

Abstract
Gas-phase modification of carboxylic acid functionalities is performed via ion/ion reactions with carbodiimide reagents [N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC) and [3-(3-Ethylcarbodiimide-1-yl)propyl]trimethylaminium (ECPT). Gas-phase ion/ion covalent chemistry requires the formation of a long-lived complex. In this instance, the complex is stabilized by an electrostatic interaction between the fixed charge quaternary ammonium group of the carbodiimide reagent cation and the analyte dianion. Subsequent activation results in characteristic loss of an isocyanate derivative from one side of the carbodiimide functionality, a signature for this covalent chemistry. The resulting amide bond is formed on the analyte at the site of the original carboxylic acid. Reactions involving analytes that do not contain available carboxylic acid groups (e.g., they have been converted to sodium salts) or reagents that do not have the carbodiimide functionality do not undergo a covalent reaction. This chemistry is demonstrated using PAMAM generation 0.5 dendrimer, ethylenediaminetetraacetic acid (EDTA), and the model peptide DGAILDGAILD. This work demonstrates the selective gas-phase covalent modification of carboxylic acid functionalities.
INTRODUCTION
Chemical derivatization of analyte molecules has a long history in analytical mass spectrometry.[1] The motivations for derivatization are varied and range from increasing volatility, improving ionization characteristics, facilitating structural characterization, etc. With the explosive growth in biological applications over the past two decades, for example, many derivatization strategies have been developed for the condensed-phase manipulation of biopolymers such as proteins, oligosaccharides, oligonucleotides, and lipids [2] for subsequent interrogation by mass spectrometry. In the case of biomolecules, solution phase bioconjugation techniques are frequently employed to facilitate analyte ionization [3], quantification [4,5], and characterization [6,7]. Early characterization experiments examined, for example, the relative reactivity of peptide primary amine groups towards N-hydroxysuccinimide (NHS) ester derivative reagents [8]. A similar study was used to characterize the reactivities of denatured and native-state ubiquitin within the context of a top-down Fourier transform ion cyclotron resonance (FTICR) experiment [9]. More recently, NHS ester derivatives have been used to perform cross-linking studies on peptides and proteins [10,11,12], to covalently anchor peptides onto solid supports [13], and to attach chromophores to peptides so as to facilitate photoactivation experiments [14]. The targeted reaction chemistries utilized in these techniques can allow for site-specific information to be obtained in the course of a mass spectrometry experiment. For example, the conversion of lysine residues to more basic homoarginine residues via guanidination chemistry alters proton mobility in peptides and can enhance preferential cleavages at aspartic acid residues [15]. Derivatization reactions are usually performed in an offline fashion but have also recently been demonstrated with reactive desorption electrospray ionization (DESI), which allows for the rapid derivatization of analyte species as part of the ionization process. [16,17,18,19,20]
The advent of electrospray ionization (ESI) and its analogues has provided for the generation of multiply charged ions [21,22], which has in turn allowed for the study of interactions between oppositely charged ions (viz. cations and anions) in the gas-phase by mass spectrometry [23,24,25,26]. These types of reactions have been shown to proceed through either long-range charged particle transfers or through relatively long-lived complexes [27]. Not surprisingly, the types of chemistries that can occur during the lifetimes of the long-lived intermediates are largely dependent on the chemical properties of the two reaction partners [28], making these ion/ion interactions amenable, in principle, to covalent bond formation. For this reason, we have embarked on an effort to develop gas-phase derivatization techniques that can be used as part of an overall MSn strategy for the characterization of analyte ions. Recently, several experiments describing bioconjugation techniques performed via ion/ion reactions in the gas-phase have been reported. For example, 4-formyl-1,3-benzene disulfonic acid (FBDSA) has been used to modify primary amine groups in peptide ions via Schiff base chemistry [29,30,31,32]. Following gas-phase complex formation between the FBDSA reagent and peptide analyte, collisional activation can result in water loss and imine bond formation. Formation of the dehydrated product is consistent with nucleophilic attack on the carbonyl carbon of the FBDSA aldehyde by an unprotonated primary amine on the peptide (e.g., the N-terminus or the ε-amino group of lysine) and represents a signature for the covalent chemistry [29]. These Schiff base modifications have been shown to enhance the sequence coverage of peptides in collision induced dissociation (CID) experiments, thereby facilitating accurate primary structure determination [30]. Similarly, ion/ion reactions involving NHS-based reagents have been used to modify primary amino groups of peptides [33,34,35]. In those cases, an amide bond is formed by nucleophilic attack by the unprotonated primary amine on the carbonyl carbon of the ester functionality. Activation of the ion/ion complex results in signature loss of either neutral NHS or neutral sulfo-NHS [33]. Gas-phase NHS chemistry has shown promise with cross-linking experiments [34]. In these instances, the strong electrostatic interactions between the sulfonate groups of the anionic reagents and the protonated sites on the polypeptides provide for the formation of long-lived electrostatically bound complexes by which covalently chemistry can then proceed. The bifunctional nature of these analytes (viz. they contain both a site which facilitates electrostatic attachment and a site which can perform some sort of chemical transformation) is key to their success as reagents for gas-phase covalent chemistry.
