

Abstract
Spartina species have a critical ecological role in salt marshes and represent an excellent system to investigate recurrent polyploid speciation. Using the 454 GS-FLX pyrosequencer, we assembled and annotated the first reference transcriptome (from roots and leaves) for two related hexaploid Spartina species that hybridize in Western Europe, the East American invasive Spartina alterniflora and the Euro-African S. maritima. The de novo read assembly generated 38 478 consensus sequences and 99% found an annotation using Poaceae databases, representing a total of 16 753 non-redundant genes. Spartina expressed sequence tags were mapped onto the Sorghum bicolor genome, where they were distributed among the subtelomeric arms of the 10 S. bicolor chromosomes, with high gene density correlation. Normalization of the complementary DNA library improved the number of annotated genes. Ecologically relevant genes were identified among GO biological function categories in salt and heavy metal stress response, C4 photosynthesis and in lignin and cellulose metabolism. Expression of some of these genes had been found to be altered by hybridization and genome duplication in a previous microarray-based study in Spartina. As these species are hexaploid, up to three duplicated homoeologs may be expected per locus. When analyzing sequence polymorphism at four different loci in S. maritima and S. alterniflora, we found up to four haplotypes per locus, suggesting the presence of two expressed homoeologous sequences with one or two allelic variants each. This reference transcriptome will allow analysis of specific Spartina genes of ecological or evolutionary interest, estimation of homoeologous gene expression variation using RNA-seq and further gene expression evolution analyses in natural populations.
Keywords: transcriptome assembly, polyploidy, invasive species, Spartina, chloridoideae
Introduction
The recent advent of next-generation sequencing (NGS) technologies has opened unique avenues to address ecological and evolutionary questions involving non-model biological systems for which there are limited genomic resources (Hudson, 2008; Ekblom and Galindo, 2010). This is particularly relevant for complex and redundant genomes of polyploid species, which represent a major fraction of eukaryotic lineages (Otto, 2007). Although sequencing performance is rapidly improving in read depth, technologies generating long-sequence fragments such as 454 Roche pyrosequencing have proven particularly useful in de novo sequencing and development of new resources for non-model species, without an available reference genome (Wheat, 2008). High-throughput transcriptome sequencing allows assembly of reference transcriptomes that may be used for various purposes in evolutionary ecology, such as functionally important gene annotation or discovery (for example, Alagna et al., 2009; Barakat et al., 2009; Sun et al., 2010; Logacheva et al., 2011), molecular marker (for example, microsatellite, single-nucleotide polymorphism (SNP)) detection (Barbazuk et al., 2007; Novaes et al., 2008; Bundock et al., 2009) or gene expression variation (Buggs et al., 2010; Swarbreck et al., 2011; Ilut et al., 2012; Yoo et al., 2012). As polyploidy is a recurrent process, many lineages exhibit superimposed traces of genome duplication. Large-scale sequencing and deep read coverage offer a unique opportunity to explore the redundant genome and transcriptome of polyploids, even when diploid progenitors are unidentified or extinct, which makes identification of duplicated homoeologous gene copies particularly challenging.
Recurrent polyploidy is particularly well illustrated in the genus Spartina (Poaceae), where all extant species are polyploids (reviewed in Ainouche et al. (2012)). The grass genus Spartina belongs to the Chloridoideae subfamily, a genomically poorly explored Poaceae lineage, contrasting with well-investigated crops, such as rice, sorghum, maize or wheat that belong to other grass subfamilies. Divergence between Spartina and these grass models is currently estimated to be 35–40 million years ago (MYA) with Panicoideae (including Sorghum and maize) and at least 50 MYA with Ehrhartoideae (including rice) (Christin et al., 2008). Spartina is composed of 13–15 perennial species (Mobberley, 1956), colonizing coastal or inland salt marshes. The basic chromosome numbers in Spartina is x=10, as in most Chloridoideae (Marchant, 1968). Spartina species exhibit various ploidy levels ranging from tetraploid to dodecaploid (Ainouche et al., 2004a). Two closely related hexaploid species, Spartina maritima (Curt.) Fern., and S. alterniflora Lois., are derived from a common hexaploid ancestor (Baumel et al., 2002a; Fortune et al., 2007); although divergence time has not been definitively ascertained, analysis of chloroplast DNA divergence suggests that they diverged less than 3 MYA. They have a critical ecological role in coastal salt marshes at the interface of land and sea, and represent classical models involved in reticulate evolution and recent polyploid speciation (Ainouche et al., 2004a, 2004b; Ainouche et al., 2009). They thus make a good model in evolutionary ecology to investigate the consequences of polyploidy at different evolutionary time scales in natural populations, and to explore the adaptive processes accompanying hybridization, polyploid species formation and expansion.
As for most Spartina species, S. alterniflora is native to the New World, where it is distributed from Canada to southern Argentina along the North and South American Atlantic coast (Mobberley, 1956), whereas S. maritima is distributed along the western European and African Atlantic coasts. Divergence between the two species across the Atlantic Ocean was accompanied by ecological and phenotypic differentiation. Spartina alterniflora has a larger distribution and displays invasive abilities in most regions where it was introduced: in California (Ayres et al., 2004; Civille et al., 2005), in China (Li et al., 2009) and in western Europe (Campos et al., 2004; Ainouche et al., 2009; Querné et al., 2011). In contrast, S. maritima populations are regressing. The recession of S. maritima in its northern range limit (southern England and Brittany) is interpreted as a consequence of climatic changes and anthropogenic habitat disturbance (Raybould et al., 1991), but may also be related to the biological and morphological differences between these two species. Spartina alterniflora exhibits strong rhizomes facilitating lateral expansion and sediment accretion, and thus has an important role in the salt marsh dynamics where it is considered as an ecosystem engineer, whereas S. maritima is a non-rhizomatous, genetically depauperate species (Yannic et al., 2004) with very low seed production (Marchant and Goodman, 1969; Castellanos et al., 1994; Castillo et al., 2008). Spartina maritima and S. alterniflora also exhibit chromosome number differences, as the former has a regular hexaploid number (2n=6x=60) whereas the latter presents aneuploidy (2n=62), and genome size differences (2C=3.8 pg for S. maritima and 2C=4.3 pg for S. alterniflora, Fortune et al., 2008). Less than 5% nucleotide divergence was encountered at 10 putative orthologous-coding loci between the two species, but consistent gene expression differences (13% of the examined genes) were detected using heterologous rice microarrays (Chelaifa et al., 2010a). Genes involved in cellular growth were found highly expressed in S. alterniflora and downregulated in S. maritima, whereas stress-related genes were highly expressed in S. maritima (Chelaifa et al., 2010a).
Spartina alterniflora and S. maritima are involved in one of the textbook examples of recent allopolyploid speciation (reviewed in Ainouche et al., 2004b; Ainouche et al., 2009). Spartina alterniflora was accidentally introduced during the 19th century in Europe, where it hybridized with the native S. maritima. In England, hybridization (with S. alternifora as maternal genome donor, Ferris et al., 1997; Baumel et al., 2001) resulted in Spartina × townsendii, a perennial sterile hybrid first recorded around 1870 (Groves and Groves, 1880), that gave rise by chromosome doubling to a fertile, vigorous and highly invasive allo-dodecapolyploid species, Spartina anglica, which has now been introduced on several continents. An independent hybridization event between S. maritima and S. alterniflora occurred also in southwest France with S. alterniflora as the maternal parent (Baumel et al., 2003), contributing to the formation of another sterile F1 hybrid, Spartina × neyrautii.
Recent studies have been aimed at examining the evolutionary fate of the homoeologous parental genomes from S. maritima and S. alterniflora in the neo-allododecaploid species to understand the genomic determinants of the ecological success of the invasive neopolyploid (Baumel et al., 2002b; Ainouche et al., 2004a; Salmon et al., 2005; Parisod et al., 2009). These studies have revealed that epigenetic reprogramming (for example, DNA methylation, Salmon et al., 2005; Parisod et al., 2009) and evolution of gene expression (Chelaifa et al., 2010b) represent important components of the speciation process in polyploid Spartina species, and are most likely having a critical role in the ecology of the species. However, the previously employed technology for transcriptome analysis (heterologous hybridization on rice microarrays) had several limitations (for example, only a fraction of the genes that hybridized on the array could be analyzed, only global gene expression variation could be evaluated, with no possible distinction of the copies duplicated by polyploidy). Developments in sequencing technology mean that there is now the potential to develop more advanced genomic resources in this important model system for understanding the ecological and evolutionary consequences of hybridization and polyploidy.
When analyzing species such as these where genomic resources are lacking, constitution of a reference transcriptome represents a first critical step to explore the genic compartment. In polyploids, assembled contigs from sequence reads represent consensus sequences among potentially different alleles at strictly orthologous loci, more or less divergent homoeologues (parental orthologues duplicated by polyploidy), or recent paralogues (resulting from individual gene duplication); thus necessitating a more complicated analytical strategy than for diploids. The goal of this study is to build a reference ‘consensus' transcriptome in the hexaploid parental species S. alterniflora and S. maritima using NGS technology, which will allow annotation and identification of specific Spartina genes, including genes of ecological or evolutionary (that is, genes whose expression is altered following speciation) interest. The strategy was then to (i) choose the appropriate high-throughput sequencing that generates long reads facilitating de novo assemblies in the absence of a reference genome (that is, the GS-FLX Roche 454 technology) and (ii) to sequence as many diverse transcripts as possible (to annotate a maximum of genes), by using different types of complementary DNA (cDNA) libraries (normalized and non-normalized) from different tissues (leaves, roots) and from different (natural or controlled) environmental conditions. Sequence heterogeneity at putative homologous loci (within ‘consensus' contigs) is discussed in the context of the (hexaploid) redundant genomes of S. maritima and S. alterniflora. Beyond the Spartina model, the procedure presented here may be applicable to any polyploid system for which no reference genome is available and whose parental species (that is, homoeologous copies) are unknown.
Materials and methods
Plant material
Samples from S. alterniflora were collected in Landerneau (Finistère, France). Spartina maritima was collected at two sites from the French Atlantic coast: Pointe du Verdon (Morbihan) and Noirmoutier (Vendée). Several individuals were collected at each site, and plants were transplanted in the greenhouse (University of Rennes 1).
