

Background: Hepatocyte growth factor (HGF) is a potent mediator of endothelial barrier enhancement, however, the mechanisms are poorly understood.
Results: HGF-induced endothelial barrier enhancement is mediated via sphingosine 1-phosphate receptor 1 (S1PR1) and integrin β4 (ITGB4) transactivation.
Conclusion: ITGB4 and S1PR1 are essential for HGF-induced endothelial barrier augmentation.
Significance: Our findings identify novel mechanisms of endothelial barrier enhancement by HGF.
Keywords: Endothelial Cell, Endothelial Dysfunction, Hepatocyte Growth Factor, Integrin, Sphingosine 1-Phosphate
Abstract
Vascular endothelial cell (EC) barrier integrity is critical to vessel homeostasis whereas barrier dysfunction is a key feature of inflammatory disorders and tumor angiogenesis. We previously reported that hepatocyte growth factor (HGF)-mediated increases in EC barrier integrity are signaled through a dynamic complex present in lipid rafts involving its receptor, c-Met (1). We extended these observations to confirm that S1PR1 (sphingosine 1-phosphate receptor 1) and integrin β4 (ITGB4) are essential participants in HGF-induced EC barrier enhancement. Immunoprecipitation experiments demonstrated HGF-mediated recruitment of c-Met, ITGB4 and S1PR1 to caveolin-enriched lipid rafts in human lung EC with direct interactions of c-Met with both S1PR1 and ITGB4 accompanied by c-Met-dependent S1PR1 and ITGB4 transactivation. Reduced S1PR1 expression (siRNA) attenuated both ITGB4 and Rac1 activation as well as c-Met/ITGB4 interaction and resulted in decreased transendothelial electrical resistance. Furthermore, reduced ITGB4 expression attenuated HGF-induced c-Met activation, c-Met/S1PR1 interaction, and effected decreases in S1P- and HGF-induced EC barrier enhancement. Finally, the c-Met inhibitor, XL880, suppressed HGF-induced c-Met activation as well as S1PR1 and ITGB4 transactivation. These results support a critical role for S1PR1 and ITGB4 transactivation as rate-limiting events in the transduction of HGF signals via a dynamic c-Met complex resulting in enhanced EC barrier integrity.
Introduction
Inflammatory vascular injury and the subsequent loss of vascular integrity is a major cause of morbidity and mortality in critically ill patients because of leakage of fluid, protein, and cells into organ parenchyma resulting in tissue edema and, potentially, multiple organ dysfunctions. Therefore, agents that protect vascular endothelial cell (EC)2 barrier properties and vascular integrity may have important therapeutic implications. In prior studies, we detailed the barrier-enhancing effects of bioregulatory molecules such as sphingosine 1-phosphate (S1P) (2, 3, 4, 5, 6), hepatocyte growth factor (HGF) (7, 1), HMW HA (high molecular weight hyaluronic acid; 8), ATP (9, 10), and APC (activated protein C) (11, 12, 13) via ligation of their cognate receptors resulting in restoration of EC barrier integrity and reduced permeability. HGF promotes angiogenesis and EC barrier function via ligation of c-Met, a cell surface receptor tyrosine kinase, as well as Rac1-dependent cytoskeleton rearrangement (7, 1). More recently, we demonstrated that HGF promotes c-Met recruitment into specialized caveolin-1-enriched plasma membrane microdomains (CEM) also known as lipid rafts with transactivation of CD44, a major glycoprotein receptor for hyaluronan, within these lipid raft structures (1).
Lipid rafts are enriched in lipids such as cholesterol, sphingomyelin, gangliosides, diacylglycerol (DAG), PIP2, and the scaffold protein, caveolin, and serve to regulate numerous EC functions including lung vascular barrier function (14, 15, 16, 4). Multiple barrier-regulatory agonists, such as S1P and HGF induce receptor recruitment to lipid rafts such as S1PR1, EPCR (endothelial protein C receptor), CD44, and c-Met, in association with key signaling components (small GTPases and cytoskeletal linker proteins, radixin and actinin) resulting in receptor activation and signaling to the EC cytoskeleton. Proteomic studies have facilitated the identification of key barrier-regulatory proteins that are recruited to lipid rafts upon S1P treatment (6). Significant increases in tyrosine phosphoproteins within lipid rafts were observed and included key cytoskeletal effectors such as cortactin, c-Abl, MLCK, and filamin (17, 6). These events collectively contribute to S1P-induced enhancement of EC barrier integrity and function.
The exact signals involved in HGF-mediated cytoskeletal rearrangement are not completely understood but involve activation of the small G protein, Rac1 (7), a critical mediator of cytoskeletal reorganization and key contributor to the barrier-protective effects of S1P (2, 3) and simvastatin (18). Both Rac1 and the specific Rho family guanine nucleotide exchange factor, Tiam1, are recruited to lipid rafts effecting EC barrier function enhancement (4, 1, 19, 20). Evidence suggests that integrin-mediated adhesion contributes to regulation of lipid raft trafficking (21, 22, 23), including both translocation of GTP-bound Rac1 and Cdc42 and effector coupling (24). Consistent with this paradigm, we previously demonstrated that simvastatin-mediated barrier enhancement occurs in a Rac-1-dependent manner (18, 25) and is highly dependent on the induced expression of integrin β4 (ITGB4), an integrin with unique structural elements and post-translational modifications. ITGB4 is the only integrin subunit modified by S-palmitoylation (cysteine residues) necessary for integrin coupling to an S-palmitoylated c-Src to promote mitogenic signaling whereas other integrins indirectly interact with c-Src through caveolin-1 (26). Notably, the large cytoplasmic tail of ITGB4 bears no significant homology to the short cytoplasmic tails of other integrin β subunits. Upon binding to extracellular matrix proteins, the ITGB4 tail is phosphorylated thereby facilitating interaction with the adaptor Shc that then triggers Ras-mediated activation of extracellular signal-regulated kinase (ERK) signaling (27, 28, 25). In addition, ITGB4 activation (ECM engagement and phosphorylation) in turn leads to the activation of phosphatidylinositol-3 kinase (PI3K) and Rac1 (29). Interestingly, mice with a targeted deletion of the ITGB4 cytoplasmic tail display defective cell proliferation in the epidermis and intestinal epithelium, highlighting the physiological significance of ITGB4 signaling in these tissues (30).