In addition to a suitable bifunctional reagent, ion/ion covalent chemistry also requires the presence of an available reactive site on the analyte, in these cases an unprotonated primary amine, for the chemistry to occur. In addition to some of the benefits that gas-phase ion/ion reactions can offer compared to solution reactions (e.g., extremely rapid reaction rates compared to solution, ready comparison between modified and unmodified analyte forms), there also exists the potential for new or altered chemistries compared to the solution phase. For example, it has recently been shown that sulfo-NHS-ester reagents can be used to modify arginine peptide residues in the gas-phase, chemistry not normally observed in solution [36]. In solution, it is difficult to establish pH conditions that prevent both protonation of the guanidine side-chain, which renders it unreactive, and hydrolysis of the reagent. However, in the gas-phase, conditions can be established in which arginine residues are not necessarily protonated, depending on the charge state and amino acid composition of the peptide. This result indicates that some gas phase ion/ion reactions may provide for the observation of reaction products not typically observed or explored in solution.
Here, we explore the use of N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC) as an ion/ion reagent with reactive specificity towards carboxylic acid moieties. CMC, which is structurally similar to the well-known 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) reagent, is a common, water-soluble crosslinking reagent. However, unlike CMC, EDC lacks the electrostatically “sticky” group necessary to form a stable long-lived ion/ion complex. Reactions utilizing these carbodiimide reagents are routinely performed in solution as activation steps in the crosslinking of carboxylic acid groups to primary amines and have been used in a wide array of bioconjugation applications, including peptide synthesis [37], probing secondary structure of nucleic acids [38], and amino acid residue quantitation in proteins [39]. In solution, the carboxyl group adds across one of the carbodiimide double bonds, forming an o-acylisourea ester. This ester is quite reactive and its formation is typically followed by subsequent nucleophilic attack by a primary amine in the vicinity [40] or by hydrolysis which regenerates a carboxylate [41] (though the ester can be stabilized via subsequent reaction by converting it to a sulfo-NHS ester [42]). However, in the gas phase, the local absence of chemical groups which could result in the facile disruption of the carbodiimide/carboxylic acid ester bond allows for the successful observation of the ion/ion complex in the context of a mass spectrometry experiment. In addition to the previously reported primary amine- and guanidine-specific reactions, the targeted carboxylic acid reactivity presented herein represents another type of selective gas-phase reaction.
EXPERIMENTAL SECTION
Materials
PAMAM dendrimer generation 0.5 (PAMAM G0.5), ethylenediaminetetraacetic acid (EDTA), and N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMC) were purchased from Sigma-Aldrich (St. Louis, MO). Methanol, ammonium hydroxide, and glacial acetic acid were purchased from Mallinckrodt (Phillipsburg, NJ). The model peptide DGAILDGAILD was synthesized and purchased from NeoBioSci (Cambridge, MA). 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide methiodide [termed [3-(3-Ethylcarbodiimide-1-yl)propyl]trimethylaminium, or ECPT, for nomenclature simplicity in these experiments] was purchased from Acros Organics (part of Thermo Fisher Scientific, Geel, Belgium). 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) was purchased from Thermo Fisher Scientific (Rockford, IL). 1 mg/mL stock solutions of CMC, ECPT, EDC, and DGAILDGAILD were all prepared in 50/50 (v/v) water/methanol solutions. A 7% (v) solution of PAMAM G0.5 was prepared in 5% (v) ammonium hydroxide. Nanoelectrospray ionization (nESI) of PAMAM G0.5 generates a distribution of charge states and, within each charge state, a distribution of Na+ and H+ counter ions associated with the carboxylate functionalities. A 1 mg/mL stock solution of EDTA was prepared in water. Solutions were diluted roughly 10 fold prior to use. Oxidation of the carbodiimide bonds of CMC and ECPT, effectively forming N-cyclohexyl-N′-(2-morpholinoethyl)urea (CMU) and [3-(3-Ethylurea-1-yl)propyl]trimethylaminium (EUPT), respectively, was performed by adding >1% (v) acetic acid to the carbodiimide solutions.