To maximize detection and annotation of various expressed Spartina genes, RNA extraction was performed on different organs (leaves and roots) from plants sampled either from wild populations and so grown in variable natural conditions (normalized cDNA libraries) or transplanted in a common greenhouse environment (non-normalized cDNA libraries) (Figure 1). Non-normalized libraries usually offer an overview of the most transcribed genes, whereas normalization facilitates the assessment of rare transcripts by decreasing the prevalence of abundant transcripts. For practical reasons, the normalized library could be done only on one species (the European native S. maritima), which was chosen because a larger population sampling was available as part of an ongoing project in our laboratory, involving genome sequencing of this species.
Figure 1.

Sampling strategy and construction of the normalized (S. maritima) and non-normalized (S. maritima and S. alterniflora) cDNA libraries.
Non-normalized cDNA libraries for both S. maritima (from Pointe du Verdon) and S. alterniflora (from Landerneau) were created from plants grown in the same conditions in the greenhouse (30 cm3 daily watered pots containing a mixture of soil, fertilizer and sand) under a day temperature of 20 °C and night temperature of 14 °C. After 21 days of acclimatization, 1–2 g of young leaves and roots per plant were collected separately from three different individuals (from the same population), frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
A normalized library (for S. maritima) was created using leaves from eight individuals collected in the population from Noirmoutier and sampled along a tidal gradient to capture subtleties in gene expression under varying environmental conditions. Two additional S. maritima individuals collected from Pointe du Verdon and transplanted in the greenhouse were also included in the normalized library. Five young leaves were selected for each individual plant, and stored in RNAlater solution (Ambion Inc., Austin, TX, USA) at −20 °C until RNA extraction. For practical reasons, the root normalized library was performed from the same plants used for the non-normalized library that were transplanted in the greenhouse. Roots were carefully washed in distilled water, and then young roots were cut and collected in liquid nitrogen.
For each sample, total RNA was extracted from frozen leaves and roots with Trizol reagent (Sigma-Aldrich Inc., St. Louis, MO, USA) using three cycles of precipitation with isopropanol (Sigma-Aldrich), according to a procedure previously described for Spartina (Chelaifa et al., 2010a, 2010b). All RNA samples were quantified using a Nanodrop Spectrophotometer ND 1000 (Nanodrop Technologies, Thermo Fisher Scientific Inc. Waltham, MA, USA) and the RNA quality (absence of degradation and DNA contamination) was checked on an Agilent 2100 Bioanalyzer (DNA 7500 Chip, Agilent Technologies, Santa Clara, CA, USA). After processing, RNA was stored at −80 °C.
cDNA preparation
cDNA synthesis was performed with 1 μg of total RNA using the SMARTer cDNA Synthesis Kit (Clontech, Mountain View, CA, USA), following the protocol recommended by manufacturers. Briefly, first-strand cDNA synthesis was primed with a modified oligo(dT) primer (the 3′SMART CDS Primer II A). When SMARTScribe RT reaches the 5′-end of the mRNA, the enzyme adds a few additional nucleotides to the 3′-end of the cDNA. After a second-strand cDNA synthesis reaction, double-stranded cDNAs were amplified (21 cycles with primer 5′ PCR Primer II A). This procedure yielded about 2–6 μg of cDNAs that were purified using the Qiaquick PCR Purification Kit (Qiagen, Hilden, Germany). An equimolar mix of samples was constituted for each organ and each species to reach 10 μg of total cDNA and stored at −20 °C until sequencing.
Normalization of S. maritima cDNA
A total of 1 μg of cDNAs from each organ (leaves and roots) of S. maritima was separately normalized as following: 4 μl 4 × hybridization buffer were added and the samples denatured at 95 °C for 5 min and then allowed to anneal at 68 °C for 5 h. The following preheated reagents from the Trimmer kit (Evrogen, Moscow, Russia) were added to the hybridization reaction at 68 °C: 3.5 μl milliQ water, 1 μl 5 × DNAse buffer, 1 μl double-strand nuclease (DSN) enzyme. After incubation at 68 °C for 25 min, the DSN enzyme was inactivated by adding 10 μl of DSN stop solution and heating at 68 °C for 5 min. The normalized cDNA samples were diluted by adding 40.5 μl milliQ water and used for two PCR amplifications. The first PCR (50 μl) contained 1 μl diluted cDNA, 5 μl 10 × Advantage 2 PCR buffer (Clontech), 1 μl 50 × dNTPs mix, 1.5 μl PCR primer M1 10 μℳ (Evrogen), 1 μl 50 × Advantage 2 Polymerase mix (Clontech) and was amplified as following: initial denaturation at 95 °C for 1 min, followed by 18 cycles (95 °C for 15 s, 66 °C for 20 s, 72 °C for 3 min). The second PCR reaction (100 μl) was performed using 2 μl of diluted normalized cDNA, 1 μl of 10 × Advantage 2 PCR Buffer (Clontech), 2 μl 50 × dNTP mix, 4 μl PCR Primer M2 10 μℳ (Evrogen), 2 μl 50 × Advantage 2 Polymerase mix (Clontech) and was amplified following an initial denaturation at 95 °C for 1 min, then 12 cycles (95 °C for 15 s, 64 °C for 20 s, 72 °C for 3 min), and a final extension step (64 °C for 15 s and 72 °C for 3 min). The normalized double-stranded cDNAs were checked on an agarose gel and on an Agilent 2100 bioanalyzer DNA chip (DNA 7500 chip), quantified with a ND 1000 Spectrophotometer (Nanodrop Technologies Inc., Wilmington, DE, USA), and stored at −20 °C.
Sequencing, cleaning and assembly
The four non-normalized cDNA libraries (roots and leaves from S. maritima and S. alterniflora) were sheared by nebulization and sequenced at the Genoscope Platform (Evry). A total of 500 ng of cDNAs were sequenced for each library in two runs on a 454 GS XLR70 Titanium Genomic Sequencer (Roche Inc., Basel, Switzerland). The tissues (leaves and roots) were distinctly distributed on two half regions of the sequencing plate.
Sequencing of the normalized S. maritima cDNA libraries was performed at the Environmental Genomic Platform of the University of Rennes 1. A total of 500 ng of each normalized cDNA library from S. maritima leaves and roots were nebulized and sequenced separately in two half-plates on a 454 GS XLR70 Titanium Genomic Sequencer (Roche Inc.).
The 454 sequence primers (Roche Inc.) and low-quality sequences were removed during signal processing. GS Assembler version 2.3 (Roche, Inc.) was employed to assemble reads into contigs; this program was already successfully used for assembly in transcriptome analyses (Bellin et al. (2009) in Vitis vinifera; Gedye et al. (2010) in S. pectinata; Sun et al. (2010) in Panax ginseng).
Different assemblies were performed for each separate library or for combined data sets per species, tissue and normalization type. Finally, a global assembly of all the obtained reads provided the reference transcriptome for both hexaploids.
As hexaploid Spartina species are expected to potentially express up to six allelic transcripts per locus (resulting from three duplicated pairs of homoeologous genes), the assembly strategy aimed at assembling potentially homologous reads (orthologues and homoeologues) with relatively low stringency to construct consensus contigs constituting the ‘hexaploid reference transcriptome' that will be used for identification and annotation of Spartina genes. In this perspective, effects of different minimum match percentages (90, 95, 96 and 97%) on the assembly process were explored. Analyses presented in this paper are based on de novo assemblies executed with 90% of minimum match on at least 100 bp and GS Assembler version 2.3 (Roche, Inc.) default parameters for cDNA. This low minimum match percentage (90%) was chosen to maximize assembly of reads corresponding to putative orthologous and homoeologous transcripts, although we cannot rule out assembling weakly divergent paralogs. Useful information (such as the number of reads used in the assembly, the number of contigs and singletons, mean length and read depth) was extracted from assembly files. Read depth is estimated by GS Assembler as the total number of included bases from all the obtained 454 sequence reads aligned to generate the consensus contig sequence, divided over the contig length. To test validity of the assembly, we aligned 10 contigs against homologous expressed sequence tags (ESTs), which were sequenced using the Sanger method (Chelaifa et al., 2010a) and sequence identities were calculated.
Contigs from S. maritima and S. alterniflora were then mapped to the Sorghum bicolor genome, the closest related species to Spartina that has a fully sequenced and annotated genome (Paterson et al., 2009), to compare the distribution and density of the identified Spartina homologous genes across the different Sorghum chromosomes. The Sorghum bicolor gene annotation was retrieved from the Sbicolor_79_gene.gff3 annotation file available at http://genome.jgi-psf.org/Sorbi1/ and gene density was estimated from the proportion of annotated genes per 100 kb intervals. Colinearity between Spartina and Sorghum has not been investigated previously, but conservation of gene colinearity is expected according to what is known from related lineages (for example, finger millet, Chloridoideae and rice, Ehrhartoideae, Srinivasachary et al. (2007)) in the grass family. The BLASTn algorithm was used with a P-value of 10−5 and Best BLAST Hit (corresponding to the highest e-value and bit score) parsed for each query sequence. The proportion of Spartina homologs was calculated by 100 kb windows (delimitated from Sorghum) and the results were represented using the Circos v.0.55 software (Krzywinski et al., 2009). To evaluate the genome-wide representation of the assembled contigs on the Sorghum genome, Pearson's correlations and linear regressions were calculated between gene densities (number of genes per 100 kb window) in Sorghum and corresponding homologs in the investigated Spartina species. Both statistics were calculated for all 10 Sorghum chromosomes and by individual chromosomes using the R software (R Development Core Team, 2011).
Annotation
BLASTn and tBLASTx (Altschul et al., 1990) analyses of contigs and singletons were conducted against two nucleotide databases: Oryza sativa ESTs database (http://rapdb.dna.affrc.go.jp), and a home-built regularly updated Poaceae database, including ESTs from Oryza sativa, Zea mays, Brachypodium distachyon and Sorghum bicolor (www.gramene.org). All BLAST searches were performed with an e-value of 10−5. Best BLAST Hit from all BLAST results were parsed for a homology-based functional annotation.
GO annotations using BLAST2Go (Conesa et al., 2005; Götz et al., 2008) were performed using tBLASTx (e-value 10−6) on assembled contigs against the Arabidopsis thaliana database from the TAIR website (www.arabidopsis.org) (with GO IDs and term assigned), with an annotation e-value hit filter of 10−6 and a cutoff of 55 (maximum similarity).
