

Background: Osmotic stress triggers RyR-mediated Ca2+ sparks that are spatially confined at the periphery of muscle fibers.
Results: Pharmacological intervention of IP3 production and genetic ablation of IP3R suppressed RyR-mediated Ca2+ sparks.
Conclusion: Activation of the type 1 IP3R is necessary to induce Ca2+ sparks.
Significance: This work highlights a potential interaction between IP3R and RyR in mediating Ca2+ signaling in adult skeletal muscle fibers.
Keywords: Calcium Imaging; Calcium Intracellular Release; Calcium Signaling; Inositol 1,4,5-Trisphosphate; Ryanodine Receptor; Skeletal Muscle
Abstract
Functional coupling between inositol (1,4,5)-trisphosphate receptor (IP3R) and ryanodine receptor (RyR) represents a critical component of intracellular Ca2+ signaling in many excitable cells; however, the role of this mechanism in skeletal muscle remains elusive. In skeletal muscle, RyR-mediated Ca2+ sparks are suppressed in resting conditions, whereas application of transient osmotic stress can trigger activation of Ca2+ sparks that are restricted to the periphery of the fiber. Here we show that onset of these spatially confined Ca2+ sparks involves interaction between activation of IP3R and RyR near the sarcolemmal membrane. Pharmacological prevention of IP3 production or inhibition of IP3R channel activity abolishes stress-induced Ca2+ sparks in skeletal muscle. Although genetic ablation of the type 2 IP3R does not appear to affect Ca2+ sparks in skeletal muscle, specific silencing of the type 1 IP3R leads to ablation of stress-induced Ca2+ sparks. Our data indicate that membrane-delimited signaling involving cross-talk between IP3R1 and RyR1 contributes to Ca2+ spark activation in skeletal muscle.
Introduction
Ca2+ sparks are localized Ca2+ release events originating from the opening of clustered ryanodine receptors (RyR)4 on the sarcoplasmic reticulum in muscle cells (1–3). In cardiac muscle, these spontaneous Ca2+ release events underlie the rhythmic contractile activity of the heart. In skeletal muscle, opening of the type 1 ryanodine receptor (RyR1) is strictly controlled by the voltage sensor located on the sarcolemmal membrane, and thus spontaneous Ca2+ sparks are rarely observed during resting conditions. We previously showed that application of transient osmotic stress led to robust Ca2+ sparks that are predominantly distributed at the periphery of the fibers from young, normal skeletal muscles (4). These Ca2+ spark events involve Ca2+ release from RyR1 as knock-out of RyR3 does not prevent the activation of Ca2+ sparks (5). Immunostaining and electron microscopy studies established that RyR1 is distributed throughout the junctional sarcoplasmic reticulum membrane in association with the transverse tubular invagination of the sarcolemmal membrane (6); thus, activation of these spatially confined Ca2+ sparks must reflect localized activation of RyR1 channels near the periphery of the muscle fiber. At present, it is not known whether physical perturbation of the junctional membrane structure and/or production of a local second messenger are essential for the initiation of stress-induced Ca2+ sparks in skeletal muscle.
It is known that application of osmotic stress can affect stretch-activated phospholipases on the plasma membrane and lead to production of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and inositol (1,4,5)-trisphosphate (IP3), which could impact intracellular Ca2+ signaling (7, 8). Several studies in smooth muscle and cardiomyocytes demonstrated that activation of IP3 receptors (IP3R) can contribute to RyR-mediated Ca2+ signaling in these cells and shape the spatial distribution and kinetic properties of Ca2+ sparks in smooth and cardiac muscle cells (9–14). Although some studies have shown that the IP3R can affect intracellular Ca2+ signaling in skeletal muscle (15–17) and potentially contribute to excitation-transcription coupling (18), the influence of IP3R on RyR-mediated Ca2+ spark signaling in skeletal muscle has not been clearly resolved.
In this study, we tested the hypothesis that cross-talk between IP3R and RyR underlies activation of stress-induced Ca2+ sparks in mammalian skeletal muscle. We found that inhibition of IP3R by pharmacological agents suppresses osmotic stress-induced Ca2+ sparks and that UV photo-release of caged IP3 could increase Ca2+ spark activity in intact skeletal muscle fibers. Selective activation of RyR1 near the periphery of the sarcolemma through coupling with IP3R appears to contribute to the onset of localized Ca2+ sparks as RNAi silencing of IP3R1 abolishes stress-induced skeletal muscle Ca2+ sparks. Our data provide direct evidence that IP3R1 modulates RyR1-mediated Ca2+ spark signaling in skeletal muscle.
EXPERIMENTAL PROCEDURES
Isolation of Flexor Digitorum Brevis (FDB) Muscle Fibers
The method was described in previous work (4, 19). Briefly, FDB muscles were surgically removed from mice in zero Ca2+ Tyrode's buffer (in mm) 140 NaCl, 5 KCl, 10 HEPES, 2 MgCl2 (pH 7.2) and incubated in Tyrode's buffer containing 2 mg/ml type I collagenase (Sigma C-5138) for 90 min at 37 °C. After two washes in isotonic Tyrode's buffer containing 2.5 mm Ca2+ (osmolality = 290 mosm), muscle fibers were gently dissociated by several passages through a series of micropipette tips of gradually decreasing diameter. Individual FDB muscle fibers were plated onto ΔTC3 glass-bottomed Petri dishes (Fisher Scientific) in Tyrode's buffer and used for experimentation within 6 h.
