

Background: ARHGAP21 is an important Rho-GAP for Cdc42 involved in vesicle trafficking and focal adhesion kinase activity.
Results: ARHGAP21 participates in cell-cell adhesion formation and cellular migration, interacts and modulates α-tubulin acetylation, and is essential for epithelial-mesenchymal transition.
Conclusion: ARHGAP21 is a novel α-tubulin partner coordinating cell-cell adhesion, migration, and epithelial-mesenchymal transition.
Significance: ARHGAP21 might be involved in cancer metastasis.
Keywords: Cell Adhesion, Cell Migration, Epithelial Mesenchymal Transition, Rho GTPases, Tubulin, ARHGAP21, α-Tubulin Acetylation
Abstract
Cell-cell adhesions and the cytoskeletons play important and coordinated roles in cell biology, including cell differentiation, development, and migration. Adhesion and cytoskeletal dynamics are regulated by Rho-GTPases. ARHGAP21 is a negative regulator of Rho-GTPases, particularly Cdc42. Here we assess the function of ARHGAP21 in cell-cell adhesion, cell migration, and scattering. We find that ARHGAP21 is localized in the nucleus, cytoplasm, or perinuclear region but is transiently redistributed to cell-cell junctions 4 h after initiation of cell-cell adhesion. ARHGAP21 interacts with Cdc42, and decreased Cdc42 activity coincides with the appearance of ARHGAP21 at the cell-cell junctions. Cells lacking ARHGAP21 expression show weaker cell-cell adhesions, increased cell migration, and a diminished ability to undergo hepatocyte growth factor-induced epithelial-mesenchymal transition (EMT). In addition, ARHGAP21 interacts with α-tubulin, and it is essential for α-tubulin acetylation in EMT. Our findings indicate that ARHGAP21 is a Rho-GAP involved in cell-cell junction remodeling and that ARHGAP21 affects migration and EMT through α-tubulin interaction and acetylation.
Introduction
Cadherin-based cell-cell adhesions play a fundamental role in cell biology, holding together single cells into diverse arrangements necessary for tissue structure and function (1). The actin cytoskeleton plays a major role in the formation and maintenance of cell-cell adhesions, underlying major morphological changes that accompany cell-cell adhesion (2). Establishment of cadherin-mediated cell-cell adhesions alters activity of Rho-GTPases and recruits them to the contact site (3, 4). These actin regulators in turn influence cadherin-mediated cell-cell adhesion (4, 5). Defective cell-cell adhesion is a hallmark of cancer progression, as it allows cells to detach from tissues and then migrate and invade distant sites (6), and it is thought to result from perturbations in cell-cell junction remodeling programs (7–10).
Migrating cells have an asymmetrical distribution of signaling molecules and the cytoskeleton (11, 12); microtubules and actin are important for establishing and maintaining the cell polarity (11, 13, 14). Microtubules are dynamically assembled polymers of α and β-tubulin (11). In addition to the actin cytoskeleton and cell-cell adhesion, Rho-GTPases also regulate microtubule dynamics (11, 12). Rho-GTPases stabilize microtubules through their effectors, leading to a polarized microtubule arrangement; in turn, microtubules affect the activities of Rho-GTPases (3, 11, 12, 15).
Cdc42, a member of the Rho-GTPase family, has been implicated in early cell-cell junction formation (2). This Rho-GTPase induces actin-based membrane protrusions that are thought to expand nascent cell-cell contacts (16–18). During cell migration, Cdc42 is activated at the leading edge, polarizing the actin and microtubule cytoskeleton (11, 19). Cdc42 is also responsible for the cell polarization and reorientation of the Golgi apparatus in the direction of the leading edge (11, 12, 19). However, it is not completely understood how Cdc42 activity is regulated by Rho-GTPase activating proteins (Rho-GAPs) during cell-cell adhesion and migration.
Rho-GAPs facilitate GTP hydrolysis by Rho-GTPases (20, 21). The Rho-GTPase family includes around 20 members, but ∼80 distinct putative proteins contain a Rho-GAP domain (21). Thus, Rho-GAPs may have specialized roles in regulating distinct Rho-GTPases at specific cellular locations (22, 23). ARHGAP21, a protein of 1958 amino acids, contains a PDZ, a pleckstrin homology (PH),3 and a Rho-GAP domain (24). ARHGAP21 functions in the negative regulation of Cdc42 (25, 26). In addition, ARHGAP21 has been reported as a partner of α-catenin that controls α-catenin recruitment to the cell-cell junctions (23). Because Rho-GTPases control cell-cell junction formation and dysregulation might lead to altered cell-cell junction remodeling (27), we sought to know if ARHGAP21 participates in cell-cell adhesion and sought to define its involvement in cell migration and epithelial-mesenchymal transition.
EXPERIMENTAL PROCEDURES
Cell Culture and Calcium Switch Assay
Madin-Darby canine kidney (MDCK), DU145 (human prostate cancer), and SW480 (human colon adenocarcinoma) cell lines were obtained from ATCC (Manassas, VA). These cells lines were not used to compare normal and malignant cells or cells from different epithelial functions according to their origin; these cell lines were used in specific assays as appropriated. MDCK and DU145 cells were maintained in DMEM with 10% fetal bovine serum (FBS). SW480 cells were cultured in McCoy's medium with 10% FBS. In calcium switch assays, MDCK and DU145 cells were seeded in low calcium medium plus 10% Dialyzed FBS (GIBCO) at high confluence on collagen-coated coverslips. After 3 h, regular medium was perfused into the culture. Cells were processed for fluorescence imaging 0, 1, 4, 7, or 24 h after calcium restoration. MDCK cells are a well established cell line to study cell-cell adhesion, and DU145 cells are a human cell line suitable to study cell-cell adhesion.
Cdc42 Activity Assay
Cdc42 WASP binding domain (kindly provided by Dr. David Sacks from Brigham and Women's Hospital, Boston) was expressed as previously described (4). MDCK cells were lysed in GST-Fish buffer (10% glycerol, 50 mm Tris, pH 7.4, 100 mm NaCl, 1% Nonidet P-40, 2 mm MgCl2, 10 μg/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, and 1 μg/ml leupeptin). Extracts containing 500 μg of total proteins were incubated with 50 μg of GST-WBD beads on a rotator for 90 min at 4 °C. Beads were precipitated by centrifugation at 12,000 rpm and washed repeatedly with GST-Fish buffer. Cdc42-WBD (Cdc42-GTP) complexes and total cell extracts were separated by SDS-PAGE and transferred to PVDF, and the blots probed with anti-Cdc42 monoclonal antibody.