In the present study, we expanded our investigation of the mechanisms underlying HGF-mediated EC barrier protection and identified a c-Met signaling complex that sequesters S1PR1 and ITGB4 to drive barrier-regulatory responses. We observed HGF-induced c-Met and S1PR1 interaction with ITGB4 that was phosphorylation-dependent and essential for HGF-mediated enhancement of vascular barrier enhancement. This increased understanding of the HGF/c-Met complex signal transduction pathway in lipid rafts provides novel opportunities for development of therapeutic strategies in inflammatory vascular injury.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Human lung microvascular EC (HLMVEC) and human pulmonary artery EC (HPAEC) were obtained from Lonza (Walkersville, MD) and cultured in ECM-2 complete medium (Lonza) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air, with cell passages 6–10 used for experimentation. Unless otherwise specified, reagents were obtained from Sigma. SDS-PAGE reagents were from Bio-Rad, Immobilon-P transfer membrane from Millipore (Bedford, MA), and gold microelectrodes from Applied Biophysics (Troy, NY). Recombinant human HGF was commercially obtained (PeproTech, Inc.). Rabbit anti-phospho-Tyr1234/Tyr1235 c-Met, rabbit anti-phospho-Tyr1349 c-Met, and mouse anti-c-Met antibodies were purchased from Cell Signaling Technology (Boston, MA). Rabbit anti-caveolin-1 or rabbit anti-S1PR1 and rabbit anti-ITGB4 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). OptiPrepTM was purchased from Sigma. Horseradish peroxidase-labeled secondary antibodies were obtained from Amersham Biosciences (Pittsburgh, PA).
Isolation of Lipid Rafts (CEM)
Lipid rafts were isolated from HPAEC as previously described (4, 5, and 1). Triton X-100-insoluble materials were mixed with 0.6 ml of cold 60% OptiPrepTM, overlaid with 0.6 ml of 40–20% OptiPrepTM; and centrifuged at 35,000 rpm for 12 h at 4 °C, then fractions were collected and analyzed.
Immunoprecipitation and Immunoblotting
Cell lysates prepared from treated or untreated EC were incubated in immunoprecipitation buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 20 mm MgCl2, 1% Nonidet P-40, 0.4 mm Na3VO4, 40 mm NaF, 50 μm okadaic acid, 0.2 mm phenylmethylsulfonyl fluoride, and Calbiochem protease inhibitor mixture III at 1:250 dilution). The immunoprecipitation was carried out with either anti-c-Met, anti-S1PR1, or anti-ITGB4 antibodies, and the precipitates separated on a gradient SDS-PAGE (4–15%), transferred onto Immobilon-P membranes, and subjected to immunoblotting using specific primary and secondary antibodies. Visualization of immunoreactive bands was achieved by enhanced chemiluminescence (Amersham Biosciences, Pittsburgh, PA). Densitometric data were obtained with ImageJ software.
Transfection of Small Interfering RNA (siRNA)
The siRNA specific for c-Met (IDT, Coralville, IA), S1PR1 and ITGB4 were commercially obtained (Dharmacon, Lafayette, CO). Human pulmonary EC were transfected with siRNA using Dharmafect according to the manufacturer's protocol (Dharmacon, Lafayette, CO). Cells (∼60% confluent) were serum-starved for 1 h and treated with 100 nm siRNA (or scrambled siRNA or no siRNA) for 6 h in serum-free medium. Serum-containing medium was then added (10% serum final concentration) for 76 h before biochemical experiments and/or functional assays were conducted.
Determination of Tyrosine or Threonine Protein Phosphorylation
Cell lysates from treated or untreated HPAEC, were analyzed by gradient SDS-PAGE (4–15%) and the proteins transferred to an Immobilon-P membrane. After blocking nonspecific sites with 5% bovine serum albumin, the blots were incubated with either mouse anti-tyrosine or mouse anti-threonine antibodies followed by incubation with horseradish peroxidase-labeled goat anti-mouse IgG. Visualization of immunoreactive bands was achieved by enhanced chemiluminescence.
Determination of Complex Formation Involving S1PR1, c-Met, and ITGB4
Cell lysates from treated or untreated EC were subjected to immunoprecipitation using rabit anti-S1PR1 or mouse anti-c-Met antibodies. The proteins were then analyzed by immunoblotting using either rabbit anti-S1PR1, anti-ITGB4, or anti-c-Met antibodies, followed by incubation with horseradish peroxidase-labeled goat anti-mouse or goat anti-rabbit IgG. Visualization of immunoreactive bands was achieved by enhanced chemiluminescence.
Measurement of Transendothelial Electrical Resistance
HPAEC were grown to confluence in polycarbonate wells containing evaporated gold microelectrodes, and transendothelial electrical resistance (TER) measurements were performed using an electrical cell substrate impedance sensing system obtained from Applied Biophysics (Troy, NY) as described previously in detail (2). TER values from each microelectrode were pooled at discrete time points and plotted versus time as the means ± S.E.
Rac1 Activation Assay
Rac1 activity assays in HPAEC were performed as described elsewhere (31). Briefly, EC were solubilized and incubated with p21-binding domain (PBD)-conjugated beads to bind activated (GTP-bound) Rac1. The PBD bead-associated material was analyzed after immunoblotting with an anti-Rac1 antibody.
Proximity Ligation Assay (PLA)
HLMVEC were used for PLA in situ analysis. In brief, an oligonucleotide-conjugated probe (used as secondary antibody) is directed against primary antibody raised against a cell surface receptor. HLMVEC grown on coverslips were starved for 18 h and treated with HGF or S1P for 5 min. Cells were washed with chilled PBS and fixed with 4% paraformaldehyde for 15 min, blocked, and permeabilized with 3% BSA containing 0.2% Triton, and incubated overnight with appropriate antibodies. Proximity ligation was performed according to the manufacturer's protocol using the Duolink Detection Kit with PLA PLUS and MINUS Probes for mouse and rabbit (Olink Bioscience, Uppsala, Sweden). DAPI stain was included in the Duolink Detection Kit while anti-Phalloidin Alexa488 at 1:5,000 (Invitrogen) was added during the detection reaction. Specimens were mounted with Vectashield mounting media (Vector Laboratories, Burlingame, CA) and examined with an epi-fluorescence microscope under a ×60 oil objective. Texas-Red signal was analyzed via BlobFinder Imaging Software, developed and optimized for the analysis of images generated by the in situ PLA (Uppsala Science Park, Sweden) (32, 33). Four fields were randomly chosen for analysis and averaged and four separate samples were examined per condition (around 60–80 cells).