Confocal Ca2+ Imaging and Spark Analysis
Imaging of intracellular Ca2+ levels used previously described methods (4, 19). Individually isolated muscle fibers were loaded with Fluo4-AM (10 μm) for 60 min at room temperature. Measurements of Ca2+ sparks were performed using a Bio-Rad Radiance-2100 confocal microscope equipped with an argon laser (488 nm) and a ×40, 1.3 NA oil immersion objective. For Ca2+ spark measurements, fibers were perfused with hypotonic solution (osmolality = 170 mosm) containing (in mm) 70 NaCl, 5 KCl, 10 Hepes, 2.5 CaCl2, 2 MgCl2 (pH 7.2), for 100 s to induce swelling before perfusion was switched back to the initial isotonic Tyrode's buffer (osmolality = 290 mosm). The osmolality of all solutions was measured using an Advanced model 3300 micro-osmometer (Advanced Instruments). Line scan images of 512 pixels in length were acquired at a sampling rate of 2 ms/line, and serial x-y images of muscle fibers were acquired at 3.08 s/frame.
Patch Clamp Electrophysiology
This approach was adapted from the methods developed by Jacquemond and colleagues (20, 21) and adapted in our recent publication (19).
Photo-release of IP3
ciIP3/PM (caged membrane-permeant derivative of IP3) was dissolved in DMSO to yield a final concentration of 10 μm. FDB fibers were loaded with 10 μm ciIP3/PM for 45 min and an additional 30 min with 10 μm Fluo-4 AM. Photo-uncaging was performed according to Zhang et al. (12). The caged IP3 was released into the fibers by photolysis of the compound using a UV lamp following the protocol previously described (12). Only one photolysis was induced in each fiber.
shRNA Design
Multiple short hairpin RNA (shRNA) probes targeting common sequences on the mRNA for IP3R1 and IP3R2 were screened for their efficacy in knocking down IP3R1 and IP3R2 protein expression using transfection in C2C12 cells. We found one shRNA IP3R probe, 5′-GATCGACTACAGGAAGAACCAGGAGTACTTCAAGAGAGTACTCCTGGTTCTTCCTGTAGTCTTTTTT-3′, that was effective in knocking down the expression of IP3R1 and IP3R2. shRNA IP3R1 for specific knockdown of IP3R1 expression was according to the published sequence (22). Bold underlined nucleotides represent the conserved sequence on the target mRNAs. shRNA control targets sequence in the luciferase cDNA as described previously (19). These shRNA probes were annealed to the pU6-mRFP expression vector (19).
Electroporation of Plasmid DNA into Adult Muscle
Plasmid delivery followed previously published methods (19). For all experimental results reported here, mice were sacrificed at 14 days after electroporation, and FDB muscles were surgically removed. IP3R2−/− mice and age-matched wild-type control mice (11) were kindly provided by Dr. Ju Chen.
Western Blot Preparation
RFP expression was used as an indicator to measure the transfection efficiency of shRNA into the FDB muscle. Untransfected muscle bundles were trimmed out from the RFP-positive muscle for preparation of tissue lysates. The lysates were separated on a 6–10% SDS-PAGE gel. Antibodies used for immunoblots were as follows: rabbit polyclonal antibodies against IP3R1 and IP3R2 (gifts from Drs. J. Maxwell and G. Mignery (Loyola University) and J. Chen (University of California, San Diego (UCSD)); polyclonal anti-CSQ1 (from Dr. E. K. Kim (Gwangju Institute of Science and Technology)); mouse monoclonal antibodies against RyR1 (34c-ABR from Affinity BioReagents), actinin (from Sigma), and sarcoplasmic reticulum Ca2+-ATPase (SERCA) (from Affinity BioReagents); and goat polyclonal antibody of phospholipase Cδ (PLCδ) (from Dr. L. Runnels (University of Medicine and Dentistry of New Jersey (UMDNJ)). HRP-conjugated RFP antibody was from Abcam. Densitometry analysis was performed using ImageJ software.
Resting and Store Ca2+ Measurement
Individual RFP-positive FDB fibers (as indicator for transfection of shRNA) were loaded with 10 μm fura-2 AM. The ratio of fura-2 fluorescence at excitation wavelength of 340 and 380 nm was measured using a PTI spectrofluorometer (Photon Technology International) to assess the resting [Ca2+]i level. Zero Ca2+ Tyrode's solution was perfused onto the fiber before adding either caffeine/ryanodine or ionomycin (Sigma) to induce Ca2+ store release.
Chemicals
U73122 and U73343 (Cayman Chemicals), xestospongin C (Enzo Life Sciences), anthracene 9-carboxylic acid (Sigma), nifedipine (Sigma, MP Biomedicals), and ciIP3/PM (siChem) were dissolved in DMSO and diluted in experimental buffer to yield final concentration of 0.1% DMSO.
Statistical Analysis
Results are presented as mean ± S.E. as tested for statistical significance by Student's t test, *, p < 0.01.
