

Abstract
Although α-1,3-glucan is one of the major cell wall polysaccharides in filamentous fungi, the physiological roles of α-1,3-glucan remain unclear. The model fungus Aspergillus nidulans possesses two α-1,3-glucan synthase (AGS) genes, agsA and agsB. For functional analysis of these genes, we constructed several mutant strains in A. nidulans: agsA disruption, agsB disruption, and double-disruption strains. We also constructed several CagsB strains in which agsB expression was controlled by the inducible alcA promoter, with or without the agsA-disrupting mutation. The agsA disruption strains did not show markedly different phenotypes from those of the wild-type strain. The agsB disruption strains formed dispersed hyphal cells under liquid culture conditions, regardless of the agsA genetic background. Dispersed hyphal cells were also observed in liquid culture of the CagsB strains when agsB expression was repressed, whereas these strains grew normally in plate culture even under the agsB-repressed conditions. Fractionation of the cell wall based on the alkali solubility of its components, quantification of sugars, and 13C-NMR spectroscopic analysis revealed that α-1,3-glucan was the main component of the alkali-soluble fraction in the wild-type and agsA disruption strains, but almost no α-1,3-glucan was found in the alkali-soluble fraction derived from either the agsB disruption strain or the CagsB strain under the agsB-repressed conditions, regardless of the agsA genetic background. Taken together, our data demonstrate that the two AGS genes are dispensable in A. nidulans, but that AgsB is required for normal growth characteristics under liquid culture conditions and is the major AGS in this species.
Introduction
The fungal cell wall is a complex structure that is essential for the maintenance of cellular shape and integrity, prevention of cell lysis, and protection against adverse environmental conditions. Fungal cells constantly remodel their rigid structure during growth and development. Proper cell wall architecture requires several cell wall components that are mainly composed of polysaccharides: α-glucans (α-1,3-glucan and α-1,4-glucan, which are polysaccharides composed of D-glucose), β-glucans (β-1,3-glucan and β-1,6-glucan), mannan, and chitin [1]–[4]. Because of the biological importance and structural specificity of the fungal cell wall, inhibitors that affect the fungal cell wall have been considered ideal drugs against both mammalian- and plant-pathogenic fungi for many years. Indeed, the group of β-1,3-glucan synthase inhibitors called “echinocandins”, which include anidulafungin, caspofungin, and micafungin, have been used commercially to control a variety of fungal pathogens [5].
Cell wall biogenesis and its regulation by cell signaling have been well characterized in the yeast Saccharomyces cerevisiae [6], [7]. The activation of the cell wall integrity signaling (CWIS) pathway leads to activation of the downstream mitogen-activated protein kinase (MAPK) cascade via Pkc1p and the small G protein Rho1p, which acts upon Pkc1p [8]. The MAPK Mpk1 protein activates the transcription factor Rlm1p, which regulates the transcription of at least 25 genes involved in cell wall biogenesis, including β-1,3-glucan synthase genes and chitin synthase genes [9]. In the model filamentous fungus Aspergillus nidulans, Fujioka et al. [10] constructed disruptants of mpkA and rlmA (orthologues of S. cerevisiae MPK1 and RLM1, respectively), as well as disruptants of Answi4 and Answi6 (orthologues of S. cerevisiae SWI4 and SWI6, which together encode the Mpk1p-activating transcription factor Swi4p–Swi6p complex). The transcription of most cell wall–related genes, except for two α-1,3-glucan synthase (AGS) genes (agsA and agsB), is transiently up-regulated in these mutants by treatment with the β-1,3-glucan synthase inhibitor micafungin; thus, transcription of the β-1,3-glucan synthase gene fksA and of several chitin synthase genes is independent of RlmA and AnSwi4/AnSwi6, suggesting that their transcription is regulated by a non-MpkA pathway. The transcription of agsA is weakly up-regulated in mpkAΔ and rlmAΔ strains, and the transcription of agsB depends mainly on MpkA–RlmA signaling [10]. Based on these results, Fujioka et al. [10] concluded that the transcriptional regulation of the α-1,3-glucan synthase genes agsA and agsB was specifically regulated by the MpkA pathway in A. nidulans, and that regulation of cell wall–related genes via CWIS in A. nidulans differed markedly from that in S. cerevisiae [10].
Among the Aspergillus species, the functions of α-1,3-glucan synthase genes have been studied in A. fumigatus and A. niger [11]–[14]. Aspergillus fumigatus contains three AGS genes, ags1 to ags3. Aspergillus fumigatus ags1, which is an orthologue of A. nidulans agsB, is involved in the formation of 50% of the cell wall α-1,3-glucan, whereas disruption of ags2, which is an orthologue of agsA, had no detectable effect on glucan levels [11]. Disruption of the third gene, ags3, which has no orthologue in A. nidulans, results in overexpression of ags1, which may serve to compensate for the lost enzyme activity and maintain normal cell wall composition [12]. In addition, disruption of ags3 in A. fumigatus causes hypervirulence, whereas the disruption of ags1 and ags2 did not affect virulence [11], [12]. A. niger has five α-1,3-glucan synthases encoded by agsA to agsE. The expression of agsA (an orthologue of A. fumigatus ags3) and agsE (an orthologue of A. fumigatus ags1 and A. nidulans agsB) was induced in the presence of cell wall stress–inducing compounds such as calcofluor white (CFW), sodium dodecyl sulfate, and caspofungin [13]. More recently, a triple-mutant strain of A. fumigatus lacking the three α-1,3-glucan synthase genes (ags1, ags2, and ags3) was generated, and growth of the triple mutant in plate culture was similar to that of the parental strain [14]. The triple mutant showed slightly decreased conidiogenesis, as did the single ags1 and ags2 mutants [11], [14], and the lack of cell wall α-1,3-glucan led to an increase in β-1,3-glucan and chitin levels in mycelia of the triple mutant [14]. Despite these studies of the functions of α-1,3-glucan synthase genes, the biological roles of α-1,3-glucan are only partly understood.
The importance of cell wall α-1,3-glucan in relation to fungal virulence has been studied in several pathogenic fungi. In the opportunistic pathogen Cryptococcus neoformans, loss of cell wall α-1,3-glucan leads to loss of the surface capsule [15]. The capsule appears to promote pathogenesis in the wild-type strain, since mutants that lack cell wall α-1,3-glucan are unable to grow in a mouse model of infection [16]. A correlation between reduction in α-1,3-glucan levels and virulence has also been observed in several dimorphic fungal pathogens, including Histoplasma capsulatum, Paracoccidioides brasiliensis, and Blastomyces dermatitidis [17], [18], [19]. Rappleye et al. [20] reported that α-1,3-glucan conceals β-glucans on the cell wall of H. capsulatum from dectin-1-mediated detection by immune cells, blocking host recognition of fungal invasion. Rappleye et al. [20] assumed that pathogenic fungi have evolved such unique stealth mechanisms because they conceal the immunostimulatory molecular patterns of the pathogen from recognition via leukocyte receptors, which disrupts host immune responses. In the rice blast fungus Magnaporthe grisea, α-1,3-glucan masks β-1,3-glucan and chitin in the cell wall of the infectious hyphae; in this way, α-1,3-glucan protects the fungal cell wall from digestive enzymes produced by plant cells during infection and inhibits recognition by host cells [21]. Moreover, expression of the MgAGS1 AGS gene of M. grisea depends fully on Mps1p MAPK, which is an orthologue of Mpk1p in S. cerevisiae, whereas the expression of genes that encode other cell wall–related enzymes, such as β-1,3-glucan synthase and chitin synthase, is Mps1p-independent in M. grisea [21], as is observed in A. nidulans [10]. Thus, the regulation of α-1,3-glucan biosynthesis by the CWIS pathway appears to be conserved in filamentous fungi.
In the present study, we carried out functional analysis of the two α-1,3-glucan synthase genes in A. nidulans: agsA and agsB. We constructed disruption strains of agsA and agsB (the DagsA and DagsB strains, respectively), an agsA agsB double-disruption mutant (the DagsA-DagsB strain), and a conditional-agsB strain (CagsB) in which agsB expression is conditionally regulated by the alcA promoter. We also created a strain in which CagsB was combined with a disruption of agsA (the CagsB-DagsA strain). Using these strains, we performed transcriptional analysis of cell wall–related genes and biochemical analyses of cell wall components. These analyses revealed that almost all of the cell wall α-1,3-glucan was lost in the agsB disruption mutants, regardless of the presence or absence of agsA. We also observed that most of the cell wall α-1,3-glucan was lost in the CagsB strain under agsB-repressing conditions, regardless of the agsA genetic background. Based on these results, we discuss the relationship of the biological roles of α-1,3-glucan and the two AGS genes in A. nidulans.
Materials and Methods
Strains and Growth Media
Throughout this study, A. nidulans ABPU1 (biA1, pyrG89, wA3, argB2, pyroA4) with ligDΔ (ligDΔ::ptrA) was used for all genetic manipulations and for the control strain (CNT). For the A. nidulans agsA and agsB disruption studies, ABPU1 cells were transformed with the gene replacement constructs described in the following section. The alcA promoter–driven agsB allele was introduced by transformation of ABPU1 with a replacement cassette containing the pyrG marker. Czapek-Dox (CD) medium was used for standard culture [10]. CD medium containing 100 mM threonine and 0.1% fructose instead of 1% glucose as a carbon source (designated CDTF medium henceforth) was used for up-regulation of alcA promoter–driven agsB [22]. To investigate gene expression, conidia (5×105 cells/mL) were inoculated into liquid medium (CD or CDTF) and cultured at 37°C for 24 h, and then mycelia were harvested. DNA extraction, RNA extraction, reverse transcriptase reactions, and transformation analysis were all performed as described previously [23].
Construction of Gene Replacement Cassettes
The gene replacement cassettes for the agsA and agsB disruption mutants (DagsA and DagsB) and the alcA promoter–driven agsB (CagsB) strain were constructed by PCR fusion following a strategy similar to that used by Izumitsu et al. [24]. The sequences of all primers used in this study are listed in Table S1. To disrupt a gene in A. nidulans, we replaced it with an A. oryzae pyrG or argB gene as the selectable marker. Fungal transformations were performed as described previously [10].
To construct the disruption cassette for the agsA gene, the first round of PCR amplified gene fragments containing the 5′ non-coding region (amplicon 1) and the coding region (amplicon 2) of agsA from the A. nidulans ABPU1 genomic DNA template, and amplified the pyrG gene (amplicon 3) from the A. oryzae genomic DNA template (Figure S1). Amplicon 1 was amplified with the primers agsA-LU and agsA-LL, amplicon 2 with agsA-RU and agsA-RL, and amplicon 3 with agsA-PU and agsA-PL. The primers agsA-LL, agsA-RU, agsA-PU, and agsA-PL were chimeric oligonucleotides; each contained a reverse-complement sequence for PCR fusion. The three resulting PCR products were gel-purified and used as substrates for a second round of PCR using the primers agsA-LU and agsA-RL to fuse the three separate fragments from the first round into a disruption cassette. All PCR reactions were performed using the GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA) using PrimeSTAR HS DNA polymerase (Takara, Tokyo, Japan). The resulting major PCR product was gel-purified and used to transform the ABPU1 strain.