Statistical Analysis
Statistical analyses were performed using a standard 2-sample Student's t test, assuming unequal variances of the data sets. Statistical significance was determined using a 2-tailed distribution assumption and was set at 5% (p < 0.05). Data are expressed as mean ± S.E.
RESULTS
Transactivation of S1PR1 Is Necessary for HGF-induced Rac1 Activation and Endothelial Cell Barrier Enhancement
We previously reported that PI3 kinase (PI3K) is required for S1P-mediated S1PR1 activation (reflected by enhanced threonine phosphorylation) and HGF-induced EC barrier enhancement (4, 5, 1). To examine the role of S1PR1 in HGF-induced EC barrier enhancement, HPAEC were challenged with HGF (25 ng/ml, 5 min) and whole cell lysates utilized to immunoprecipitate S1PR1 under non-denaturing conditions. HGF-induced threonine but not serine phosphorylation of S1PR1 was completely abrogated by the PI3K inhibitor, LY294002 (Fig. 1A). Having established that S1PR1 activation is downstream of HGF/c-Met/PI3K signaling, we next investigated whether Rac1, the small GTPase activated by S1PR1 ligation, also participates in HGF-induced barrier enhancement (1). Reduced S1PR1 (>85% reduction, siRNA) significantly attenuated HGF-induced Rac1 activation indicating that S1PR1 lies upstream of HGF-mediated Rac1 activation (Fig. 1B). Furthermore, S1PR1 silencing resulted in significant loss of HGF-induced increases in TER, a measure of EC barrier integrity, consistent with the essential nature of S1PR1 in HGF-induced EC barrier enhancement (Fig. 1C).
FIGURE 1.

S1PR1 receptor transactivation is required for HGF-induced Rac1 activation and EC barrier enhancement. A, HPAEC were treated with a PI3K inhibitor (LY 294002, 10 μm, 1 h) prior to treatment with HGF (25 ng/ml, 5 min). Whole cell lysates were then immunoprecipitated with an anti-S1PR1 antibody and immunoblotted with an anti-phospho-serine, anti-phospho-threonine, or anti-S1PR1 antibody. HGF-induced S1PR1 threonine phosphorylation was abrogated by PI3K inhibition. B, in separate experiments, nonspecific (ns) siRNA or siRNA against S1PR1 were transfected into HPAEC prior to treatment with HGF (25 ng/ml, 5 min) and cell lysates then analyzed for acativated Rac1 (GTP-Rac1). Silencing of S1PR1 inhibited HGF-induced Rac1 activation. Densitometry analysis of Rac1 activation is expressed as a percentage relative to control cells (ns siRNA, untreated) (n = 3/group, *, p < 0.05). C, maximal TER responses measured in cells transfected with siRNA specific for S1PR1 and then treated with HGF (25 ng/ml) confirmed a marked reduction in HGF-induced TER increases compared with controls cells transfected with ns siRNA. (n = 3/condition, *, p < 0.05).
c-Met Forms a Complex with S1PR1 and ITGB4
ITGB4 is known to be involved in GTP-bound Rac1 and Cdc42 translocation to lipid rafts and the plasma membrane (21, 24). Accordingly, we investigated the potential association of ITGB4 with c-Met after S1P or HGF ligation. Initial studies determined the phosphorylation status of ITGB4, c-Met, and S1PR1 as a reflection of activation (Fig. 2, A and B). Cell lysates prepared from untreated and HGF-treated HPAEC were utilized for immunoprecipitation studies with anti-ITGB4, anti-p-Tyr or anti-S1PR1 antibodies under non-denaturing conditions. Our results indicate a constitutively present complex comprised of c-Met, S1PR1, and ITGB4 (Fig. 2, A–E). As predicted HGF stimulates tyrosine phosphorylation of c-Met with both HGF and S1P evoking rapid S1PR1 activation (increased threonine phosphorylation) consistent with HGF-mediated S1PR1 transactivation (Fig. 2A). Interestingly, S1P also induced c-Met activation, consistent with S1P-mediated c-Met transactivation (Fig. 2A), while ITGB4 was also identified in S1PR1 immunoprecipitates with evidence of increased phosphorylation following either HGF or S1P challenge (Fig. 2, D and E). We found the same HGF- or S1P-induced complex formation c-Met, S1PR1 and ITGB4 in HLMVEC cells as well (data not shown). Subsequently, HLMVEC were used for immunofluorescence imaging and PLA analyses due to technical limitations related to the use of HPAEC which form less confluent monolayers with more abundant stress fibers under basal conditions. These studies confirmed marked colocalization of complex components in response to HGF or S1P (Fig. 2, C and E). Although the kinetics of these post- translational modifications is agonist-specific, these data support increased complex formation of these three proteins after receptor ligation.
FIGURE 2.

c-Met/S1PR1/ITGB4 complex modulation by HGF and S1P. A, HPAEC were treated with either S1P (1 μm) or HGF (25 ng/ml) for 1 and 5 min. Cell lysates were subjected to immunoprecipitation with an anti-ITGB4 antibody and tyrosine or threonine phosphorylation corresponding to the same molecular weight was determined via repeat immunoblotting on the same membrane (representative blots shown, densitometric data expressed as fold change phosphorylation relative to control and normalized to total protein, n = 3/group, *, p < 0.05 compared with untreated contols). B, in separate experiments, HPAEC were treated with HGF (25 ng/ml, 5 min) and immunoprecipitation performed with an antibody specific for tyrosine phosphorylation prior to immunoblotting for c-Met and ITGB4 (representative blots shown). C, associations of c-Met with ITGB4 induced by HGF (upper panels) and S1PR1 (middle panels), as well as HGF-induced associations between c-Met and S1PR1 were detected by in situ proximity ligation assay (red dots) in untreated HLMVEC as well as both cells transfected with specific siRNA (ITGB4 or S1PR1) and control cells (ns siRNA) prior to treatment with HGF (25 ng/ml, 5 min) or S1P (1 μm, 5 min). Average signal intensity for each condition was quantified as described under “Experimental Procedures” (n = 4/condition, *, p < 0.05). D, HPAEC were treated with either S1P (1 μm) or HGF (25 ng/ml) and lysates then immunoprecipitated with an anti-S1PR1 antibody followed by immunoblotting with an anti-ITGB4 or anti-S1PR1 antibody. Representative blots and densitometry of the association between S1PR1 and ITGB4 are shown. (n = 3/condition, *, p < 0.05 compared with untreated controls). E, interaction of S1PR1 and ITGB4 in response to HGF treatment (25 ng/ml, 5 min) was also as assessed by PLA in situ.