RESULTS
Interference with IP3R Signaling Affects Stress-induced Ca2+ Sparks in Skeletal Muscle
We showed in our previous publication (4) that a characteristic feature of osmotic stress-induced Ca2+ sparks in intact skeletal muscle is their spatial distribution at the periphery of the muscle fiber directly underneath the sarcolemma (Fig. 1a). These osmotic stress-induced Ca2+ sparks are mediated by activation of RyR channel because preincubation of the FDB muscle fibers with 10 μm ryanodine could completely abolish the appearance of Ca2+ sparks (4). To test whether changes in sarcolemmal membrane potential could influence the onset of Ca2+ sparks in skeletal muscle, we used electrophysiological means to clamp the membrane potential at −70 mV following the method developed by Jacquemond and colleagues (20, 21). As shown in Fig. 1b, an apparently normal Ca2+ spark response was observed when the muscle fibers were clamped at −70 mV during application of osmotic stress. This suggests that changes in sarcolemmal membrane potential are not required in activation of osmotic stress-induced Ca2+ sparks in muscle fibers. Additional studies show that changes in Cl− flux associated osmotic stress did not appear to contribute to the onset of Ca2+ sparks (supplemental Fig. S1a).
FIGURE 1.
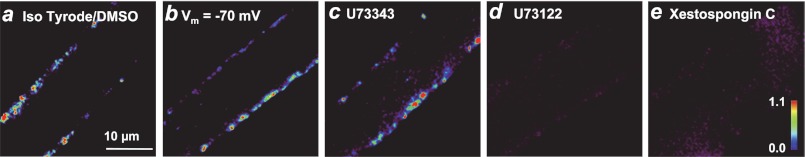
Interference of IP3R function affects osmotic stress-induced Ca2+ sparks in skeletal muscle. A binary plot of Fluo-4 fluorescence in skeletal muscle fibers, illustrating the peripheral distribution of Ca2+ sparks triggered by osmotic stress is shown (for details, see Refs. 4, 5, and 19). a, spatially confined Ca2+ sparks near the sarcolemmal region were observed in FDB muscle fiber following transient exposure to a hypo-osmotic stress (170 mosm) with a normal Tyrode's solution containing 2.5 mm Ca2+ (290 mosm) and 0.1% DMSO (n = 20 fibers). b, clamping of FDB fiber at −70 mV did not affect osmotic stress-induced Ca2+ sparks in a normal Tyrode's solution supplemented with 0.1 mm anthracene 9-carboxylic acid (n = 10 fibers). c, FDB fibers treated with U73343 (as a negative control) showed abundant Ca2+ sparks similar to untreated controls (n = 15 fibers). d, application of PLC inhibitor (5 μm U73122) prevented onset of osmotic stress-induced Ca2+ sparks (n = 12 fibers). e, application of 10 μm xestospongin C, an IP3R antagonist, prevented appearance of Ca2+ sparks (n = 15 fibers).
Exposure of cells to osmotic stress has been shown to increase PI(4,5)P2 production (8) and to activate PLC that can hydrolyze membrane-delimited PI(4,5)P2 to IP3 and diacylglycerol (7). Given the well known contribution of this signaling cascade to Ca2+ spark activation in various tissues (9–14), we used pharmacological inhibitors of PLC to test whether IP3R was involved in the generation of osmotic stress-induced Ca2+ sparks. Preincubation of FDB fibers with U73122 (5 μm), a pharmacological inhibitor of PLC, led to complete suppression of the osmotic stress-induced Ca2+ spark activity (Fig. 1d). As a control, FDB fibers treated with U73343 (an inactive chemical analog for U73122) displayed normal onset of osmotic stress Ca2+ sparks (Fig. 1c). Moreover, pretreatment of FDB fibers with xestospongin C (10 μm) (Fig. 1e) that specifically inhibited IP3R channel activation (18) greatly suppressed the Ca2+ spark activity following osmotic stress. Quantification analysis showed an average of 0.5 ± 0.1 Ca2+ spark events/min when compared with 4 ± 0.2 Ca2+ spark events/min in DMSO-treated fibers (Fig. 1a). Kinetic analysis of the amplitude (ΔF/F0) and full duration at half-maximum (FDHM) of the remaining Ca2+ sparks showed similar Ca2+ spark amplitude (0.85 ± 0.02; 0.92 ± 0.03) but reduced FDHM (67 ± 21.0ms; 102.2 ± 7.6 ms) in xestospongin C-treated fibers (Fig. 1e) when compared with DMSO-treated fibers (Fig. 1a). These findings provide initial evidence that activation of IP3R might be required for RyR-mediated Ca2+ sparks in skeletal muscle.
Facilitation of Ca2+ Spark Activity through Uncaging of IP3
To test whether local activation of IP3R could produce a Ca2+ spark response in intact skeletal muscle fibers, we used UV flash photo-uncaging to produce local elevation of IP3 by photo-uncaging of a caged IP3 analog, ciIP3/PM (12). A pulse of UV flash photo-uncaging of IP3 was applied at 20 s after the start of a 60-s confocal line scan of the fibers. Application of UV photo-uncaging in the DMSO-treated fibers did not affect the resting cytosolic Ca2+ level (Fig. 2a, top panel). Uncaging of IP3 in a resting muscle fiber bathed in an isotonic solution also did not produce any immediate Ca2+ release (Fig. 2a, middle panel).