The annotated Spartina transcriptome was examined to identify genes of potential ecological interest (for example, genes involved in salt stress response, oxidative stress, heavy metal tolerance or growth). Genes whose expression was previously found altered following hybridization and genome duplication from a rice microarray-based study on these species (Chelaifa et al., 2010a) were investigated. The corresponding accession numbers of the rice oligos spotted on Agilent microarrays (44 K Agilent G2519F) employed in that study were used to retrieve putative homologs in our Spartina reference transcriptome using BLASTn (e-value 10−5).
Sequence heterogeneity at homologous gene copies
As both Spartina species studied here are hexaploid, sequence read heterogeneity is expected in the assembled contigs, resulting from both genome duplication and allelic variation within homoeologues (heterozygosity at orthologous loci). In this study, we chose the 454 technology because it generates long read sequences to facilitate de novo assembly, but this sequencing method offers less read depth than alternative technologies generating short reads to capture all the allelic variants that may be transcribed at each locus. As a preliminary evaluation of sequence heterogeneity among assembled reads obtained with the 454 pyrosequencing technique, we have selected contigs with relatively good coverage (at least 50 reads) that were present in both S. maritima and S. alterniflora data sets.
We looked at polymorphisms within contigs by mapping the corresponding reads (using Genome Assembler v 2.5.3, Roche) to a subset of selected homologous contigs between the two species. We then scanned the resulting alignments for SNPs using the Ace.py program from the biopython package (http://biopython.org/). Rare SNPs or SNPs detected within homopolymeric regions were removed from the analysis to avoid putative false-positive SNPs. We then assembled reads presenting 100% similarity (using at least one shared SNP) to maximize the consensus sequence length. This consensus sequence was then considered as a haplotype, representing a particular copy in the corresponding contig.
Results
De novo assemblies and contig annotation
Spartina maritima
Sequencing of the non-normalized and normalized cDNA libraries from roots and leaves resulted in 425 274 reads (average length 314±147.3 bp) and 558 732 reads (average length 203±102.8 bp), respectively. Data are available in Genbank under accession references SRP015701 and SRP015702 for S. maritima and S. alterniflora, respectively.
Assemblies and annotations were first performed separately on the sequences obtained from the non-normalized and normalized cDNA libraries for each tissue, respectively, then on the pooled reads from both normalized and non-normalized libraries. A total of nine different assemblies (as presented in Table 1) were performed using individual (by tissue and normalization) or combined data sets, allowing the comparison of annotated contigs by tissue and evaluation of the normalization process efficiency.
Table 1. Summary of assemblies and annotations of the Spartina maritima and Spartina alterniflora complementary DNA libraries.
| Analysis |
Assemblies |
Annotations |
|||
|---|---|---|---|---|---|
| Number of reads used in the assembly | Number of contigs | Number of singletons | tBLASTX Oryza sativa | tBLASTX Poaceae | |
| S. maritima (non-normalized) | |||||
| Leaves | 273 659 | 5866 | 63 064 | 5237 (3143) | 5505 (3825) |
| Roots | 131 727 | 5910 | 43 945 | 5275 (3029) | 5551 (3824) |
| Leaves and roots | 405 386 | 12 309 | 83 878 | 11 118 (5705) | 11 718 (7290) |
| S. maritima (normalized) | |||||
| Leaves | 95 045 | 3654 | 43 993 | 1797 (1589) | 2069 (1938) |
| Roots | 264 114 | 13 315 | 74 418 | 9948 (7193) | 10 550 (9115) |
| Leaves and roots | 359 159 | 17 182 | 89 765 | 10 805 (8195) | 12 518 (10 629) |
| S. maritima total (non-normalized+normalized) | |||||
| Leaves | 371 111 | 10 098 | 79 436 | 8517 (4821) | 9002 (6100) |
| Roots | 398 991 | 19 069 | 84 789 | 17 409 (8485) | 18 162 (11 149) |
| Total (all organs and all cDNAs) | 755 309 | 25 239 | 114 857 | 16 137 (9958) | 17 307 (13 786) |
| S. alterniflora | |||||
| Leaves | 140 733 | 3217 | 30 480 | 2995 (1806) | 3127 (2169) |
| Roots | 203 990 | 11 155 | 43 904 | 10 805 (5 281) | 11 201 (6811) |
| Leaves and roots | 344 723 | 14 317 | 58 298 | 13 919 (6430) | 14 123 (8370) |
| S. maritima and S. alterniflora | |||||
| Leaves | 511 844 | 13 824 | 93 274 | 12 246 (5999) | 12 910 (7773) |
| Roots | 602 981 | 29 187 | 102 638 | 28 164 (10 268) | 29 054 (14 135) |
| Total | 1 114 825 | 38 478 | 153 409 | 36 549 (11 776) | 38 089 (16 753) |
In brackets are numbers of non-redundant gene annotations.
After trimming the adapter sequences and removing sequences shorter than 50 bases, 405 386 and 359 159 reads remained for the S. maritima non-normalized library and S. maritima normalized library, respectively. Assembly of the trimmed reads resulted in 12 309 contigs for the non-normalized library and 17 182 contigs for the normalized library. The mean contig length was 617 bp (s.d.=540.3, range=50–8036) and 415 bp (s.d.=246.9, range=50–2252) for the non-normalized and normalized libraries, respectively.
Separate assemblies for roots and leaves were also processed for each library, as well as global assembly of all the reads from S. maritima to get a global gene annotation for this species. Unequal read numbers were obtained for leaf and root cDNA sequencing in both the normalized and non-normalized libraries. In the non-normalized cDNA library, the read number in leaves was twice that of roots. In the root normalized library, read number was three times larger than the number obtained in the non-normalized library (Table 1). Equivalent number of contigs were assembled for leaves (5866) and roots (5910) in the non-normalized cDNA library, but many more contigs were assembled for roots (13 315) than for leaves (3654) in the normalized library. When pooling all reads from S. maritima (normalized and non-normalized for both organs), 25 239 contigs were assembled. Separate assemblies of roots and leaves resulted in 19 069 and 10 098 contigs, respectively.
Functional annotation was performed by sequence comparisons with public databases. The different S. maritima data sets (from non-normalized and normalized cDNAs in each tissue) were first compared with the Oryza sativa EST database, then to a larger database including four sequenced Poaceae genomes. As expected, the use of this homemade Poaceae database improved the number of annotated genes (Table 1). In the non-normalized library, 5705 different genes were annotated with the O. sativa database and 7290 with the Poaceae database. In the normalized library, 8195 were annotated with the O. sativa database and 10 629 with the Poaceae database. The normalization of the cDNA library significantly increased the number of annotated genes, as among these 10 629 annotated genes, 3620 were common to both libraries and 6642 genes were specific to the normalized data set (Figure 2a).
Figure 2.
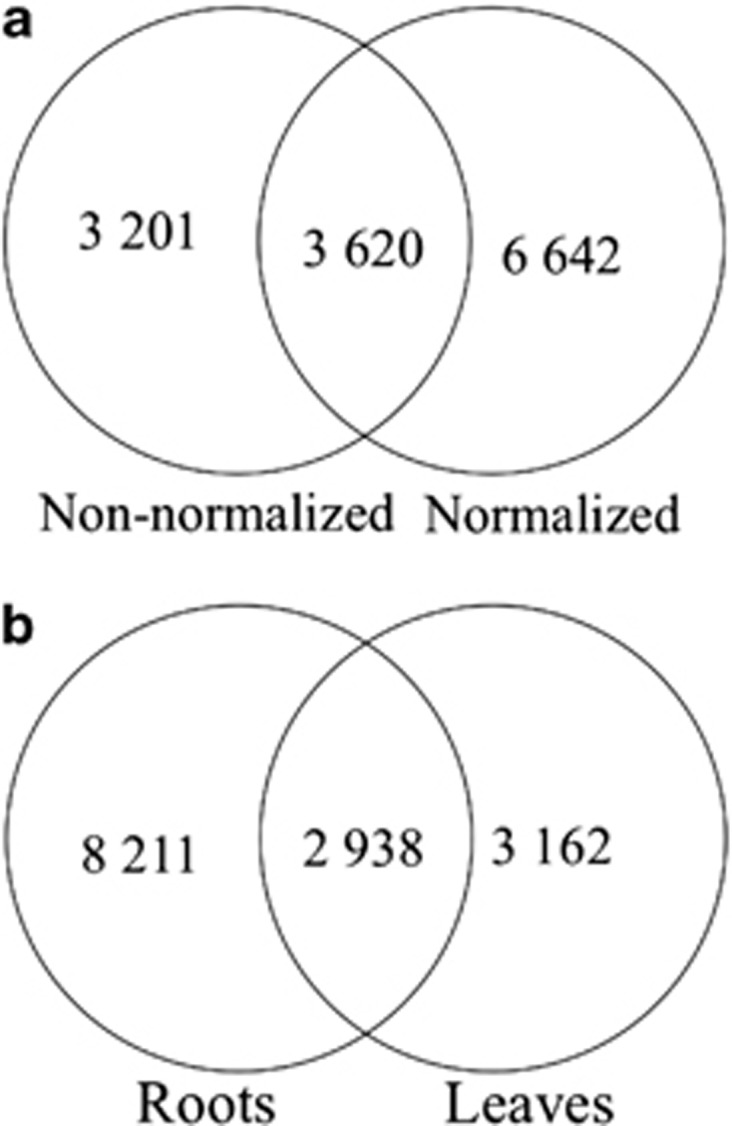
Common annotated contigs (using the Poaceae database) of S. maritima (a) between non-normalized and normalized cDNA libraries (leaves+roots) (b) between roots and leaves.
The Poaceae database allowed annotation of 6100 different genes for S. maritima leaves and 11 149 genes for roots (Table 1). Among these, 2938 genes were found in both root and leaf transcriptomes, (Figure 2b). When pooling all the read data sets (both tissues and both normalization types), 13 786 genes were annotated in total for S. maritima with the Poaceae database (Table 1).
Spartina alterniflora
Sequencing of the S. alterniflora non-normalized cDNA library from roots and leaves resulted in 495 749 reads, with an average length of 285±160.6 bp. After trimming, 344 723 reads were used for the assembly, which resulted in 14 137 contigs (Table 1). The S. alterniflora contigs have an average length of 759 bp (s.d.=637.1, range=50–12 334) and a mean read depth of 14.3. Separate assemblies of roots and leaves were processed as for S. maritima and resulted in 3217 contigs for leaves and 11 155 contigs for roots. More reads and more contigs were obtained for roots than for leaves, as observed in S. maritima (Table 1).