To construct the disruption cassette for agsB, we used the same PCR fusion strategy. In the first round of PCR, the primers agsB-LU and agsB-LL (for amplicon 1), agsB-RU and agsB-RL (for amplicon 2), and argB-F and argB-R (for amplicon 3) were used (Table S1). The primers agsB-LU and agsB-RL were used for the second round of PCR. The resulting major PCR product was gel-purified and used to transform the ABPU1 strain (Figure S2).
To construct the gene replacement cassette for the alcA promoter–driven agsB (CagsB) strain, two fragments were amplified by PCR using A. nidulans ABPU1 genomic DNA as a template together with the primers listed in Table S1. The pyrG-alcA(p) cassette, which contains the A. nidulans pyrG marker and alcA promoter, was used as the PCR template with the primers CagsB-PU and CagsB-PL. The three fragments were used as substrates for the second round of PCR fusion. The resulting major PCR product was gel-purified and used to transform the ABPU1 strain (Figure S3).
To construct the disruption cassette for agsA in the CagsB strain, we used the same PCR fusion strategy. In the first round of PCR, the primers agsA-F and agsA-LL2 (for amplicon 1), agsA-RU2 and agsA-R (for amplicon 2), and argB-F and argB-R (for amplicon 3) were used (Table S1). The primers agsA-F and agsA-R were used for the second round of PCR. The resulting major PCR product was gel-purified and used to transform the ABPU1 strain.
Construction of the Complementation Strain of the agsB Disruptant
To construct the complementation strain of the agsB disruptant, we amplified the full-length agsB gene, including its native promoter region (2 kb upstream of the 5′ end of the open reading frame [ORF]) and the terminator region (0.4 kb downstream of the 3′ end of the ORF), by means of PCR using the primers agsB-LU and agsB-T400R-HindIII (Table S1). The restriction enzyme sites for HindIII were found 1.4 kb upstream of the 5′ end of the agsB ORF and were amplified using the primer agsB-T400R-HindIII. The PCR fragment was digested with HindIII, and then introduced into the HindIII site of pUCpyroA (kindly provided by H. Horiuchi of the University of Tokyo), which contained the A. nidulans pyroA gene as the selectable marker. The resulting plasmid (pPAagsB) was used to transform the agsB disruptant strain.
Quantitative RT- PCR
Real-time RT-PCR was performed using the Mini Opticon real-time PCR system (Bio-Rad Laboratories, Hercules, CA, USA) with SYBR Green detection, according to the manufacturer’s instructions. For reaction mixture preparation, the DyNAmo SYBR Green qPCR kit (Finnzymes Oy, Espoo, Finland) was used. We used the primer sets described previously [10] and listed in Table S1 for quantifying cell wall–related gene expression. An equivalent amount of cDNA, obtained from reverse transcription reactions using an equivalent amount of total RNA, was applied to each reaction mixture. The histone H2B gene was used as a normalization reference (an internal control) for target gene expression ratios. Statistical analyses were based on Welch’s t-test, and significance was defined as a P-value of <0.05.
Growth Inhibition Testing of Mutants
We evaluated the sensitivity of the strains to the cell wall stress–inducing compounds micafungin, CFW, and Congo Red (CR) by plotting the dose response for colony growth using the plate dilution method. The chemicals were added from 1000-fold concentrated stock solutions in water (micafungin and CR) or in 0.1 M NaOH (CFW). Conidial suspensions of each mutant (a total of 1×103 cells) were spotted on the centers of CD plates containing various concentrations of micafungin (0.001 and 0.01 µg/mL), CFW (5 and 10 µg/mL), and CR (1, 5, 20, 40, and 80 µg/mL). The dose response was determined 4 days after incubation at 37°C by plotting the mean diameters of the colonies on the treated media as a percentage of those on the control medium. Each experiment was performed in triplicate.
Assay for Adsorption of CR to Hyphae
Mycelia cultured in CD medium for 24 h (500 mg fresh weight) were suspended in 50 mL of CR solution (40 µg/mL), and the solutions were rotated for 1 h at room temperature. After the reaction, the mycelia were removed by filtration through a 70 µm cell strainer (BD Biosciences, San Jose, CA, USA), and the absorbance of the supernatants was measured (UVmini-1240, Shimazu Co., Ltd., Tokyo, Japan) at 500 nm; in this assay, high absorbance indicates that most of the CR remained in the supernatant instead of being adsorbed to the mycelia. Each experiment was performed in triplicate. Statistical analyses were based on Welch’s t-test, and significance was defined as a P-value of <0.05.
Assay for Cell Wall Susceptibility to Lysing Enzymes
Susceptibility of the fungal cell wall to Lysing Enzymes, a commercial preparation containing β-1,3-glucanase and chitinase (Sigma, St. Louis, MO, USA), was assayed according to the modified methods described in Fujikawa et al. [21]. Washed 1-d-old mycelia (30 mg fresh weight) grown in CD or CDTF medium at 37°C were resuspended in 1 mL of 0.8 M NaCl in sodium phosphate buffer (10 mM, pH 6.0) containing 10 mg/mL Lysing Enzymes. To determine the effect of additional α-1,3-glucanase on lysis of the fungal cell wall by Lysing Enzymes, 20 µg of purified α-1,3-glucanase from Bacillus circulans KA-304 [25] was added to the enzyme–buffer mixture. After 1, 2, and 4 h of incubation at 30°C, the number of protoplasts generated from the mycelia was counted using a hemocytometer (A106, SLGC, Tokyo, Japan). Mycelia incubated in 0.8 M NaCl in sodium phosphate buffer (10 mM, pH 6.0) containing no enzyme were used as controls.
Fractionation of Cell Walls by Alkali Treatment
The steps used for cell wall fractionation are summarized in Figure S4. Mycelia cultivated in liquid medium were collected by filtration, freeze-dried, and pulverized in liquid nitrogen, and then 2 g of the mycelial powder was suspended in 80 mL of 0.1 M phosphate buffer (pH 7.0). The mycelial suspension was autoclaved at 120°C for 60 min and centrifuged at 10,000×g for 15 min. The supernatant was retained, and the pellet was resuspended in 80 mL of 0.1 M phosphate buffer (pH 7.0), autoclaved, and centrifuged again at 10,000×g for 15 min. The supernatants from the first two centrifugations were combined, dialyzed against water, and lyophilized, resulting in the hot-water-soluble (HW) fraction. The pellet was treated with 2 M NaOH for 24 h at 4°C. After centrifugation of the reaction products (10,000×g, 15 min), the supernatant was designated as the alkali-soluble fraction (AS) and the pellet was designated as the alkali-insoluble fraction (AI). The alkali-soluble fraction was neutralized with acetic acid (17 M), dialyzed against water, and centrifuged (10,000×g, 15 min). The supernatant was designated AS1, and the precipitate as AS2. The AI fraction was suspended in water, neutralized (as above), and dialyzed against water. All fractions were freeze-dried and the weight of each fraction was measured.
Quantitative Determination of the Carbohydrate Composition of the Cell Wall Fractions
Each freeze-dried cell wall fraction (10 mg dry weight) was incubated in 0.2 mL of 12.5 M H2SO4 at 4°C for 16 h. Distilled water (4.8 mL) was added to the cell wall solution, resulting in a final concentration of 0.5 M H2SO4. After the cell wall was hydrolyzed at 100°C for 12 h, the hydrolysate was neutralized with barium carbonate and centrifuged to remove the resulting barium sulfate. The carbohydrate composition of the supernatant was determined using high-performance anion-exchange chromatography (HPAC) with a pulsed electrochemical detector and an anion-exchange column (Carbo PAC PA-1, 4×25 mm, Dionex, Sunnyvale, CA, USA) at a flow rate of 1 mL/min for 16 min. Isocratic elution was performed with 18 mM NaOH. The column was stabilized for 20 min before injection. To quantify the monosaccharides, we used glucose (Wako Pure Chemical Industries, Ltd., Osaka, Japan), mannose (Wako), galactose (Wako), glucosamine hydrochloride (Sigma), and galactosamine hydrochloride (Sigma) as standards.
13C-NMR Analysis of Cell Wall Fractions
The bacterial polysaccharide mutan, which was enzymatically synthesized by GTF1 of Streptococcus mutans and mainly composed of α-1,3-glucan [S. Yano et al., unpublished data], and the AS2 fractions of the control (wild-type) and agsA mutant strains (each 2 mg) were suspended in 540 µL of D2O and autoclaved at 120°C for 60 min. Then, 60 µL of 5 M NaOH was added to each suspension, and the fractions were sonicated and dissolved. One drop of Me2SO-d6 (deuterated dimethyl sulfoxide, DMSO-d6) was added to each solution, and the solutions were centrifuged (3000×g, 5 min). 13C-NMR spectra of the supernatants were obtained using a JEOL (Tokyo, Japan) JNM-ECX400P spectrometer at 400 MHz, at 35°C for 60 h, with an external standard (tetramethylsilane).
Treatment of the AS2 and AI Fractions with α-1,3-glucanase or β-1,3-glucanase
The AS2 and AI fractions (5 mg pure powder) from the control (CNT), agsB disruption, and CagsB strains were dissolved in 1 mL of potassium phosphate buffer (50 mM, pH 6.5) containing 10 µg of purified α-1,3-glucanase from Bacillus circulans KA-304 [25] or in 1 mL of acetate buffer (50 mM, pH 5.0) containing 500 µg of purified β-1,3-glucanase from A. niger (Sigma). The reaction was performed at 37°C for 12 h (α-1,3-glucanase) or at 55°C for 12 h (β-1,3-glucanase). After the reaction, the samples were centrifuged at 10,000×g for 10 min. The supernatants were retained, and each pellet was resuspended in the same buffer and enzyme mixture as before, incubated under the same conditions, and centrifuged as described above. The supernatants were removed and the procedure was performed a third time. The supernatants from the three centrifugations were combined and hydrolyzed with 2 M H2SO4. The liberated saccharides were quantified by means of HPAC with a pulsed electrochemical detector and an anion exchange column (Carbo PAC PA-1, 4×25 mm, Dionex) using glucose as the standard.
Linkage Analysis of Glucan
Pachyman, which is a β-1,3-glucan, the bacterial mutan, and the alkali-soluble (AS2) fraction of the control strain were methylated using the modified method of Ciucanu and Kerek [26]. Each fraction (0.5 mg) was suspended in NaOH/dimethyl sulfoxide (DMSO) solution (10 mg NaOH in 0.5 mL of DMSO), and 0.2 mL of methyl iodide was added. The mixture was stirred for 15 min in a closed vial at room temperature. Water (2 mL) and chloroform (0.5 mL) were then added, the contents of the vial were mixed, and the aqueous phase was removed. The chloroform layer was mixed again with 2 mL water, and the aqueous phase was removed. Then, 1 mL of methylbenzene was added to the chloroform phase and the mixture was dried down. The dried material was resuspended in 0.5 mL of the NaOH/DMSO solution, and the methylation procedure was repeated.