RNA Interference
Stable SW480 ARHGAP21-knockdown cell lines were established with the shRNA GTATTCCGGCCATGGAAACA inserted in pSilencer 2.1-U6/neo (Ambion). SW480 cells were transfected using Effectene (Qiagen) and selected with G418 (0.8 mg/ml). Stable transfectants were screened for ARHGAP21 expression levels by Western blot analysis. Transient inhibition of ARHGAP21 was performed in DU145 cells with silencing RNA SMARTpools from Dharmacon. DU145 cells were transfected with ARHGAP21 and siCONTROL nontargeting siRNA using Lipofectamine (Invitrogen) following the manufacturer's instructions. The silencing efficiency of siRNA for the targeted mRNA was tested by quantitative RT-PCR and Western blot 72 h post-transfection.
DNA Constructs
The PCR fragments, encoding PDZ-ARHGAP21 (571 bp), PH+GAP-ARHGAP21 (1221 bp), or Cter-ARHGAP21 (1865 bp), were subcloned into the mammalian expression vector pEGFP-N2 (Clontech). The constructs were verified by double-strand sequencing. MDCK cells were transfected with the plasmids constructs using Lipofectamine (Invitrogen) following the manufacturer's instructions. Sequences of the primers are available upon request. DU145 cells were unsuccessfully transfected.
Quantitative RT-PCR
Total RNA was extracted using TRIzol® reagent (Invitrogen) as per the manufacturer's instructions, and cDNA was synthesized from 1 μg of RNA using SuperScript® III Reverse Transcriptase kit (Invitrogen). Primers used were 5′-GTCCATCCTGCTATTGCTCTG and 5′-AAGCCGCACATATCCAACAT. GAPDH and β-actin primers were used as housekeeping genes. Quantitative RT-PCR was performed in a 7500 Sequence Detector System (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems). Samples were run in quadruplicate, and gene expression was calculated using the equation 2−ΔΔCt (28).
Hanging Drop Aggregation Assay
The assay was performed as described (29) with minor modifications. Briefly, trypsinized SW480 cells were resuspended at 2.5 × 105 cells/ml in the appropriate medium. Drops (20 μl) of cell suspension were placed onto the inner surface of the lid of a Petri dish and incubated for 0–3 h. At each time point, drops were either placed directly onto a glass slide or placed on the slide after being triturated 5 times through a 20-ml pipette. Three random fields of cells were photographed for each condition (with and without trituration), and cells in aggregates of different sizes (1–10, 11–50, and >50 cells) were counted. This experiment was performed three times for each cell line condition.
Migration Assay
Oris Cell Migration Assays (Platypus Technologies) were performed in triplicate following the manufacturer's instructions. Briefly, 6 × 104 cells were seeded into each assay. Migration barriers were removed after 24 h and then incubated for another 18 h. Cells were washed with PBS, fixed with 4% paraformaldehyde for 15 min, and stained with crystal violet. The area covered by the migration barrier was captured using a microscope and analyzed with ImageJ software. Transwell migration assays were performed in sextuplicate using a 96-well format (8 micron pore, Corning). 7 × 104 cells were seeded onto the filter and incubated for 24 h. HGF-conditioned medium (0, 10 and 20%) was placed in the bottom chamber, and cells incubated an additional 20 h. Filters were washed with PBS (2.7 mm KCl, 1.5 mm KH2PO4, 137 mm NaCl, 8.1 mm Na2HPO4), fixed with 4% paraformaldehyde for 15 min, and stained with crystal violet. Remaining cells were removed from the upper side of the filter. Photometry analyses were performed using a FluorChem photometer. For the wound-healing assay, confluent cells were seeded in coverslips and serum-starved for 12 h. A linear wound was made using a pipette tip. Coverslips were fixed in various times.
Immunofluorescence
Cells were washed repeatedly with ice-cold PBS, fixed for 10 min on ice in 4% paraformaldehyde, and blocked with PBS supplemented with 0.4% bovine serum albumin, 50 mm NH4Cl, 0.5% Triton X-100, and 0.5% goat serum. Cells were stained with primary antibodies against ARHGAP21 (Sigma), α-catenin, β-catenin (BD Transduction Laboratories), E-cadherin, JAM-A (Santa Cruz Biotechnology), or TRITC-phalloidin. Alexa Fluor 488-conjugated anti-rabbit and Alexa Fluor 633-conjugated antibodies were used as secondary antibodies. Coverslips were mounted in VectaShield (Vector Labs) and viewed under an Olympus inverted IX-8I microscope with a 60× or 100× oil immersion objective lens. Images were acquired with a Hamamatsu Orca-ER digital camera and analyzed using Slidebook software.
Immunoprecipitation
DU145 cell lysates were prepared in radioimmune precipitation assay buffer containing protease inhibitors, as previously described (30). Briefly, 500 μg of total DU145 cell extracts were incubated overnight with 20 μl of anti-ARHGAP21, α-tubulin antibody (Santa Cruz Biotechnology), or with normal rabbit immunoglobulin (IgG) as a negative control. The immune complexes were precipitated with protein-A-Sepharose 50% slurry (GE Healthcare), washed in radioimmune precipitation assay buffer to remove unspecific proteins, and then analyzed by Western blotting with the antibodies of interest.
Epithelial-Mesenchymal Transition (EMT)
To observe HGF-induced scattering, 1 × 105 cells were seeded on collagen-coated DeltaT imaging dishes (Bioptechs) and allowed to form colonies. Cells were maintained in 10% FBS for 24 h, then starved for 24 h in 0.5% of FBS. Cells were then stimulated with 20% conditioned medium containing HGF. Phase contrast images were captured every 2 min for 10 h using a 10× (0.30 aperture) objective and 1.6× slider. Data were acquired from >20 colonies for each condition over 3 separate experiments. Scattering behavior was quantified as performed by Sperry et al. (31); no scattering: hardly any intracolony spaces (ICS) and retraction fibers (RF); light scattering: showing ICS/RF but no cell break away; semi-scattering: ICS/RF with at least one cell that completely breaks away; full scattering: ICS/RF with two or more cells breaking from colony. To protein extraction, 1.5 × 106 DU145 cells were plated and maintained in 10% FBS for 24 h followed by starving for 24 h in 0.5% of FBS. Then, 20 ng/ml HGF was added to the cells. Cells were collected at 10 and 30 min and 1, 2, 3, and 4 h after HGF addition. DU145 cells were chosen here because that is a human cell line that undergoes EMT by HGF, and the siRNA experiments could not be performed in MDCK cells because ARHGAP21 canine gene is only predicted in a database.