FIGURE 2.

Uncaging of IP3 elicits additional Ca2+ sparks following osmotic stress in skeletal muscle. a, exposure of FDB fibers with UV laser did not induce changes in cytosolic Ca2+ at resting condition (top panel, n = 10 fibers). Fibers loaded with ciIP3/PM did not show localized Ca2+ sparks following UV flash photo-uncaging at resting condition (middle panel, n = 30 fibers). Fibers pretreated with osmotic stress showed significant elevation of Ca2+ sparks following photo-uncaging of ciIP3/PM. (bottom panel, n = 35 fibers); the line scan image was conducted at 7 min after exposure to osmotic stress. b, quantification of Ca2+ spark frequency before and after photo-uncaging of IP3 (calculated from the experiments shown in the bottom panel of a (n = 20 fibers). c, statistical analysis of the amplitude of Ca2+ sparks before and after photo-uncaging of IP3. These analyses were conducted on the first 20 s following photo-uncaging. Results are presented as mean ± S.E.
Other muscle fibers were challenged with osmotic stress, and IP3 uncaging was performed at 7 min after osmotic stress, a point when Ca2+ spark activity has greatly subsided. In greater than 50% of the fibers tested, we observed a significant increase in Ca2+ spark events within 20 s of the uncaging of IP3 in fibers that had previously been exposed to osmotic stress (Fig. 2a, lower panel, and 2b). The average amplitude of Ca2+ sparks was similar before and after uncaging of IP3 (Fig. 2c). In both resting (Fig. 2a, middle panel) and osmotic stress- (Fig. 2a, bottom panel) treated fibers, we observed diffuse Ca2+ release along with gradual elevation of cytosolic Ca2+ at later stages of the experiment (beyond 20 s after UV flash). This Ca2+ release could result from diffusion of IP3 to other IP3R targets (18, 23) rather than direct photo-damage to the muscle fiber as application of UV flash to the DMSO only-treated fibers did not result in any distinct Ca2+ release (Fig. 2a). Considering the immediate effect of IP3R in activating RyR-associated Ca2+ sparks in smooth muscle cells (12), we therefore focused our analysis on the production of Ca2+ sparks immediately following uncaging of IP3 by analyzing the characteristics of Ca2+ sparks appearing within 20 s of photo-uncaging of IP3. In this experiment, line scans were conducted at the periphery of the fiber as we normally observed the Ca2+ sparks in this region. Additional line scan recording at the middle of fiber upon photo-uncaging of IP3 did not reveal any increase in Ca2+ release events over base line (not shown). These findings provide direct evidence that IP3 can act as a diffusible second messenger to facilitate activation of Ca2+ sparks in skeletal muscle.
Our results are consistent with early studies from Pozzan and colleagues (16), who demonstrated that injection of IP3 in skinned muscle preparation could elicit intracellular Ca2+ release from skeletal muscle. Many subsequent studies from other investigators have revealed a role for IP3R in facilitating the RyR-mediated Ca2+ release in smooth muscle and cardiac muscle. Recent studies from Jaimovich and colleagues (18) demonstrated a role for IP3R in modulation of excitation-transcription coupling in skeletal muscle; however, controversy exists in this topic as Blaauw et al. (24) did not find a significant role for IP3 in skeletal muscle Ca2+ signaling. The observation that uncaging of IP3 alone could not produce Ca2+ spark events in muscle fibers without pre-exposure to osmotic stress suggests the possibility that other factors such as physical uncoupling of voltage sensor from RyR during osmotic stress could be required for induction of the robust Ca2+ spark events (4, 25).
Knockdown of IP3R Ablates Stress-induced Ca2+ Sparks in Skeletal Muscle
Toward understanding the role of IP3Rs in regulation of Ca2+ spark signaling in skeletal muscle, we examined the distribution of IP3R isoforms in skeletal muscle. Western blots showed that both IP3R1 and IP3R2 were present in the skeletal muscle and that IP3R3 was absent (Fig. 3a). Immunolocalization revealed staining of IP3R1 and IP3R2 in the perinuclear and subsarcolemmal region where Ca2+ sparks appear (supplemental Fig. S2). To directly test whether IP3R is required for induction of Ca2+ sparks in skeletal muscle, we used shRNA to simultaneously knock down both IP3R1 and IP3R2 in the FDB muscle using a probe targeting conserved sequences in the two mRNAs (Fig. 3b). As a control, we used an shRNA sequence targeting the luciferase cDNA (19). Plasmid DNA was delivered to FDB muscles in mice using electroporation-mediated transfection (19), and after 14 days, FDB muscles were removed. Western blotting showed that both IP3R1 and IP3R2 expression was significantly reduced in these muscles, whereas the expression of other Ca2+ regulatory proteins was not affected (Fig. 3c). The shRNA sequence was cloned into an RFP expression vector, allowing for identification of FDB muscle fibers with targeted knockdown of IP3R1 and IP3R2 (Fig. 3d). Muscle fibers with knockdown of both IP3R1 and IP3R2 (based on the presence of RFP fluorescence) were challenged with osmotic stress. Although robust Ca2+ sparks were observed in the shRNA control fibers (Fig. 4a, top panel), shRNA IP3R fibers displayed very few, if any, Ca2+ sparks following osmotic stress (Fig. 4a, bottom panel). Quantification of Ca2+ spark frequency showed that these events were extremely rare in the shRNA IP3R when compared with the shRNA control fibers (Fig. 4c). This result was in accordance with our data shown in Fig. 1 where pharmacological interference of IP3R signaling had a significant impact on the activation of Ca2+ sparks in skeletal muscle. Kinetic analysis of the remaining Ca2+ sparks showed that knockdown of IP3R1 and IP3R2 did not affect the Ca2+ spark amplitude (ΔF/F0) between shRNA control (0.96 ± 0.04, n = 20) and shRNA IP3R (0.91 ± 0.04, n = 30), whereas FDHM changed from 88.3 ± 5.5 ms in the shRNA control to 44.9 ± 21.0 ms in the shRNA IP3R fibers. The reduction in frequency of Ca2+ sparks following knockdown of IP3R was not the result of changes in the resting intracellular Ca2+ levels or loading of the internal Ca2+ stores. fura-2 ratiometric measurements showed that the resting [Ca2+]i levels were similar between shRNA IP3R (0.47 ± 0.01, n = 15) and shRNA control (0.43 ± 0.02, n = 15) (Fig. 4c). Ionomycin and caffeine plus ryanodine-induced release of intracellular Ca2+ stores showed no significant difference between shRNA control and shRNA IP3R fibers (Fig. 4c).