HGF-mediated Increases in c-Met, S1PR1, and ITGB4 Interactions in Lipid Rafts
We previously reported that HGF promotes c-Met movement into EC lipid rafts, in concert with CD44 association and recruitment of key signaling molecules such as Tiam1 (a Rac1 guanine exchange factor), cortactin, and dynamin (1). Accordingly, we examined whether the c-Met/S1PR1/ITGB4 complex is localized to lipid rafts after HGF challenge. Immunoblotting of enriched lipid raft fractions (30% Optiprep fraction) revealed significant increases in abundance of c-Met, S1PR1, and ITGB4 in response to HGF with increased phosphorylation (tyrosine or threonine), albeit with varying kinetics of recruitment (Fig. 3B). This marked recruitment of ITGB4, c-Met, and S1PR1 to the lipid raft fractions after HGF in conjunction with phosphorylation, despite unchanged levels of caveolin, indicates that the c-Met/S1PR1/ITGB4 complex is dynamically regulated within the lipid rafts in response to HGF.
FIGURE 3.

HGF recruits c-Met, ITGB4, and S1PR1 to lipid rafts. A, Optiprep fractions were isolated and subjected to immunoblotting with an anti-cav-1 antibody. The 30% Optiprep fraction represents the lipid rafts. B, HPAEC were treated with HGF (25 ng/ml) for 1 and 5 min and the 30% Optiprep fraction was isolated and subjected to immunoblotting with antibodies specific for c-Met, ITGB4, or S1PR1. Phosphorylation corresponding to the same molecular weight was determined via repeat immunoblotting on the same membrane with specific antibodies as shown (representative blots shown, densitometry data expressed as fold change phosphorylation relative to control and normalized to total protein, n = 3/condition, *, p < 0.05 compared with untreated controls).
Reduced Expression of c-Met, or ITGB4 or S1PR1 Attenuates HGF- and S1P-induced EC Barrier Enhancement
We next investigated whether each complex component contributes to HGF- and S1P-mediated EC barrier enhancement. Reductions in the expression levels of each receptor by specific siRNA (∼60–85% reductions) significantly attenuated HGF-enhanced TER by ∼50–80% when compared with control cells transfected with scrambled siRNA (Fig. 4, A and B). Similar results were also observed after S1P challenge after receptor silencing (Fig. 4C). Maximal reductions in HGF-induced EC barrier enhancement was observed in c-Met-silenced EC whereas maximal reductions in S1P-induced EC barrier enhancement was observed in S1PR1-silenced EC providing support for the role of this complex in cell surface receptor crosstalk in EC barrier regulation.
FIGURE 4.
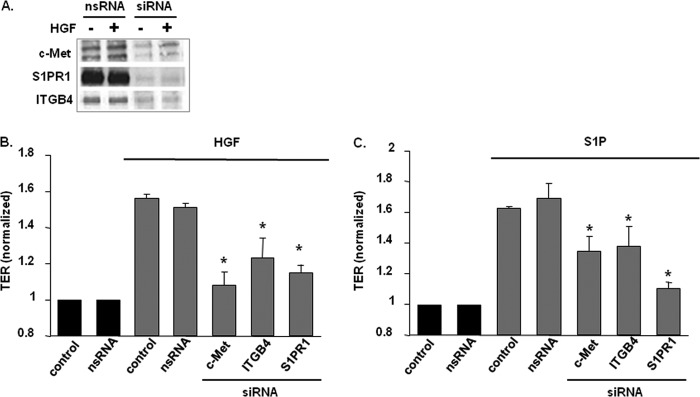
Role of c-Met/S1PR1/ITGB4 complex components on HGF- and S1P-induced EC barrier enhancement. A, HPAEC were transfected with either nonspecific (ns) siRNA or siRNA specific for c-Met, S1PR1, or ITGB4 prior to treatment with HGF (25 ng/ml, 5 min). Cell lysates were then immunoblotted with anti-c-Met, anti-S1PR1 or anti-ITGB4 antibodies to confirm silencing. B and C, maximal TER responses after HGF (25 ng/ml) or S1P (1 μm) are shown for control cells and cells transfected with specific siRNA (n ≥ 5/condition, results normalized to control, untreated cells, *, p < 0.05 compared with control cells transfected with nsRNA and stimulated with HGF or S1P, respectively).
Inhibition of c-Met Kinase Activity Abrogates S1PR1 and ITGB4 Activation and Barrier-regulatory Function
To address whether c-Met tyrosine kinase activity is required for HGF-mediated increases in EC barrier function, HPAEC were pretreated with a c-Met kinase inhibitor, XL880 (0.4 nmol/liter, 1 h) (34, 35), followed by HGF challenge (25 ng/ml, 5 min). Basal as well as HGF-induced increases in levels of tyrosine phosphorylation in c-Met immunoprecipitates were completely abolished by XL880 (Fig. 5A). Although the abrogation of c-Met tyrosine kinase activity failed to affect S1PR1 levels, XL880 elicited decreased ITGB4 levels within the c-Met complex and S1PR1 and ITGB4 transactivation (threonine and tyrosine phosphorylation status, respectively) was dramatically suppressed indicating that c-Met tyrosine kinase activity is a critical and a potentially rate-limiting step for active complex formation. The importance of c-Met tyrosine kinase activity was further evidenced by abrogation of HGF-induced increases in EC barrier enhancement (Fig. 5B) associated with kinase inhibition although basal TER levels were unaffected.
FIGURE 5.

c-Met tyrosine kinase inhibition abrogates HGF-induced activation of S1PR1 and ITGB4 and EC barrier enhancement. A, HPAEC were pretreated with a c-Met inhibitor, XL880 (0.4 nmol/liter, 1 h) prior to HGF treatment (25 ng/ml, 5 min). Cell lysates were immunoprecipitated with an anti-c-Met antibody and then subjected to immunoblotting with antibodies specific for c-Met, ITGB4, or S1PR1. Phosphorylation corresponding to the same molecular weight was determined via repeat immunoblotting on the same membrane with specific antibodies as shown (representative blots shown, densitometry data expressed as fold change phosphorylation relative to control and normalized to total protein, n = 3/condition, *, p < 0.05 compared with HGF-treated control cells). B, maximal TER responses to HGF (25 ng/ml) were measured in cells pretreated with XL880 (0.4 nmol/liter, 1 h) and compared with untreated control cells (n ≥ 5/condition, *, p < 0.05).