Functional annotation of the S. alterniflora contigs using the Oryza and Poaceae databases, respectively, resulted in 1806 and 2169 different genes annotated in leaves. For roots, 5281 (Oryza database) and 6811 (Poaceae database) genes were annotated. When pooling root and leaf data sets, 6430 genes were annotated when using the Oryza database, and 8370 genes were annotated in total for S. alterniflora, when using the Poaceae database (Table 1).
Spartina leaf and root transcriptomes
To maximize the number of contigs and annotated genes per tissue, S. maritima and S. alterniflora reads were pooled, which resulted in 13 824 and 29 187 assembled contigs for leaves and roots, respectively (Table 1). When using the Poaceae database for functional annotation, 7773 and 14 135 different genes were annotated for leaves and roots, respectively. Among these, 4019 (22.5%) genes were common to root and leaf Spartina transcriptomes (Figure 3d).
Figure 3.

Common annotated contigs (using the Poaceae database) between S. maritima and S. alterniflora. (a) Comparison between S. maritima and S. alterniflora leaves. (b) Comparison between S. maritima and S. alterniflora roots. (c) Comparison between S. maritima and S. alterniflora (combined data from leaves and roots). (d) Comparison between roots and leaves (combined data from both species).
When examining leaf and root transcriptomes between species, 978 and 3251 annotated genes were found common to S. maritima and S. alterniflora for leaves and roots, respectively (Figures 3a and b). Overall, S. maritima and S. alterniflora share 4298 expressed genes (pooled leaf and root data sets) with 9488 genes annotated only in S. maritima and 4072 genes only in S. alterniflora (Figure 3c). The total data set (both species and organs) resulted in 38 478 contigs and 16 753 annotated Spartina genes (Table 1), which represent the first reference transcriptome for the hexaploid Spartina species.
Distribution of the contigs on the Sorghum genome
The number of homologous genes sequenced in Spartina hexaploid species was about half the number found in Sorghum bicolor per 100 kb sliding window. Mapping of the Spartina contigs to the Sorghum genome revealed similar relative gene densities for both Spartina EST libraries among the 10 chromosomes (Figure 4b, Supplementary Figure 1). High correlation between Sorghum gene densities along chromosomes and the number of homologous Spartina genes in a 100-kb Sorghum window were encountered for most chromosomes. A relatively lower correlation was found for chromosomes 5 and 8 (Supplementary Figure 1), which could suggest more extensive rearrangements during evolution of these taxa. Furthermore, we observed that Spartina gene densities were higher in the corresponding subtelomeric Sorghum chromosome positions than in pericentromeric ones, as expected from gene distributions in Sorghum (Paterson et al., 2009).
Figure 4.

(a) Spartina contigs mapped to the Sorghum genome. The 10 individual chromosomes are shown in the outer circle. Relative gene densities on each chromosome are displayed successively inward as following: (i) gene density in Sorghum bicolor, (ii) gene density in Spartina maritima, (iii) Spartina maritima gene density relative to Sorghum gene density, (iv) gene density in Spartina alterniflora, (v) Spartina alterniflora gene density relative to Sorghum gene density (by 100 kb region). (b) Correlations between Sorghum density and S. maritima and S. alterniflora homologous gene densities by 100 kb region (P-value<2.2 e−16).
Most-represented genes in the normalized and non-normalized Spartina data sets
The 20 most-represented transcripts (according to read depth) in the non-normalized libraries appear very similar in S. alterniflora and S. maritima (Supplementary Table 1). In both leaves and roots, they are mainly involved in respiratory pathways (for example, cytochrome c oxidase, ATP synthase), and in RNA and ribosomal protein synthesis. In roots, NADH-ubiquinone oxidoreductase and acylCoA-binding protein were also well-represented. Genes involved in stress responses were observed mainly in root transcripts. Among the most represented are the metallothionein and zinc finger (A20 and AN1) domains involved in metal binding and control of oxidative stress. A transcription elongation factor (EF) was also well represented in the root transcriptome of S. maritima and S. alterniflora (Supplementary Table 1); this gene is involved in protein elongation during translation (Andersen et al., 2003) and is also found highly represented in the roots of other grass species (for example, in Zea mais, Poroyko et al. (2005) or Avena barbata, Swarbreck et al. (2011)). The chaperone protein DnaJ gene was also encountered in the root transcriptome of S. maritima. This gene is induced by heat shock and prevents apoptosis (Gotoh et al., 2004). In addition, in S. alterniflora, two contigs annotated with a pathogenesis-related Bet V family protein were highly represented. This gene can be induced by different pathogens, such as viruses, bacteria and fungi (Liu and Ekramoddoullah, 2006).
The most abundant sequences annotated from the normalized cDNA data set in S. maritima belong to a larger set of gene categories compared with those encountered in the non-normalized data sets for both S. alterniflora and S. maritima. In leaves, all of the important functions are represented: we encountered genes involved in flowering control (tetratricopeptide repeat protein 1), in cell wall structure (glycine-rich protein), in the C4 assimilation process (phosphoenol-pyruvate carboxykinase, carbonic anhydrase) and in fatty acid metabolism (Acyl coA-binding protein). The thioredoxin gene has a critical role in redox regulation in the apoplast, which regulates cell division (Tian et al., 2009), cell differentiation (Takeda et al., 2003), pollen germination (Ge et al., 2011) and stress responses (Song et al., 2011).
In the normalized root cDNA data set, apart from three highly represented contigs annotated as ribosomal genes, all others were genes and proteins involved in primary metabolism, such as cell transport (ADP-ribosylation factor, ranBP1 domain-containing protein), cell organization (mps 1 binder kinase activator-like 1A, steroid-binding protein, FYVE zinc finger domain-containing protein), plant growth (peptidase T1 family, tetratricopeptide repeat protein 1) and stress response (calreticulin precursor protein, phosphatase 2C, cytosolic ascorbate peroxidase gene, peroxiredoxin).
GO (Gene ontology) annotation and biological process analyses
Functional annotation
Using the A. thaliana protein database of the TAIR website, GO functions could be assigned to Spartina transcripts. Among the various biological processes, cellular (5865) and metabolic (5660) processes, as well as biological regulations (2125) were most highly represented (Figure 5). Important functions were also identified, such as response to stimulus, protein localization and transport and developmental process. Similarly, cell and organelle were most represented between the cellular component and binding and catalytic activities among the various molecular functions (Figure 5).
Figure 5.

Functional classification of the leaf transcriptome of S. maritima and S. alterniflora. GO annotations were used for classification for GO cellular component, GO molecular function and GO biological process.
Identification of ecologically relevant genes
Annotated Spartina genes with potential ecological relevance are listed in Supplementary Table 2, with the corresponding number of putative homologous regions identified in the Sorghum genome. Transcription factors, such as zinc finger proteins, anti-oxidants (for example, gdp-mannose pyrophosphorylase) and osmolyte synthetic transporters were identified. Heat shock proteins, such as zeaxanthin epoxidase, a precursor of abscisic acid (ABA), which is involved in response to abiotic stress (including salt and heavy metal tolerance), were also encountered.
Among the known genes of the lignin biosynthetic pathway (Humphreys and Chapple, 2002), we were able to identify the cinnamoyl-CoA reductase and cinnamyl alcohol dehydrogenase genes. Gene families associated with the production of cellulose, such as cellulose synthases (CesA) and glycosyl transferases, were also identified in the reference Spartina transcriptome (Supplementary Table 2).
Identification of genes whose expression is altered following speciation in Spartina
When searching for the differentially expressed genes between the parental species (S. maritima and S. alterniflora) and between the parents and their hybrid (Spartina × townsendii) or allopolyploid (S. anglica) derivatives detected using rice microarrays by Chelaifa et al. (2010a, 2010b) we found 409 Spartina contigs exhibiting similarities to rice sequences (Supplementary Table 3). A BLAST2Go analysis was performed on these 409 sequences, of which 271 were found to have different functional annotation. Sequences whose expression is altered following speciation according to Chelaifa et al. (2010a, 2010b) such as transcription factors, retrotransposons, peptide transport system genes, glutathione transferases, peroxidases and cytochrome c oxidase were parsed to provide a sequence database. This database now constitutes a reference for future studies regarding genomic and transcriptomic consequences of polyploidy speciation in Spartina.
Polymorphism analysis at homologous genes
Because of the polyploid nature of these highly redundant genomes, up to three duplicated homoeologs may be encountered at each locus, leading to sequence heterogeneity among reads. Contigs from four genes (phosphoenol-pyruvate carboxykinase, HECT domain-containing protein, homeobox domain-containing protein and a heat shock protein) were analyzed in detail to identify homologous sequences, polymorphic sites and putative haplotypes. In these contigs, three to four haplotypes could be distinguished within individuals when comparing the homologous sequences for each gene (Table 2). The polymorphism analysis is illustrated in Table 3, for a 200-bp region of the HECT domain-containing protein gene. In this window, seven haplotypes (over the two species) were aligned. Six polymorphic sites were detected in each species, including four polymorphic sites shared between S. maritima and S. alterniflora, and two species-specific polymorphic sites. The shared polymorphisms allow distinction of two divergent haplotypes (where all six polymorphic sites are different) present in both hexaploids, and one (in S. maritima) or two (in S. alterniflora) additional less divergent variants (one or two nucleotide difference). Although the number of polymorphic sites defining haplotypes is variable among the other analyzed contigs, we observed the same pattern distinguishing two divergent haplotypes and one or two less divergent variants within individuals (Table 2).
