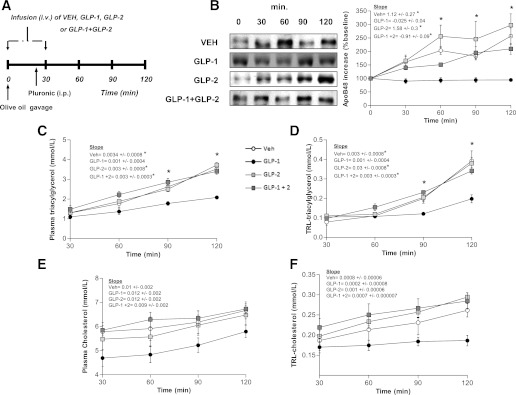
Effects of GLP-1 and GLP-2 coinfusion on postprandial lipemia in FF insulin-resistant hamsters. A: Hamsters were fed for 2 weeks with a high-fructose diet to induce insulin resistance and then received an oral fat load followed by a 30-min infusion (intravenous) of either VEH, GLP-1, GLP-2, or GLP-1 and GLP-2; poloxamer was given 20 min post–fat load (intraperitoneal). Plasma was collected at 30, 60, 90, and 120 min to assess lipemia. B: A representative blot of TRL-apoB48 is shown along with a graph of TRL-apoB48 levels as determined by densitometry. Plasma (C) and TRL-TG (D) levels and plasma (E) and TRL-cholesterol (F) levels were quantified and shown in the graphs. Each graph represents the mean ± SEM at each time point for the given parameter. Calculated slopes are shown within each graph (n = 4–5; *P < 0.05 vs. GLP-1, #P < 0.05 vs. GLP-2, ^P < 0.05 vs. GLP-1 + GLP-2).
GLP-1 effect increases in response to oral glucose load in chow-fed hamsters.
GLP-1 augments insulin secretion after oral intake of glucose (26) via glucose-sensing mechanisms that increase the GLP-1 release from gut endocrine L cells (27). To test the oral glucose response in GLP-1/GLP-2 coinfusion on intestinal lipid metabolism and CM production, chow-fed hamsters received glucose (1 mg/kg) plus 200 µL olive oil by oral gavage, followed by a 30-min intravenous infusion of VEH, GLP-1, GLP-2, or both GLP-1/GLP-2. The rate of entry of TRL-apoB48, TRL-TG, and TRL-cholesterol into plasma significantly decreased by 90 and 120 min post–fat load in GLP-1–treated hamsters (P < 0.05). Moreover, although the GLP-2 infusion did cause increases in these parameters, they were not as pronounced as those seen without glucose pretreatment (data not shown). Coinfusion of both peptides showed similar TG and apoB48 levels as those achieved in VEH-treated hamsters (Fig. 5B–D). GLP-1 infusion lowered total plasma and TRL-cholesterol by 120 min postgavage, and a slight increase in total cholesterol by 120 min postgavage was observed in GLP-2–treated hamsters (Fig. 5E and F). These data suggest that the GLP-1 effect is potentiated with oral glucose gavage enhancing the GLP-1–mediated inhibition of CM release.
FIG. 5.

Glucose attenuates stimulation of postprandial lipemia by GLP-1 and GLP-2 coinfusion. A: Chow-fed hamsters received an oral fat load (200 µL) mixed with glucose (1 mg/kg) followed by an infusion (intravenous, 30 min) of either VEH, GLP-1, GLP-2, or GLP-1 and GLP-2; poloxamer was given 20 min post–fat load (intraperitoneal). Plasma was collected to assess lipemia. B: A representative blot of TRL-apoB48 is shown along with a graph showing TRL-apoB48 levels as determined by densitometry. Plasma (C) and TRL-TG (D) levels and plasma (E) and TRL-cholesterol (F) levels are shown in graphs. Each graph represents the mean ± SEM at each time point for the given parameter. Calculated slopes are shown within each graph (n = 4–5; *P < 0.05 vs. GLP-1, #P < 0.05 vs. GLP-2).
DISCUSSION
Previous findings from our group and others indicate that two gut peptides that are cosecreted in equimolar amounts paradoxically have completely opposite acute effects on intestinal apoB48 secretion. GLP-2 increases both apoB48 particle number and size (7), whereas GLP-1 appears to completely blunt apoB48 particle secretion (8). In the current study, we confirmed our previously published results but also observed that the net effect of GLP-1/GLP-2 coinfusion is enhanced postprandial TG secretion; although this enhancement is dampened during an extended coinfusion. Moreover, the inhibitory effect of GLP-1 on CM secretion becomes more evident when DPP-4 is inhibited or in the presence of glycemia but is less apparent in insulin-resistant FF hamsters.