Reduced S1PR1 or ITGB4 Expression Attenuates c-Met/S1PR1/ITGB4 Complex Activation
In subsequent experiments, reduced S1PR1 expression (siRNA) was associated with marked reductions in HGF-induced phosphorylation of ITGB4 but not c-Met (Fig. 6) whereas ITGB4 silencing was associated with reductions in both tyrosine phosphorylation of c-Met and S1PR1 transactivation by HGF consistent with the idea that c-Met/S1PR1/ITGB4 complex is critical to HGF barrier effects.
FIGURE 6.
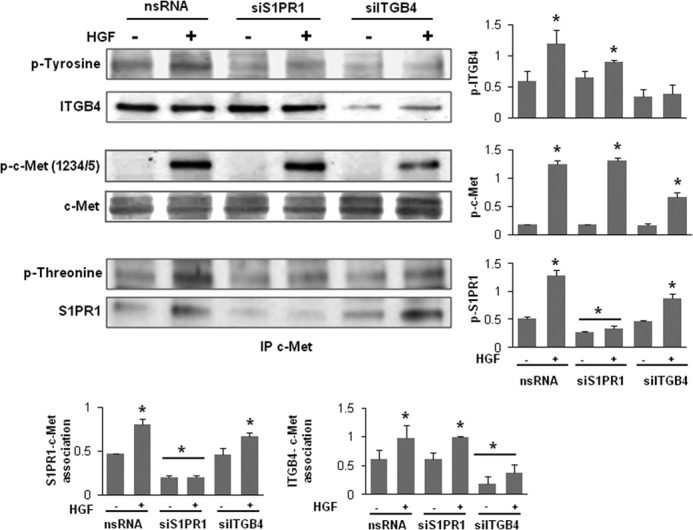
Reductions in S1PR1 or ITGB4 expression inhibits c-Met/S1PR1/ITGB4 complex activation. HPAEC were transfected with nonspecific (ns) siRNA or siRNA specific for S1PR1 or ITGB4 prior to treatment with HGF (25 ng/ml, 5 min). Cells lysates were immunoprecipitated with an anti-c-Met antibody and then subjected to immunoblotting with antibodies specific for c-Met, ITGB4, or S1PR1. Phosphorylation corresponding to the same molecular weight was determined via repeat immunoblotting on the same membrane with specific antibodies as shown (representative blots shown, densitometry data expressed as fold change phosphorylation relative to control and normalized to total protein, n = 3/condition, *, p < 0.05 compared with untreated nsRNA controls).
HGF-induced Rac1 Activation Is Mediated by ITGB4
We have previously reported Rac1 activation is required for the barrier-enhancing effects of a number of agonists including S1P, simvastatin, HMW HA, ATP, and HGF. However, the mechanism of HGF-induced Rac1 activation in human EC remains poorly defined. As we have previously linked the up-regulation of ITGB4 to Rac1 activation by simvasatin (18), we investigated the role of ITGB4 in Rac1 activation by HGF. Knockdown of ITGB4 (siRNA) was associated with an abrogation of HGF-induced Rac1 activation in EC while total Rac1 levels were unchanged (Fig. 7, A and B). These data confirm a critical role for ITGB4 in Rac1 activation by HGF.
FIGURE 7.
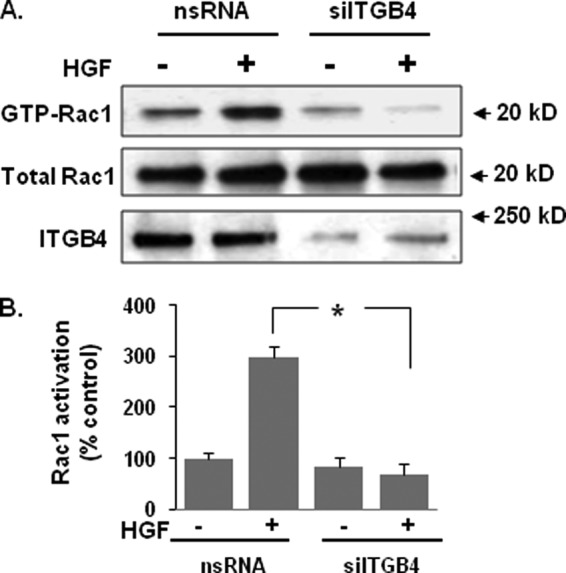
HGF-induced Rac1 activation involves ITGB4-mediated signaling. A, HPAEC were transfected with nonspecific (ns) siRNA or ITGB4 siRNA prior to treatment with HGF (25 ng/ml, 5 min). Cell lysates were then prepared and analyzed for Rac1 activation (GTP-Rac1). Representative blots are shown. B, densitometry analysis of Rac1 activation is expressed as a percentage relative to control cells (ns siRNA, untreated) (n = 3/group, *, p < 0.05).
DISCUSSION
The loss of EC barrier integrity is a hallmark of inflammatory syndromes, including acute lung injury, and is centrally involved in the development of tumor metastases (36, 37). Inflammatory agonists, such as thrombin, disrupt EC barrier integrity whereas a number of protective agonists, such as S1P, conversely promote barrier enhancement (7). In the current study, we extend our previous findings that the HGF/c-Met signaling axis augments vascular barrier integrity in vitro and in vivo (1) and now have identified the dependence of these barrier-regulatory events on a dynamic complex consisting of c-Met, S1PR1, and ITGB4 localized to EC lipid rafts. Our results indicate that HGF challenge increases the recruitment of c-Met and other receptors to lipid rafts, induces c-Met activation, and promotes the transactivation of both S1PR1 and ITGB4. Similarly, we found that S1P also induces increased association of c-Met and S1PR1 with ITGB4. This c-Met/S1PR1/ITGB4 complex serves as a common portal for the transduction of EC barrier protection signals for both HGF and S1P ligands. In addition, c-Met tyrosine kinase activity is essential for HGF-induced effects as its inhibition fully abrogates transactivation of both S1PR1 and ITGB4 by HGF as well as HGF-mediated EC barrier enhancement. The functional links within the c-Met/S1PR1/ITGB4 complex were evident after silencing of S1PR1 or ITGB4 expression as knock down of either (siRNA) significantly blunted HGF-induced activation of the other two subunits as well as HGF-induced EC barrier enhancement, thus providing evidence of a structural link and critical cross-talk among the three receptors. Finally, we determined that ITGB4 contributes to HGF-induced Rac1 activation, an important regulator of actin cytoskeletal rearrangement. This observation is notable as there are no prior reports of ITGB4 interactions with the EC actin cytoskeleton.