Table 2. Nucleotide polymorphisms detected among reads within four annotated contigs from S. maritima and S. alterniflora.
| Gene annotation |
Contig length |
Number of reads |
Number of polymorphisms |
Number of haplotypes |
||||
|---|---|---|---|---|---|---|---|---|
| S. maritima | S. alterniflora | S. maritima | S. alterniflora | S. maritima | S. alterniflora | S. maritima | S. alterniflora | |
| Phosphoenol-pyruvate Carboxykinase (LOC_Os03g15050.4|13103.m01762|cDNA) | 632 | 470 | 132 | 50 | 8 | 3 | 4 | 3 |
| HECT domain-containing protein, expressed (LOC_Os12g24080.1|13112.m02448|cDNA) | 4294 | 3961 | 127 | 85 | 113 | 103 | 4 | 4 |
| Homeobox domain-containing protein, expressed—Transcription factor MEIS1 and related HOX domain proteins (LOC_Os12g43950.4|13112.m08878|cDNA) | 3031 | 2777 | 185 | 129 | 6 | 4 | 4 | 3 |
| Heat shock protein, putative, expressed (LOC_Os06g50300.1|13106.m05403|cDNA) | 3019 | 3391 | 67 | 263 | 42 | 5 | 4 | 4 |
Table 3. Single-nucleotide polymorphisms among assembled reads of the gene coding the HECT domain-containing protein in Spartina maritima and Spartina alterniflora.
| HECT domain-containing protein, expressed | ||||||
|---|---|---|---|---|---|---|
| S. alterniflora contig 03059 (length=3961, reads=85) | ||||||
| Nucleotide position | 1034 | 1085 | 1100 | 1119 | 1130 | 1167 |
| Haplotype 1 | C | T | C | A | A | T |
| Haplotype 2 | T | T | T | A | A | C |
| Haplotype 3 | T | C | T | A | A | C |
| Haplotype 4 | C | T | C | G | C | T |
| S. maritima contig 02799 (length=4294, reads=127) | ||||||
| Nucleotide position | 1344 | 1352 | 1371 | 1382 | 1401 | 1419 |
| Haplotype 1 | C | C | G | C | C | T |
| Haplotype 2 | T | C | G | C | C | T |
| Haplotype 3 | T | T | A | A | A | C |
Analysis of a 200-bp window, including two species-specific polymorphic sites (positions 1304, 1085 in S. alterniflora and positions 1344 and 1401 in S. maritima) and four polymorphic sites shared between the two species. These shared polymorphic sites are vertically aligned in the table.
Discussion
We have explored the transcriptome of two related Spartina species (S. maritima and S. alterniflora) using 454 sequencing technology. Before this study, only a limited number of Spartina ESTs were deposited in the NCBI EST database. If we exclude a recent transcriptome analysis in the tetraploid Spartina pectinata that generated 556 198 ESTs (Gedye et al., 2010), a few hundred sequences only were available for S. maritima (Chelaifa et al., 2010a) and S. alterniflora (Baisakh et al., 2008). Our work represents the first effort to analyze the transcriptome of the hexaploid Spartina species, resulting in a reference transcriptome of more than 16 700 annotated genes from leaves and roots.
De novo transcriptome assembly using 454 sequencing technology
Compared with other NGS technologies, the Roche platform offers long read lengths that facilitate assembly and annotation (Morozova et al., 2009) and for this reason it is the most widely used technology for de novo EST sequencing (Sun et al., 2010). In total, 25 239 (normalized and non-normalized libraries) and 14 317 contigs were assembled for S. maritima and S. alterniflora, respectively, representing 65.1% and 57.8% of the reads, the remaining of the reads left as singletons. Using a similar technology and assembly software, Gedye et al. (2010) assembled 65% of the reads into contigs for S. pectinata. The contig lengths found for both species are comparable to the length range reported in similar studies on other species (for example 299 bp in Oryza longistaminata, Yang et al. (2010); 394 bp in S. pectinata, Gedye et al. (2010); 526 bp in Panax quinquefolius, Sun et al. (2010)). From this data set, 17 307 contigs were annotated for S. maritima and 14 123 contigs were annotated for S. alterniflora (38 089 total annotated contigs for both species) corresponding to 16 753 different genes. These results are situated in the range of reported studies in non-model species (69.8% in ginseng; 72.6% in S. pectinata; 82% in amaranth; 85.5% in Cicer). Functional annotation could be assigned to 68.6% of the S. maritima contigs and 98.6% of the S. alterniflora contigs. Nonetheless, a large number of unique reads (singletons) were found, that is, 15% for our data set compared with other studies using the same assembler: 13% in S. pectinata (Gedye et al., 2010); 10–25% in Mytilus galloprovincialis (Craft et al., 2010); 8.8% in Palomero maize (Vega-Arreguin et al., 2009) and 7% in Amaranthus and Ginseng (Sun et al., 2010; Délano-Frier et al., 2011). This could result from various causes such as the presence of rare transcripts from lowly expressed genes. The 454 sequencing technology also has some limitations resulting mainly from sequencing errors associated with homopolymers (Margulies et al., 2005; Moore et al., 2006; Wicker et al., 2006), A/T bias (Moore et al., 2006; Wicker et al., 2006) or random nucleotide misincorporation (Huse et al., 2007; Holt and Jones, 2008). The error rate for 454 sequencing is higher than the rate usually observed with Sanger sequencing (0.04 and 0.01%, respectively (Ewing and Green, 1998; Margulies et al., 2005; Moore et al., 2006)). Nevertheless, the error rate drops significantly to 0.4 bp errors per 10 kb after assembly (Margulies et al., 2005; Moore et al., 2006). We checked the quality of our sequence assemblies from 454 sequencing by comparing 10 assembled contigs to their putative homologs in S. maritima ESTs sequenced with the Sanger method (Chelaifa et al., 2010a, 2010b). The identity between the sequences was found very high (99.5%), which validates the procedures employed.
As there is no reference genome for Spartina, we used information from several EST and protein databases for gene annotation, a procedure successfully employed for other non-model species (for example, Barakat et al., 2009; Gedye et al., 2010; Franssen et al., 2011; Garg et al., 2011). In de novo sequencing projects transcriptome coverage efficiency has been evaluated by comparing the number of unique genes to the nearest transcriptome available (Parchman et al., 2010). We compared our data to the nearest sequenced grass genomes: Oryza sativa (51 258 protein-coding transcripts, Yu et al., 2005 and the Rice Genome Annotation project, http://rice.plantbiology.msu.edu//) and Sorghum bicolor (36 338 protein-coding transcripts, Paterson et al., 2009). Using combined cDNA libraries, we identified 16 753 putative (non-redundant) genes by homology searches, which represent more than half of the genes found in fully sequenced related plant genomes. Interestingly, these genes appear distributed among the different Sorghum chromosomes, particularly in high gene density subtelomeric regions. Global gene colinearity is known to be well conserved among grass genomes (Feuillet and Keller, 2002; Srinivasachary et al., 2007) and the comparison here between hexaploid Spartina and Sorghum bicolor validates the utilization of Sorghum as a comparative model, as first observed in Gedye et al. (2010) for S. pectinata. The percentage of contigs without a BLAST hit in our study is quite low (1.01%), with 389 contigs that did not match any putative homolog in the Poaceae database. This fraction varies among other studies fluctuating from 14.5% in Cicer (Garg et al., 2011) to 30.2% in Panax (Sun et al., 2010), for instance. These sequences without homology hit can be attributed to technical biases, such as low-quality data, inaccurate assembly, assembly parameters and contamination by genomic DNA. The causes can also be biological: some cDNAs are non-coding, lineage-specific or highly variable (Logacheva et al., 2011). Specific Spartina (or Chloridoideae) sequences also might be too divergent from the grass model species used.
In this study, among the 13 786 genes annotated in S. maritima, 6642 were retrieved in the normalized library, 3201 genes in the non-normalized and only 3620 genes overlapping both libraries, which indicates that normalization significantly improved the number of annotated genes. The normalization reduces oversampling of abundant transcripts and maximizes the potential to sequence less abundant transcripts (Zhulidov et al., 2004). RNA-Seq studies on Zebra finch and rice have reported a higher efficiency in gene discovery using normalized cDNA libraries compared with non-normalized libraries (Yang et al., 2010; Ekblom et al., 2012). In contrast, Hale et al. (2009) demonstrated that normalization has a limited influence on increasing sequenced gene number. Ekblom et al. (2012) suggest that differences in technologies used and sequencing efforts can affect the outcome of the comparison between normalized and non-normalized libraries. In our present study, the normalized library was constructed from plants grown under natural conditions along a tidal gradient, which might also have increased the number of transcripts annotated. The transcriptome size, unknown in most non-model species may also affect the coverage and the sequencing effort. Therefore, it can affect indirectly the efficiency of normalization: normalized libraries show less efficiency when the non-normalized library already covers the whole transcriptome. This suggests that the combination of both normalized and non-normalized libraries is essential for gene discovery in non-model species, particularly in species exhibiting redundant genomes such as hexaploid Spartina.
Functional aspects: biology and ecology of Spartina
The 16 753 Spartina unigenes annotated in this study represent an important resource to explore genes involved in functions of ecological and adaptive interest. The genus Spartina exhibits a C4-type photosynthesis, which evolved in the Chloridoideae between 25 and 32 MYA (Christin et al., 2008), and which uses the ATP-dependent phosphoenolpyruvate carboxykinase (PCK) as decarboxylating enzyme (Christin et al., 2009). C4 metabolism confers high plant productivity under warm, arid and saline conditions, although Spartina species (and most particularly the hexaploids) colonize temperate regions (Long et al., 1975). In the study conducted by Christin et al. (2009), one PCK-sequence-type was found in S. maritima, whereas two sequence types were found in S. anglica, one being sister to the maritima-type sequence and the other one most likely originating from the other parent of S. anglica (S. alterniflora, which was not analyzed by these authors). When analyzing an 830-bp partial PCK-coding region in S. maritima and S. alterniflora, Chelaifa et al. (2010a) found high nucleotide identity (99.7%) between S. maritima and S. alterniflora. In our study, a fragment of the PCK gene was found well represented in the leaf transcriptome of both S. maritima (623 bp) and S. alterniflora (470 bp), which is less than 25% of the total CDS length of O. sativa being 2820 bp but provides an indication of levels of heterogeneity. SNPs examined in this region revealed the presence of up to two haplotypes for each species. The identity between the two most divergent haplotypes of S. maritima was 98.5%, whereas the two less divergent sequences exhibited 99.4% identity. Our results then indicate that at least two different, putative homoeologous PCK sequences are expressed in the leaves of the hexaploid S. maritima and S. alterniflora species.