Given that GLP-1 and GLP-2 are secreted in equimolar amounts, it may be expected that their net physiological effect in vivo would be no change in postprandial lipemia. It is important to consider that the GLP-2R is not expressed by the absorptive enterocytes (28), meaning that GLP-2’s hyperlipidemic action proceeds indirectly; hence, GLP-2 stimulates a GLP-2R–positive cell to secrete factors that stimulate the absorptive enterocyte, providing an opportunity for amplification of the GLP-2 signal in the intestine. In contrast, such biological amplification does not occur when GLP-1 may directly antagonize its receptor on the absorptive enterocyte. The diminution of the hyperlipidemic effect with the inclusion of glucose in the oral fat load suggests that glucose-dependent insulinotropism, at least in hamsters, is one pathway that mediates GLP-1’s hypolipidemic function. Insulin’s involvement may also explain the delayed peak postprandial TG concentration observed with glucose ingestion in humans during a fat tolerance test where ostensibly both GLP-1 and GLP-2 were secreted (29). This would have pathophysiological implications in states of defective insulin secretion or compromised insulin sensitivity, where one aspect of GLP-1 influence is unavailable and would thus allow the GLP-2 effect to outweigh. In streptozotosin-treated rats that are hyperphagic and oversecrete GLPs but unable to produce insulin, the elevated plasma apoB48 levels (30) could be attributed to a dominance of GLP-2 action. In B6D2F1 mice fed a high-fat diet for 3 weeks to induce insulin resistance, there is greater transport capacity of linoleic acid, along with elevated CD36 expression and higher intestinal mitotic index (31). This is consistent with a preponderance of GLP-2 action. The potentiation of GLP-1’s hypolipidemic action with the addition of glucose to the oral gavage (Fig. 5) suggests that the incretin effect of GLP-1 contributes to its inhibition of CM secretion in insulin-sensitive states. In diseases with dampened GLP secretion, such as type 2 diabetes (32,33), decreased insulin sensitivity implies that the reduced GLP-1 levels would have an especially compromising acute hypolipidemic effect. In addition to preventing GLP-1 from exerting insulin-mediated reductions in CM production, insulin resistance might also prevent GLP-1 from having potential stimulatory effects on CM clearance. Exenatide, a GLP-1R agonist, is thought to potentially promote CM clearance by reducing postprandial levels of apoCIII (34), which inhibits lipoprotein lipase (35) and hinders hepatic TRL clearance (36). However, insulin resistance is associated with the impaired uptake of LDLR-related protein 1 ligands, thus opposing an important clearance mechanism for CM remnants (37) and resulting in a lipidemic profile more characteristic of GLP-2 dominance. The greater magnitude of GLP-2–stimulated CM production in insulin-resistant states may possibly explain the exacerbated postprandial apoB48 secretion in type 2 diabetic individuals (38).
A major contribution of DPP-4–mediated GLP degradation to the net physiological effect on postprandial TRL production is consistent with the conclusions that can be inferred from the sitagliptin-treated FF hamsters and mice. When neither the hamsters nor mice were injected with an exogenous GLP-1R agonist, sitagliptin administration alone attenuated postprandial CM secretion (8). Although both GLP-1 and GLP-2 are substrates for DPP-4 with similar Km values, DPP-4’s kcat for GLP-1 is 7.1 s−1 as opposed to 0.87 s−1 for GLP-2; thus, the former is degraded nine times faster (39). Extending the bioactivity of endogenous GLP-2 with valine-pyrrolidide did not affect intestinal morphology in rats (40); thus GLP-2 functions likely intensify minimally with protection from DPP-4–mediated degradation. The ability of sustained GLP-1 action to limit postprandial lipemia is also evident in this current study, given that a 120-min infusion of both GLPs has relatively tempered CM secretion compared with the 30-min infusion (Fig. 2). Therefore, in the physiological setting, the hypolipidemic effect of GLP-1 is limited largely by its susceptibility to DPP-4, and this presents implications for therapeutic strategies to address aberrant postprandial lipemia. Furthermore, reduced CM secretion during prolonged coinfusion may be due to decreased gastric emptying and impaired intestinal motility. These two phenomena are associated with GLP-1 treatment and limit the availability of lipid for uptake (41), as reflected in the reduced lipid absorption seen with GLP-1 infusion. Once the short 30-min coinfusion ended, intestinal motility was likely restored shortly after. However, during a prolonged coinfusion, GLP-1 may have maintained its effects, thus limiting the amount of lipid available for GLP-2–stimulated uptake.