Mass Spectrometry
All experiments were performed on a QTRAP 4000 hybrid triple quadrupole/linear ion trap mass spectrometer (AB SCIEX, Concord, ON, Canada) previously modified for ion/ion reactions [43]. Briefly, cation (singly charged carbodiimide reagent or a urea form of the reagent) and anion (doubly deprotonated analyte) populations are sequentially injected into the q2 reaction cell using alternately pulsed nanoelectrospray ionization (nESI) [44,45]. The precursor cations and anions are isolated prior to reaction using the Q1 mass filter. After a defined mutual storage reaction period in q2 (typically 300 to 1000 milliseconds), the charge reduced anionic product complex is transferred to the Q3 linear ion trap where MSn and mass analysis via mass-selective axial ejection is performed [46].
RESULTS AND DISCUSSION
To date, it has been observed that the formation of a long-lived complex is a necessary condition for covalent ion/ion chemistry to proceed in the gas-phase. Typically, the generation of such complexes has been facilitated by employing bifunctional reagents. In addition to the reactive functionality (e.g., the ester functionality of a sulfo-NHS ester derivative), an electrostatically “sticky” group is also present on the reagent (e.g., the sulfonate group of a sulfo-NHS ester derivative). This group serves to anchor the reagent onto the peptide cation, thereby providing sufficient time for covalent chemistry to occur. In the case of multiply deprotonated analytes, it has been shown that covalent chemistry is facilitated using fixed charge cationic NHS-based ester reagents [33]. Rather than sulfonate groups, these bifunctional reagents employ fixed charge quaternary ammonium groups to promote strong electrostatic interactions with the anionic analyte.
PAMAM G0.5
CMC contains a fixed charge quaternary ammonium group that can promote stable long-lived complex formation (Scheme 1). This can be illustrated using an ion/ion reaction between CMC and the doubly deprotonated form of PAMAM generation 0.5 dendrimer (Figure 1(b)). PAMAM G0.5 is a carboxylic acid-terminated dendrimer comprised of eight carboxylic acid sites. Upon generation of the -2 charge state via nESI, two of these eight sites are converted to carboxylate anions. Since the gas-phase carbodiimide chemistry requires a neutral carboxylic acid reactive site, six available reaction sites remain on the dendrimer dianion. Following isolation of the two reactants (Figure 1(a)) shows the isolated dendrimer dianion), mutual storage of the two ion populations leads to prominent complex formation, denoted as [M-2H+CMC]− (see Figure 1(b)). In this instance, proton transfer, as reflected by the [M-H]− signal, represents a minor competitive pathway. As it is neutralized during the reaction process, the structure of the CMC reagent following proton transfer has not been experimentally determined. X is a common PAMAM fragment ion [47] and its losses in Figure 1(b) are due to CID resulting from energetic ion transfer conditions between the q2 reaction cell and the Q3 linear ion trap. An ion/ion reaction performed using a carbodiimide containing reagent without a fixed charge (i.e., EDC) results exclusively in proton transfer (Online Resource 1), which highlights the importance of the fixed charge present in CMC for generating the stable ion/ion complex noted in Figure 1(b). Following complex formation between a multiply deprotonated analyte and CMC, a rearrangement occurs that, upon CID, gives a signature neutral loss of 125 Da corresponding to cyclohexyl isocyanate (see Scheme 1). A loss of 125 Da from the dendrimer/CMC ion/ion complex is extremely unlikely without first invoking a rearrangement involving the carboxylic acid and carbodiimide functionalities. The remaining charge-reduced product contains a new amide bond between the original analyte carboxylic acid group and one of the original carbodiimide nitrogens.
Scheme 1.