Protein Analysis by Immunoblotting
Pelleted cells were resuspended in radioimmune precipitation assay buffer, incubated for 45 min at 4 °C, and centrifuged for 30 min at 12000 rpm at 4 °C. The same amount of protein was loaded on SDS-PAGE, and blots were probed with anti-ARHGAP21 (Sigma), anti-β-actin, anti-α-tubulin, anti-ZEB1, anti-Slug, anti-vimentin, anti-E-cadherin (Santa Cruz Biotechnology), anti-acetylated α-tubulin, anti-Twist1 (Abcam), anti-Snail, anti-Cdc42 (Cell Signaling) antibodies.
HDAC Activity
Cytoplasmic HDAC activity was measured using the EpiQuik HDAC Activity/Inhibition Assay kits (Epigentek) according to the manufacturer's instructions. In brief, triplicates of DU145 cytoplasmic extracts were incubated with specific substrate for 1 h at 37 °C followed by capture antibody for 1 h on an orbital shaker and then detection antibody for 30 min at room temperature. Absorbance was read on a microplate spectrophotometer at 450 nm.
Statistical Analysis
The number of cells obtained from the aggregation assay and the EMT scattering was analyzed using a χ2 test. The migration values between ARHGAP21 inhibition and control cells were compared using a Mann-Whitney U test. A p value of ≤0.05 was considered statistically significant.
RESULTS
ARHGAP21 Appears at Cell-Cell Contacts during Cell-Cell Adhesion Formation
ARHGAP21 localizes to the nucleus, cytoplasm, and perinuclear region of the cells depending on the cell type (25, 26). Given the role of Rho-GTPases in cell-cell junction formation, we sought to determine whether ARHGAP21 might participate in regulation of Rho-GTPases at cell-cell contacts. To test this we analyzed ARHGAP21 localization in MDCK and DU145 epithelial cells. To detect a transient localization of ARHGAP21 at cell-cell contacts during cell-cell junction formation, we subjected those cell lines to a calcium switch and fixed the cells at various times after initiation of adhesion.
In MDCK cells (Fig. 1A), ARHGAP21 was localized in the nucleus and in the cytoplasm at time 0 and 1 h after initiation of cell-cell adhesion. However, 4 h after calcium restoration, ARHGAP21 was present at cell-cell junctions (arrow), with a decreasing expression at the junctions in the latter hours (arrow at 7 h). After 24 h of calcium restoration, ARHGAP21 are no longer seen at MDCK cell-cell junctions. ARHGAP21 co-localized with actin at cell-cell contacts (Fig. 1B). Cdc42 activity was high for the first 4 h after calcium restoration, but a significant decrease was observed at 5 h (Fig. 1C), right after ARHGAP21 appeared at cell-cell junctions.
FIGURE 1.

Localization of ARHGAP21 at cell-cell junctions during adhesion formation. A, shown is immunofluorescence analysis of MDCK cells stained with an antibody against ARHGAP21 (green) after calcium restoration at 0, 1, 4, 7 and 24 h; images were taken using a 60× oil immersion objective. The arrow indicates ARHGAP21 localization at cell-cell adhesion sites. Note that ARHGAP21 was detected at cell-cell junctions 4 h after calcium restoration in MDCK cells. B, shown is an enlarged image of MDCK cells 4 h after calcium restoration stained with an antibody against ARHGAP21 (green) and F-actin (phalloidin-TRITC); co-localization of ARHGAP21 and actin is observed in the merged image. C, shown is Cdc42 Rho-GTPase activity during cell-cell adhesion formation at 0, 1, 2, 3, 4, 5, and 7 h after calcium restoration in MDCK cells. Cdc42 Rho-GTPase activity drastically reduced after ARHGAP21 localization at the cell-cell junctions. IB, immunoblot. D, shown is a schematic representation of ARHGAP21 and its domains and of the ARHGAP21 regions cloned in fusion to GFP. E, F, and G, MDCK cells were transfected with plasmids encoding ARHGAP21 constructs fused to GFP (PDZ-ARHGAP21, PH+GAP-ARHGAP21, Cter-ARHGAP21); the PDZ-ARHGAP21 domain was at the cell-cell contacts 4 h after calcium addition. H, shown is immunofluorescence analysis of DU145 cells stained with antibody against ARHGAP21 (green) 4 and 24 h after calcium restoration; images were acquired with 100× oil immersion objective. Note that ARHGAP21 was detected at cell-cell junctions 4 h after calcium restoration in DU145 cells. I, shown is an enlarged image of DU145 cells 4 h after calcium restoration stained with antibody against ARHGAP21 (green) and E-cadherin (red) which shows that both proteins are not co-localized at cell-cell contacts. J, shown is an enlarged image of DU145 cells 4 h after calcium restoration stained with antibody against ARHGAP21 (green) and JAM-A (red). K, shown is an enlarged image of DU145 cells 4 h after calcium restoration stained with antibody against ARHGAP21 (green) and α-catenin (red); the arrows indicate that both proteins were not co-localized at cell-cell contacts. L, shown is immunofluorescence analysis of DU145 ARHGAP21 siRNA cells stained with antibodies against ARHGAP21 (green) and α-catenin (red), indicating that α-catenin is still located at the cell membrane in ARHGAP21-depleted cells. M and N, quantitative RT-PCR and Western blotting analyses of ARHGAP21 expression in DU145 cells submitted to control siRNA or to ARHGAP21 siRNA transfection show efficiency of ARHGAP21 inhibition. O, shown is endogenous ARHGAP21 co-immunoprecipitated with Cdc42. Total extracts from 4 and 24 h after calcium restoration in DU145 cells were submitted to immunoprecipitation with an anti-ARHGAP21 antibody followed by Western blot analysis using anti-Cdc42 and anti-ARHGAP21 antibody. Bar, 10 μm.
To evaluate how ARHGAP21 protein is localized to cell-cell junctions during adhesion formation, we performed the calcium switch assay with MDCK cells transfected with plasmids encoding ARHGAP21 fragments fused to GFP (Fig. 1D). PDZ-ARHGAP21 localized to cell-cell junctions (arrows) 4 h after calcium addition (Fig. 1E). The localization of PH+GAP-ARHGAP21 is predominantly perinuclear (arrow). Cells expressing this ARHGAP21 fragment exhibited considerable protrusions (Fig. 1F). Interestingly, Cter-ARHGAP21 predominantly localizes in the nucleus (arrow, Fig. 1G). Cter-ARHGAP21 includes the nuclear localization signal (RKRKK amino acids) but also includes the α-catenin binding site. These results indicate that ARHGAP21 is not targeted to cell-cell junctions by binding α-catenin.