FIGURE 3.

shRNA-mediated knockdown of IP3R1 and IP3R2 in skeletal muscle. a, Western blots were performed using lysates from gastrocnemius muscle, lung, and brain tissues derived from WT mice (n = 6 mice). 10-fold of muscle lysates when compared with brain and lung lysates were loaded. IP3R1 and IP3R2, but not IP3R3, were present in the skeletal muscle. b, shRNA IP3R was designed to target a common sequence on IP3R1 and IP3R2. Knocking down both IP3Rs did not alter the expression of other Ca2+ regulatory proteins. c, densitometry analysis showed that shRNA IP3R resulted in significant knockdown of IP3R1 and IP3R2 in FDB muscle (n = 8 mice). A. U., arbitrary units. d, FDB fibers with knockdown of IP3Rs could be identified by the appearance of red fluorescence.
FIGURE 4.

Silencing of IP3R abolishes osmotic stress-induced Ca2+ sparks in skeletal muscle. a, robust peripheral Ca2+ sparks following osmotic stress was observed in the shRNA control fibers (n = 20 fibers), but not in the shRNA IP3R fibers (bottom panel) (n = 30 fibers). b, quantification analysis showed reduced Ca2+ spark activity after hypotonic perfusion in the shRNA IP3R fibers (green square) when compared with the shRNA control fibers (black square). c, representative fura-2 fluorescence in FDB fibers following treatment with 5 μm ionomycin to release the total intracellular [Ca2+]i store (left panel, n = 15 fibers) or following the addition of 20 mm caffeine + 5 μm ryanodine to release the sarcoplasmic reticulum Ca2+ store (middle panel, n = 15 fibers). Fiber was previously perfused with zero Ca2+ Tyrode's solution before application of either caffeine/ryanodine or ionomycin to induce store Ca2+release. Data are presented as mean ± S.E.
Activation of IP3R1, but Not IP3R2, Is Involved in RyR1-mediated Ca2+ Spark in Skeletal Muscle
Previous studies have shown that different IP3R isoforms exhibit different functional properties in Ca2+ signaling (22, 26, 27). We next asked whether a specific IP3R isoform is required for RyR-mediated Ca2+ sparks. Genetic ablation of IP3R2 produced a viable mouse model (11), allowing for examination of stress-induced Ca2+ sparks in adult skeletal muscle. When examined under our experimental conditions, we found that the IP3R2−/− skeletal muscle fibers displayed robust peripheral Ca2+ sparks upon osmotic stress stimulation (Fig. 5b), which was similar to the results in wild-type (WT) control or fibers transfected with the shRNA control (Fig. 5a).
FIGURE 5.

IP3R1 but not IP3R2 contributes to regulation of RyR-mediated Ca2+ spark activity. FDB muscle derived from the IP3R2−/− mice displayed robust peripheral Ca2+ sparks following osmotic stress (b, n = 18 fibers), similar to those observed in age-matched WT muscle (a, n = 18 fibers). c, knockdown of IP3R1 alone suppressed Ca2+ sparks in FDB muscle following osmotic stress (n = 22 WT/shRNA IR3P1 fibers). d, compromised Ca2+ spark response was also observed in the IP3R2−/− muscle fiber following electroporation of shRNA IP3R1 (n = 16 fibers). e, Western blots showed the specificity of shRNA IP3R1 in knocking down IP3R1, but not IP3R2 (n = 4 mice). f, time-dependent changes in Ca2+ spark activity in FDB fibers following hypo-osmotic stress were quantified. Knocking down of IP3R1 from WT muscle led to near complete suppression of Ca2+ spark activity (red) when compared with robust Ca2+ spark events in WT muscle that decline with time (black). Muscle fibers derived from IP3R2−/− mice showed normal Ca2+ spark response (blue), and knockdown of IP3R1 led to complete ablation of Ca2+ events (aqua).