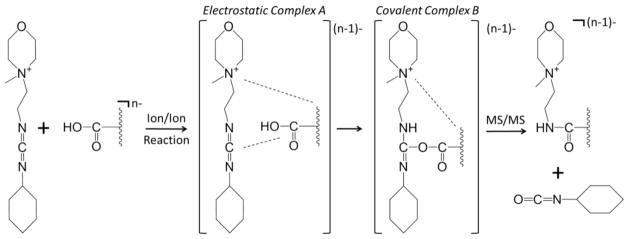
Sequence of events in the ion/ion reaction between a multiply deprotonated analyte and fixed charge CMC cation resulting in the gas-phase covalent modification of the analyte at a carboxylic acid functional group.
Figure 1.

Spectra illustrating gas-phase covalent modification of PAMAM G0.5, including (a) isolation of the double deprotonated PAMAM G0.5 precursor, (b) ion/ion reaction between dendrimer dianion and CMC cation (structure inset), and (c) CID of isolated ion/ion complex showing a 125 Da signature loss. The lightning bolt (
 ) is used to denote the ion that has been subjected to CID.
) is used to denote the ion that has been subjected to CID.
As the carbodiimide reactivity depends on the availability of a neutral carboxylic acid group on the analyte (see Scheme 1), removal of this functionality should block the reaction. This hypothesis can be tested by converting all of the neutral carboxylic acid groups on the dendrimer dianion to sodium salts (viz. –COONa). As before, the ion/ion reaction between the sodiated PAMAM dendrimer dianion and CMC results in complex formation (Figure 2(a)). However, subsequent CID of the isolated complex does not lead to the 125 Da signature loss (Figure 2(b)). In this instance, proton transfer and a loss of 101 Da from the complex dominate. The 101 Da loss likely arises from cleavage of the N-C bond between the quaternary nitrogen in the morpholino ring and the first carbon of the ethyl alky chain. Whereas a nominal loss of 125 Da from the ion/ion complex requires a rearrangement and covalent bond formation at the carboxylic acid group, the loss of 101 Da does not and is therefore not indicative of a covalent reaction. X-type (−72 Da) and Y-type (−280 Da) neutral loss fragments are common fragments of PAMAM dendrimers associated with retro-Michael addition at an interior and the most exterior tertiary nitrogen, respectively [47].
Figure 2.

The covalent chemistry described requires both a carboxylic acid and a carbodiimide to occur. (a) An ion/ion reaction between a PAMAM G0.5 sodium salt analyte and CMC produces an ion/ion complex. (b) However, CID of the complex does not result in a signature loss of 125 Da. (c) Similarly, oxidation of the carbodiimide functionality to a urea functionality renders the carbodiimide reagent (now CMU, structure inset) unreactive towards the original PAMAM G0.5 dendrimer.
Another control experiment for testing carbodiimide reactivity is to use the urea form of CMC, which is referred to as CMU. An ion/ion reaction between the original PAMAM G0.5 dianion (viz. with –COOH functionalities instead of –COONa functionalities) and the CMU reagent cation again produces a stable complex (data not shown). However, CID of the complex produces only proton transfer and X-type fragments (Figure 2(c)). Similar to the loss of 101 Da observed earlier, this X-type fragmentation does not signify the formation of a covalent bond. The nominal addition of 14 Da to the proton transfer product is believed to result from a methyl cation transfer, denoted as [M-2H+15]−, from the quaternary ammonium on the reagent to a carboxylate anion. This phenomenon is undergoing further study and will be described in more detail elsewhere.