S. alterniflora and S. maritima are low-marsh species that have developed particular adaptation to tolerate several hours of immersion under seawater at high tide (Adams and Bate, 1995; Daehler and Strong, 1996). Survival of low-marsh Spartina species in anoxic sediments is facilitated by their ability to develop aerenchyma systems (studied particularly in S. alterniflora) that supply the submerged plants with atmospheric oxygen and efficiently transport oxygen to the roots (Maricle and Lee, 2002). High salinity can be damaging by salt toxicity and dehydration caused by low water potential. Thus, plants living in saline, high-light environments are adapted to minimize water loss to prevent dehydration, and have developed particular adaptive anatomical features with this regard (Maricle et al., 2007). Salt marsh Spartina species have thick leaves with pronounced ridges on the adaxial side. They are adapted to controlling water loss by having stomata on the adaxial side and by having large leaf ridges that fit together as the leaf rolls during water stress (Maricle et al., 2009). To prevent salt toxicity, Spartina have large vacuoles for salt storage (Munns and Tester, 2008) and salt-secreting glands to excrete inorganic ions (Zhu, 2001). Phenotypic adaptations are well documented but little is known about genes involved in these responses. The first Spartina transcriptome analyses under salt stress were performed in S. alterniflora using cDNA amplified fragment length polymorphism (Baisakh et al., 2006) and EST analyses (Baisakh et al., 2008); these analyses identified various transcripts involved in ion transport and compartmentalization, osmolyte production, cell division, metabolism and protein synthesis, as well as previously unknown genes induced by salt stress. Although our transcriptome analysis of S. maritima and S. alterniflora was not performed under salt stress, we retrieved 937 (4642 contigs) of the 1266 ESTs Baisakh et al. (2008) and Subudhi and Baisakh (2011) generated. Using A. thaliana as a functional reference transcriptome, we were also able to annotate 130 genes (305 contigs) involved in salt stress response. These genes include transcription factors, heat shock protein and cytochrome c oxidase that have been found to respond to salt and oxidative stress by balancing ion concentrations in Spartina (Maricle et al., 2006).
We also annotated 71 genes (190 contigs) involved in heavy metal tolerance. Spartina species are of high interest regarding their ecological role in polluted coastal environment, where they exhibit particular tolerance to oil spill and where they are considered for phytoremediation purposes (Maricle and Lee, 2002; Martinez-Dominguez et al., 2008; Mateo-Naranjo et al., 2008). Ramana Rao et al. (2011) found 28 differentially expressed genes following experimental petroleum hydrocarbon exposure in S. alterniflora. We retrieved in our data set 8 of these genes (52 contigs).
Genes involved in stress response or in developmental and cellular growth were found to be differentially expressed in controlled conditions in the two species, the former being overexpressed in S. maritima, whereas the latter were overexpressed in S. alterniflora (Chelaifa et al., 2010a). Most of these genes have also been found to be predominantly affected following hybridization between these species (that is, in Spartina × townsendii) and subsequent genome duplication in S. anglica (Chelaifa et al., 2010b). Here, we identified 409 contigs corresponding to 271 different genes matching the putative homologous rice probes described in Chelaifa et al. (2010b). Our Spartina sequence data set may provide useful information to target genes of ecological and evolutionary interest (that is, whose expression is affected by divergent and reticulate speciation). Specific primers may be now designed to explore gene expression evolution in natural conditions and under various ecological situations.
Sequence polymorphism at homologous loci in hexaploid Spartina
The contigs assembled from the 454 reads in each of these hexaploid species actually represent a consensus sequence among strictly homologous (that is, orthologous) sequences but may also include homoeologous sequences (generated by polyploidy). Within homoelogues (at strictly orthologous loci), levels of heterozygosity have been poorly investigated in S. maritima, although this species is well known for its predominant clonal propagation and weak inter-individual genetic variation (Yannic et al., 2004). Spartina alterniflora has a mixed, predominantly outcrossing mating system (Travis et al., 2004); thus, more allelic variation within homoeologues might be expected than for S. maritima. Reads were assembled using a 90% identity threshold, to avoid potential comparisons involving divergent paralogs, but homoeologous sequences are expected to exhibit more similarity at each locus, and thus will most likely be aligned in the same contig. Homology assessment requires sequence comparison examined in a phylogenetic context. Such an analysis was performed for Spartina for the granule-bound starch synthase I (Waxy) gene (Fortune et al., 2007). Molecular cloning, sequencing, and phylogenetic analyses allowed detection of paralogous, homoeologous and orthologous copies. In S. alterniflora, three homoeologous waxy copies were detected, exhibiting substitution rates ranging from 0.0218 to 0.0479. When analyzing sequence polymorphism among the assembled reads at four putative homologous loci between S. maritima and S. alterniflora, we found at each of these loci four different haplotypes that include two divergent sequences and two other less divergent variants. These results suggest the presence of two expressed homoeologous sequences with, respectively, two allelic variants; complementary phylogenetic analyses involving tetraploid Spartina species and outgroups will help to elucidate the evolutionary origin of these different sequences. As S. maritima and S. alterniflora are hexaploid, up to three duplicated homoeologs may be expected per locus. The fact that only two homoeologs were encountered in the analyzed transcripts might result from either homoeologous silencing as observed in the various cases of subfunctionalization reported in allopolyploids (reviewed in Osborn et al., 2003; Adams and Wendel, 2005; Doyle et al., 2008), from physical loss of the duplicated copies that may occur more or less rapidly following polyploid speciation (for example, Gaeta et al., 2007; Tate et al., 2009; Koh et al., 2010) or from homoeologous recombination (Cifuentes et al., 2010; Salmon et al., 2010; Gaeta and Pires, 2010). For the Waxy gene mentioned above, Fortune et al. (2007) found a variable number of retained copies per homologous locus. Two paralogs (A and B) were identified in the genus Spartina, only one B copy was found in S. maritima, whereas three distinct B copies were encountered in S. alterniflora. The A copy was apparently lost in these two species but is maintained in the hexaploid S. foliosa, which is sister species to S. alterniflora.
Conclusions
NGS technologies open new opportunities to screen large sets of genes and their evolution in polyploid species (Buggs et al., 2012). This first reference transcriptome, coupled with ongoing studies in our laboratory, involving deeper coverage from (Illumina INC., San Diego, CA, USA) RNA-Seq, and high-throughput genomic DNA sequencing, will facilitate a more accurate estimate of the level of duplicated homoeologous gene retention and relative expression in the hexaploid Spartina species and their hybrid and allopolyploid derivatives, in controlled and natural conditions. The analysis of the retained gene copies will also shed light into the origin of the hexaploid lineage and improve our understanding of the deepest Spartina history.
Data archiving
Data have been deposited at Genbank (Sequence Read Archive SRA) under accession references SRP015701 and SRP015702 for Spartina maritima and Spartina alterniflora, respectively.
Acknowledgments
This work is being developed in the frame of the International Associated Laboratory ‘Ecological Genomics of Polyploidy' supported by CNRS (INEE, UMR CNRS 6553 Ecobio), University of Rennes 1 and Iowa State University (Ames, USA). Sequencing was supported by Genoscope funds (GENOSPART Project). This work benefited from BioGenouest (Environmental and Functional Genomics) and Genouest (Bioinformatics) Plateform facilities. J Ferreira de Carvalho benefited from a PhD grant (ARED EVOSPART) from the Regional Council of Brittany. B Mable, JF Wendel and one anonymous reviewer are thanked for their helpful comments on an earliest version of this manuscript.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Adams JB, Bate GC. Ecological implications of tolerance of salinity and inundation by Spartina maritima. Aquat Bot. 1995;52:183–191. [Google Scholar]
- Adams KL, Wendel JF. Novel patterns of gene expression in polyploid plants. Trends Genet. 2005;21:539–543. doi: 10.1016/j.tig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Ainouche ML, Baumel A, Salmon A. Spartina anglica C. E. Hubbard: a natural model system for analysing early evolutionary changes that affect allopolyploid genomes. Biol J Linn Soc Lond. 2004a;82:475–484. [Google Scholar]
- Ainouche ML, Baumel A, Salmon A, Yannic G. Hybridization, polyploidy and speciation in Spartina (Poaceae) New Phytol. 2004b;161:165–172. [Google Scholar]
- Ainouche ML, Chelaifa H, Ferreira de Carvalho J, Bellot S, Ainouche AK, Salmon A.2012Polyploid evolution in Spartina: dealing with highly redundant genomesIn: Soltis PS, Soltis DE (eds).Polyploidy and Genome Evolution Springer: Berlin, Heidelberg; pp 225–244. [Google Scholar]
- Ainouche ML, Fortune PM, Salmon A, Parisod C, Grandbastien M-A, Fukunaga K, et al. Hybridization, polyploidy and invasion: lessons from Spartina (Poaceae) Biol Invasions. 2009;11:1159–1173. [Google Scholar]
- Alagna F, D'Agostino N, Torchia L, Servili M, Rao R, Pietrella M, et al. Comparative 454 pyrosequencing of transcripts from two olive genotypes during fruit development. BMC Genomics. 2009;10:399. doi: 10.1186/1471-2164-10-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andersen GR, Nissen P, Nyborg J. Elongation factors in protein biosynthesis. Trends Biochem Sci. 2003;28:434–441. doi: 10.1016/S0968-0004(03)00162-2. [DOI] [PubMed] [Google Scholar]