We have previously shown that HGF signaling in human lung EC promotes activation of several kinases, including PI3K, ERK, and p38 that are necessary for the prevention of agonist-induced EC barrier leakiness (7). We also found that HGF recruited Tiam1 (a Rac1 exchange factor) to lipid rafts in a CD44-dependent fashion to promote Rac1 activation (1). In the present study, we have now identified a strong connection between transactivation of S1PR1 and HGF/c-Met signaling-induced PI3K activation, as PI3K inhibition abolished HGF-mediated transactivation of S1PR1. The interdependence between HGF/c-Met signaling and S1PR1 is further evidenced by the fact that knockdown of S1PR1 prevented HGF-mediated Rac1 activation.
The mechanism by which c-Met transactivates S1PR1 is unknown. However, sphingosine kinase (SPK) is a key enzyme that catalyzes the formation of sphingosine-1-phosphate (S1P). HGF induces not only tyrosine phosphorylation of c-Met but also activation of SPK in a concentration-dependent manner (38). We speculate that this may serve as a separate mechanism of HGF-induced transactivation of S1PR1. In addition, S1P is known to have diverse properties. For example, S1P can transactivate a number of EC growth factor receptors including the VEGF receptor, VEGF2, and Flk-1/KDR (39, 40, 41). S1P activation of VEGF2 is both Gi protein and Src family kinase dependent and is not dependent on the synthesis of VEGF. Thus, it is possible that S1P has additional effects relevant to the bidirectional interactions of S1P/S1PR1 with HGF/c-Met.
Integrin β subunits are transmembrane proteins and typically have a relatively short cytoplasmic tail of 40–70 amino acids. However, ITGB4 is an important exception with its particularly long cytoplasmic tail (1088 amino acids) suggesting unique signaling properties (42, 43, 44, 45). ITGB4 interacts with multiple oncogenic receptor tyrosine kinases, such as EGF receptor (EGF-R), ErbB2/Neu, and c-Met, and is a mediator of cell survival, proliferation, invasion, and migration (46, 47, 48). In addition, integrins control translocation of GTP-bound Rac1 and Cdc42 to the plasma membrane and their coupling to effectors (24, 49). Here, we have shown for the first time that ITGB4 is associated with c-Met and S1PR1 in EC lipid rafts and is transactivated by HGF. The functional relationship between c-Met and ITGB4 is evidenced by the fact that inhibition of c-Met tyrosine kinase activity resulted in attenuation of HGF-induced ITGB4 activation and knockdown of ITGB4 expression resulted in decreased c-Met activation in response to HGF. In epithelial cells, upon dephosphorylation, the ITGB4 cytoplasmic tail associates with the keratin cytoskeleton, leading to assembly of hemidesmosomes (30, 42). In this context, ITGB4 also interacts with multiple receptor protein tyrosine kinases. For example, activation of EGF-R and c-Met enhances ITGB4 phosphorylation and Shc signaling, causing disruption of hemidesmosomes and increased cell motility and proliferation (46, 47). However, ITGB4 does not form hemidesmosomes in EC (46), and until recently little was known about the functional role of ITGB4 in normal EC physiology (44, 51).
The importance of lipid rafts in the regulation of EC barrier integrity was demonstrated in our previous work with S1PR1 transactivated within lipid rafts by other membrane-spanning receptors such as APC/EPCR, HA/CD44, and OxPAPC, potentially representing a major focus of EC barrier regulation (11, 52, 5, 1, 53). In the current study, we have shown the presence of a c-Met/S1PR1/ITGB4 complex in lipid rafts with each receptor protein playing a significant and interlinked role in maintaining EC barrier integrity. In a previous study, we also showed the presence of a c-Met/CD44 complex in EC lipid rafts wherein CD44 was found to be critical for HGF-enhanced EC barrier integrity (1). It is therefore possible that there is a larger c-Met signalosome in lipid rafts that is structurally and functionally associated with S1PR1, CD44, ITGB4, and other receptor tyrosine kinases and adapter molecules. Our future studies will focus on the isolation and characterization of the c-Met signalosome using FPLC and mass spectrometry techniques.
In summary, our data support the presence of a large c-Met signaling complex in EC lipid rafts that includes c-Met, S1PR1, and ITGB4 with direct physical interaction and a functional linkage (Fig. 8). HGF mediates dynamic changes in the complex formation, the transactivation of S1PR1 and ITGB4; and the importance of each of the three components in the regulation of EC barrier integrity attests to a significant role for this complex in disease states such as ALI, tumorogenesis, and inflammation.
FIGURE 8.

Model for HGF-mediated c-Met signalosome formation resulting in EC barrier enhancement. Based on our previous work and current study, an HGF-mediated, EC barrier-enhancing signalosome model is proposed. A constitutive c-Met/S1PR1/ITGB4 complex is present in lipid rafts of the plasma membrane. Treatment with HGF promotes further recruitment of complex components and strengthens the c-Met signalosome. Ligation of c-Met results in auto-activation and subsequent activation of key downstream targets such as PI3K and Akt, which drive phosphorylation of the S1PR1 cytoplasmic tail and its activation. Activated c-Met may also phosphorylate ITGB4, either directly or indirectly through c-Src, which leads to the activation of Rac1 through the adapter protein, Tiam1, resulting in cytoskeletal changes. We envisage the coordinated signals from the receptors in the c-Met complex bring about cytoskeletal reorganization leading to cortactin/actin interaction leading to EC barrier enhancement.
Acknowledgment
We thank Lakshmi Natarajan for expert technical assistance.
Footnotes
- EC
- endothelial cell
- HGF
- hepatocyte growth factor
- S1PR
- sphingosine 1-phosphate receptor
- ITGB4
- integrin β4
- CEM
- caveolin-1-enriched plasma membrane microdomains
- DAG
- diacylglycerol
- TER
- transendothelial electrical resistance
- PLA
- proximity ligation assay
- HLMVEC
- human lung microvascular EC
- HPAEC
- human pulmonary artery EC.