Given an analyte has an available carboxylic acid, the carbodiimide group should show some degree of reactivity regardless of the identities of the R-groups on either side of the carbodiimide functionality (provided the fixed charge quaternary ammonium to stabilize complex formation is present). ECPT, a carbodiimide reagent similar to CMC but with the morpholino ring replaced by two methyl substituents and the cyclohexyl group replaced by an ethyl group, contains both the necessary fixed charge site and carbodiimide to perform this covalent chemistry. As expected, an ion/ion reaction between ECPT and the dianion of PAMAM readily gives complex formation (data not shown). Though less abundant than the signature loss observed with the CMC reagent, subsequent CID of the complex does show the expected signature loss of one side of the carbodiimide functionality (Figure 3(a)). In this case, this characteristic loss is a 71 Da loss corresponding to ethyl isocyanate. As with CMC, this loss comes from the side of the reagent that does not contain the fixed charge group. The covalent bond fragmentation pathway appears to be substantially less favorable with the ECPT reagent when compared to the CMC reagent. This could be due to the relative stabilities of the isocyanate leaving groups and/or the relative strengths of the electrostatic interactions between the fixed charged quaternary ammonium groups on the reagents and the carboxylate groups on the analytes, both being properties which may be altered with the new R-groups on the ECPT reagent. Proton transfer, methyl cation transfer, and common neutral losses are much more prevalent than with the CMC reagent. In this instance, the characteristic neutral loss is close but not identical to loss of a neutral X fragment of PAMAM G0.5 from the complex (viz. 71 Da for the characteristic loss versus 72 Da for the X fragment, Figure 3(a) inset). The 71 Da loss is not merely an isotope related to the 72 Da loss. During the CID step, care was taken to activate only the monoisotopic precursor ion. Additionally, the abundance of the 71 Da loss relative to that of the 72 Da loss is not consistent with an expected isotope pattern. An ion/ion reaction with the sodium salt form of the PAMAM dendrimer and subsequent CID does not show a loss of 71 Da (see Figure 3(b)), which is again consistent with this loss of 71 Da being a unique signature of the covalent chemistry. A loss of 59 Da corresponds to the loss of trimethylamine, which is analogous to the 101 Da loss from CMC (see Figure 2(b)). Employing EUPT, the urea form of ECPT, the characteristic loss is further confirmed. CID of the PAMAM/EUPT complex does not give a characteristic 71 Da loss, but still does show proton transfer, methyl cation transfer, and X-type fragmentation of PAMAM (Figure 3(c)).
Figure 3.

CID of the ion/ion complexes for reactions between (a) PAMAM G0.5 and ECPT, (b) PAMAM G0.5 sodium salt and ECPT (structure inset), and (c) PAMAM G0.5 and EUPT.
EDTA
While carbodiimide reactions with carboxylic acid-rich analytes, such as PAMAM G0.5, may be expected to be quite efficient, covalent carbodiimide chemistry is also observed for analytes that contain fewer sites available for reaction. The polyamino carboxylic acid EDTA contains four carboxylic acids, two of which are deprotonated upon ionization, leaving two neutral carboxylic acid functionalities available for reaction with a carbodiimide reagent. Upon CID of the EDTA/CMC ion/ion complex, the signature 125 Da neutral loss is observed, indicating that covalent amide bond formation has again taken place (Figure 4(a)). Competitive 101 Da neutral loss, X-type fragmentation, proton transfer, and methyl cation transfer are also apparent. When the urea reagent CMU is employed, CID of the resulting complex shows very similar fragmentation, but with much less relative contribution from the 125 Da loss channel (Figure 4(b)). We note that the loss of cyclohexyl isocyanate from CMU can take place without covalent reaction. The loss of 125 Da from the EDTA/CMU complex is therefore not completely unexpected, although it appears to be a much less abundant pathway than for the CMC reaction complex. In any case, the 125 Da loss from a CMC-containing ion/ion complex represents a signature for covalent reaction, as a 125 Da loss from CMC is not readily rationalized without invoking rearrangement and covalent bond formation. Similar results are obtained with the other carbodiimide reagent and its urea analogue, ECPT and EUPT, respectively (Online Resource 2). Again, the covalent bond formation pathway with the ECPT reagent is much less efficient when compared to the CMC reagent, as viewed by the lower relative abundance of the signature loss.
Figure 4.

CID of the ion/ion complexes for reactions between (a) EDTA and CMC and (b) EDTA and CMU.
DGAILDGAILD
Many of the solution phase carbodiimide reactions to date have performed on biopolymers such as peptides and proteins [2]. The gas-phase carbodiimide reactivity noted here can also be applied to these types of analytes. An ion/ion reaction between CMC and the aspartic acid-containing model peptide DGAILDGAILD produces a complex (data not shown). Though this peptide has four carboxylic acid sites (the side chains of the three aspartic acid residues and the C-terminus), two are presumably converted to carboxylate anions during the ionization process, which leaves up to two sites available for reaction (though the C-terminal aspartic acid side chain may participate in a salt-bridge-like structure with the C-terminus upon deprotonation of one of these functionalities, rendering both unreactive and leaving only one reactive site on the peptide). CID of the ion/ion complex shows abundant signature loss of 125 Da as well as competitive 101 Da loss and many water losses, a common CID fragment of acidic peptides in the negative mode [48] (Figure 5(a)). CID of the DGAILDGAILD/CMU complex, on the other hand, does not give the characteristic loss (Figure 5(b)). CID of the ion/ion complex of the peptide and ECPT results in a signature loss of 71 Da, though this is again less abundant than the characteristic loss from the CMC reagent (Online Resource 3(a)). CID of the ion/ion complex of the peptide and EUPT does not exhibit the characteristic ethyl isocyanate loss (Online Resource 3(b)). Collectively, these results are all fully consistent with reaction of the carbodiimide reagents with a carboxylic acid group in the peptide.