A transient localization of ARHGAP21 at cell-cell contacts was also observed in DU145 cells 4 h after initiation of adhesion formation, in contrast to only cytoplasmic and perinuclear localization at 24 h (Fig. 1H). Importantly, ARHGAP21 colocalized with neither E-cadherin nor JAM 4 h after calcium restoration (Fig. 1, I and J). ARHGAP21 was not co-localized with α-catenin at cell-cell junctions (Fig. 1K). Furthermore, DU145 cells depleted of ARHGAP21 were still present α-catenin at cell-cell contacts (Fig. 1L), indicating that ARHGAP21 is not required for recruitment of α-catenin to junctions in DU145 cells. Efficacy of ARHGAP21 siRNA was confirmed by RT-PCR and Western blotting (Fig. 1, M and N).
We then tested whether ARHGAP21 associates with Cdc42 during cell-cell junction formation. Co-immunoprecipitation assays demonstrate that ARHGAP21 interacts with Cdc42 4 and 24 h after calcium restoration in DU145 cells (Fig. 1O). Taken together, these data suggest that ARHGAP21 transiently localizes to actin structures at cell-cell junctions during cell-cell junction formation, where it interacts with Cdc42.
Knockdown of ARHGAP21 Generates Weaker Cell-Cell Adhesions
To ascertain the function of ARHGAP21 in cell-cell adhesion, we measured the effects of ARHGAP21 protein depletion on cell-cell junction formation. We chose SW480 cells to ensure that ARHGAP21 function was independent of α-catenin, as α-catenin is not expressed at cell-cell contacts of these cells (32). SW480 cells were transfected with plasmids expressing shRNAs directed against ARHGAP21, and knockdown was confirmed by Western blot analysis. ARHGAP21 levels in cells expressing the ARHGAP21 shRNA were decreased to 48% that of the levels observed in parental cells (Fig. 2A). The control plasmid does not affect ARHGAP21 levels compared with parental cells.
FIGURE 2.

ARHGAP21 role in cell aggregation. Shown is analysis of ARHGAP21 knockdown SW480 cells and control-transfected SW480 cells in a quantitative and functional adhesion assay, the hanging drop aggregation assay, at time points of 0, 1, and 3 h with triturated and non-triturated cells. A, shown is Western blot analysis of SW480 cells submitted to control shRNA or to ARHGAP21 shRNA transfection immunoblotted (IB) with anti-ARHGAP21 and anti-actin antibodies (as a control for equal sample loading). B and C, graphs show the percentage of cells in clusters of 0–10 cells (gray), 11–50 cells (dark gray), and >50 cells (white) at the time points indicated with and without trituration, respectively. D—K, photographs are representative fields at 1 and 3 h with and without trituration. D, control cells at 1 h not triturated. E, shRNA cells at 1 h not triturated. F, control cells at 1 h triturated. G, shRNA cells at 1 h triturated. H, control cells at 3 h not triturated. I, shRNA cells at 3 h not triturated. J, control cells at 3 h triturated. K, shRNA cells at 3 h triturated. Note that ARHGAP21 shRNA cells do not present clusters after trituration. Objective 20×.
Formation and strengthening of cell-cell adhesions in SW480 cells expressing shARHGAP21 or control shRNAs were quantified using a hanging drop assay, which measures the ability of cells to aggregate by forming cell-cell junctions in suspension culture. In this assay the size of aggregates is measured with and without trituration, which disrupts weak adhesions between aggregated cells. SW480 cells with ARHGAP21 knockdown exhibited similar levels of aggregation to those observed in control cells, but aggregates were more easily disrupted by trituration forces (Fig. 2, B–K). At both 1 and 3 h, aggregates were only partially disrupted in SW480 cells expressing control shRNAs (Fig. 2, C, F, and J). However, trituration disrupted all cell-cell adhesion of SW480 ARHGAP21 knockdown cells; these cells were observed as a single cell suspension after trituration, even after 3 h of cell-cell junction formation (Fig. 2, C, G, and K). Quantitative analysis confirms this observation: 21.9% of control cells remained in clusters of 11–50 cells, and 5.1% remained in clusters of >50 cells after trituration at 3 h compared with none for the ARHGAP21 knockdown cells (p < 0.001, Fig. 2C). This indicates that these cells initiate cell-cell adhesion at a normal rate but that reinforcement of initial cell-cell contacts is disrupted. Consistent with this idea, aggregates of control shRNA-expressing cells appeared tight, whereas aggregates of ARHGAP21 knockdown cells appeared in looser groups (Fig. 2, H and I).
ARHGAP21 Knockdown Altered Cell Migration Rates, and ARHGAP21 Interacts with α-Tubulin
Because reductions in cell-cell adhesion are often associated with increased cell migration, we hypothesized that cells depleted of ARHGAP21 would show increased cell migration. To test this, we measured the rate of cell migration in SW480 cells expressing ARHGAP21 or control shRNAs (Fig. 3A). The rate of migration of ARHGAP21 knockdown cells was increased about 2-fold compared with that of control cells (Fig. 3B, p = 0.0035).
FIGURE 3.
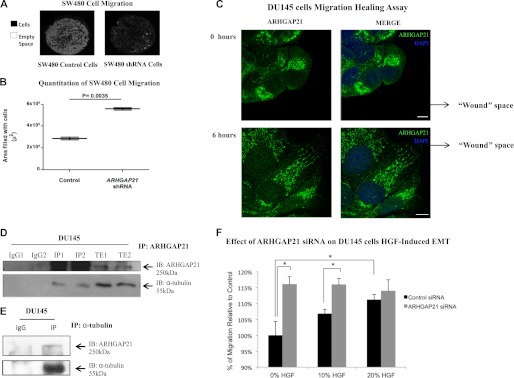
ARHGAP21 knockdown increases cell migration and ARHGAP21 interacts with α-tubulin. A, SW480 ARHGAP21 shRNA and control shRNA cell migration were detected using Oris Cell Migration 96-well plate. Images from the central well region were captured; cells are visualized in black with the empty area in white. B, the graph shows the migration rates of control and ARHGAP21 shRNA cells, with an increased migration in the shRNA cells (p = 0.0035). C, shown is an immunofluorescence analysis of DU145 cells in a wound-healing assay stained with antibody against ARHGAP21 (green) at 0 and 6 h of migration. DAPI staining indicates nuclear area. D, shown is endogenous ARHGAP21 co-immunoprecipitated (IP) with α-tubulin. Total extracts from independent samples of DU145 cells were submitted to immunoprecipitation with an anti-ARHGAP21 antibody (lanes IP1 and IP2) followed by Western blot (IB) analysis using anti-α-tubulin and anti-ARHGAP21 antibody. Lanes TE1 and TE2 correspond to a Western blot performed on total cell extracts. E, DU145 cell extracts were submitted to immunoprecipitation with an anti-α-tubulin antibody followed by Western blot analysis using anti-ARHGAP21 and anti-α-tubulin antibody. F, DU145 cells were transfected with the ARHGAP21 siRNA or control siRNA. After 24 h cells were seeded in sextuplicate onto a 96-well filter plate. HGF was added below the filter as a chemo-attractant 24 h later. After 20 h the migration was stopped, and the cells were fixed and analyzed. The percentage of migration was calculated relative to control. An increased migration of DU145 ARHGAP21 siRNA compared with the control cells was observed. The migration of control siRNA cells was increased proportionally to HGF amount. However, the migration of ARHGAP21 siRNA cells was not modulated by HGF treatment. *, p < 0.05. Bar, 10 μm.