Next, 0.5 mL of 2 M trifluoroacetic acid was added to the dried material (the methylated sample), which was hydrolyzed at 90°C for 1 h. Subsequently, 1 mL of methylbenzene was added to the sample and evaporated to dryness. The hydrolyzed sample was mixed with 0.5 mL of 0.25 M NaBH4 and reduced by incubation for 16 h at room temperature. Acetic acid and methylbenzene (approximately 0.4 mL and 1 mL, respectively) were added to the reduced sample and the mixture was evaporated to dryness. Methanol (1 mL) was added to the dried material and the mixture was evaporated to dryness; this step was performed three times. Pyridine (0.2 mL) and acetic anhydride (0.2 mL) were added to the sample, which was then acetylated by heating at 90°C for 20 min. Subsequently, 1 mL of methylbenzene was added to the acetylated sample and evaporated to dryness. The sample was mixed with chloroform (0.5 mL) and water (2 mL×3 times); each time, the aqueous phase was removed. Then, 1 mL of methylbenzene was added to the final chloroform layer and evaporated to dryness, and the sample was resuspended in 50 mL of chloroform.
The methylated sugars were analyzed using a gas chromatography system (GC-2010/GCMS-QP2010; Shimazu) with an HP-5 column (Agilent Technologies, Inc., Santa Clara, CA, USA). The identification of the peaks was confirmed by comparison with information in the Shimazu mass spectral libraries.
Freeze-Substitution Electron Microscopy
Cells were collected from liquid media and snap-frozen by plunging them into a melting propane–isopentane mixture cooled with liquid nitrogen [27]. Specimens were then freeze-substituted in acetone containing 2% osmium tetroxide at –80°C, and embedded in epoxy resin. Ultrathin sections were cut to a thickness of 70 to 80 nm, stained with uranyl acetate and lead citrate [28], covered with Super support film (Nisshin EM, Tokyo, Japan), and examined using a JEM-1200EX transmission electron microscope (JEOL) at 80 kV.
Results
Isolation and Characterization of an A. nidulans agsA Disruptant (DagsA)
To investigate the role of the α-1,3-glucan synthase genes in A. nidulans, we first generated DagsA mutants with a disruption of the agsA gene. The agsA gene of the parental strain was disrupted using the targeted replacement method by selecting double-crossover events from homologous recombination. We constructed the disruption cassettes, which replaced the region between part of the promoter region of agsA and part of the coding region (including the start codon) of the A. oryzae pyrG gene as the selectable marker in A. nidulans, by means of PCR fusion and introduced the cassette into the parental strain (Figure S1). Correct integration of the cassettes and gene disruption were confirmed by means of PCR and Southern blot analysis (Figure S1). The results of quantitative RT-PCR revealed that the level of transcription of the agsA gene in the control strain was very low (Figure S5A and described below). Moreover, the transcripts of the agsA gene were scarcely detected in the agsA disruption strain (Figure S5A). The hyphal morphology, conidiation, phialide formation, and colony diameter of the agsA disruptants on CD plates were similar to those of the control strain (Figure S5B, S5C; data not shown). No significant differences in sensitivities to the cell wall stress–inducing compounds micafungin, CFW, and CR were observed between the agsA disruption strain and the control strain (Figure S5D). The susceptibility of the mycelia of the agsA disruption strain to Lysing Enzymes, a mixture that contains β-1,3-glucanase and chitinase, was not significantly different from that of the control strain (Figure S5E). In addition, the transcription levels of cell wall–related genes, such as the genes encoding β-glucan synthase and chitin synthase (data not shown), and the cell wall components that we examined were not altered in the agsA disruption strain compared with the control strain (described below). These results suggested that AgsA is not responsible for the biosynthesis of cell wall α-1,3-glucan under normal growth conditions.
Isolation of the agsB Disruption Strains, agsA agsB Double-disruption Strains, Conditional-agsB Strains, and Conditional-agsB with agsA Disruption Strains of A. nidulans
To analyze the functions of the agsB gene in A. nidulans, we constructed two types of strains: strains with disruption of the agsB gene (DagsB strains), and conditional-agsB (CagsB) strains in which expression of agsB was regulated by the A. nidulans alcA promoter. We isolated the agsB disruption strains by disrupting the agsB gene of the parental strain using the targeted replacement method. The disruption cassette, which replaced an agsB region containing part of the promoter region and part of the coding region (including the start codon) with the A. oryzae argB gene as the selectable marker, were constructed by means of PCR fusion and introduced into the control strain (Figure S2). Correct integration of the disruption cassette and successful gene disruption were confirmed by means of PCR and Southern blot analysis (Figure S2). To confirm whether the agsA gene could compensate for the disruption of agsB, we generated an agsA agsB double-disruption strain (DagsA-DagsB). The agsB gene of the agsA disruption strain was also disrupted using the targeted replacement method. Correct integration of the cassettes and gene disruption were confirmed by means of PCR and Southern blot analysis (data not shown).
Next, we isolated several CagsB strains. We constructed a replacement cassette that replaced the native agsB promoter (–1000 bp from the start codon) with the alcA promoter and that also contained the pyrG selectable marker, and introduced the cassette into the parental strain (Figure S3). The genomic structure of the resulting CagsB strains was confirmed by means of PCR and Southern blot analysis (Figure S3). Three independent mutant lines were established to provide replication in the experiments. To confirm the regulation of agsB expression in the CagsB strains, we performed transcriptional analysis by means of quantitative RT-PCR under several medium conditions. The alcA promoter has been reported to activate gene expression in the presence of carbon sources such as ethanol, glycerol, and threonine, and to repress it in the presence of glucose [29]. First, we tested the CagsB strains on CD (1% glucose) medium as the agsB-repressing conditions. After 24 h of culture in CD liquid medium, the CagsB strains exhibited significantly lower agsB expression than the control strain (Figure S6A, left). When the CagsB strains were cultured in CDTF medium (0.1 M threonine, 0.1% fructose to replace the glucose), agsB expression in the CagsB strains was considerably (and significantly) higher than that of the control strain (Figure S6A, right). These results showed that the alcA promoter in the CagsB strains successfully regulated agsB expression in response to differences in the carbon source in the medium. We also generated a CagsB strain with an agsA disruption (CagsB-DagsA). We constructed a disruption cassette that replaced an agsA region that included part of the promoter region and part of the coding region (including the start codon) by the A. oryzae argB gene as the selectable marker by means of PCR fusion and introduced the cassette into the CagsB strain. Correct integration of the cassette and gene disruption were confirmed by means of PCR and Southern blot analysis (data not shown).
The Growth Characteristics of the agsB Mutants
To understand the role of agsB, we examined the growth characteristics of the agsB mutant strains. The colonial growth rate and conidiation of the agsB disruption (DagsB) and double-disruption (DagsA-DagsB) strains on CD plates were similar to those of the control strain (Figure 1A; data not shown). In contrast to the results obtained with plate media, the growth characteristics of the agsB disruption and control strains were easily distinguishable in liquid culture (Figure 1B). The hyphae of the agsB disruption strain were obviously dispersed in liquid CD medium, whereas the control strain formed hyphal pellets (Figure 1B). The control strain showed an aggregation of hyphae in liquid culture (Figure 2, dark area), whereas the hyphae of the agsB disruption strain were scarcely aggregated (Figure 2). In contrast to the differences in the growth characteristics, the microscopic hyphal tip morphology and the phialide formation of the agsB disruption strain were similar to those of the control strain (Figure 2; data not shown). Also, the hyphae of the double-disruption strain were dispersed in liquid CD medium (Figure 1B).
Figure 1. Phenotypes of the agsB disruption and agsA agsB double-disruption strains.

(A) Mycelial growth of the control (CNT), agsB disruption (DagsB), and double-disruption (DagsA-DagsB) strains cultured on solid CD medium for 4 d. (B) Growth characteristics of the control (CNT), agsB disruption (DagsB), and double-disruption (DagsA-DagsB) strains. Conidia (final concentration, 5×105/mL) of each strain were inoculated into CD liquid medium and rotated at 160 rpm at 37°C for 18 h.
Figure 2. Hyphal morphology of the control and agsB disruption strains grown in CD liquid medium for 12 h at 37°C.

(A) Hyphal pellets of the control strain, showing hyphal aggregation (dark areas). Scale bar = 200 µm. (B) Hyphal tip morphology of the control strain. High magnification of the hyphal image in the white frame in panel A. Scale bar = 50 µm. (C) Dispersed hyphae of the agsB disruption strain (DagsB). Scale bar = 200 µm. (D) Hyphal tip morphology of the DagsB strain. High magnification of the hyphal image in the white frame in panel A. Scale bar = 50 µm.
To further confirm whether the phenotypic alterations in the agsB disruption strain were caused by disruption of the agsB gene, the growth characteristics of the CagsB strains were observed under agsB-repressing and -inducing conditions. No differences in colonial growth were observed between the CagsB and control strains on either CD (repressing conditions) or CDTF (inducing conditions) plates (Figure S6B). Conidiation on CD plates was not significantly different between the control and CagsB strains (Figure S6C). In liquid CD medium (agsB-repressing conditions), the hyphae of the CagsB strain were dispersed (Figure S6D, left), as in the case of liquid culture of the agsB disruption strains (Figure 1B). Under agsB-inducing conditions (liquid CDTF medium), both the CagsB and control strains formed hyphal pellets resembling those produced by the control strain cultured in liquid CD medium (Figure S6D, right). In addition, complementation of the agsB disruptants with the authentic agsB gene, including its native promoter, rescued the agsB disruptant and restored the wild-type phenotype (data not shown). These data indicated that the dispersed growth characteristic observed in the agsB disruption mutants was caused by disruption of the agsB gene.
The freeze-substitution electron microscopy assay revealed that the ultrastructures of the CagsB strains under agsB-repressing and agsB-inducing conditions were similar to those of the control strain (Figure S7). No significant differences in the thickness of the cell wall were observed between the control and CagsB strains (Table S2). In the CagsB with agsA disruptant strain (CagsB-DagsA), no obvious phenotypic alterations were observed compared with CagsB. The colonial growth rate and conidiation were indistinguishable between the CagsB-DagsA strain and the parental CagsB strain on both CD (agsB-repressing conditions) and CDTF (agsB-inducing conditions) plates (data not shown). The hyphae of the CagsB-DagsA strain were dispersed in liquid CD medium (agsB-repressing conditions in the CagsB-DagsA strain), similar to those seen in the parental CagsB, agsB disruption, and double-disruption strains (data not shown). These observations suggested that the agsA gene did not compensate for disruption or repression of agsB under our experimental conditions.
Expression Profiles of Genes Involved in Cell Wall Biosynthesis
To test whether the expression of genes associated with cell wall biogenesis was affected by the disruption of agsB or by the level of agsB expression, we analyzed the levels of transcription of several cell wall–related genes. We analyzed the transcriptional levels of agsA and agsB (α-1,3-glucan synthase genes); fksA (a β-1,3-glucan synthase gene); gelA and gelB (β-1,3-glucanosyl transferase genes); chsA, chsB, chsC, chsD, csmA, and csmB (chitin synthase genes); and gfaA (a glutamine-fructose-6-phosphate amidotransferase gene) by means of quantitative RT-PCR in the agsB disruption strain (DagsB; Figure 3) or in the CagsB strain (Figure 4) after treatment with agsB-repressing and inducing conditions. In the agsB disruption strain, in which agsB expression was scarcely detected (Figure 3), the expression of agsA increased significantly, although the basal level of agsA expression was still less than 1% of that of agsB in the control (Figures 3, S5). In addition, levels of transcription of several cell wall–related genes (gelA, chsA, and gfaA) were significantly up-regulated in the agsB disruption strain (Figure 3); however, transcription of gelB and csmB were significantly down-regulated in the disruptant. In the CagsB strain, the expression of agsA, fksA, gelA, chsA, and gfaA were significantly up-regulated under the agsB-repressing conditions (CD medium; Figure 4), as was the case in the agsB disruption strain for all except fksA. In contrast, under the agsB-inducing conditions (CDTF medium), the expression levels of fksA, gelB, chsD, and gfaA were significantly reduced in the CagsB strain (Figure 4); however, csmA was significantly up-regulated under these conditions. These results suggested that alteration of the α-1,3-glucan amount was counterbalanced by alterations in the amount of another cell wall component such as β-1,3-glucan or chitin, so we examined this possibility.