Figure 5.

CID of the ion/ion complexes for reactions between (a) DGAILDGAILD and CMC and (b) DGAILDGAILD and CMU. Degree symbols (°) denote water losses while asterisks (*) denote ammonia losses.
CONCLUSIONS
This work extends the recent development of ion/ion reactions for functional group-specific gas-phase conjugation to the carboxylic acid functionality. Bifunctional reagent cations containing a fixed charged quaternary ammonium group as well as a carbodiimide functionality are shown to modify carboxylic acid sites of dianion analytes in the gas-phase. The quaternary ammonium moiety is a “sticky” group which serves to anchor the reagent to the dianion reaction partner, stabilizing a long-lived complex. From there, covalent chemistry involving the carbodiimide and the carboxylic acid functionality can proceed via a rearrangement. A signature for this covalent chemistry is a loss of an isocyanate derivative from one side of the carbodiimide reagent upon collisional activation. In the absence of a carboxylic acid reactive site (i.e., all carboxylic acid sites have been converted to carboxylate anions or carboxylate salts), the covalent chemistry cannot proceed and no signature loss is observed. Similarly, if the carbodiimide group of the reagent is converted to the urea functionality, no covalent chemistry is apparent. These findings are significant for the further development of reagents for specific purposes in that they demonstrate that carbodiimide reagents are selective for carboxylic acids in the gas-phase.
Supplementary Material
Acknowledgments
The Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy under Award DE-FG02-00ER15105 is acknowledged for the work with PAMAM and EDTA and the National Institutes of Health under Grant GM 45372 is acknowledged for extension of the work to peptide ions. B.M.P. wishes to acknowledge receipt of an American Chemical Society, Division of Analytical Chemistry (ACS DAC) Fellowship sponsored by the Society for Analytical Chemists of Pittsburgh (SACP).
References
- 1.Halket JM, Zaikin VG. Derivatization in mass spectrometry – 1. Silylation. Eur J Mass Spectrom. 2003;9:1–21. doi: 10.1255/ejms.527. [DOI] [PubMed] [Google Scholar]
- 2.Hermanson GT. Bioconjugate techniques. 2. Academic Press; Amsterdam: 1998. [Google Scholar]
- 3.Yang WC, Mirzaei H, Liu XP, Regnier RE. Enhancement of amino acid detection and quantification by electrospray ionization mass spectrometry. Anal Chem. 2006;78:4702–4708. doi: 10.1021/ac0600510. [DOI] [PubMed] [Google Scholar]
- 4.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 5.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Beardsley RL, Sharon LA, Reilly JP. Peptide de novo sequencing facilitated by a dual-labeling strategy. Anal Chem. 2005;77:6300–6309. doi: 10.1021/ac050540k. [DOI] [PubMed] [Google Scholar]
- 7.Madsen JA, Brodbelt JS. Simplifying fragmentation patterns of multiply charged peptides by N-terminal derivatization and electron transfer collision activated dissociation. Anal Chem. 2009;81:3645–3653. doi: 10.1021/ac9000942. [DOI] [PubMed] [Google Scholar]
- 8.Knock SL, Miller BT, Blankenship JE, Nagle GT, Smith JS, Kurosky A. N-acylation of Aplysia egg-laying hormone with biotin. Characterization of bioactive and inactive derivatives. J Biol Chem. 1991;266:24413–24419. [PubMed] [Google Scholar]
- 9.Novak P, Kruppa GH, Young MM, Schoeniger J. A top-down method for the determination of residue-specific solvent accessibility in proteins. J Mass Spectrom. 2004;39:322–328. doi: 10.1002/jms.587. [DOI] [PubMed] [Google Scholar]
- 10.Tubb MR, Silva RAGD, Fang J, Tso P, Davidson WS. Lipids and lipoproteins: metabolism, regulation, and signaling. J Biol Chem. 2008;283:17314–17323. doi: 10.1074/jbc.M800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh P, Panchaud A, Goodlett DR. Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal Chem. 2010;82:2636–2642. doi: 10.1021/ac1000724. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowsk-Willis JC, Nilges M, Cramer P, Rappsilber J. Architecture of the RNA polymerase II–TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang P, Hadjar O, Gassman P, Laskin J. Reactive landing of peptide ions on self-assembled monolayer surfaces: an alternative approach for covalent immobilization of peptides on surfaces. Phys Chem Chem Phys. 2008;10:1512–1522. doi: 10.1039/b717617a. [DOI] [PubMed] [Google Scholar]