We then tested whether IP3R1 was required for the onset of Ca2+ sparks in skeletal muscle. We used electroporation to deliver an shRNA probe that specifically targets knockdown of IP3R1 (shRNA IP3R1) (22), without affecting the expression of IP3R2 (Fig. 5e). We found that knocking down IP3R1 alone in FDB muscle derived from WT mice could completely suppress Ca2+ sparks following osmotic stress stimulation (Fig. 5c). Similar results were observed with electroporation of shRNA IP3R1 into the IP3R2−/− FDB muscle, where Ca2+ spark response was nearly completely ablated (Fig. 5, d and f). No significant differences were observed in the average Ca2+ spark amplitude (ΔF/F0) of WT (0.97 ± 0.05, n = 18), IP3R2−/− (0.93 ± 0.04, n = 18), shRNA IP3R1 (0.89 ± 0.06, n = 22), and shRNA IP3R1 in IP3R2−/− background (0.85 ± 0.08, n = 16). Although the average FDHM was similar between WT (105 ± 10.7 ms) and IP3R2−/− (100 ± 11.3 ms), it was significantly reduced in the shRNA IP3R1 in WT (45.7 ± 10.9 ms) or in IP3R2−/− fibers (42.5 ± 15.4 ms). Overall, our findings indicate that activation of IP3R1, and not IP3R2, was required to trigger RyR-mediated Ca2+ sparks in skeletal muscle.
DISCUSSION
We present evidence for a role for IP3R in regulation of RyR-mediated Ca2+ sparks in skeletal muscle. The Ca2+ spark response in skeletal muscle was absent with pharmacological intervention that inhibited activation of the IP3R channels. It was also absent in muscle fibers following knockdown of IP3R1; thus, activation of IP3R1 is required for the onset of osmotic stress-induced Ca2+ sparks in mammalian skeletal muscle. Based on our findings, we propose that initial Ca2+ release through IP3R1 channel coupled with transient membrane deformation activates neighboring RyR1, leading to the production of peripherally localized Ca2+ sparks in intact skeletal muscle fibers.
Although transient elevation of IP3 through photo-uncaging could amplify RyR-mediated Ca2+ release, the frequency of Ca2+ sparks triggered by IP3 was much less when compared with the osmotic stress-induced Ca2+ sparks. Moreover, significant elevation of Ca2+ sparks associated with photo-uncaging of IP3 was only observed in muscle fibers following exposure to osmotic stress. Thus, stress-induced Ca2+ sparks in mammalian skeletal muscle would likely require two cellular events, one involving production of a local IP3 second messenger and the other involving structural changes at the triad junction that would lead to uncoupling of RyR1 channel inhibition by the voltage sensor on the sarcolemmal membrane (4, 25). Our study added additional evidence that osmotic stress-induced Ca2+ sparks in adult skeletal muscle does not involve changes in ion flux across the sarcolemmal membrane or changes in membrane potential.
A distinct feature of the stress-induced Ca2+ sparks in skeletal muscle is their confinement to the periphery of the muscle fiber. We show that the IP3R channels are preferentially localized near the subsarcolemmal region of the muscle fiber, which could potentially facilitate local activation of the RyR1 channel through Ca2+ as an intermediate messenger. The asymmetrical distribution of phospholipids on the sarcolemmal membrane (28, 29) could result in localized activation of PLC and IP3 production, which could represent a peripherally confined signal for activation of the RyR1 channel in skeletal muscle. Although we showed that IP3R2 is not required for the osmotic stress-induced Ca2+ sparks in skeletal muscle, a specific role for IP3R1 in the activation of Ca2+ sparks is established through our shRNA silencing approaches. Although the different Ca2+ signaling properties of the different IP3R isoforms (26, 27) could contribute to the specificity of IP3R1 in regulating RyR1-mediated Ca2+ sparks, it is not clear why IP3R2 is not required for Ca2+ spark activation. Future studies are necessary to test whether a direct interaction between the different isoforms of IP3R and RyR plays a role in their functional cross-talk in modulating the spatial and temporal aspects of Ca2+ signaling in skeletal muscle or whether other intermediate messengers that favor the IP3R1 activation pathway could participate in modulation of the stress-induced changes in Ca2+ signaling in skeletal muscle.
The role for IP3 in Ca2+ signaling in skeletal muscle was discovered over 25 years ago (15, 16); however, cross-talk between IP3R and RyR in modulation of Ca2+ signaling has mostly been studied in cardiac and smooth muscles (9–13). Altered Ca2+ release mediated by cross-talk between IP3R and RyR has been linked to hypertrophic cardiomyopathy and arrhythmia (9, 11, 13, 14, 30–33), indicating the role of such cross-talk in physiology and pathophysiology. Peripherally confined Ca2+ sparks in smooth muscle have been linked to activation of Ca2+-dependent K+ channel, which is important for vasodilation (2, 34). Numerous studies in other cell types have shown the physiological relevance of localized Ca2+ signals in vesicle fusion (28) and cytoskeletal reorganization (35). Our findings of IP3R cross-talk with RyR1 in skeletal muscle suggest that this may be a mechanism governing normal Ca2+ regulation in skeletal muscle, and these findings also open a new line of investigation into potential pathophysiologic mechanism during muscle diseases. Several studies showed that the pathological consequence of altered Ca2+ signals observed in the dystrophic skeletal muscle fibers was associated with abnormal IP3R distribution (36, 37). Knockdown of IP3R1 could minimize cell death in dystrophin-deficient muscle fibers (38). Our previous studies showed that Ca2+ spark in the dystrophic (mdx) muscle fibers was irreversible and no longer peripherally confined (4). Hence, aberrant Ca2+ sparks observed in the dystrophic skeletal muscle could be associated with altered IP3R1 expression, localization, or activation.