Figure 3. Expression of cell wall–related genes in the control (CNT) and agsB disruption (DagsB) strains.

Strains were grown in CD liquid medium at 37°C. Levels of transcription of the indicated genes were determined by means of quantitative RT-PCR of total RNA using the gene-specific primers reported previously by Fujioka et al. [10] (Table S1). Each value represents the ratio of expression relative to the histone H2B gene in each strain. Error bars represent the standard error of the mean calculated for three replicates (*P<0.05, **P<0.01).
Figure 4. Expression of cell wall–related genes in the control (CNT) and CagsB strains.

Strains were grown in CD liquid medium (agsB-repressing conditions) or CDTF liquid medium (agsB-inducing conditions) at 37°C. Levels of transcription of the indicated genes were determined by means of quantitative RT-PCR of total RNA using the gene-specific primers reported previously by Fujioka et al. [10] (Table S1). Each value represents the ratio of expression relative to the histone H2B gene in each strain. Bars represent the standard error of the mean calculated for at least three replicates (*P<0.05, **P<0.01).
Susceptibility to Cell Wall–Stress Compounds and Cell Wall–Degrading Enzymes in the agsB Mutants
To investigate the consequences of cell wall alteration caused by the disruption of agsB or by the control of agsB gene expression, we first tested the sensitivities of the mutant strains to cell wall stress–inducing compounds. The agsB disruption strain showed greater sensitivity than the control to CR, which interferes with cell wall assembly (Figure 5A). Under the agsB-repressing conditions, the CagsB strain also showed increased sensitivity to CR (Figure 5A). The DagsB and CagsB strains were barely able to grow on CD plates containing 80 µg/mL of CR, whereas the growth rate of the control strain was still approximately 20% of normal on CD containing 80 µg/mL of CR (Figure 5A). The median effective concentrations (EC50) of CR for the control, DagsB, and CagsB strains were 18.1, 11.8, and 8.9 µg/mL, respectively. No significant differences in the sensitivity to CR were observed in the DagsA-DagsB double-disruption strain compared with the DagsB strain or the CagsB-DagsA strain compared with the CagsB strain (Figure 5A). On CDTF plate medium (agsB-inducing conditions for the CagsB strain), no obvious differences in the sensitivity to CR were detected between the control and the CagsB strain. The minimum inhibitory concentration (MIC) for the CagsB strain was the same level as for the control strain (MIC >100 µg/mL; data not shown). Next, we measured the adsorption of CR to the hyphae of the control, DagsB, DagsA-DagsB, CagsB, and CagsB-DagsA strains. The amount of CR adsorbed to the hyphae of these agsB mutants grown in liquid CD culture medium was significantly greater than that of the control strain (i.e., low absorbance by the supernatant; Figure 5B). In contrast to the inhibitory effect seen with CR, the DagsB and DagsA-DagsB strains were not sensitive to either micafungin (an inhibitor of β-glucan synthase) or CFW (a chitin binding agent) (data not shown). Similarly, the CagsB and CagsB-DagsA strain were not sensitive to micafungin or CFW under either agsB-repressing or -inducing conditions (data not shown).
Figure 5. Sensitivity of the control (CNT), agsB disruption (DagsB), agsA agsB double disruption (DagsA-DagsB), CagsB, and CagsB with agsA disruption (CagsB-DagsA) strains to Congo Red (CR).
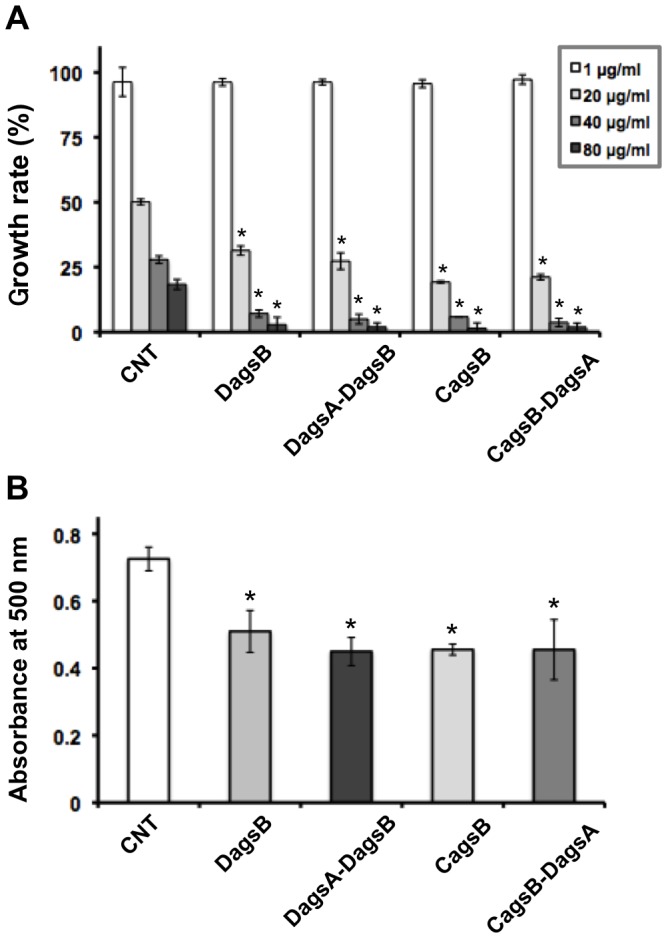
(A) Growth rate after 4 days of growth on CD medium at the indicated concentration of added CR. Error bars represent the standard deviations (n = 3). *, significantly different from the control (P<0.01). (B) Adsorption of CR to the hyphae of each strain. Mycelia cultured in CD for 24 h (500 mg fresh weight) were suspended in 50 mL of CR solution (40 µg/mL) and rotated (160 rpm) for 1 h at room temperature. After the reaction, the mycelia were removed by filtration, and the absorbance of the supernatant was measured at 500 nm. Absorbance levels indicate the levels of CR remaining in the supernatants (i.e., not adsorbed to the hyphae). Error bars represent the standard error of the mean calculated for three replicates (*P<0.05).
To examine whether agsB expression contributes to protection of the cell wall from cell wall–degrading enzymes, we tested the susceptibility of mycelia to Lysing Enzymes, which contains both β-1,3-glucanase and chitinase. For each strain, the number of protoplasts produced from the mycelia by lysis was measured after treatment with Lysing Enzymes. When mycelia cultured in CD liquid medium were treated with Lysing Enzymes, few protoplasts were generated from the control strain even after 4 h (Figure 6A); in contrast, large numbers of protoplasts were generated from the DagsB, DagsA-DagsB, CagsB, and CagsB-DagsA mycelia cultured in CD liquid medium (agsB-repressing conditions for the CagsB and CagsB-DagsA strains) after 4 h. On the other hand, when mycelia cultured in CDTF medium (agsB-inducing conditions for the CagsB and CagsB-DagsA strains) were incubated with Lysing Enzymes, the number of protoplasts generated from the CagsB strain was not significantly different from the number generated from the control strain (Figure 6B). The number of protoplasts may have increased for the CNT strain in CDTF, relative to the number in CD, as a result of the production of a weak cell wall in CDTF media. This weakness would result from changes in the alkali-insoluble cell wall fraction, which is composed of chitin and β-glucan (see below), which was reduced by up to 30% of the total cell wall fractions in the CNT strains cultured in CDTF media (data not shown). When mycelia were digested with both Lysing Enzymes and α-1,3-glucanase (Figure 6C), the numbers of protoplasts generated from the control strain increased significantly compared with those after treatment with the Lysing Enzymes alone, and the numbers of protoplasts generated during each incubation period were comparable to those generated from these agsB mutant strains (Figure 6B). These results suggested that the loss of α-1,3-glucan caused by the disruption or repression of agsB increased the sensitivity of the mycelial cell wall to Lysing Enzymes, suggesting the possibility that cell wall β-1,3-glucan and chitin are more accessible to β-1,3-glucanase and chitinase in mycelial cells with a decreased α-1,3-glucan content.
Figure 6. Cell wall susceptibility of the control (CNT), agsB disruption (DagsB), agsA agsB double disruption (DagsA-DagsB), CagsB, and CagsB with agsA disruption (CagsB-DagsA) strains to Lysing Enzymes.

(A) Susceptibility of mycelia cultured in CD liquid medium (agsB-repressing conditions). Mycelia cultured in CD medium for 24 h (30 mg fresh weight) were digested in reaction buffer (10 mM phosphate buffer, pH 6.0) containing 10 mg/mL Lysing Enzymes. After 1, 2, and 4 h of incubation at 30°C, the number of protoplasts in each sample was determined using a hemocytometer. Error bars represent the standard deviations (n = 3). All values of the mutants differed significantly from that of the control strain. (B) Susceptibility of the control and CagsB strains cultured in CDTF liquid medium (agsB-inducing conditions). Mycelia cultured in CDTF medium for 24 h (30 mg fresh weight) were digested, and the number of protoplasts in each sample was determined according to the method described above. None of the differences were statistically significant. (C) Effect of combined treatment with α-1,3-glucanase and Lysing Enzymes on protoplast formation from fungal mycelia. Mycelia cultured in CD medium for 24 h were digested in reaction buffer containing both Lysing Enzymes and α-1,3-glucanase. The number of protoplasts generated in each sample was determined according to the method described above. None of the differences were statistically significant.
Comparison of Cell Wall Components in the Control Strain and the ags Mutant Strains
Cell wall components of the control and ags mutant strains were fractionated based on their solubility in alkali ([30]; O. Mizutani et al., unpublished data). It has been reported that the alkali-soluble fraction in A. fumigatus is composed mainly of α-1,3-glucan with some galactomannan [31] and that the alkali-insoluble fraction is composed of chitin, β-1,6-branched β-1,3-glucan, and galactomannan [1], [30]. The cell wall components from the A. nidulans control and ags mutant strains were separated into four fractions (Figure S4), and the weight ratios of the fractions were measured (Table 1). The total recovery rate of the four fractions during the fractionation was 55% to 65% of the total cell dry weight in each strain. We determined the sugar compositions in the four fractions for each strain (Figure 7). The hot-water-soluble (HW) fraction was mainly composed of glucose, which accounted for 25 to 34% of the HW fraction (by weight) in each strain (Figure 7A). The AS1 fraction, which contained the water-soluble components after dialysis of the alkali-soluble fraction (Figure S4), was composed of glucose, galactose, and mannose (Figure 7B). However, these sugars together accounted for 8 to 11% of the total AS1 fraction weight in each strain. Glucose again dominated the sugars in the AS2 fraction, although it was only dramatically greater than the other sugars in the control and DagsA strains (Figure 7C). The alkali-insoluble (AI) fraction contained glucose, galactose, mannose, and glucosamine in both the control and ags mutant strains (Figure 7D). In each strain, the glucose and glucosamine contents of the AI fraction each accounted for about 20% of the AI fraction dry weight, and the galactose and mannose contents each accounted for about 5% of the fraction dry weight.