- 14.Vasicek L, Brodbelt JS. Enhancement of ultraviolet photodissociation efficiencies through attachment of aromatic chromophores. Anal Chem. 2010;82:9441–9446. doi: 10.1021/ac102126s. [DOI] [PubMed] [Google Scholar]
- 15.Pitteri SJ, Reid GE, McLuckey SA. Affecting proton mobility in activated whole protein ions via lysine guanidination. J Proteome Res. 2004;3:46–54. doi: 10.1021/pr034054u. [DOI] [PubMed] [Google Scholar]
- 16.Venter A, Sojka PE, Cooks RG. Droplet dynamics and ionization mechanisms in desorption electrospray ionization mass spectrometry. Anal Chem. 2006;78:8549–8555. doi: 10.1021/ac0615807. [DOI] [PubMed] [Google Scholar]
- 17.Girod M, Moyano E, Campbell DI, Cooks RG. Accelerated bimolecular reactions in microdroplets studied by desorption electrospray ionization mass spectrometry. Chem Sci. 2011;2:501–510. [Google Scholar]
- 18.Miao Z, Chen H. Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS) J Am Soc Mass Spectrom. 2009;20:10–19. doi: 10.1016/j.jasms.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Perry RH, Splendore M, Chien A, Davis NK, Zare RN. Detecting Reaction Intermediates in Liquids on the Millisecond Time Scale Using Desorption Electrospray Ionization. Angew Chem Int Ed. 2011;50:250–254. doi: 10.1002/anie.201004861. [DOI] [PubMed] [Google Scholar]
- 20.Badu-Tawiah AK, Li A, Jjunju FPM, Cooks RG. Peptide cross-linking at ambient surfaces by reactions of nanosprayed molecular cations. Angew Chem Int Ed. 2012;51 doi: 10.1002/anie.201205044. in press. DOI: 10.1002. [DOI] [PubMed] [Google Scholar]
- 21.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 22.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization–principles and practice. Mass Spectrom Rev. 1990;9:37–70. [Google Scholar]
- 23.Loo RRO, Udseth HR, Smith RD. A New Approach For The Study Of Gas-Phase Ion-Ion Reactions Using Electrospray Ionization. J Am Soc Mass Spectrom. 1992;3:695–705. doi: 10.1016/1044-0305(92)87082-A. [DOI] [PubMed] [Google Scholar]
- 24.Pitteri SJ, McLuckey SA. Recent developments in the ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev. 2005;24:931–958. doi: 10.1002/mas.20048. [DOI] [PubMed] [Google Scholar]
- 25.McLuckey SA, Stephenson JL., Jr Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev. 1998;17:369–407. doi: 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Scalf M, Westphall MS, Krause J, Kaufman SL, Smith LM. Controlling charge states of large ions. Science. 1999;283:194–197. doi: 10.1126/science.283.5399.194. [DOI] [PubMed] [Google Scholar]
- 27.Wells JM, Chrisman PA, McLuckey SA. Formation and characterization of protein-protein complexes in vacuo. J Am Chem Soc. 2003;125:7238–7249. doi: 10.1021/ja035051l. [DOI] [PubMed] [Google Scholar]
- 28.McLuckey SA. The emerging role of ion/ion reactions in biological mass spectrometry: considerations for reagent ion selection. Eur J Mass Spectrom. 2010;16:144–153. doi: 10.1255/ejms.1031. [DOI] [PubMed] [Google Scholar]
- 29.Han H, McLuckey SA. Selective covalent bond formation in polypeptide ions via gas-phase ion/ion reaction chemistry. J Am Chem Soc. 2009;131:12884–12885. doi: 10.1021/ja904812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hassell KM, Stutzman JR, McLuckey SA. Gas-phase bioconjugation of peptides via ion/ion charge inversion: Schiff base formation on the conversion of cations to anions. Anal Chem. 2010;82:1594–1597. doi: 10.1021/ac902732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stutzman JR, Hassel KM, McLuckey SA. Dissociation behavior of tryptic and intramolecular disulfide-linked peptide ions modified in the gas phase via ion/ion reactions. Int J Mass Spectrom. 