Acknowledgments
We thank Dr. Ju Chen (UCSD) for the kind gifts of IP3R2−/− mice and age-matched wild-type control mice; Drs. J. Maxwell and G. Mignery (Loyola University) and J. Chen (UCSD) for IP3R1 and IP3R2 antibodies; Dr. E. K. Kim (Gwangju Institute of Science and Technology) for CSQ1 antibody; Dr. L Runnels (UMDNJ) for PLCδ antibody; and Dr. Jerome Parness (Children's Hospital of Pittsburgh) for comments and critical reading of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants AG28614, AG28856, and HL69000 (to J. M.) and AR054793 (to N. W.).
This article was selected as a Paper of the Week.

This article contains supplemental Figs. S1 and S2.
- RyR
- ryanodine receptor(s)
- IP3
- inositol (1,4,5)-trisphosphate
- IP3R
- IP3 receptor(s)
- FDB
- flexor digitorum brevis
- PLC
- phospholipase C
- DMSO
- dimethyl sulfoxide
- FDHM
- full duration at half-maximum.
REFERENCES
- 1. Cheng H., Lederer W. J., Cannell M. B. (1993) Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262, 740–744 [DOI] [PubMed] [Google Scholar]
- 2. Nelson M. T., Cheng H., Rubart M., Santana L. F., Bonev A. D., Knot H. J., Lederer W. J. (1995) Relaxation of arterial smooth muscle by calcium sparks. Science 270, 633–637 [DOI] [PubMed] [Google Scholar]
- 3. Klein M. G., Cheng H., Santana L. F., Jiang Y. H., Lederer W. J., Schneider M. F. (1996) Two mechanisms of quantized calcium release in skeletal muscle. Nature 379, 455–458 [DOI] [PubMed] [Google Scholar]
- 4. Wang X., Weisleder N., Collet C., Zhou J., Chu Y., Hirata Y., Zhao X., Pan Z., Brotto M., Cheng H., Ma J. (2005) Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat. Cell Biol. 7, 525–530 [DOI] [PubMed] [Google Scholar]
- 5. Weisleder N., Ferrante C., Hirata Y., Collet C., Chu Y., Cheng H., Takeshima H., Ma J. (2007) Systemic ablation of RyR3 alters Ca2+ spark signaling in adult skeletal muscle. Cell Calcium 42, 548–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Block B. A., Imagawa T., Campbell K. P., Franzini-Armstrong C. (1988) Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107, 2587–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujii M., Ohtsubo M., Ogawa T., Kamata H., Hirata H., Yagisawa H. (1999) Real-time visualization of PH domain-dependent translocation of phospholipase C-δ1 in renal epithelial cells (MDCK): response to hypo-osmotic stress. Biochem. Biophys. Res. Commun. 254, 284–291 [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto M., Chen M. Z., Wang Y. J., Sun H. Q., Wei Y., Martinez M., Yin H. L. (2006) Hypertonic stress increases phosphatidylinositol 4,5-bisphosphate levels by activating PIP5KIβ. J. Biol. Chem. 281, 32630–32638 [DOI] [PubMed] [Google Scholar]
- 9. Mackenzie L., Roderick H. L., Berridge M. J., Conway S. J., Bootman M. D. (2004) The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J. Cell Sci. 117, 6327–6337 [DOI] [PubMed] [Google Scholar]
- 10. Gordienko D. V., Bolton T. B. (2002) Crosstalk between ryanodine receptors and IP3 receptors as a factor shaping spontaneous Ca2+-release events in rabbit portal vein myocytes. J. Physiol. 542, 743–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X., Zima A. V., Sheikh F., Blatter L. A., Chen J. (2005) Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ. Res. 96, 1274–1281 [DOI] [PubMed] [Google Scholar]
- 12. Zhang W. M., Yip K. P., Lin M. J., Shimoda L. A., Li W. H., Sham J. S. (2003) ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am. J. Physiol. Lung Cell Mol. Physiol. 285, L680–L690 [DOI] [PubMed] [Google Scholar]
- 13. Zima A. V., Blatter L. A. (2004) Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation-contraction coupling and arrhythmias. J. Physiol. 555, 607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Domeier T. L., Zima A. V., Maxwell J. T., Huke S., Mignery G. A., Blatter L. A. (2008) IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H596–H604 [DOI] [PubMed] [Google Scholar]
- 15. Vergara J., Tsien R. Y., Delay M. (1985) Inositol 1,4,5-trisphosphate: a possible chemical link in excitation-contraction coupling in muscle. Proc. Natl. Acad. Sci. U.S.A. 82, 6352–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Volpe P., Salviati G., Di Virgilio F., Pozzan T. (1985) Inositol 1,4,5-trisphosphate induces calcium release from sarcoplasmic reticulum of skeletal muscle. Nature 316, 347–349 [DOI] [PubMed] [Google Scholar]