Table 1. Yield of each fraction obtained by alkali treatment of the control (CNT), agsA disruption (DagsA), agsB disruption (DagsB), double-disruption (DagsA-DagsB), conditional-agsB (CagsB), and CagsB with agsA disruption (CagsB-DagsA) strains.
| Yielda (% of dry weight)a | ||||
| Strain | HW | AS1 | AS2 | AI |
| CNT | 31.8 | 6.9 | 14.7 | 46.7 |
| DagsA | 31.7 | 5.7 | 14.4 | 48.2 |
| DagsB | 33.8 | 7.0 | 9.2 | 50.1 |
| DagsA-DagsB | 31.4 | 6.8 | 9.1 | 52.7 |
| CagsB | 36.9 | 10.3 | 7.1 | 45.7 |
| CagsB-DagsA | 40.3 | 7.0 | 6.6 | 46.2 |
The yield of each fraction is expressed as the percentage of the total dry weight. Values represent the mean of triplicate measurements. Fractions: HW, hot-water-soluble; AS, alkali-soluble; AI, alkali-insoluble. Figure S4 shows the fractionation process.
Figure 7. Cell wall composition of each cell wall fraction.

The control (CNT), agsA disruption (DagsA), agsB disruption (DagsB), double-disruption (DagsA-DagsB), CagsB, and CagsB with agsA disruption (CagsB-DagsA) strains were cultured in CD liquid medium (agsB-repressing conditions for the CagsB and CagsB-DagsA strains). Monosaccharide compositions are reported for the (A) hot-water-soluble (HW), (B) alkali-soluble 1 (AS1), (C) alkali-soluble 2 (AS2), and (D) alkali-insoluble (AI) cell wall fractions (see Figure S4) as a percentage of the total dry weight of each fraction. Error bars represent the standard deviations calculated for at least three replicates. *, significantly different from the control (P<0.01). Glc, glucose; Gal, galactose; Man, mannose; GlcN, glucosamine.
No quantitatively significant differences were observed in the sugar compositions of the HW, AS1, and AI fractions between the control and ags mutant strains. On the other hand, statistically significant differences were visible in the sugar compositions in the alkali-soluble AS2 fraction, which was the precipitate that remained after dialysis of the alkali-soluble fraction, between the control and agsB mutant strains (Figure 7C). The AS2 fraction of the control and agsA disruption strains was mainly composed of glucose (i.e., the monosaccharide that makes up α-1,3-glucan), which represented 45% and 41% of the AS2 fraction, respectively. The results from the fractionation and the quantification of sugar contents revealed that the cell wall composition of the agsA disruption strain did not differ greatly from that of the control strain. Typical signals for α-1,3-glucan in 13C-NMR spectroscopic analysis were detected in the AS2 fraction of the control and agsA disruption strains (Figure S8; data not shown) [32]. The chemical shifts in the 13C-NMR corresponded to that of the bacterial mutan (Figure S8), which is composed primarily of α-1,3-glucan [S. Yano et al., unpublished data].
In addition, we performed a methylation (Me) analysis for the AS2 fraction of the control strain to determine the linkage structure of the glucans (Table S3). The presence of 2,4,6-tri-O-Me-Glc (1,3,5-triacetyl-2,4,6-tri-O-methyl-D-glucitol), which indicates 1,3-glucoside bonds, was mainly detected in the AS2 fraction of the control strain as in the case of the standard β-1,3-glucan pachyman and the bacterial mutan (Table S3). In contrast, we detected little 2,3,4,6-tetra-O-Me-Glc, 2,3,4-tri-O-Me-Glc, and 2,4-di-O-Me-Glc, which indicate (respectively) a non-reducing terminal, 1,6-glucoside bonds, and 3,6-branching points (Table S3). These data indicated that the AS2 fraction of the control strain contained mainly linear glucan consisting of 1,3-glucoside bonds. Taken together, these results from the 13C-NMR and the methylation analysis indicated that the AS2 fraction of the control strain was mainly composed of linear α-1,3-glucan.
In contrast, the glucose content of the AS2 fractions accounted for less than 5.0% by weight of the AS2 fraction in the DagsB, DagsA-DagsB, CagsB, and CagsB-DagsA strains when grown in CD liquid medium (agsB-repressing conditions for the CagsB and CagsB-DagsA strains) (Figure 7C). There was very little remaining glucan in the AS2 fraction derived from the DagsB and CagsB strains. Glucose was liberated from the AS2 fraction of these two strains by hydrolysis with β-1,3-glucanase but not with α-1,3-glucanase (Figure S9), indicating that the little glucan remaining in the AS2 fraction derived from the two mutants was β-1,3-glucan. These data suggested that the disruption of agsB and agsB-repressing conditions resulted in a marked decrease in the α-1,3-glucan content of the cell wall in the agsB disruption and CagsB strains. The cell wall components of the DagsA-DagsB and CagsB-DagsA strains were not distinguishable from those of the DagsB and parental CagsB strains, indicating that the agsA gene does not make a major contribution to α-1,3-glucan production (Table 1, Figure 7). The amount of the AS2 fraction and the remaining glucose content of the AS2 fraction of the DagsB strains were at the same low levels as those from the CagsB strains under the agsB-repressing conditions (Table 1, Figure 7C), suggesting that most of the cell wall α-1,3-glucan was lost in the CagsB strain.
Discussion
Aspergillus nidulans possesses two α-1,3-glucan synthase genes, agsA and agsB. In the present study, we constructed disruption mutants of the agsA and agsB genes (DagsA and DagsB, respectively) and a conditional-agsB (CagsB) strain in which expression of agsB is regulated by the conditional promoter alcA. The disruption mutants of agsA did not show obvious phenotypic alterations under normal growth conditions. The hyphae of the DagsB strain were obviously dispersed in liquid CD medium (Figure 1B), and the hyphae of the DagsB strain were scarcely aggregated (Figure 2). Fontaine et al. [33] showed that latex beads coated with α-1,3-glucan bound to the swollen conidia of A. fumigatus and that α-1,3-glucan was solely responsible for conidial aggregation in A. fumigatus. Therefore, the dispersed hyphal growth of the A. nidulans DagsB strains appears to be attributable to the loss of α-1,3-glucan [33]. In the CagsB strain, agsB down-regulation also resulted in abnormal growth characteristics in liquid culture, whereas the strain showed normal growth on plate media regardless of the level of agsB expression. All phenotypes observed in the agsB disruption strains were consistent with those of the CagsB strain under the agsB-repressing conditions. In addition, the DagsA-DagsB and CagsB-DagsA strains showed no obvious phenotypic alteration compared with their respective parental strains (i.e., the DagsB and CagsB strains, respectively). Biochemical analysis of the cell wall polysaccharides revealed that both the disruption of agsB and the down-regulation of agsB expression in the CagsB strain led to almost complete loss of cell wall α-1,3-glucan. These results suggested that AgsB plays a crucial role in biosynthesis of cell wall α-1,3-glucan in A. nidulans, whereas AgsA plays only a small role.
Aspergilli generally possess several genes that encode an α-1,3-glucan synthase (AGS). A. niger is reported to have five AGS genes (agsA to agsE) [13], and A. fumigatus has three such genes (AGS1, AGS2, and AGS3) [11], [12]. A. fumigatus AGS1 (AfAGS1) and AGS2 (AfAGS2) are the counterparts of A. nidulans agsB and agsA, respectively. Disruption of AfAGS1 led to a 50% decrease in the level of cell wall α-1,3-glucan, whereas deletion of AfAGS2 had no detectable effect on glucan levels [11]. Deletion of either AfAGS1 or AfAGS2 yielded cells with altered hyphal morphology, altered phialide formation, and reduced conidiation [11]. During the preparation of this manuscript, a new paper appeared [14] in which a triple α-1,3-glucan synthase mutant of A. fumigatus was generated by deletions of the three α-1,3-glucan synthase genes, AfAGS1, AfAGS2, and AfAGS3. The triple AGS mutant of A. fumigatus was totally deficient in α-1,3-glucans, whereas the growth and conidial germination of the triple mutant were similar to those of the parental strain [14], and the conidiation of the triple mutant was slightly decreased compared to the parental strain, as was found for the single AfAGS1 and AfAGS2 mutants [11], [14]. In the present study, A. nidulans agsA disruptants did not show obviously altered phenotypes under normal growth conditions, and the α-1,3-glucan contents (i.e., the AS2 fraction) were the same as those of the control strain (Figure 7C). Although the colonial growth, conidiation, hyphal tip morphology, and formation of phialides were normal in the DagsB and CagsB strains, the disruption or down-regulation of agsB resulted in abnormal growth characteristics in liquid culture. The results of cell wall fractionation, determination of sugar compositions of the cell wall fractions, 13C-NMR analysis, and methylation analysis indicated that the AS2 fractions of the DagsB and CagsB strains contained very little α-1,3-glucan (Figures 7, S9; Tables 1, S3). Because the nearly complete loss of cell wall α-1,3-glucan caused by the disruption or down-regulation of agsB was greater than the 50% loss of cell wall α-1,3-glucan in the AfAGS1 deletion strain [11], the contribution of agsB to α-1,3-glucan biosynthesis in A. nidulans seems to be larger than that of AfAGS1 in A. fumigatus. These observations suggest that the agsB class of genes is important for the biosynthesis of α-1,3-glucan in both A. nidulans and A. fumigatus, but that the individual agsB-class genes seem to have marked differences in function.
Disruption of the third A. fumigatus AGS gene, AfAGS3, which is absent in A. nidulans, results in overexpression of AfAGS1, which may compensate for the loss of AfAGS3 to maintain normal cell wall composition [12]. In addition, the lack of cell wall α-1,3-glucan in the A. fumigatus triple AGS mutant was accompanied by an increase in the β-1,3-glucan and chitin content of the mycelia [14]. In A. nidulans, the disruption and down-regulation of agsB led to a slight increase in agsA expression, but the expression level of agsA was significantly lower than that of agsB in the control strain (Figures 3, 4, S5). In addition, the phenotypes of the DagsA-DagsB and CagsB-DagsA strains did not differ from those of their parental strains (DagsB and CagsB, respectively), and the amount of each cell wall fraction derived from the DagsA-DagsB and CagsB-DagsA strains was the same as that of the respective parental strains (Table 1, Figure 7). These results suggested that agsA did not compensate for the loss of agsB expression in A. nidulans under our experimental conditions.