2012;312:195–200. doi: 10.1016/j.ijms.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stutzman JR, Luongo CA, McLuckey SA. Covalent and non-covalent binding in the ion/ion charge inversion of peptide cations with benzene-disulfonic acid anions. J Mass Spectrom. 2012;47:669–675. doi: 10.1002/jms.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mentinova M, McLuckey SA. Covalent modification of gaseous peptide ions with N-hydroxysuccinimide ester reagent ions. J Am Chem Soc. 2010;132:18248–18257. doi: 10.1021/ja107286p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mentinova M, McLuckey SA. Intra- and inter-molecular cross-linking of peptide ions in the gas-phase: reagents and conditions. J Am Soc Mass Spectrom. 2011;22:912–921. doi: 10.1007/s13361-011-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mentinova M, Barefoot NZ, McLuckey SA. Solution versus gas-phase modification of peptide cations with NHS-ester reagents. J Am Soc Mass Spectrom. 2011;23:282–289. doi: 10.1007/s13361-011-0291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGee WM, Mentinova M, McLuckey SA. Gas-phase conjugation to arginine residues in polypeptide ions via N-hydroxysuccinimide ester-based reagent ions. J Am Chem Soc. 2012;134:11412–11414. doi: 10.1021/ja304778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheen JC, Hlavka JJ. The use of water-soluble and basic carbodiimides in peptide synthesis. J Org Chem. 1956;21:439–441. [Google Scholar]
- 38.Metz DH, Brown GL. The investigation of nucleic acid secondary structure by means of chemical modification with a carbodiimide reagent. I The reaction between N-cyclohexyl-N′-beta-(4-methylmorpholinium)ethylcarbodiimide and model nucleotides. Biochemistry. 1969;8:2312–2328. doi: 10.1021/bi00834a012. [DOI] [PubMed] [Google Scholar]
- 39.Hoare DG, Koshland DE., Jr A method for the quantitative modification and estimation of carboxylic acid groups in proteins. J Biol Chem. 1967;242:2447–2453. [PubMed] [Google Scholar]
- 40.Williams A, Ibrahim IA. A new mechanism involving cyclic tautomers for the reaction with nucleophiles of the water-soluble peptide coupling reagent 1-ethyl-3-(3′-(dimethylamino)propyl)carbodiimide (EDC) J Am Chem Soc. 1981;103:7090–7095. [Google Scholar]
- 41.Giles MA, Hudson AQ, Borders CL. Stability of water-soluble carbodiimides in aqueous solution. Anal Biochem. 1990;184:244–248. doi: 10.1016/0003-2697(90)90675-y. [DOI] [PubMed] [Google Scholar]
- 42.Staros JV, Wright RW, Swinghe DM. Enhancement of N-hydroxysulfosuccinimide of water-soluble carbodiimide-mediated coupling reactions. Anal Biochem. 1986;156:220–222. doi: 10.1016/0003-2697(86)90176-4. [DOI] [PubMed] [Google Scholar]
- 43.Xia Y, Wu J, Londry FA, Hager JW, McLuckey SA. Mutual storage mode ion/ion reactions in a hybrid linear ion trap. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 44.Liang X, Xia Y, McLuckey SA. Alternately pulsed nano-electrospray ionization/atmospheric pressure chemical ionization for ion/ion reactions in an electrodynamic ion trap. Anal Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang X, Han H, Xia Y, McLuckey SA. A pulsed triple ionization source for sequential ion/ion reactions in an electrodynamic ion trap. J Am Soc Mass Spectrom. 2007;18:369–376. doi: 10.1016/j.jasms.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 46.Londry FA, Hager JW. Mass selective axial ion ejection from a linear quadrupole ion trap. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 47.He M, McLuckey SA. Tandem mass spectrometry of half generation PAMAM dendrimer anions. Rapid Commun Mass Spectrom. 2004;18:960–972. doi: 10.1002/rcm.1431. [DOI] [PubMed] [Google Scholar]
- 48.Bowie JH, Brinkworth CS, Dua S. Collision-induced fragmentations of the (M-H)− parent anions of underivatized peptides: An aid to structure determination and some unusual negative ion cleavages. Mass Spectrom Rev. 2002;21:87–107. doi: 10.1002/mas.10022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.