REFERENCES
- 1. Singleton P. A., Salgia R., Moreno-Vinasco L., Moitra J., Sammani S., Mirzapoiazova T., Garcia J. G. (2007) CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J. Biol. Chem. 282, 30643–30657 [DOI] [PubMed] [Google Scholar]
- 2. Garcia J. G., Liu F., Verin A. D., Birukova A., Dechert M. A., Gerthoffer W. T., Bamberg J. R., English D. (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 108, 689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dudek S. M., Jacobson J. R., Chiang E. T., Birukov K. G., Wang P., Zhan X., Garcia J. G. (2004) Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J. Biol. Chem. 279, 24692–24700 [DOI] [PubMed] [Google Scholar]
- 4. Singleton P. A., Dudek S. M., Chiang E. T., Garcia J. G. (2005) Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1PR1 receptor, PI3 kinase, Tiam1/Rac1, and α-actinin. Faseb J. 19, 1646–1656 [DOI] [PubMed] [Google Scholar]
- 5. Singleton P. A., Dudek S. M., Ma S. F., Garcia J. G. (2006) Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J. Biol. Chem. 281, 34381–34393 [DOI] [PubMed] [Google Scholar]
- 6. Zhao J., Singleton P. A., Brown M. E., Dudek S. M., Garcia J. G. (2009) Phosphotyrosine protein dynamics in cell membrane rafts of sphingosine-1-phosphate-stimulated human endothelium: role in barrier enhancement. Cell Signal. 21, 1945–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu F., Schaphorst K. L., Verin A. D., Jacobs K., Birukova A., Day R. M., Bogatcheva N., Bottaro D. P., Garcia J. G. (2002) Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3β. FASEB J. 16, 950–962 [DOI] [PubMed] [Google Scholar]
- 8. Singleton P. A., Mirzapoiazova T., Guo Y., Sammani S., Mambetsariev N., Lennon F. E., Moreno-Vinasco L., Garcia J. G. (2010) High-molecular-weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. Am. J. Physiol. Lung Cell Mol. Physiol. 299, 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jacobson J. R., Dudek S. M., Singleton P. A., Kolosova I. A., Verin A. D., Garcia J. G. (2006) Endothelial cell barrier enhancement by ATP is mediated by the small GTPase Rac and cortactin. Am. J. Physiol. Lung Cell Mol. Physiol. 291, 289–295 [DOI] [PubMed] [Google Scholar]
- 10. Kolosova I. A., Mirzapoiazova T., Moreno-Vinasco L., Sammani S., Garcia J. G., Verin A. D. (2008) Protective effect of purinergic agonist ATPγS against acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 294, 319–324 [DOI] [PubMed] [Google Scholar]
- 11. Finigan J. H., Dudek S. M., Singleton P. A., Chiang E. T., Jacobson J. R., Camp S. M., Ye S. Q., Garcia J. G. (2005) Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J. Biol. Chem. 280, 17286–17293 [DOI] [PubMed] [Google Scholar]
- 12. Finigan J. H., Boueiz A., Wilkinson E., Damico R., Skirball J., Pae H. H., Damarla. M., Hasan E., Pearse D. B., Reddy S. P., Grigoryev D. N., Cheadle C., Esmon C. T., Garcia J. G., Hassoun P. M. (2009) Activated protein C protects against ventilator-induced pulmonary capillary leak. Am. J. Physiol. Lung Cell Mol. Physiol. 296, 1002–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jacobson J. R., Garcia J. G. (2007) Novel therapies for microvascular permeability insepsis. Curr. Drug Targets 8, 509–514 [DOI] [PubMed] [Google Scholar]
- 14. Singleton P. A., Bourguignon L. Y. (2004) CD44 interaction with ankyrin and IP3 receptor in lipid rafts promotes hyaluronan-mediated Ca2+ signaling leading to nitric oxide production and endothelial cell adhesion and proliferation. Exp. Cell Res. 295, 102–118 [DOI] [PubMed] [Google Scholar]
- 15. Schubert W., Frank P. G., Woodman S. E., Hyogo H., Cohen D. E., Chow C. W., Lisanti M. P. ( 2002) Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J. Biol. Chem. 277, 40091–40098 [DOI] [PubMed] [Google Scholar]
- 16. Minshall R. D., Sessa W. C., Stan R. V., Anderson R. G., Malik A. B. (2003) Caveolin regulation of endothelial function. Am. J. Physiol. Lung. Cell Mol. Physiol. 285, 1179–1183 [DOI] [PubMed] [Google Scholar]
- 17. Dudek S. M., Chiang E. T., Camp S. M., Guo Y., Zhao J., Brown M. E., Singleton P. A., Wang L., Desai A., Arce F. T., Lal R., Van Eyk J. E., Imam S. Z., Garcia J. G. (2010) Abl tyrosine kinase phosphorylates nonmuscle Myosin light chain kinase to regulate endothelial barrier function. Mol. Biol. Cell 21, 4042–4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobson J. R., Dudek S. M., Birukov K. G., Ye S. Q., Grigoryev D. N., Girgis R. E., Garcia J. G. (2004) Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am. J. Respir. Cel.l Mol. Biol. 30, 662–670 [DOI] [PubMed] [Google Scholar]
- 19. Mertens A. E., Roovers R. C., Collard J. G. (2003) Regulation of Tiam1-Rac signalling. FEBS Lett. 546, 11–16 [DOI] [PubMed] [Google Scholar]
- 20. Burridge K., Wennerberg K. (2004) Rho and Rac take center stage. Cell 116, 167–179 [DOI] [PubMed] [Google Scholar]
- 21. del Pozo M. A., Alderson N. B., Kiosses W. B., Chiang H. H., Anderson R. G., Schwartz M. A. (2004) Integrins regulate Rac targeting by internalization of membrane domains. Science 303, 839–842 [DOI] [PubMed] [Google Scholar]
- 22. Palazzo A. F., Eng C. H., Schlaepfer D. D., Marcantonio E. E., Gundersen G. G. (2004) Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science 303, 836–839 [DOI] [PubMed] [Google Scholar]
- 23. Echarri A., Del Pozo M. A. (2006) Caveolae internalization regulates integrin-dependent signaling pathways. Cell Cycle 19, 2179–2182 [DOI] [PubMed] [Google Scholar]