Migrating cells are polarized, displaying front and back sides, with activated Rac and Cdc42 Rho-GTPases at the front of the cell, a site of fast actin polymerization, whereas RhoA is located at the back (12, 19). Microtubules and actin are crucial for establishing and maintaining the polarity (11, 12). Because Rho-GTPases regulate the dynamics and organization of microtubules during cell migration (15, 19), we assessed ARHGAP21 localization during cell migration. DU145 cell monolayers were wounded, and migration into the wound was allowed to proceed. Cells were then fixed, and ARHGAP21 localization was evaluated in cells at the wound front. Immediately after wounding, migration was not initiated, and ARHGAP21 localization was predominately perinuclear (Fig. 3C). After initiation of migration into the wound (6 h after wounding), ARHGAP21 localized to the front of the polarized cells (Fig. 3C).
Because there is a polarized microtubule arrangement at the front of migrating cells (11, 12), we evaluated the possible interaction of ARHGAP21 and α-tubulin. ARHGAP21 antibody immunoprecipitates were isolated from DU145 cell extracts and analyzed for α-tubulin (Fig. 3D). Based on the co-immunoprecipitation in multiple experiments, we conclude that ARHGAP21 interacted with α-tubulin (Fig. 3D). This interaction was confirmed by the reverse co-immunoprecipitation; ARHGAP21 was precipitated from DU145 extracts with an antibody against α-tubulin (Fig. 3E).
Epithelial cells disassemble cell-cell junctions during development and cancer progression, a process termed epithelial-mesenchymal transition (EMT). During EMT, disassembly of cell-cell contacts is associated with increased cell migration. HGF induces EMT in DU145 prostate cancer cells in vitro, and a high level of HGF receptor (c-Met) expression is observed in prostate cancer cells (33). Because depletion of ARHGAP21 results in weaker cell-cell adhesion and increased migration, we decided to study the effects of HGF induced scattering in these cells using Transwell filter migration assays. ARHGAP21 siRNA and control siRNA DU145 cells were seeded onto Transwell filters. Even in the absence of HGF treatment, a markedly increased number of ARHGAP21-depleted cells was observed on the underside of the filter compared with DU145 cells transfected with control siRNA (Fig. 3F, p = 0.0139). This indicates that ARHGAP21 depletion facilitates cell detachment and migration, consistent with our findings in earlier experiments. We expected that HGF would induce scattering and increase the number of cells that had detached and migrated through the filter. An expected HGF-dependent increase in cell migration was observed for control siRNA-expressing DU145 cells, and the difference is statistically significant (p = 0.0397, Fig. 3F). Surprisingly, HGF treatment did not increase Transwell migration of ARHGAP21 siRNA cells (Fig. 3F); the rate of migration of ARHGAP21 siRNA cells was increased to the same extent as control cells treated with HGF, both with and without HGF stimulation (Fig. 3F).
ARHGAP21 Is Required for HGF-induced Scattering
In an attempt to confirm the observed effects of HGF in Transwell migration assays and understand ARHGAP21 function in EMT, we performed time-lapse imaging of DU145 cells undergoing HGF-induced scattering. Similar to colonies of parental DU145 cells, control siRNA colonies are fully scattered within 6 h of HGF treatment, with full disruption of cell-cell junctions and increased cell migration. However, ARHGAP21 depletion results in reduced scattering, particularly the ability of cells to detach cell-cell adhesions (Fig. 4A). Only a few ARHGAP21 siRNA cells were able to undergo scattering; the number of colonies that did not dissociate was significantly increased compared with control cells (p = 0.01, Fig. 4B). This was surprising given weaker cell-cell adhesion and increased migration observed in ARHGAP21 knockdown cells in previous experiments but consistent with our findings in Transwell filter migration assays.
FIGURE 4.

ARHGAP21 knockdown inhibits EMT induced by HGF. A, live cell imaging shows colonies of DU145 cells on collagen responding to HGF stimulation. ARHGAP21 siRNA and control siRNA cells were starved in medium with 0.5% of FBS for 24 h before the HGF stimulation. Colonies were filmed for 10 h. Images from DU145 cells not stimulated with HGF and ARHGAP21/control siRNA-transfected cells stimulated with HGF are exemplified here. Those images were obtained from two independent experiments for each condition (ARHGAP21 siRNA and control siRNA) and used as representative data. B, the colonies were graduated according to the intensity of scattering response. No scattering, hardly any ICS and RF; Light scattering, showing ICS/RF but no cells break away; Semi-scattering, ICS/RF with at least one cell that completely breaks away; Full scattering, ICS/RF with two or more cells breaking from colony. Data were collected from 24 colonies for ARHGAP21 siRNA and 28 colonies for control-siRNA over 3 independent experiments. ARHGAP21 inhibition results in a diminishing of cell ability to undergo EMT in response to HGF signaling. Bar, 50 μm.
This difference in scattering between ARHGAP21 depletion and control cells was confirmed in quantitative analysis; 37.5% of ARHGAP21 siRNA colonies showed no scattering in contrast to 7.1% of the control siRNA colonies. Light scattering (with intracolony spaces and retraction fibers but with no cell-cell detachment) was observed in 41.6% of ARHGAP21 siRNA colonies compared with 17.9% of control siRNA colonies. Partial scattering (with only a single cell detaching) occurred in 16.7% of ARHGAP21 siRNA colonies versus 42.86% of control siRNA colonies. Only 4.2% (1 colony) of ARHGAP21 siRNA colonies underwent complete scattering (with multiple cells breaking from the colony) in contrast to 32.14% of the control siRNA colonies (Fig. 4B).
In an attempt to validate that the defect in cellular scattering here-described was the result of an effect on EMT and not just on epithelial scattering, we examined EMT biomarker expression in DU145 cells after stimulation with HGF. Transcription factors ZEB1, Snail1, and TWIST1 were expressed upon HGF treatment of control cells (Fig. 5A and B). In contrast, the HGF-induced increase in Twist1 expression was blocked in ARHGAP21 siRNA cells (Fig. 5B).