Despite the completely opposite effects on dietary lipid transport, the biological functions of GLP-1 and GLP-2 may in fact be complimentary. One example of this potential complimentary action would be the ileal break (42). The ileal brake is a primary function ascribed to GLP-1 (43) and is thought to be the driving force behind GLP-1–induced satiety (44). This phenomenon is evident in the increased GLP-1 levels observed in high fat–fed, MGAT2-deficient mice, which have delayed kinetics of dietary fatty-acid absorption (45). This effect is also present in rats fed a fat-enriched diet and the intestine-specific microsomal triglyceride transfer protein (MTP) inhibitor (46). Although the bulk of dietary fatty acid is absorbed by passive diffusion across the brush border membrane, by the time dietary lipids have reached the distal intestine the concentration of lipids in the lumen of the proximal intestine would be considerably lower. In this case, increased absorption could be facilitated by the GLP-2–induced increase in expression of CD36 at the apical membrane to allow complete and facilitated absorption of dietary lipids (7). The stimulatory effect of GLP-2 on lipid transport may also serve to coordinate with GLP-1 in mediating satiety. The GLP-2–increased presence of CD36 at the apical membrane may be instrumental in transporting oleic acid as a substrate for the synthesis of oleoylethanolamide, a satiety factor (47).
In conclusion, the relative contributions of GLP-1 and GLP-2 to the regulation of intestinal CM secretion depend on the duration of their presence, their susceptibility to DPP-4–mediated degradation, and glycemia. These modulators have implications for postprandial lipemia observed in physiological and pathophysiological circumstances when GLP-1 and GLP-2 are cosecreted in equimolar amounts. However, these findings also propose that tipping the balance in favor of GLP-1, either by GLP-1R agonist-based therapies or DPP-4 inhibitors, is a viable approach to help correct dyslipidemia in insulin-resistant/diabetic states.
ACKNOWLEDGMENTS
This work was supported by an operating grant from the Canadian Institutes of Health Research, to K.A.
No potential conflicts of interest relevant to this article were reported.
G.J.H. and C.B. researched data and wrote the manuscript. J.H. researched data and reviewed and edited the manuscript. S.F. reviewed and edited the manuscript and contributed to discussion. K.A. designed the study and reviewed and edited the manuscript. K.A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in poster form at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0202/-/DC1.
See accompanying commentary, p. 336.
REFERENCES
- 1.Roth J, Qiang X, Marbán SL, Redelt H, Lowell BC. The obesity pandemic: where have we been and where are we going? Obes Res 2004;12(Suppl. 2):88S–101S [DOI] [PubMed] [Google Scholar]
- 2.Nakano T, Nakajima K, Niimi M, et al. Detection of apolipoproteins B-48 and B-100 carrying particles in lipoprotein fractions extracted from human aortic atherosclerotic plaques in sudden cardiac death cases. Clin Chim Acta 2008;390:38–43 [DOI] [PubMed] [Google Scholar]
- 3.Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 2003;46:733–749 [DOI] [PubMed] [Google Scholar]
- 4.Haidari M, Leung N, Mahbub F, et al. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J Biol Chem 2002;277:31646–31655 [DOI] [PubMed] [Google Scholar]
- 5.Zoltowska M, Ziv E, Delvin E, et al. Cellular aspects of intestinal lipoprotein assembly in Psammomys obesus: a model of insulin resistance and type 2 diabetes. Diabetes 2003;52:2539–2545 [DOI] [PubMed] [Google Scholar]
- 6.Federico LM, Naples M, Taylor D, Adeli K. Intestinal insulin resistance and aberrant production of apolipoprotein B48 lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia: evidence for activation of protein tyrosine phosphatase-1B, extracellular signal-related kinase, and sterol regulatory element-binding protein-1c in the fructose-fed hamster intestine. Diabetes 2006;55:1316–1326 [DOI] [PubMed] [Google Scholar]