Our analysis of the transcriptional responses of the cell wall–related genes revealed that genes encoding β-1,3-glucan synthase and chitin synthases showed significantly increased expression levels in the DagsB strain and in the CagsB strain under agsB-repressing conditions (Figures 3, 4), suggesting that the loss of α-1,3-glucan was counterbalanced by alterations in the amount of another cell wall component such as β-1,3-glucan or chitin. In other fungi, compensatory changes in cell wall components have been reported. For example, the human pathogen C. neoformans possesses only one AGS gene (AGS1) involved in α-1,3-glucan synthesis [15]. A strain of C. neoformans in which the AGS1 gene is disrupted shows a slow growth rate and a temperature-sensitive phenotype, and disruption of the AGS1 gene results in a lack of capsule formation, which is one of the most important virulence factors in this pathogen [15]. In the AGS1-deleted strain, the loss of α-1,3-glucan is accompanied by an increase in chitin and chitosan contents and the redistribution of β-1,3-glucan from the alkali-soluble glucan fraction to the alkali-insoluble glucan fraction [16]. In the fission yeast Schizosaccharomyces pombe, the α-1,3-glucan synthase gene mok1 (synonym ags1) is essential for the synthesis of cell wall α-1,3-glucan [34], [35]. A strain containing a mutated mok1 gene shows slightly decreased α-glucan levels, but β-glucan levels are slightly increased. In contrast to the observations in other fungi, our analysis of cell wall polysaccharides showed that the cell wall chitin and β-1,3-glucan contents were not markedly altered in the ags mutants (Figures 7, S9; Table 1). In addition, no significant differences in staining of the cell wall with CFW or in the sensitivity to CFW were observed among the control and agsB disruption strains (data not shown). Although the agsB mutants showed a loss of cell wall α-1,3-glucan and up-regulation of transcription levels of several cell wall–related genes, the transcription levels of some cell wall–related genes, such as gelB and csmB, was down-regulated in the DagsB strain (Figure 3). In addition, the transcription levels of gelB, chsC, and, csmB tended to be down-regulated in the CagsB strain under the agsB-repressing conditions (Figure 4). Although the contribution of gelB, chsC and, csmB to biosynthesis of cell wall β-1,3-glucan and chitin is unclear in this experimental conditions, the lack of compensatory changes in the cell wall components in the agsB mutants might have been caused by differences in the contribution of each cell wall–related gene to the biosynthesis of cell wall polysaccharides.
Moreover, no marked differences in the ultrastructure or thickness of the cell wall were detected between the control and CagsB strains under either agsB-inducing or agsB-repressing conditions (Figure S7, Table S2). These observations suggested that the proportions of β-1,3-glucan and chitin in the cell wall were regulated appropriately because of the importance of the cell wall for fungal viability, regardless of the presence of cell wall α-1,3-glucan in A. nidulans.
Cell wall stress–inducing compounds often induce the expression of cell wall–related genes [10], [13], [36], [37]. In A. nidulans, levels of agsB transcripts are transiently increased by micafungin treatment, whereas agsA transcripts are maintained at low levels even after micafungin treatment [10]. As in the case of A. nidulans, the expression of A. niger agsA (which is absent from A. nidulans) and agsE (an orthologue of agsB in A. nidulans) is induced in the presence of CFW, sodium dodecyl sulfate, and caspofungin [13]. In addition, a disruptant of A. niger agsA was more sensitive than its parental (non-disrupted) strain to CFW [13]. Based on these observations, we predicted that the perturbation of cell wall α-1,3-glucan synthesis would affect the sensitivity to such cell wall stress–inducing compounds. Unexpectedly, the A. nidulans agsB disruption and CagsB strains did not show increased sensitivity to CFW or micafungin under any experimental conditions in the present study. Our analysis of cell wall polysaccharides revealed that the contents of cell wall chitin and β-1,3-glucan were not markedly altered in the DagsB and CagsB strains. Thus, the sensitivity of A. nidulans to CFW or micafungin is not dependent on the level of cell wall α-1,3-glucan. In contrast, the agsB disruption strain and the CagsB strain under agsB-repressed conditions both showed increased sensitivity to CR (Figure 5A), and the amount of CR adsorption to the hyphae of the DagsB and CagsB strains was significantly greater than that of the control strain (Figure 5B). Both CR and CFW interact with various polysaccharides, although β-1,3-glucan shows a strong interaction with CR but a weak interaction with CFW [38]. Although the amount of cell wall α-1,3-glucan was significantly lower in the DagsB and CagsB strains than in the control strain, the content of the AI fraction was the same (Table 1, Figure 7C, 7D). When we examined the adsorption of CR to each cell wall component (α-1,3-glucan, β-1,3-glucan, or chitin), the amount of CR adsorbed to α-1,3-glucan was significantly less than the amount adsorbed to β-1,3-glucan or chitin (A. Inaba et al., unpublished data). It is reasonable to hypothesize that the loss of cell wall α-1,3-glucan in the DagsB and CagsB strains led to increased exposure of β-1,3-glucan on the cell surface and to the resulting increased sensitivity to CR.
The outermost layer of the cell wall in many fungi consists primarily of α-1,3-glucan [16], [20], [21], [39], [40]. In C. neoformans, ags1Δ cells show a disorganization of the cell wall that accompanies the loss of α-1,3-glucan, and the perturbed cell wall can no longer serve as an attachment site for capsule fibers [16]. Ultrastructural studies of the dimorphic pathogen H. capsulatum revealed that the outermost layer of the cell wall consists primarily of α-1,3-glucan [39]. Rappleye et al. [20] also showed that α-1,3-glucan formed the outermost surface of H. capsulatum cells, based on cytological observations by immunofluorescence and immunoelectron microscopy. Fujikawa et al. [21] demonstrated a dynamic change in the composition of fungal cell wall polysaccharides during infection by the plant pathogen M. grisea. In M. grisea, α-1,3-glucan accumulates on the surface of germ tubes during plant infection, and the accumulation is induced by a plant cue [21]. In the infectious hyphae, the α-1,3-glucan was localized more outwardly than the β-1,3-glucan, resulting in increased tolerance to cell wall–digesting enzymes [21]. Our results of the susceptibility test against digestive enzymes and the biochemical analysis of cell wall polysaccharides are in good agreement with these observations. In A. nidulans, the disruption or repression of agsB resulted in increased susceptibility to cell wall–digesting enzymes (Figure 6A), and cell wall α-1,3-glucan was barely detected in hyphae of the DagsB strain or the CagsB strain cultured under the agsB-repressing conditions (Figure 7). Therefore, α-1,3-glucan seems to be localized to the outermost layer of the cell wall in A. nidulans.
In conclusion, our functional analysis of the agsA and agsB genes demonstrated that the two AGS genes were dispensable in A. nidulans; however, agsB was involved in determining the growth characteristics in liquid culture, although the colonial growth, conidiation, hyphal tip morphology, and formation of phialides were normal in the ags mutants. We were able to control the cell wall α-1,3-glucan content by creating several AGS mutants and by incorporating the conditional alcA promoter in the CagsB strain. Biochemical analysis of the cell wall of the DagsA, DagsB, DagsA-DagsB, CagsB, and CagsB-DagsA strains revealed that most of the cell wall α-1,3-glucan in A. nidulans was synthesized by AgsB under our experimental conditions and that the cell wall β-1,3-glucan and chitin composition was not markedly affected by the loss of cell wall α-1,3-glucan in the agsB mutants. This suggests that cell wall α-1,3-glucan is not responsible for fungal viability, but that it seems to protect the cell wall from some cell wall stresses, such as cell wall–degrading enzymes and environmental chemicals. It may also play a role in aggregation of hyphae, particularly in liquid media.
Supporting Information
Construction of the agsA gene disruption strains in Aspergillus nidulans. (A) Schematic illustration of agsA gene disruption. The first round of PCR was done to amplify the fragments containing the right and left arms and the selectable marker for the disruption cassette. The second round of PCR was done to fuse the three separate fragments from the first round of PCR. The resulting disruption cassette was used for fungal transformation. Primer agsA-F (Table S1) was derived from the sequences of non-coding regions of A. nidulans agsA outside the disruption cassette. Primer agsA-R (Table S1) is specific for the A. nidulans agsA coding region. The restriction enzyme sites and the point at which the probes hybridized are indicated. (B) PCR results for agsA gene disruption in A. nidulans. Lane M, λ/StyI digest (molecular weight marker); lane C, control strain (ABPU1); lanes 1–3, DagsA strains (three independently isolated mutant strains). (C) Southern analysis of the agsA locus in the control and disruption (DagsA) strains using the probe indicated in (A). Chromosomal DNA of the control strain (lane C) and of the DagsA strains (lanes 1, 2, and 3) was digested with HindIII.
(TIF)
Construction of the agsB gene disruption strains in Aspergillus nidulans. (A) Schematic illustration of agsB gene disruption. The first round of PCR was done to amplify the fragments containing the right and left arms and the selectable marker for the disruption cassette. The second round of PCR was done to fuse the three separate fragments from the first round of PCR. The resulting disruption cassette was used for fungal transformation. Primer agsB-F (Table S1) was derived from the sequences of non-coding regions of A. nidulans agsB outside the disruption cassette. Primer agsB-R1 (Table S1) is specific for the A. nidulans agsB coding region. The restriction enzyme sites and the point at which the probes hybridized are indicated. (B) PCR results for agsB gene disruption in A. nidulans. Lane M, λ/StyI digest (molecular weight marker); lane C, control strain (ABPU1); lanes 1 and 2, agsB disruption strains (two independently isolated mutant strains). (C) Southern analysis of the agsB locus in the control and agsB disruption (DagsB) strains using the probe indicated in (A). Chromosomal DNA of the control strain (lanes C) and of the DagsB strains (lanes 1 and 2) was digested with SphI.
(TIF)
Construction of the conditional- agsB (CagsB) strains in Aspergillus nidulans . (A) Schematic illustration of conditional-agsB gene construction in Aspergillus nidulans. First line, the control (wild-type) gene; second line, the gene replacement cassette; third line, the conditional-agsB gene. Primers PalcA-F and agsB-R2 (Table S1) are specific for the A. nidulans alcA promoter and agsB coding regions, respectively. The restriction enzyme sites and the point at which the probes hybridized are indicated. (B) PCR results for the conditional-agsB gene mutation in A. nidulans. Lane M, λ/StyI digest (molecular weight marker); lane C, control strain (ABPU1); lane 05, CagsB strain (CagsB05); lane 06, CagsB strain (CagsB06); lane 07, CagsB strain (CagsB07). (C) Southern analysis of the agsB locus in the control and CagsB strains using the probe indicated in (A). Chromosomal DNAs of the control strain (lane C) and the CagsB strains (lanes 05, 06, and 07) were digested with BamHI.
(TIF)
Fractionation scheme for the A. nidulans cell wall using alkali. Centrifugation steps were performed at 10,000×g for 15 min. Neutralization was done with 17 M acetic acid. HW, AS, and AI indicate the hot-water-soluble, alkali-soluble, and alkali-insoluble fractions, respectively.
(TIF)
Phenotypic analysis of the agsA disruption strain (DagsA). (A) Expression of agsA in the control (CNT) and DagsA strains cultured in CD medium. Conidia (final concentration, 5×105/mL) of the CNT and DagsA strains were inoculated into the indicated liquid medium and cultured for 24 h. RNA samples from each strain were prepared, and the expression of agsA was quantified by means of RT-PCR. Each value represents the ratio of the expression relative to the histone H2B gene in each strain. Error bars represent the standard error of the mean calculated for three replicates. (B) Hyphal morphology of the control (CNT) and DagsA strains grown in CD liquid medium for 24 h at 37°C. (C) Number of conidia of the control (CNT) and DagsA strains obtained from the colonies after 1 week of growth on CD medium at 37°C. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant. (D) Sensitivities to cell wall–stress compounds in the control (CNT) and DagsA strains. Growth rate (% of the control’s growth) was measured after 4 days of growth on CD medium containing the indicated compound. Left panel, micafungin; center panel, calcofluor white (CFW); right panel, Congo Red. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant. (E) Susceptibility to Lysing Enzymes of mycelia cultured in CD liquid medium. Mycelia cultured in CD medium for 24 h (30 mg fresh weight) were digested in reaction buffer (10 mM phosphate buffer, pH 6.0) containing 10 mg/mL Lysing Enzymes. After 1, 2, and 4 h of incubation at 30°C, the number of protoplasts in each sample was determined using a hemocytometer. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant.