- Ayres DR, Smith DL, Zaremba K, Klohr S, Strong DR. Spread of exotic cordgrasses and hybrids (Spartina sp.) in the tidal marshes of San Francisco Bay, California, USA. Biol Invasions. 2004;6:221–231. [Google Scholar]
- Baisakh N, Subudhi PK, Parami NP. cDNA-AFLP analysis reveals differential gene expression in response to salt stress in a halophyte Spartina alterniflora Loisel. Plant Sci. 2006;170:1141–1149. [Google Scholar]
- Baisakh N, Subudhi PK, Varadwaj P. Primary responses to salt stress in a halophyte, smooth cordgrass (Spartina alterniflora Loisel.) Funct Integr Genomics. 2008;8:287–300. doi: 10.1007/s10142-008-0075-x. [DOI] [PubMed] [Google Scholar]
- Barakat A, DiLoreto D, Zhang Y, Smith C, Baier K, Powell W, et al. Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biol. 2009;9:51. doi: 10.1186/1471-2229-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS. SNP discovery via 454 transcriptome sequencing. Plant J. 2007;51:910–918. doi: 10.1111/j.1365-313X.2007.03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumel A, Ainouche M, Kalendar R, Schulman AH. Retrotransposons and genomic stability in populations of the young allopolyploid species Spartina anglica CE Hubbard (Poaceae) Mol Biol Evol. 2002a;19:1218–1227. doi: 10.1093/oxfordjournals.molbev.a004182. [DOI] [PubMed] [Google Scholar]
- Baumel A, Ainouche ML, Bayer RJ, Ainouche AK, Misset MT. Molecular phylogeny of hybridizing species from the genus Spartina Schreb. (Poaceae) Mol Phylogenet Evol. 2002b;22:303–314. doi: 10.1006/mpev.2001.1064. [DOI] [PubMed] [Google Scholar]
- Baumel A, Ainouche ML, Levasseur JE. Molecular investigations in populations of Spartina anglica C.E. Hubbard (Poaceae) invading coastal Brittany (France) Mol Ecol. 2001;10:1689–1701. doi: 10.1046/j.1365-294x.2001.01299.x. [DOI] [PubMed] [Google Scholar]
- Baumel A, Ainouche ML, Misset MT, Gourret JP, Bayer RJ. Genetic evidence for hybridization between the native Spartina maritima and the introduced Spartina alterniflora (Poaceae) in South-West France: Spartina x neyrautii re-examined. Plant Syst Evol. 2003;237:87–97. [Google Scholar]
- Bellin D, Ferrarini A, Chimento A, Kaiser O, Levenkova N, Bouffard P, et al. Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC genomics. 2009;10:555. doi: 10.1186/1471-2164-10-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggs RJA, Chamala S, Wu W, Gao L, May GD, Schnable PS, et al. Characterization of duplicate gene evolution in the recent natural allopolyploid Tragopogon miscellus by next-generation sequencing and Sequenom iPLEX MassARRAY genotyping. Mol Ecol. 2010;19:132–146. doi: 10.1111/j.1365-294X.2009.04469.x. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Renny-Byfield S, Chester M, Jordon-Thaden IE, Viccini LF, Chamala S, et al. Next-generation sequencing and genome evolution in allopolyploids. Am J Bot. 2012;99:372–382. doi: 10.3732/ajb.1100395. [DOI] [PubMed] [Google Scholar]
- Bundock PC, Eliott FG, Ablett G, Benson AD, Casu RE, Aitken KS, et al. Targeted single nucleotide polymorphism (SNP) discovery in a highly polyploid plant species using 454 sequencing. Plant Biotechnol J. 2009;7:347–354. doi: 10.1111/j.1467-7652.2009.00401.x. [DOI] [PubMed] [Google Scholar]
- Campos JA, Herrera M, Biurrun I, Loidi J. The role of alien plants in the natural coastal vegetation in central-northern Spain. Biodivers Conserv. 2004;13:2275–2293. [Google Scholar]
- Castellanos E, Figueroa M, Davy A. Nucleation and facilitation in salt-marsh succession—interactions between Spartina maritima and Arthrocnemum perenne. J Ecol. 1994;82:239–248. [Google Scholar]
- Castillo JM, Leira-Doce P, Rubio-Casal AE, Figueroa E. Spatial and temporal variations in aboveground and belowground biomass of Spartina maritima (small cordgrass) in created and natural marshes. Estuar Coast Shelf Sci. 2008;56:2037–2042. [Google Scholar]
- Chelaifa H, Mahé F, Ainouche M. Transcriptome divergence between the hexaploid salt-marsh sister species Spartina maritima and Spartina alterniflora (Poaceae) Mol Ecol. 2010a;19:2050–2063. doi: 10.1111/j.1365-294X.2010.04637.x. [DOI] [PubMed] [Google Scholar]
- Chelaifa H, Monnier A, Ainouche M. Transcriptomic changes following recent natural hybridization and allopolyploidy in the salt marsh species Spartina × townsendii and Spartina anglica (Poaceae) New Phytol. 2010b;186:161–174. doi: 10.1111/j.1469-8137.2010.03179.x. [DOI] [PubMed] [Google Scholar]
- Christin P-A, Petitpierre B, Salamin N, Büchi L, Besnard G. Evolution of C4 phosphoenolpyruvate carboxykinase in grasses, from genotype to phenotype. Mol Biol Evol. 2009;26:357–365. doi: 10.1093/molbev/msn255. [DOI] [PubMed] [Google Scholar]
- Christin PA, Besnard G, Samaritani E, Duvall MR, Hodkinson TR, Savolainen V, et al. Oligocene CO2 decline promoted C4 photosynthesis in grasses. Curr Biol. 2008;18:37–43. doi: 10.1016/j.cub.2007.11.058. [DOI] [PubMed] [Google Scholar]
- Cifuentes M, Grandont L, Moore G, Chèvre AM, Jenczewski E. Genetic regulation of meiosis in polyploid species: new insights into an old question. New Phytol. 2010;186:29–36. doi: 10.1111/j.1469-8137.2009.03084.x. [DOI] [PubMed] [Google Scholar]
- Civille JC, Sayce K, Smith SD, Strong DR. Reconstructing a century of Spartina alterniflora invasion with historical records and contemporary remote sensing. Ecoscience. 2005;12:330–338. [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Craft JA, Gilbert JA, Temperton B, Dempsey KE, Ashelford K, Tiwari B, et al. Pyrosequencing of Mytilus galloprovincialis cDNAs: tissue-specific expression patterns. PLoS One. 2010;5:e8875. doi: 10.1371/journal.pone.0008875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daehler CC, Strong DR. Status, prediction and prevention of introduced cordgrass Spartina spp. invasions in Pacific estuaries, USA. Biol Conserv. 1996;78:51–58. [Google Scholar]
- Délano-Frier J, Aviles-Arnaut H, Casarrubias-Castillo K, Casique-Arroyo G, Castrillon-Arbelaez P, Herrera-Estrella L, et al. Transcriptomic analysis of grain amaranth (Amaranthus hypochondriacus) using 454 pyrosequencing: comparison with A. tuberculatus, expression profiling in stems and in response to biotic and abiotic stress. BMC genomics. 2011;12:363. doi: 10.1186/1471-2164-12-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, et al. Evolutionary genetics of genome merger and doubling in plants. Annu Rev Genet. 2008;42:443–461. doi: 10.1146/annurev.genet.42.110807.091524. [DOI] [PubMed] [Google Scholar]
- Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2010;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom R, Slate J, Horsburgh GJ, Birkhead T, Burke T. Comparison between normalised and unnormalised 454-sequencing libraries for small-scale RNA-Seq studies. Comp Funct Genomics. 2012;2012:1–8. doi: 10.1155/2012/281693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred II error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ferris C, King RA, Gray AJ. Molecular evidence for the maternal parentage in the hybrid origin of Spartina anglica. Mol Ecol. 1997;6:185–187. [Google Scholar]
- Feuillet C, Keller B. Comparative genomics in the grass family: molecular characterization of grass genome structure and evolution. AoB Plants. 2002;89:3–10. doi: 10.1093/aob/mcf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune PM, Schierenbeck K, Ayres D, Bortolus A, Catrice O, Brown S, et al. The enigmatic invasive Spartina densiflora: A history of hybridizations in a polyploidy context. Mol Ecol. 2008;17:4304–4316. doi: 10.1111/j.1365-294X.2008.03916.x. [DOI] [PubMed] [Google Scholar]
- Fortune PM, Schierenbeck KA, Ainouche AK, Jacquemin J, Wendel JF, Ainouche ML. Evolutionary dynamics of Waxy and the origin of hexaploid Spartina species (Poaceae) Mol Phylogenet Evol. 2007;43:1040–1055. doi: 10.1016/j.ympev.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Franssen S, Shrestha R, Brautigam A, Bornberg-Bauer E, Weber A. Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics. 2011;12:227. doi: 10.1186/1471-2164-12-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeta RT, Pires JC. Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytol. 2010;186:18–28. doi: 10.1111/j.1469-8137.2009.03089.x. [DOI] [PubMed] [Google Scholar]
- Gaeta RT, Pires JC, Iniguez-Luy F, Leon E, Osborn TC. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell. 2007;19:3403–3417. doi: 10.1105/tpc.107.054346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W, Song Y, Zhang C, Zhang Y, Burlingame AL, Guo Y. Proteomic analyses of apoplastic proteins from germinating Arabidopsis thaliana pollen. Biochim Biophys Acta. 2011;1814:1964–1973. doi: 10.1016/j.bbapap.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedye K, Gonzalez-Hernandez J, Ban Y, Ge X, Thimmapuram J, Sun F, et al. Investigation of the transcriptome of prairie cord grass, a new cellulosic biomass crop. Plant Genome. 2010;3:69. [Google Scholar]
- Gotoh T, Terada K, Oyadomari S, Mori M. Hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- Groves H, Groves J. Spartina townsendii Nobis. Rep Bot Soc Exch Club Bri Id. 1880;1:37. [Google Scholar]
- Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale MC, McCormick CR, Jackson JR, DeWoody JA. Next-generation pyrosequencing of gonad transcriptomes in the polyploid lake sturgeon (Acipenser fulvescens): the relative merits of normalization and rarefaction in gene discovery. BMC Genomics. 2009;10:203. doi: 10.1186/1471-2164-10-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RA, Jones SJM. The new paradigm of flow cell sequencing. Genome Res. 2008;18:839–846. doi: 10.1101/gr.073262.107. [DOI] [PubMed] [Google Scholar]
- Hudson ME. Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol Ecol Resour. 2008;8:3–17. doi: 10.1111/j.1471-8286.2007.02019.x. [DOI] [PubMed] [Google Scholar]
- Humphreys JM, Chapple C. Rewriting the lignin roadmap. Curr Opin Plant Biol. 2002;5:224–229. doi: 10.1016/s1369-5266(02)00257-1. [DOI] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch D. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilut DC, Coate JE, Luciano AK, Owens TG, May GD, Farmer A, et al. A comparative transcriptomic study of an allotetraploid and its diploid progenitors illustrates the unique advantages and challenges of RNA-seq in plant species. Am J Bot. 2012;99:383–396. doi: 10.3732/ajb.1100312. [DOI] [PubMed] [Google Scholar]