- 17. Valdivia C., Vaughan D., Potter B. V., Coronado R. (1992) Fast release of 45Ca2+ induced by inositol 1,4,5-trisphosphate and Ca2+ in the sarcoplasmic reticulum of rabbit skeletal muscle: evidence for two types of Ca2+ release channels. Biophys. J. 61, 1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Casas M., Figueroa R., Jorquera G., Escobar M., Molgó J., Jaimovich E. (2010) IP3-dependent, post-tetanic calcium transients induced by electrostimulation of adult skeletal muscle fibers. J. Gen. Physiol. 136, 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tjondrokoesoemo A., Park K. H., Ferrante C., Komazaki S., Lesniak S., Brotto M., Ko J. K., Zhou J., Weisleder N., Ma J. (2011) Disrupted membrane structure and intracellular Ca2+ signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS ONE 6, e25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacquemond V. (1997) Indo-1 fluorescence signals elicited by membrane depolarization in enzymatically isolated mouse skeletal muscle fibers. Biophys. J. 73, 920–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Collet C., Csernoch L., Jacquemond V. (2003) Intramembrane charge movement and L-type calcium current in skeletal muscle fibers isolated from control and mdx mice. Biophys. J. 84, 251–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hattori M., Suzuki A. Z., Higo T., Miyauchi H., Michikawa T., Nakamura T., Inoue T., Mikoshiba K. (2004) Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J. Biol. Chem. 279, 11967–11975 [DOI] [PubMed] [Google Scholar]
- 23. Wu X., Zhang T., Bossuyt J., Li X., McKinsey T. A., Dedman J. R., Olson E. N., Chen J., Brown J. H., Bers D. M. (2006) Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Invest. 116, 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blaauw B., Del Piccolo P., Rodriguez L., Hernandez Gonzalez V. H., Agatea L., Solagna F., Mammano F., Pozzan T., Schiaffino S. (2012) No evidence for inositol 1,4,5-trisphosphate-dependent Ca2+ release in isolated fibers of adult mouse skeletal muscle. J. Gen. Physiol. 140, 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Apostol S., Ursu D., Lehmann-Horn F., Melzer W. (2009) Local calcium signals induced by hyper-osmotic stress in mammalian skeletal muscle cells. J. Muscle Res. Cell Motil. 30, 97–109 [DOI] [PubMed] [Google Scholar]
- 26. Foskett J. K., White C., Cheung K. H., Mak D. O. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyakawa T., Maeda A., Yamazawa T., Hirose K., Kurosaki T., Iino M. (1999) Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18, 1303–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glitsch M. D. (2008) Spontaneous neurotransmitter release and Ca2+–how spontaneous is spontaneous neurotransmitter release? Cell Calcium 43, 9–15 [DOI] [PubMed] [Google Scholar]
- 29. Vicogne J., Vollenweider D., Smith J. R., Huang P., Frohman M. A., Pessin J. E. (2006) Asymmetric phospholipid distribution drives in vitro reconstituted SNARE-dependent membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 103, 14761–14766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harzheim D., Movassagh M., Foo R. S., Ritter O., Tashfeen A., Conway S. J., Bootman M. D., Roderick H. L. (2009) Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 106, 11406–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakayama H., Bodi I., Maillet M., DeSantiago J., Domeier T. L., Mikoshiba K., Lorenz J. N., Blatter L. A., Bers D. M., Molkentin J. D. (2010) The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ. Res. 107, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Proven A., Roderick H. L., Conway S. J., Berridge M. J., Horton J. K., Capper S. J., Bootman M. D. (2006) Inositol 1,4,5-trisphosphate supports the arrhythmogenic action of endothelin-1 on ventricular cardiac myocytes. J. Cell Sci. 119, 3363–3375 [DOI] [PubMed] [Google Scholar]
- 33. Mackenzie L., Bootman M. D., Laine M., Berridge M. J., Thuring J., Holmes A., Li W. H., Lipp P. (2002) The role of inositol 1,4,5-trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J. Physiol. 541, 395–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pérez G. J., Bonev A. D., Patlak J. B., Nelson M. T. (1999) Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J. Gen. Physiol. 113, 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wei C., Wang X., Chen M., Ouyang K., Song L. S., Cheng H. (2009) Calcium flickers steer cell migration. Nature 457, 901–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Balghi H., Sebille S., Mondin L., Cantereau A., Constantin B., Raymond G., Cognard C. (2006) Mini-dystrophin expression down-regulates IP3-mediated calcium release events in resting dystrophin-deficient muscle cells. J. Gen. Physiol. 128, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cárdenas C., Juretić N., Bevilacqua J. A., García I. E., Figueroa R., Hartley R., Taratuto A. L., Gejman R., Riveros N., Molgó J., Jaimovich E. (2010) Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells. FASEB J. 24, 3210–3221 [DOI] [PubMed] [Google Scholar]
- 38. Mondin L., Balghi H., Constantin B., Cognard C., Sebille S. (2009) Negative modulation of inositol 1,4,5-trisphosphate type 1 receptor expression prevents dystrophin-deficient muscle cells death. Am. J. Physiol. Cell Physiol 297, C1133–C1145 [DOI] [PubMed] [Google Scholar]