- 24. del Pozo M. A., Price L. S., Alderson N. B., Ren X. D., Schwartz M. A. (2000) Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 19, 2008–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen W., Pendyala S., Natarajan V., Garcia J. G., Jacobson J. R. (2008) Endothelial cell barrier protection by simvastatin: GTPase regulation and NADPH oxidase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 295, 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gagnoux-Palacios L., Dans M., van't Hof W., Mariotti A., Pepe A., Meneguzzi G., Resh M. D., Giancotti F. G. (2003) Compartmentalization of integrin α6β4 signaling in lipid rafts. J. Cell Biol. 162, 1189–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mainiero F., Pepe A., Wary K. K., Spinardi L., Mohammadi M., Schlessinger J., Giancotti F. G. (1995) Signal transduction by the α6β4 integrin: distinct β4 subunit sites mediate recruitment of Shc/Grb2 and association with the cytoskeleton of hemidesmosomes. EMBO J. 14, 4470–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mainiero F., Murgia C., Wary K. K., Curatola A. M., Pepe A., Blumemberg M., Westwick J. K., Der C. J., Giancotti F. G. (1997) The coupling of α6β4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 16, 2365–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shaw L. M., Rabinovitz I., Wang H. H., Toker A., Mercurio A. M. (1997) Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell 91, 949–960 [DOI] [PubMed] [Google Scholar]
- 30. Murgia C., Blaikie P., Kim N., Dans M., Petrie H. T., Giancotti F. G. (1998) Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin β4 cytoplasmic domain. EMBO J. 17, 3940–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ren X. D., Kiosses W. B., Schwartz M. A. (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 33. Allalou A., Wählby C. (2009) BlobFinder, a tool for fluorescence microscopy image cytometry. Comput. Methods Programs Biomed. 94, 58–65 [DOI] [PubMed] [Google Scholar]
- 34. Eder J. P., Shapiro G. I., Appleman L. J., Zhu A. X., Miles D., Keer H., Cancilla B., Chu F., Hitchcock-Bryan S., Sherman L., McCallum S., Heath E. I., Boerner S. A., LoRusso P. M. (2010) A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin. Cancer Res. 16, 3507–3516 [DOI] [PubMed] [Google Scholar]
- 35. Qian F., Engst S., Yamaguchi K., Yu P., Won K. A., Mock L., Lou T., Tan J., Li C., Tam D., Lougheed J., Yakes F. M., Bentzien F., Xu W., Zaks T., Wooster R., Greshock J., Joly A. H. (2009) Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 69, 8009–8016 [DOI] [PubMed] [Google Scholar]
- 36. Danilkovitch-Miagkova A., Zbar B. (2002) Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J. Clin. Invest. 109, 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeffers M., Schmidt L., Nakaigawa N., Webb C. P., Weirich G., Kishida T., Zbar B., Vande Woude G. F. (1997) Activating mutations for the met tyrosine kinase receptor inhuman cancer. Proc. Natl. Acad. Sci. U.S.A. 94, 11445–11450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duan H. F., Wu C. T., Lu Y., Wang H., Liu H. J., Zhang Q. W., Jia X. X., Lu Z. Z., Wang L. S. (2004) Sphingosine kinase activation regulates hepatocyte growth factor induced migration of endothelial cells. Exp. Cell Res. 298, 593–601 [DOI] [PubMed] [Google Scholar]
- 39. Tanimoto T., Jin Z. G., Berk B. C. (2002) Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS). J. Biol. Chem. 277, 42997–43001 [DOI] [PubMed] [Google Scholar]
- 40. Endo A., Nagashima K., Kurose H., Mochizuki S., Matsuda M., Mochizuki N. (2002) Sphingosine 1-phosphate induces membrane ruffling and increases motility of human umbilical vein endothelial cells via vascular endothelial growth factor receptor and CrkII. J. Biol. Chem. 277, 23747–23754 [DOI] [PubMed] [Google Scholar]
- 41. Lebman D. A., Spiegel S. (2008) Cross-talk at the crossroads of sphingosine-1-phosphate, growth factors, and cytokine signaling. J. Lipid Res. 49, 1388–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dans M., Gagnoux-Palacios L., Blaikie P., Klein S., Mariotti A., Giancotti F. G. (2001) Tyrosine phosphorylation of the β4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J. Biol. Chem. 276, 1494–1502 [DOI] [PubMed] [Google Scholar]
- 43. Abdel-Ghany M., Cheng H. C., Elble R. C., Pauli B. U. (2002) Focal adhesion kinase activated by β4 integrin ligation to mCLCA1 mediates early metastatic growth. J. Biol. Chem. 277, 34391–34400 [DOI] [PubMed] [Google Scholar]
- 44. Chen W., Garcia J. G., Jacobson J. R. (2010) Integrin β4 attenuates SHP-2 and MAPK signaling and reduces human lung endothelial inflammatory responses. J. Cell Biochem. 110, 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giancotti F. G. (2007) Targeting integrin β4 for cancer and anti-angiogenic therapy. Trends Pharmacol. Sci. 28, 506–511 [DOI] [PubMed] [Google Scholar]
- 46. Mariotti A., Kedeshian P. A., Dans M., Curatola A. M., Gagnoux-Palacios L., Giancotti F. G. ( 2001) EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 155, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trusolino L., Bertotti A., Comoglio P. M. (2001) A signaling adapter function for α6β4 integrin in the control of HGF-dependent invasive growth. Cell 107, 643–654 [DOI] [PubMed] [Google Scholar]
- 48. Guo W., Pylayeva Y., Pepe A., Yoshioka T., Muller W. J., Inghirami G., Giancotti F. G. (2006) β4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126, 489–502 [DOI] [PubMed] [Google Scholar]
- 49. Del Pozo M. A., Kiosses W. B., Alderson N. B., Meller N., Hahn K. M., Schwartz M. A. (2002) Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat. Cell Biol. 4, 232–239 [DOI] [PubMed] [Google Scholar]
- 50.Deleted in proof
- 51. Chen W., Sammani S., Mitra S., Ma S. F., Garcia J. G., Jacobson J. R. (2012) Critical Role for Integrin β4 in the Attenuation of Murine Acute Lung Injury by Simvastatin. Am. J. Physiol. Lung Cell Mol. Physiol. 303, L279–L285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Becker P. M., Verin A. D., Booth M. A., Liu F., Birukova A., Garcia J. G. (2001) Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 281, 1500–1511 [DOI] [PubMed] [Google Scholar]
- 53. Singleton P. A., Chatchavalvanich S., Fu P., Xing J., Birukova A. A., Fortune J. A., Klibanov A. M., Garcia J. G., Birukov K. G. ( 2009) Akt-mediated transactivation of the S1PR1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ. Res. 104, 978–986 [DOI] [PMC free article] [PubMed] [Google Scholar]