FIGURE 5.

ARHGAP21 depletion affects the expression of proteins EMT markers during EMT induced by HGF. A, Western blotting (IB) shows ZEB1 and Snail1 expression in DU145 cells non-treated or induced with HGF for 30 min, 2 h, and 4 h. B, shown is expression of Twist1 in DU145 cells non-treated or induced with HGF for 10 min, 30 min, 2 h, and 4 h and control and ARHGAP21 siRNA stimulated with 4 h HGF. C, Western blotting shows E-cadherin, vimentin, and slug expression in control and ARHGAP21 DU145 cells non-treated or induced with HGF for 4 h. The Western blotting confirm that ARHGAP21 inhibition results in a diminishing of cells ability to undergo EMT in response to HGF signaling. Anti-β-actin blotting was used as loading control in A, B, and C.
E-cadherin down-regulation and Vimentin up-regulation are also considered hallmarks of EMT. The expression of those proteins was evaluated in ARHGAP21 siRNA cells. E-cadherin was down-regulated in control siRNA cells treated with HGF but not in ARHGAP21 siRNA cells (Fig. 5C). Vimentin expression increased dramatically in control cells treated with HGF but only lightly in ARHGAP21 siRNA cells treated with HGF (Fig. 5C). The transcriptional factor Slug was up-regulated only in the control siRNA cells treated with HGF (Fig. 5C). These results provide direct evidence that the participation of ARHGAP21 in EMT goes beyond the effect on cell migration and cell-cell adhesion.
HGF Treatment Translocates ARHGAP21 to the Nucleus
Because our results describe the involvement of ARHGAP21 in HGF-induced scattering, we decided to observe if ARHGAP21 localization was affected by HGF treatment. To test this, we analyzed ARHGAP21 localization in colonies of DU145 cells before and after 2 h of HGF simulation. ARHGAP21 translocated to the nucleus after HGF stimulation (Fig. 6A).
FIGURE 6.

ARHGAP21 translocates to the nucleus after HGF stimulus. A, shown is immunofluorescence analysis of DU145 cells stained with antibody against ARHGAP21 (blue) and F-actin (phalloidin-TRITC) with or without HGF 2 h stimulation. Note that ARHGAP21 localizes at perinuclear areas in the absence of HGF and in the nucleus after HGF stimulation. B, shown is an immunofluorescence image of DU145 cells with no HGF and 2 h after HGF stimulus stained with antibody against β-catenin (green) and F-actin (phalloidin-TRITC). Images were acquired with a 60× oil immersion objective lens.
Colonies of DU145 cells were stained with β-catenin and actin. In control siRNA cells with no HGF, β-catenin was predominantly localized at the cell-cell contacts (Fig. 6B). After 2 h of HGF treatment, a partial disruption of cell-cell contacts and an increased β-catenin localization at the perinuclear area was observed (arrow Fig. 6B), in accordance with the report of β-catenin translocation to the nucleus after HGF treatment (34). In ARHGAP21 siRNA cells, β-catenin remained at cell-cell contacts after HGF treatment (Fig. 6B).
ARHGAP21 Was Necessary for α-Tubulin Acetylation
Tubulin acetylation influences microtubule stability and interactions with microtubule binding proteins. To test whether the role of ARHGAP21 in migration and EMT is related to α-tubulin acetylation, we investigated tubulin acetylation in cells with ARHGAP21 depletion. The extent of tubulin acetylation in DU145 cells treated with HGF was measured. An increase in α-tubulin acetylation occurred during EMT in parental (Fig. 7A) and control siRNA-treated DU145 cells, but this was not observed in ARHGAP21 siRNA-treated DU145 cells (Fig. 7B).
FIGURE 7.

ARHGAP21 knockdown inhibits α-tubulin acetylation. A, Western blotting (IB) shows acetylated-α-tubulin expression in DU145 cells, no HGF, induced with HGF for 10 min and 3 h; total α-tubulin was used as a control of the reaction. B, shown is expression of acetylated and total α-tubulin proteins in DU145 siRNA control and siRNA ARHGAP21 cells with no HGF and induced with HGF for 3 h. C, the graph shows HDAC activity in cytoplasmic extracts of control and ARHGAP21 siRNA DU145 cells, with no difference in HDAC activity between the samples (p > 0.05); bars represent analysis of colorimetric assay as mean ± S.D. of three independent experiments versus control, set as 100%.
A member of the histone deacetylase family, HDAC6, functions as a tubulin deacetylase, and HDAC6 is localized exclusively in the cytoplasm (35). In attempt to understand if the effect of ARHGAP21 in α-tubulin acetylation involved HDAC6 activation, we measure HDAC activity of cytoplasmic extracts from control and ARHGAP21 siRNA DU145 cells treated with HGF for 3 h. There is no significant difference in cytoplasmic HDAC activity between control and ARHGAP21 siRNA cells (Fig. 7C), indicating that ARHGAP21 probably affects α-tubulin acetylation through tubulin organization or stabilization, not through this deacetylase.
DISCUSSION
During metastasis, tumor cells reduce cell-cell adhesion and increase migration (7). Rho-GTPases regulate these processes, and changes in Rho-GTPase activity are related to tumor progression (3). Connections between specific Rho-GAPs and specific Rho-GTPases in different cellular functions are necessary to better comprehend tumor development. Here we studied the role of the Rho-GAP ARHGAP21 in cell-cell adhesion and cell migration. We show that ARHGAP21 transiently localizes to nascent cell-cell contacts. ARHGAP21 targets Cdc42 (23, 25, 26), which is implicated in cadherin-mediated cell-cell adhesion. We observed a significant decrease in Cdc42 activity during cell-cell adhesion formation, precisely when ARHGAP21 appears at cell-cell junctions; in addition, we demonstrated that ARHGAP21 interacts with Cdc42 at this moment, suggesting that ARHGAP21 is a Rho-GAP that targets Cdc42 at cell-cell contacts. This might reduce membrane protrusions after the cell-cell contacts have been established.
Importantly, ARHGAP21 function in cell-cell adhesion is independent of α-catenin recruitment. ARHGAP21 localization to cell-cell contacts occurs by its PDZ domain rather than its α-catenin binding site (23). Several proteins that regulate Rho-GTPases contain PDZ domains, including Tiam1, STEF, and PDZ-Rho-GEF1 (3), which are recruited to cell-cell contact by interacting with the β-catenin C-terminal PDZ binding motif (36). PDZ domain interactions could represent a mechanism for selective regulation of Rho-GTPase activity at sites of cell-cell contact.