- 7.Hsieh J, Longuet C, Maida A, et al. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology 2009;137:997–1005, 1005.e1–4 [DOI] [PubMed]
- 8.Hsieh J, Longuet C, Baker CL, et al. The glucagon-like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion in hamsters and mice. Diabetologia 2010;53:552–561 [DOI] [PubMed] [Google Scholar]
- 9.Iakoubov R, Izzo A, Yeung A, Whiteside CI, Brubaker PL. Protein kinase Czeta is required for oleic acid-induced secretion of glucagon-like peptide-1 by intestinal endocrine L cells. Endocrinology 2007;148:1089–1098 [DOI] [PubMed] [Google Scholar]
- 10.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007;132:2131–2157 [DOI] [PubMed] [Google Scholar]
- 11.Meier JJ, Nauck MA. Incretins and the development of type 2 diabetes. Curr Diab Rep 2006;6:194–201 [DOI] [PubMed] [Google Scholar]
- 12.Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ. Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. Am J Physiol 1996;271:E458–E464 [DOI] [PubMed] [Google Scholar]
- 13.Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999;140:5356–5363 [DOI] [PubMed] [Google Scholar]
- 14.Hansen L, Hartmann B, Bisgaard T, Mineo H, Jørgensen PN, Holst JJ. Somatostatin restrains the secretion of glucagon-like peptide-1 and -2 from isolated perfused porcine ileum. Am J Physiol Endocrinol Metab 2000;278:E1010–E1018 [DOI] [PubMed] [Google Scholar]
- 15.Matikainen N, Mänttäri S, Schweizer A, et al. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 2006;49:2049–2057 [DOI] [PubMed] [Google Scholar]
- 16.Yusta B, Huang L, Munroe D, et al. Enteroendocrine localization of GLP-2 receptor expression in humans and rodents. Gastroenterology 2000;119:744–755 [DOI] [PubMed] [Google Scholar]
- 17.Guan X, Karpen HE, Stephens J, et al. GLP-2 receptor localizes to enteric neurons and endocrine cells expressing vasoactive peptides and mediates increased blood flow. Gastroenterology 2006;130:150–164 [DOI] [PubMed] [Google Scholar]
- 18.Ørskov C, Hartmann B, Poulsen SS, Thulesen J, Hare KJ, Holst JJ. GLP-2 stimulates colonic growth via KGF, released by subepithelial myofibroblasts with GLP-2 receptors. Regul Pept 2005;124:105–112 [DOI] [PubMed] [Google Scholar]
- 19.Tang-Christensen M, Larsen PJ, Thulesen J, Rømer J, Vrang N. The proglucagon-derived peptide, glucagon-like peptide-2, is a neurotransmitter involved in the regulation of food intake. Nat Med 2000;6:802–807 [DOI] [PubMed] [Google Scholar]
- 20.Cheeseman CI. Upregulation of SGLT-1 transport activity in rat jejunum induced by GLP-2 infusion in vivo. Am J Physiol 1997;273:R1965–R1971 [DOI] [PubMed] [Google Scholar]
- 21.Au A, Gupta A, Schembri P, Cheeseman CI. Rapid insertion of GLUT2 into the rat jejunal brush-border membrane promoted by glucagon-like peptide 2. Biochem J 2002;367:247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meier JJ, Nauck MA, Pott A, et al. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology 2006;130:44–54 [DOI] [PubMed] [Google Scholar]
- 23.Rocca AS, LaGreca J, Kalitsky J, Brubaker PL. Monounsaturated fatty acid diets improve glycemic tolerance through increased secretion of glucagon-like peptide-1. Endocrinology 2001;142:1148–1155 [DOI] [PubMed] [Google Scholar]
- 24.Taghibiglou C, Carpentier A, Van Iderstine SC, et al. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem 2000;275:8416–8425 [DOI] [PubMed] [Google Scholar]
- 25.Yoder SM, Yang Q, Kindel TL, Tso P. Stimulation of incretin secretion by dietary lipid: is it dose dependent? Am J Physiol Gastrointest Liver Physiol 2009;297:G299–G305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mcintyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet 1964;2:20–21 [DOI] [PubMed] [Google Scholar]
- 27.Jang HJ, Kokrashvili Z, Theodorakis MJ, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci USA 2007;104:15069–15074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubé PE, Brubaker PL. Frontiers in glucagon-like peptide-2: multiple actions, multiple mediators. Am J Physiol Endocrinol Metab 2007;293:E460–E465 [DOI] [PubMed] [Google Scholar]