(TIF)
Phenotypes of the agsB disruption (CagsB) strains. (A) Expression of agsB in the control (CNT) and CagsB strains cultured in CD medium (agsB-repressing conditions; left) and CDTF medium (agsB-inducing conditions; right). Conidia (final concentration, 5×105/mL) of the CNT and CagsB strains were inoculated into the indicated liquid medium and cultured for 24 h. RNA samples of each strain were prepared, and expression of the agsB gene was quantified by means of RT-PCR analysis. Each value represents the ratio of expression relative to the histone H2B gene in each strain. Error bars represent the standard error of the mean calculated for three replicates (*P<0.05, **P<0.01). (B) Colonial growth of the control (CNT) and CagsB strains. Conidia (a total of 1×103) of each strain were inoculated on the indicated medium and cultured at 37°C for 4 days. (C) Number of conidia of the control and CagsB strains obtained from the colonies after 1 week of growth on CD medium at 37°C. Error bars indicate standard deviations of the mean (n = 3). None of the differences were statistically significant. (D) Growth characteristics of the control (CNT) and CagsB strains in liquid media. Conidia (final concentration, 5×105/mL) of the CNT and CagsB strains were inoculated into the indicated liquid medium and rotated at 160 rpm at 37°C for 18 h.
(TIF)
Ultrathin sections of the control and CagsB strains of Aspergillus nidulans . Cells of each strain were cultured in CD liquid medium (CNT_CD for the control and CagsB_CD for the conditional-agsB panels) or CDTF liquid medium (CNT_CDTF for the control and CagsB_CDTF for the conditional-agsB panels) at 37°C for 24 h, collected by centrifugation, and snap-frozen by plunging into a melting propane/isopentane mixture cooled with liquid nitrogen. Cells were then freeze-substituted in acetone, embedded in resin, sectioned, and examined by transmission electron microscopy. A medial gray band in the cell wall, which might be attributable to alterations in the cell wall components, was sometimes observed in the CagsB strain cultured in CDTF medium. However, no significant differences in the thickness of the cell wall were observed between the control and CagsB strains (Table S2). CW, cell wall; PM, plasma membrane. Scale bar = 500 nm.
(TIF)
13C-NMR spectra of bacterial mutan and of the AS2 fraction from the control strain. (A) The 13C-NMR spectrum of mutan predominantly contained six signals (at 101.3, 83.5, 73.6, 71.9, 71.5, and 62.0 ppm) that were attributable to the presence of α-1,3-glucan [32]. (B) The 13C NMR spectrum of the AS2 fraction from the control strain predominantly contained six signals (at 101.2, 83.4, 73.5, 71.9, 71.4, and 61.9 ppm) that were also attributable to α-1,3-glucan.
(TIF)
Saccharides liberated from the AS2 and AI fractions after treatment with α-1,3-glucanase or β-1,3-glucanase. (A) The amount of glucose liberated from the AS2 and AI fractions derived from the control (CNT), agsB disruption (DagsB), and conditional-agsB (CagsB) strains after treatment with purified α-1,3-glucanase from Bacillus circulans KA-304 [25]. Error bars represent the standard deviations of the mean (n = 3). *, significantly different from the control (P<0.01). (B) The amount of glucose liberated from the AS2 and AI fractions derived from the CNT, DagsB, and CagsB strains after treatment with the purified β-1,3-glucanase from A. niger. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant.
(TIF)
PCR primers used in this study.
(DOCX)
Cell wall thickness of the control (CNT) and conditional- agsB (CagsB) strains.
(DOCX)
Ratios of methyl (Me) ethers obtained after methanolysis of the permethylated bacterial mutan and the AS2 fraction (see Figure S4) of the control strain in A. nidulans .
(DOCX)
Funding Statement
This study was supported in part by grants from the Bio-oriented Technology Research Advancement Institution (BRAIN), from the Japan Science and Technology Agency (JST), and from the Tohoku Bureau of Economy, Trade and Industry. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Latgé J-P (2007) The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol 66: 279–290. [DOI] [PubMed] [Google Scholar]
- 2. Latgé J-P (2010) Tasting the fungal cell wall. Cellular Microbiol 12: 863–872. [DOI] [PubMed] [Google Scholar]
- 3. Gastebois A, Clavaud C, Aimanianda V, Latgé J-P (2009) Aspergillus fumigatus: cell wall polysaccharides, their biosynthesis and organization. Future Microbiol 4: 583–595. [DOI] [PubMed] [Google Scholar]
- 4. Klis FM (1994) Review: cell wall assembly in yeast. Yeast 10: 851–869. [DOI] [PubMed] [Google Scholar]
- 5. Denning DW (2002) Echinocandins: a new class of antifungal. J Antimicrob Chemother 49: 889–891. [DOI] [PubMed] [Google Scholar]
- 6. Levin DE (2005) Cell wall integrity signaling in Saccharomyces cerevisiae . Microbiol Mol Biol Rev 69: 262–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smits GJ, Kapteyn JC, van den Ende H, Klis FM (1999) Cell wall dynamics in yeast. Curr Opin Microbiol 2: 348–352. [DOI] [PubMed] [Google Scholar]
- 8. Levin DE, Fields FO, Kunisawa R, Bishop JM, Thorner J (1990) A candidate protein kinase C gene, PKC1, is required for the S. cerevisiae cell cycle. Cell 62: 213–224. [DOI] [PubMed] [Google Scholar]
- 9. Jung US, Levin DE (1999) Genome-wide analysis of gene expression regulated by the yeast cell wall integrity signaling pathway. Mol Microbiol 34: 1049–1057. [DOI] [PubMed] [Google Scholar]
- 10. Fujioka T, Mizutani O, Furukawa K, Sato N, Yoshimi A, et al. (2007) MpkA-Dependent and -independent cell wall integrity signaling in Aspergillus nidulans . Eukaryot Cell 6: 1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beauvais A, Maubou D, Park S, Morelle W, Tanguy M, et al. (2005) Two α(1–3) glucan synthases with different functions in Aspergillus fumigatus . Appl Environ Microbiol 71: 1531–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maubou D, Park S, Taugy M, Huerre M, Schmitt C, et al. (2006) AGS3, an α(1–3)glucan synthase gene family member of Aspergillus fumigatus, modulates mycelium growth in the lung of experimentally infected mice. Fungal Gen Biol 43: 366–375. [DOI] [PubMed] [Google Scholar]
- 13. Damveld RA, vanKuyk PA, Arentshorst M, Klis FM, van den Hondel CAMJJ, et al. (2005) Expression of agsA, one of five 1,3-α-d-glucan synthase-encoding genes in Aspergillus niger, is induced in response to cell wall stress. Fungal Gen Biol 42: 165–177. [DOI] [PubMed] [Google Scholar]
- 14. Henry C, Latgé J-P, Beauvais A (2011) α1,3 glucans are dispensable in Aspergillus fumigatus . Eukaryot Cell 11: 26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reese AJ, Doering TL (2003) Cell wall alpha-1,3-glucan is required to anchor the Cryptococcus neoformans capsule. Mol Microbiol 50: 1401–1409. [DOI] [PubMed] [Google Scholar]
- 16. Reese AJ, Yoneda A, Breger JA, Beauvais A, Liu H, et al. (2007) Loss of cell wall alpha (1–3) glucan affects Cryptococcus neoformans from ultrastructure to virulence. Mol Microbiol 63: 1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. San-Blas G, San-Blas F, Serrano LE (1977) Host-parasite relationships in the yeastlike form of Paracoccidioides brasiliensis strain IVIC Pb9. Infect Immun 15: 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hogan LH, Klein BS (1994) Altered expression of surface α-1,3-glucan in genetically related strains of Blastomyces dermatitidis that differ in virulence. Infect Immun 62: 3543–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rappleye CA, Goldman WE (2006) Defining virulence genes in the dimorphic fungi. Annu Rev Microbiol 60: 281–303. [DOI] [PubMed] [Google Scholar]
- 20. Rappleye CA, Groppe Eissenberg L, Goldman WE (2007) Histoplasma capsulatum α-(1,3)-glucan blocks innate immune recognition by the β-glucan receptor. Proc Natl Acad Sci USA 104: 1366–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujikawa T, Kuga Y, Yano S, Yoshimi A, Tachiki T, et al. (2009) Dynamics of cell wall components of Magnaporthe grisea during infectious structure development. Mol Microb 73: 553–570. [DOI] [PubMed] [Google Scholar]
- 22. Ichinomiya M, Uchida H, Koshi Y, Ohta A, Horiuchi H (2007) A protein kinase C-encoding gene. pkcA, is essential to the viability of the filamentous fungus Aspergillus nidulans . Biosci Biotechnol Biochem 71: 2787–2799. [DOI] [PubMed] [Google Scholar]
- 23. Hagiwara D, Kondo A, Fujioka T, Abe K (2008) Functional analysis of C2H2 zinc finger transcription factor CrzA involved in calcium signaling in Aspergillus nidulans . Curr Genet 54: 325–338. [DOI] [PubMed] [Google Scholar]
- 24. Izumitsu K, Yoshimi A, Kubo D, Morita A, Saitoh Y, et al. (2009) The MAPKK kinase ChSte11 regulates sexual/asexual development, melanization, pathogenicity, and adaptation to oxidative stress in Cochliobolus heterostrophus . Curr Genet 55: 439–448. [DOI] [PubMed] [Google Scholar]
- 25. Yano S, Wakayama M, Tachiki T (2006) Cloning and expression of an α-1,3-glucanase gene from Bacillus circulans KA-304: The enzyme participates in protoplast formation of Schizophyllum commune . Biosci Biotechnol Biochem 70: 1754–1763. [DOI] [PubMed] [Google Scholar]
- 26. Ciucanu I, Kerek F (1984) A simple and rapid method for the permethylation of carbohydrates. Carbohydr Res 131: 209–217. [Google Scholar]
- 27. Yamaguchi M, Okada H, Namiki Y (2009) Smart specimen preparation for freeze-substitution and serial ultrathin sectioning of yeast cells. J Electron Microsc 58: 261–266. [DOI] [PubMed] [Google Scholar]
- 28. Yamaguchi M, Shimizu M, Yamaguchi T, Ohkusu M, Kawamoto S (2005) Repeated use of uranyl acetate solution for section staining in transmission electron microscopy. Plant Morphology 17: 57–59. [Google Scholar]
- 29. Gwynne DI, Buxton FP, Sibley S, Davies RW, Lockington RA, et al. (1987) Comparison of the cis-acting control regions of two coordinately controlled genes involved in ethanol utilization in Aspergillus nidulans . Gene 51: 205–216. [DOI] [PubMed] [Google Scholar]
- 30. Fontaine T, Simenel C, Dubreucq G, Adam O, Delepierre M, et al. (2000) Molecular organization of the alkali-insoluble fraction of Aspergillus fumigatus cell wall. J Biol Chem 275: 27594–27607. [DOI] [PubMed] [Google Scholar]
- 31. Bernard M, Latgé J-P (2001) Aspergillus fumigatus cell wall: composition and biosynthesis. Med Mycol 39 suppl. 1 9–17. [PubMed] [Google Scholar]
- 32. Sugawara T, Takahashi S, Osumi M, Ohno N (2004) Refinement of the structures of cell-wall glucans of Schizosaccharomyces pombe by chemical modification and NMR spectroscopy. Carbohydr Res 339: 2255–2265. [DOI] [PubMed] [Google Scholar]
- 33. Fontaine T, Beauvais A, Loussert C, Thevenard B, Fulgsang CC, et al. (2010) Cell wall α1–3glucans induce the aggregation of germinating conidia of Aspergillus fumigatus. Fungal Gen Biol 47: 707–712. [DOI] [PubMed] [Google Scholar]