In the present study we show that ARHGAP21 functions in strengthening of cell-cell adhesions. The increased cellular migration observed here in ARHGAP21 knockdown cells is likely connected to weaker cell-cell adhesion, as cell migration and cell-cell adhesion are often in opposition. Increased migration was also described in gliobastoma-derived cells in which ARHGAP21 was depleted, where it was suggested that ARHGAP21 decreases tumor cell migration by regulating focal adhesion kinase activity (25). We found that ARHGAP21 interacts with α-tubulin and that ARHGAP21 functions in α-tubulin acetylation. We propose that alterations in migratory behavior in cells lacking ARHGAP21 likely result from a combination of different factors, such as changes in cell-cell adhesion, cell-substrate adhesion, and α-tubulin modification.
The microtubules and Golgi apparatus are oriented toward the leading edge of polarized cells during migration (11, 19). Cdc42 is found on the Golgi complex and at the leading edge, regulating the formation of cell polarity through the control of vesicular traffic (11). The reorganization of the actin cytoskeleton and microtubules are essential for cell migration (13, 14, 37); ARHGAP21 localization at the front of the polarized cell and its interaction with microtubules and Cdc42 indicates a role of ARHGAP21 in Cdc42 and actin/microtubule management in migration. The report that ARHGAP21 down-regulates activity of Arp2/3 through the control of Cdc42 in the Golgi membranes (26) supports the idea that ARHGAP21 is in the control of actin and microtubules dynamics through different mechanisms.
HGF induces membrane ruffling, cell-cell dissociation, and cell scattering (38), which is determined by an alteration in the balance between cell-cell adhesion and cell motility. Because cellular ARHGAP21 depletion decreases cell-cell adhesion and increases cellular migration, focal adhesion kinase phosphorylation states, and the production of metalloproteinase 2 (MMP-2) (25), we thought at first that cells lacking ARHGAP21 expression would readily undergo EMT. However, a significant reduction in ARHGAP21-depleted cell ability to undergo EMT was observed. This lack of EMT may be explained by the absence of acetylated α-tubulin in ARHGAP21-depleted cells, although the role of α-tubulin acetylation in EMT remains unclear. Tubulin post-translational modifications are related to the regulation of microtubule dynamics and binding of microtubule-associated proteins (39). Tubulin acetylation controls cellular signaling and mechanisms important to cell differentiation, cell stress, and survival (40). Acetylation of microtubules leads to recruitment of motor proteins that enable transport of proteins and other cellular components (39, 41, 42), which may explain recently reported functions of ARHGAP21 in transport (43, 44). Sorting out which of these mechanisms, or which other mechanisms, might explain a potential role for microtubule acetylation in EMT will prove interesting.
Coordination of cell-cell adhesion and cell migration is the foundation of morphogenetic rearrangements of epithelial tissues. During tumor progression, alterations in cell-cell adhesion and migration play a fundamental role in metastasis. We have implicated ARHGAP21 in cellular decisions about maintenance of cell-cell adhesion and initiation of cell migration, including a role in driving epithelial scattering. We here identify ARHGAP21 as a protein that associates with and regulates microtubules, which suggests that this protein may play fundamental roles in development and cancer progression.
Acknowledgments
We thank Janine Sabino for manipulating the confocal microscopic and Dr. Hernandes Carvalho (State University of Campinas, UNICAMP) and Dr. Fernando Soares (Cancer Hospital AC Camargo) for providing important antibodies used here.
This work was supported by Instituto Nacional de Ciência e Tecnologia do Sangue (INCT), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Fundação de Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
- PH
- pleckstrin homology
- MDCK
- Madin-Darby canine kidney
- HGF
- hepatocyte growth factor
- TRITC
- tetramethylrhodamine isothiocyanate
- EMT
- epithelial-mesenchymal transition
- ICS
- intracolony spaces
- RF
- retraction fibers
- HDAC
- histone deacetylase.
REFERENCES
- 1. Niessen C. M., Gottardi C. J. (2008) Molecular components of the adherens junction. Biochim. Biophys. Acta 1778, 562–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arthur W. T., Noren N. K., Burridge K. (2002) Regulation of Rho family GTPases by cell-cell and cell-matrix adhesion. Biol. Res. 35, 239–246 [DOI] [PubMed] [Google Scholar]
- 3. Fukata M., Kaibuchi K. (2001) Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2, 887–897 [DOI] [PubMed] [Google Scholar]
- 4. Kim S. H., Li Z., Sacks D. B. (2000) E-cadherin-mediated cell-cell attachment activates Cdc42. J. Biol. Chem. 275, 36999–37005 [DOI] [PubMed] [Google Scholar]
- 5. Van Aelst L., Symons M. (2002) Role of Rho family GTPases in epithelial morphogenesis. Genes Dev. 16, 1032–1054 [DOI] [PubMed] [Google Scholar]
- 6. Kümper S., Ridley A. J. (2010) p120ctn and P-cadherin but not E-cadherin regulate cell motility and invasion of DU145 prostate cancer cells. PLoS One 5, e11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perl A. K., Wilgenbus P., Dahl U., Semb H., Christofori G. (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392, 190–193 [DOI] [PubMed] [Google Scholar]
- 8. Bagutti C., Speight P. M., Watt F. M. (1998) Comparison of integrin, cadherin, and catenin expression in squamous cell carcinomas of the oral cavity. J. Pathol. 186, 8–16 [DOI] [PubMed] [Google Scholar]
- 9. Beavon I. R. (2000) The E-cadherin-catenin complex in tumour metastasis. Structure, function, and regulation. Eur. J. Cancer 36, 1607–1620 [DOI] [PubMed] [Google Scholar]
- 10. Shiozaki H., Iihara K., Oka H., Kadowaki T., Matsui S., Gofuku J., Inoue M., Nagafuchi A., Tsukita S., Mori T. (1994) Immunohistochemical detection of α-catenin expression in human cancers. Am. J. Pathol. 144, 667–674 [PMC free article] [PubMed] [Google Scholar]
- 11. Watanabe T., Noritake J., Kaibuchi K. (2005) Regulation of microtubules in cell migration. Trends Cell Biol. 15, 76–83 [DOI] [PubMed] [Google Scholar]