- 29.Cohen JC, Berger GM. Effects of glucose ingestion on postprandial lipemia and triglyceride clearance in humans. J Lipid Res 1990;31:597–602 [PubMed] [Google Scholar]
- 30.Lally S, Owens D, Tomkin GH. Genes that affect cholesterol synthesis, cholesterol absorption, and chylomicron assembly: the relationship between the liver and intestine in control and streptozotosin diabetic rats. Metabolism 2007;56:430–438 [DOI] [PubMed] [Google Scholar]
- 31.Petit V, Arnould L, Martin P, et al. Chronic high-fat diet affects intestinal fat absorption and postprandial triglyceride levels in the mouse. J Lipid Res 2007;48:278–287 [DOI] [PubMed] [Google Scholar]
- 32.Vilsbøll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes 2001;50:609–613 [DOI] [PubMed] [Google Scholar]
- 33.Lugari R, Dei Cas A, Ugolotti D, et al. Evidence for early impairment of glucagon-like peptide 1-induced insulin secretion in human type 2 (non insulin-dependent) diabetes. Horm Metab Res 2002;34:150–154 [DOI] [PubMed] [Google Scholar]
- 34.Schwartz EA, Koska J, Mullin MP, Syoufi I, Schwenke DC, Reaven PD. Exenatide suppresses postprandial elevations in lipids and lipoproteins in individuals with impaired glucose tolerance and recent onset type 2 diabetes mellitus. Atherosclerosis 2010;212:217–222 [DOI] [PubMed] [Google Scholar]
- 35.Wang CS, McConathy WJ, Kloer HU, Alaupovic P. Modulation of lipoprotein lipase activity by apolipoproteins. Effect of apolipoprotein C-III. J Clin Invest 1985;75:384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clavey V, Lestavel-Delattre S, Copin C, Bard JM, Fruchart JC. Modulation of lipoprotein B binding to the LDL receptor by exogenous lipids and apolipoproteins CI, CII, CIII, and E. Arterioscler Thromb Vasc Biol 1995;15:963–971 [DOI] [PubMed] [Google Scholar]
- 37.Laatsch A, Merkel M, Talmud PJ, Grewal T, Beisiegel U, Heeren J. Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis 2009;204:105–111 [DOI] [PubMed] [Google Scholar]
- 38.Hogue JC, Lamarche B, Tremblay AJ, Bergeron J, Gagné C, Couture P. Evidence of increased secretion of apolipoprotein B-48-containing lipoproteins in subjects with type 2 diabetes. J Lipid Res 2007;48:1336–1342 [DOI] [PubMed] [Google Scholar]
- 39.Lambeir AM, Proost P, Scharpé S, De Meester I. A kinetic study of glucagon-like peptide-1 and glucagon-like peptide-2 truncation by dipeptidyl peptidase IV, in vitro. Biochem Pharmacol 2002;64:1753–1756 [DOI] [PubMed] [Google Scholar]
- 40.Hartmann B, Thulesen J, Kissow H, et al. Dipeptidyl peptidase IV inhibition enhances the intestinotrophic effect of glucagon-like peptide-2 in rats and mice. Endocrinology 2000;141:4013–4020 [DOI] [PubMed] [Google Scholar]
- 41.MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002;51(Suppl. 3):S434–S442 [DOI] [PubMed] [Google Scholar]
- 42.Maljaars PW, Peters HP, Mela DJ, Masclee AA. Ileal brake: a sensible food target for appetite control. A review. Physiol Behav 2008;95:271–281 [DOI] [PubMed] [Google Scholar]
- 43.Nauck MA, Niedereichholz U, Ettler R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol 1997;273:E981–E988 [DOI] [PubMed] [Google Scholar]
- 44.Schirra J, Göke B. The physiological role of GLP-1 in human: incretin, ileal brake or more? Regul Pept 2005;128:109–115 [DOI] [PubMed] [Google Scholar]
- 45.Yen CL, Cheong ML, Grueter C, et al. Deficiency of the intestinal enzyme acyl CoA:monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nat Med 2009;15:442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hata T, Mera Y, Ishii Y, et al. JTT-130, a novel intestine-specific inhibitor of microsomal triglyceride transfer protein, suppresses food intake and gastric emptying with the elevation of plasma peptide YY and glucagon-like peptide-1 in a dietary fat-dependent manner. J Pharmacol Exp Ther 2011;336:850–856 [DOI] [PubMed] [Google Scholar]
- 47.Schwartz GJ, Fu J, Astarita G, et al. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab 2008;8:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]