- Koh J, Soltis P, Soltis D. Homeolog loss and expression changes in natural populations of the recently and repeatedly formed allotetraploid Tragopogon mirus (Asteraceae) BMC Genomics. 2010;11:97. doi: 10.1186/1471-2164-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Liao C-h, Zhang X-d, Chen H-l, Wang Q, Chen Z-y, et al. Spartina alterniflora invasions in the Yangtze River estuary, China: an overview of current status and ecosystem effects. Ecol Eng. 2009;35:511–520. [Google Scholar]
- Liu J-J, Ekramoddoullah AKM. The family 10 of plant pathogenesis-related proteins: their structure, regulation, and function in response to biotic and abiotic stresses. Physiol Mol Plant Pathol. 2006;68:3–13. [Google Scholar]
- Logacheva M, Kasianov A, Vinogradov D, Samigullin T, Gelfand M, Makeev V, et al. De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum) BMC Genomics. 2011;12:30. doi: 10.1186/1471-2164-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SP, Incoll LD, Woolhouse HW. C4 photosynthesis in plants from cool temperate regions, with particular reference to Spartina × townsendii. Nature. 1975;257:622–624. [Google Scholar]
- Marchant C, Goodman P. Spartina maritima (Curtis) Fernald. J Ecol. 1969;57:287–291. [Google Scholar]
- Marchant CJ. Evolution in Spartina (Gramineae). II. Chromosomes, basic relationships and the problem of Spartina × townsendii. Biol J Linn Soc Lond. 1968;60:381–409. [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricle BR, Cobos DR, Campbell CS. Biophysical and morphological leaf adaptations to drought and salinity in salt marsh grasses. Environ Exp Bot. 2007;60:458–467. [Google Scholar]
- Maricle BR, Crosier JJ, Bussiere BC, Lee RW. Respiratory enzyme activities correlate with anoxia tolerance in salt marsh grasses. J Exp Mar Bio Ecol. 2006;337:30–37. [Google Scholar]
- Maricle BR, Koteyeva NK, Voznesenskaya EV, Thomasson JR, Edwards GE. Diversity in leaf anatomy, and stomatal distribution and conductance, between salt marsh and freshwater species in the C4 genus Spartina (Poaceae) New Phytol. 2009;184:216–233. doi: 10.1111/j.1469-8137.2009.02903.x. [DOI] [PubMed] [Google Scholar]
- Maricle BR, Lee RW. Aerenchyma development and oxygen transport in the estuarine cordgrasses Spartina alterniflora and S. anglica. Aquat Bot. 2002;74:109–120. [Google Scholar]
- Martinez-Dominguez D, Heras MAdelas, Navarro F, Torronteras R, Cordoba F. Efficiency of antioxidant response in Spartina densiflora: an adaptative success in a polluted environment. Environ Exp Bot. 2008;62:69–77. [Google Scholar]
- Mateos-Naranjo E, Redondo-Gomez S, Cambrolle J, Luque T, Figueroa ME. Growth and photosynthetic responses to zinc stress of an invasive cordgrass, Spartina densiflora. Plant Biol. 2008;10:754–762. doi: 10.1111/j.1438-8677.2008.00098.x. [DOI] [PubMed] [Google Scholar]
- Mobberley DG. Taxonomy and distribution of the genus Spartina. Iowa State Coll J Sci. 1956;30:471–574. [Google Scholar]
- Moore MJ, Dhingra A, Soltis PS, Shaw R, Farmerie WG, Folta KM, et al. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2006;6:17. doi: 10.1186/1471-2229-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova O, Hirst M, Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet. 2009;10:135–151. doi: 10.1146/annurev-genom-082908-145957. [DOI] [PubMed] [Google Scholar]
- Munns R, Tester M. Mechanisms of salinity tolerance. Annu Rev Plant Biol. 2008;59:651–681. doi: 10.1146/annurev.arplant.59.032607.092911. [DOI] [PubMed] [Google Scholar]
- Novaes E, Drost D, Farmerie W, Pappas G, Grattapaglia D, Sederoff R, et al. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn TC, Pires JC, Birchler JA, Auger DL, Chen ZJ, Lee H-S, et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003;19:141–147. doi: 10.1016/s0168-9525(03)00015-5. [DOI] [PubMed] [Google Scholar]
- Otto SP. The evolutionary consequences of polyploidy. Cell. 2007;131:452–462. doi: 10.1016/j.cell.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Parchman T, Geist K, Grahnen J, Benkman C, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11:180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisod C, Salmon A, Zerjal T, Tenaillon M, Grandbastien M, Ainouche M. Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol. 2009;184:1003–1015. doi: 10.1111/j.1469-8137.2009.03029.x. [DOI] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- Poroyko V, Hejlek LG, Spollen WG, Springer GK, Nguyen HT, Sharp RE, et al. The maize root transcriptome by serial analysis of gene expression. Plant Physiol. 2005;138:1700–1710. doi: 10.1104/pp.104.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querné J, Ragueneau O, Poupart N. In situ biogenic silica variations in the invasive salt marsh plant, Spartina alterniflora: A possible link with environmental stress. Plant Soil. 2011;352:157–171. [Google Scholar]
- R Development Core Team 2011R: A language and environment for statistical computing R Foundation for Statistical Computing: Vienna, Austria; ISBN 3-900051-07-0, Available at http://www.R-project.org/ . [Google Scholar]
- Ramana Rao MV, Weindorf D, Breitenbeck G, Baisakh N. Differential expression of the transcripts of Spartina alterniflora Loisel. (smooth cordgrass) induced in response to petroleum hydrocarbon. Mol Biotechnol. 2011;51:18–26. doi: 10.1007/s12033-011-9436-0. [DOI] [PubMed] [Google Scholar]
- Raybould AF, Gray AJ, Lawrence MJ, Marshall DF. The evolution of Spartina anglica CE Hubbard (Gramineae): Origin and genetic-variability. Biol J Linn Soc Lond. 1991;43:111–126. [Google Scholar]
- Salmon A, Ainouche ML, Wendel JF. Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae) Mol Ecol. 2005;14:1163–1175. doi: 10.1111/j.1365-294X.2005.02488.x. [DOI] [PubMed] [Google Scholar]
- Salmon A, Flagel L, Ying B, Udall JA, Wendel JF. Homoeologous nonreciprocal recombination in polyploid cotton. New Phytol. 2010;186:123–134. doi: 10.1111/j.1469-8137.2009.03093.x. [DOI] [PubMed] [Google Scholar]
- Song Y, Zhang C, Ge W, Zhang Y, Burlingame AL, Guo Y. Identification of NaCl stress-responsive apoplastic proteins in rice shoot stems by 2D-DIGE. J Proteomics. 2011;74:1045–1067. doi: 10.1016/j.jprot.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasachary S, Dida M, Gale M, Devos K. Comparative analyses reveal high levels of conserved colinearity between the finger millet and rice genomes. Theor Appl Genet. 2007;115:489–499. doi: 10.1007/s00122-007-0582-5. [DOI] [PubMed] [Google Scholar]
- Subudhi PK, Baisakh N. Spartina alterniflora Loisel, a halophyte grass model to dissect salt stress tolerance in vitro. Cell Dev Biol Plant. 2011;47:441–457. [Google Scholar]
- Sun C, Li Y, Wu Q, Luo H, Sun Y, Song J, et al. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics. 2010;11:262. doi: 10.1186/1471-2164-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarbreck SM, Lindquist EA, Ackerly DD, Andersen GL. Analysis of leaf and root transcriptomes of soil-grown Avena barbata plants. Plant Cell Physiol. 2011;52:317. doi: 10.1093/pcp/pcq188. [DOI] [PubMed] [Google Scholar]
- Takeda H, Kotake T, Nakagawa N, Sakurai N, Nevins DJ. Expression and function of cell wall-bound cationic peroxidase in asparagus somatic embryogenesis. Plant Physiol. 2003;131:1765–1774. doi: 10.1104/pp.102.014654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate J, Joshi P, Soltis K, Soltis P, Soltis D. On the road to diploidization? Homoeolog loss in independently formed populations of the allopolyploid Tragopogon miscellus (Asteraceae) BMC Plant Biol. 2009;9:80. doi: 10.1186/1471-2229-9-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Zhang L, Zhang J, Song Y, Guo Y. Differential proteomic analysis of soluble extracellular proteins reveals the cysteine protease and cystatin involved in suspension-cultured cell proliferation in rice. Biochim Biophys Acta. 2009;1794:459–467. doi: 10.1016/j.bbapap.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Travis SE, Proffitt CE, Ritland K. Population structure and inbreeding vary with successional stage in created Spartina alterniflora marshes. Ecol Appl. 2004;14:1189–1202. [Google Scholar]
- Vega-Arreguin J, Ibarra-Laclette E, Jimenez-Moraila B, Martinez O, Vielle-Calzada J, Herrera-Estrella L, et al. Deep sampling of the Palomero maize transcriptome by a high throughput strategy of pyrosequencing. BMC Genomics. 2009;10:299. doi: 10.1186/1471-2164-10-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheat CW. Rapidly developing functional genomics in ecological model systems via 454 transcriptome sequencing. Genetica. 2008;138:433–451. doi: 10.1007/s10709-008-9326-y. [DOI] [PubMed] [Google Scholar]
- Wicker T, Schlagenhauf E, Graner A, Close T, Keller B, Stein N. 454 sequencing put to the test using the complex genome of barley. BMC Genomics. 2006;7:275. doi: 10.1186/1471-2164-7-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Hu L, Hurek T, Reinhold-Hurek B. Global characterization of the root transcriptome of a wild species of rice, Oryza longistaminata, by deep sequencing. BMC Genomics. 2010;11:705. doi: 10.1186/1471-2164-11-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannic G, Baumel A, Ainouche M. Uniformity of the nuclear and chloroplast genomes of Spartina maritima (Poaceae), a salt-marsh species in decline along the Western European Coast. Heredity. 2004;93:182–188. doi: 10.1038/sj.hdy.6800491. [DOI] [PubMed] [Google Scholar]
- Yoo M-J, Szadkowski E, Wendel JF.2012Homoeolog expression bias and expression level dominance in allopolyploid cotton Hereditydoi: 10.1038/hdy.2012.94 [DOI] [PMC free article] [PubMed]
- Yu J, Wang J, Lin W, Li S, Li H, Zhou J, et al. The genomes of Oryza sativa: a history of duplications. PLoS Biol. 2005;3:e38. doi: 10.1371/journal.pbio.0030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JK. Plant salt tolerance. Trends Plant Sci. 2001;6:66–71. doi: 10.1016/s1360-1385(00)01838-0. [DOI] [PubMed] [Google Scholar]
- Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32:e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.