- 12. de Forges H., Bouissou A., Perez F. (2012) Interplay between microtubule dynamics and intracellular organization. Int. J. Biochem. Cell Biol. 44, 266–274 [DOI] [PubMed] [Google Scholar]
- 13. Rodriguez O. C., Schaefer A. W., Mandato C. A., Forscher P., Bement W. M., Waterman-Storer C. M. (2003) Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 5, 599–609 [DOI] [PubMed] [Google Scholar]
- 14. Goode B. L., Drubin D. G., Barnes G. (2000) Functional cooperation between the microtubule and actin cytoskeletons. Curr. Opin. Cell Biol. 12, 63–71 [DOI] [PubMed] [Google Scholar]
- 15. Raftopoulou M., Hall A. (2004) Cell migration. Rho GTPases lead the way. Dev. Biol. 265, 23–32 [DOI] [PubMed] [Google Scholar]
- 16. Vasioukhin V., Bauer C., Yin M., Fuchs E. (2000) Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100, 209–219 [DOI] [PubMed] [Google Scholar]
- 17. Raich W. B., Agbunag C., Hardin J. (1999) Rapid epithelial-sheet sealing in the Caenorhabditis elegans embryo requires cadherin-dependent filopodial priming. Curr. Biol. 9, 1139–1146 [DOI] [PubMed] [Google Scholar]
- 18. Jacinto A., Martinez-Arias A., Martin P. (2001) Mechanisms of epithelial fusion and repair. Nat. Cell Biol. 3, E117–E123 [DOI] [PubMed] [Google Scholar]
- 19. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003) Cell migration. Integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 20. Aznar S., Lacal J. C. (2001) Rho signals to cell growth and apoptosis. Cancer Lett. 165, 1–10 [DOI] [PubMed] [Google Scholar]
- 21. Moon S. Y., Zheng Y. (2003) Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 13, 13–22 [DOI] [PubMed] [Google Scholar]
- 22. Peck J., Douglas G., 4th, Wu C. H., Burbelo P. D. (2002) Human RhoGAP domain-containing proteins. Structure, function. and evolutionary relationships. FEBS Lett. 528, 27–34 [DOI] [PubMed] [Google Scholar]
- 23. Sousa S., Cabanes D., Archambaud C., Colland F., Lemichez E., Popoff M., Boisson-Dupuis S., Gouin E., Lecuit M., Legrain P., Cossart P. (2005) ARHGAP10 is necessary for α-catenin recruitment at adherens junctions and for Listeria invasion. Nat. Cell Biol. 7, 954–960 [DOI] [PubMed] [Google Scholar]
- 24. Bassères D. S., Tizzei E. V., Duarte A. A., Costa F. F., Saad S. T. (2002) ARHGAP10, a novel human gene coding for a potentially cytoskeletal Rho-GTPase activating protein. Biochem. Biophys. Res. Commun. 294, 579–585 [DOI] [PubMed] [Google Scholar]
- 25. Bigarella C. L., Borges L., Costa F. F., Saad S. T. (2009) ARHGAP21 modulates FAK activity and impairs glioblastoma cell migration. Biochim. Biophys. Acta 1793, 806–816 [DOI] [PubMed] [Google Scholar]
- 26. Dubois T., Paléotti O., Mironov A. A., Fraisier V., Stradal T. E., De Matteis M. A., Franco M., Chavrier P. (2005) Golgi-localized GAP for Cdc42 functions downstream of ARF1 to control Arp2/3 complex and F-actin dynamics. Nat. Cell Biol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 27. Fukata M., Nakagawa M., Itoh N., Kawajiri A., Yamaga M., Kuroda S., Kaibuchi K. (2001) Involvement of IQGAP1, an effector of Rac1 and Cdc42 GTPases, in cell-cell dissociation during cell scattering. Mol. Cell. Biol. 21, 2165–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 29. Kim J. B., Islam S., Kim Y. J., Prudoff R. S., Sass K. M., Wheelock M. J., Johnson K. R. (2000) N-cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J. Cell Biol. 151, 1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren X. D., Kiosses W. B., Schwartz M. A. (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sperry R. B., Bishop N. H., Bramwell J. J., Brodeur M. N., Carter M. J., Fowler B. T., Lewis Z. B., Maxfield S. D., Staley D. M., Vellinga R. M., Hansen M. D. (2010) Zyxin controls migration in epithelial-mesenchymal transition by mediating actin-membrane linkages at cell-cell junctions. J. Cell. Physiol. 222, 612–624 [DOI] [PubMed] [Google Scholar]
- 32. El-Bahrawy M., Talbot I., Poulsom R., Alison M. (2002) Variable nuclear localization of α-catenin in colorectal carcinoma. Lab. Invest. 82, 1167–1174 [DOI] [PubMed] [Google Scholar]
- 33. van Leenders G., van Balken B., Aalders T., Hulsbergen-van de Kaa C., Ruiter D., Schalken J. (2002) Intermediate cells in normal and malignant prostate epithelium express c-MET. Implications for prostate cancer invasion. Prostate 51, 98–107 [DOI] [PubMed] [Google Scholar]
- 34. Li J., Zhou B. P. (2011) Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC cancer 11, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 [DOI] [PubMed] [Google Scholar]
- 36. Kawajiri A., Itoh N., Fukata M., Nakagawa M., Yamaga M., Iwamatsu A., Kaibuchi K. (2000) Identification of a novel β-catenin-interacting protein. Biochem. Biophys. Res. Commun. 273, 712–717 [DOI] [PubMed] [Google Scholar]
- 37. Pollard T. D., Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 38. Hartmann G., Weidner K. M., Schwarz H., Birchmeier W. (1994) The motility signal of scatter factor/hepatocyte growth factor mediated through the receptor tyrosine kinase met requires intracellular action of Ras. J. Biol. Chem. 269, 21936–21939 [PubMed] [Google Scholar]
- 39. Wynshaw-Boris A. (2009) Elongator bridges tubulin acetylation and neuronal migration. Cell 136, 393–394 [DOI] [PubMed] [Google Scholar]
- 40. Janke C., Bulinski J. C. (2011) Post-translational regulation of the microtubule cytoskeleton. Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773–786 [DOI] [PubMed] [Google Scholar]
- 41. Hammond J. W., Cai D., Verhey K. J. (2008) Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 20, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perdiz D., Mackeh R., Poüs C., Baillet A. (2011) The ins and outs of tubulin acetylation. More than just a post-translational modification? Cell. Signal. 23, 763–771 [DOI] [PubMed] [Google Scholar]
- 43. Wang S., Li H., Chen Y., Wei H., Gao G. F., Liu H., Huang S., Chen J. L. (2012) Transport of influenza virus neuraminidase (NA) to host cell surface is regulated by ARHGAP21 and Cdc42 proteins. J. Biol. Chem. 287, 9804–9816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hehnly H., Longhini K. M., Chen J. L., Stamnes M. (2009) Retrograde Shiga toxin trafficking is regulated by ARHGAP21 and Cdc42. Mol. Biol. Cell 20, 4303–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]