- 34. Hochstenbach F, Klis FM, van den Ende H, van Donselaar E, Peters PJ, et al. (1998) Identification of a putative alpha-glucan synthase essential for cell wall construction and morphogenesis in fission yeast. Proc Natl Acad Sci USA 95: 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Katayama S, Hirata D, Arellano M, Perez P, Toda T (1999) Fission yeast alpha-glucan synthase Mok1 requires the actin cytoskeleton to localize the sites of growth and plays an essential role in cell morphogenesis downstream of protein kinase C function. J Cell Biol 144: 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Damveld RA, Arentshorst M, Franken A, vanKuyk PA, Klis FM, et al. (2005) The Aspergillus niger MADS-box transcription factor RlmA is required for cell wall reinforcement in response to cell wall stress. Mol Microbiol 58: 305–319. [DOI] [PubMed] [Google Scholar]
- 37. Meyer V, Damveld RA, Arentshorst M, Stahl U, van den Hondel CAMJJ, et al. (2007) Survival in the presence of antifungals: genome-wide expression profiling of Aspergillus niger in response to sublethal concentrations of caspofungin and fenpropimorph. J Biol Chem 282: 32935–32948. [DOI] [PubMed] [Google Scholar]
- 38. Wood PJ (1980) Specificity in the interaction of direct dyes with polysaccharides. Carbohydr Res 85: 271–287. [Google Scholar]
- 39. Kanetsuna F, Carbonell LM, Gil F, Azuma I (1974) Chemical and ultrastructural studies on the cell walls of the yeast and mycelial forms of Histoplasma capsulatum . Mycopathologia 54: 1–13. [DOI] [PubMed] [Google Scholar]
- 40. Gastebois A, Mouyna I, Simenel C, Clavaud C, Coddeville B, et al. (2010) Characterization of a new β(1–3)-glucan branching activity of Aspergillus fumigatus . J Biol Chem 285: 2386–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Construction of the agsA gene disruption strains in Aspergillus nidulans. (A) Schematic illustration of agsA gene disruption. The first round of PCR was done to amplify the fragments containing the right and left arms and the selectable marker for the disruption cassette. The second round of PCR was done to fuse the three separate fragments from the first round of PCR. The resulting disruption cassette was used for fungal transformation. Primer agsA-F (Table S1) was derived from the sequences of non-coding regions of A. nidulans agsA outside the disruption cassette. Primer agsA-R (Table S1) is specific for the A. nidulans agsA coding region. The restriction enzyme sites and the point at which the probes hybridized are indicated. (B) PCR results for agsA gene disruption in A. nidulans. Lane M, λ/StyI digest (molecular weight marker); lane C, control strain (ABPU1); lanes 1–3, DagsA strains (three independently isolated mutant strains). (C) Southern analysis of the agsA locus in the control and disruption (DagsA) strains using the probe indicated in (A). Chromosomal DNA of the control strain (lane C) and of the DagsA strains (lanes 1, 2, and 3) was digested with HindIII.
(TIF)
Construction of the agsB gene disruption strains in Aspergillus nidulans. (A) Schematic illustration of agsB gene disruption. The first round of PCR was done to amplify the fragments containing the right and left arms and the selectable marker for the disruption cassette. The second round of PCR was done to fuse the three separate fragments from the first round of PCR. The resulting disruption cassette was used for fungal transformation. Primer agsB-F (Table S1) was derived from the sequences of non-coding regions of A. nidulans agsB outside the disruption cassette. Primer agsB-R1 (Table S1) is specific for the A. nidulans agsB coding region. The restriction enzyme sites and the point at which the probes hybridized are indicated. (B) PCR results for agsB gene disruption in A. nidulans. Lane M, λ/StyI digest (molecular weight marker); lane C, control strain (ABPU1); lanes 1 and 2, agsB disruption strains (two independently isolated mutant strains). (C) Southern analysis of the agsB locus in the control and agsB disruption (DagsB) strains using the probe indicated in (A). Chromosomal DNA of the control strain (lanes C) and of the DagsB strains (lanes 1 and 2) was digested with SphI.
(TIF)
Construction of the conditional- agsB (CagsB) strains in Aspergillus nidulans . (A) Schematic illustration of conditional-agsB gene construction in Aspergillus nidulans. First line, the control (wild-type) gene; second line, the gene replacement cassette; third line, the conditional-agsB gene. Primers PalcA-F and agsB-R2 (Table S1) are specific for the A. nidulans alcA promoter and agsB coding regions, respectively. The restriction enzyme sites and the point at which the probes hybridized are indicated. (B) PCR results for the conditional-agsB gene mutation in A. nidulans. Lane M, λ/StyI digest (molecular weight marker); lane C, control strain (ABPU1); lane 05, CagsB strain (CagsB05); lane 06, CagsB strain (CagsB06); lane 07, CagsB strain (CagsB07). (C) Southern analysis of the agsB locus in the control and CagsB strains using the probe indicated in (A). Chromosomal DNAs of the control strain (lane C) and the CagsB strains (lanes 05, 06, and 07) were digested with BamHI.
(TIF)
Fractionation scheme for the A. nidulans cell wall using alkali. Centrifugation steps were performed at 10,000×g for 15 min. Neutralization was done with 17 M acetic acid. HW, AS, and AI indicate the hot-water-soluble, alkali-soluble, and alkali-insoluble fractions, respectively.
(TIF)
Phenotypic analysis of the agsA disruption strain (DagsA). (A) Expression of agsA in the control (CNT) and DagsA strains cultured in CD medium. Conidia (final concentration, 5×105/mL) of the CNT and DagsA strains were inoculated into the indicated liquid medium and cultured for 24 h. RNA samples from each strain were prepared, and the expression of agsA was quantified by means of RT-PCR. Each value represents the ratio of the expression relative to the histone H2B gene in each strain. Error bars represent the standard error of the mean calculated for three replicates. (B) Hyphal morphology of the control (CNT) and DagsA strains grown in CD liquid medium for 24 h at 37°C. (C) Number of conidia of the control (CNT) and DagsA strains obtained from the colonies after 1 week of growth on CD medium at 37°C. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant. (D) Sensitivities to cell wall–stress compounds in the control (CNT) and DagsA strains. Growth rate (% of the control’s growth) was measured after 4 days of growth on CD medium containing the indicated compound. Left panel, micafungin; center panel, calcofluor white (CFW); right panel, Congo Red. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant. (E) Susceptibility to Lysing Enzymes of mycelia cultured in CD liquid medium. Mycelia cultured in CD medium for 24 h (30 mg fresh weight) were digested in reaction buffer (10 mM phosphate buffer, pH 6.0) containing 10 mg/mL Lysing Enzymes. After 1, 2, and 4 h of incubation at 30°C, the number of protoplasts in each sample was determined using a hemocytometer. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant.
(TIF)
Phenotypes of the agsB disruption (CagsB) strains. (A) Expression of agsB in the control (CNT) and CagsB strains cultured in CD medium (agsB-repressing conditions; left) and CDTF medium (agsB-inducing conditions; right). Conidia (final concentration, 5×105/mL) of the CNT and CagsB strains were inoculated into the indicated liquid medium and cultured for 24 h. RNA samples of each strain were prepared, and expression of the agsB gene was quantified by means of RT-PCR analysis. Each value represents the ratio of expression relative to the histone H2B gene in each strain. Error bars represent the standard error of the mean calculated for three replicates (*P<0.05, **P<0.01). (B) Colonial growth of the control (CNT) and CagsB strains. Conidia (a total of 1×103) of each strain were inoculated on the indicated medium and cultured at 37°C for 4 days. (C) Number of conidia of the control and CagsB strains obtained from the colonies after 1 week of growth on CD medium at 37°C. Error bars indicate standard deviations of the mean (n = 3). None of the differences were statistically significant. (D) Growth characteristics of the control (CNT) and CagsB strains in liquid media. Conidia (final concentration, 5×105/mL) of the CNT and CagsB strains were inoculated into the indicated liquid medium and rotated at 160 rpm at 37°C for 18 h.
(TIF)
Ultrathin sections of the control and CagsB strains of Aspergillus nidulans . Cells of each strain were cultured in CD liquid medium (CNT_CD for the control and CagsB_CD for the conditional-agsB panels) or CDTF liquid medium (CNT_CDTF for the control and CagsB_CDTF for the conditional-agsB panels) at 37°C for 24 h, collected by centrifugation, and snap-frozen by plunging into a melting propane/isopentane mixture cooled with liquid nitrogen. Cells were then freeze-substituted in acetone, embedded in resin, sectioned, and examined by transmission electron microscopy. A medial gray band in the cell wall, which might be attributable to alterations in the cell wall components, was sometimes observed in the CagsB strain cultured in CDTF medium. However, no significant differences in the thickness of the cell wall were observed between the control and CagsB strains (Table S2). CW, cell wall; PM, plasma membrane. Scale bar = 500 nm.
(TIF)
13C-NMR spectra of bacterial mutan and of the AS2 fraction from the control strain. (A) The 13C-NMR spectrum of mutan predominantly contained six signals (at 101.3, 83.5, 73.6, 71.9, 71.5, and 62.0 ppm) that were attributable to the presence of α-1,3-glucan [32]. (B) The 13C NMR spectrum of the AS2 fraction from the control strain predominantly contained six signals (at 101.2, 83.4, 73.5, 71.9, 71.4, and 61.9 ppm) that were also attributable to α-1,3-glucan.
(TIF)
Saccharides liberated from the AS2 and AI fractions after treatment with α-1,3-glucanase or β-1,3-glucanase. (A) The amount of glucose liberated from the AS2 and AI fractions derived from the control (CNT), agsB disruption (DagsB), and conditional-agsB (CagsB) strains after treatment with purified α-1,3-glucanase from Bacillus circulans KA-304 [25]. Error bars represent the standard deviations of the mean (n = 3). *, significantly different from the control (P<0.01). (B) The amount of glucose liberated from the AS2 and AI fractions derived from the CNT, DagsB, and CagsB strains after treatment with the purified β-1,3-glucanase from A. niger. Error bars represent the standard deviations (n = 3). None of the differences were statistically significant.
(TIF)
PCR primers used in this study.
(DOCX)
Cell wall thickness of the control (CNT) and conditional- agsB (CagsB) strains.
(DOCX)
Ratios of methyl (Me) ethers obtained after methanolysis of the permethylated bacterial mutan and the AS2 fraction (see Figure S4) of the control strain in A. nidulans .
(DOCX)